Passive Transfer of Sera from ALS Patients with Identified Mutations Evokes an Increased Synaptic Vesicle Number and Elevation of Calcium Levels in Motor Axon Terminals, Similar to Sera from Sporadic Patients

 , ,
, ,
Abstract
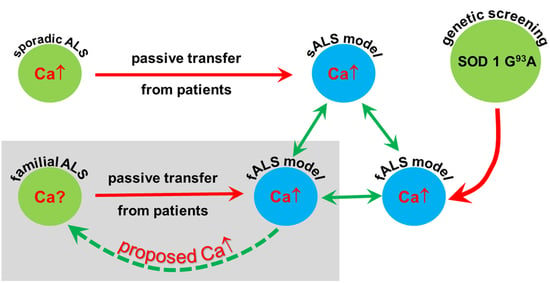
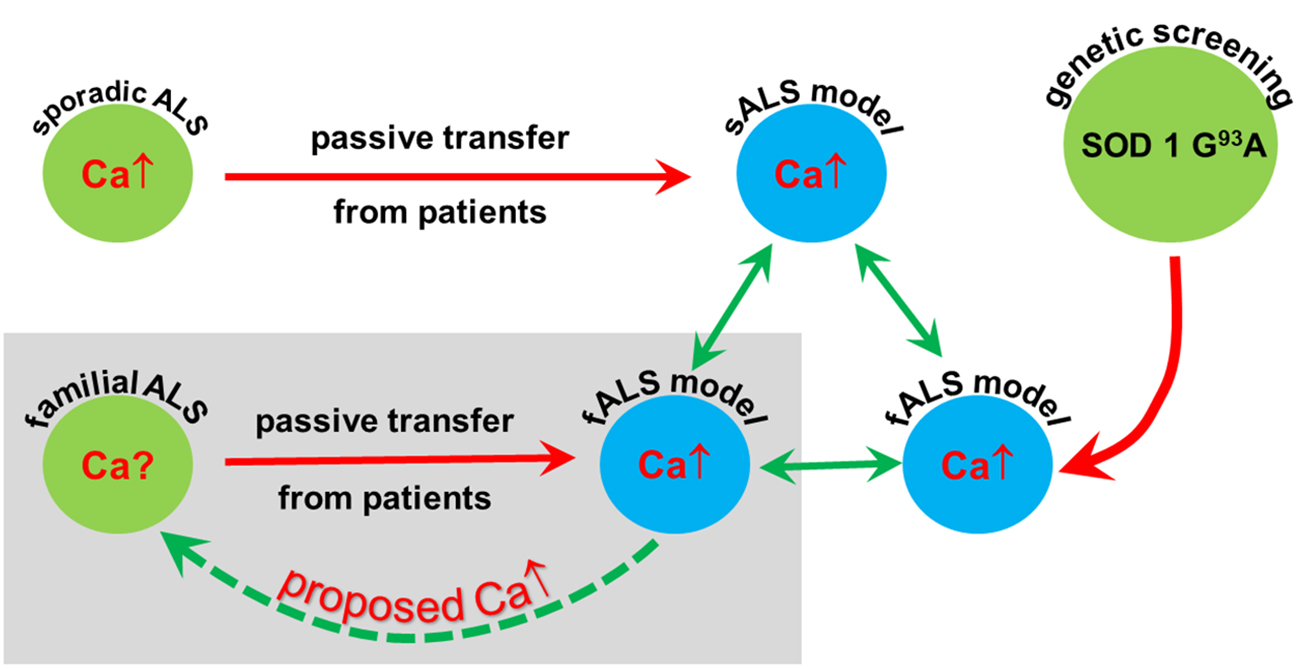
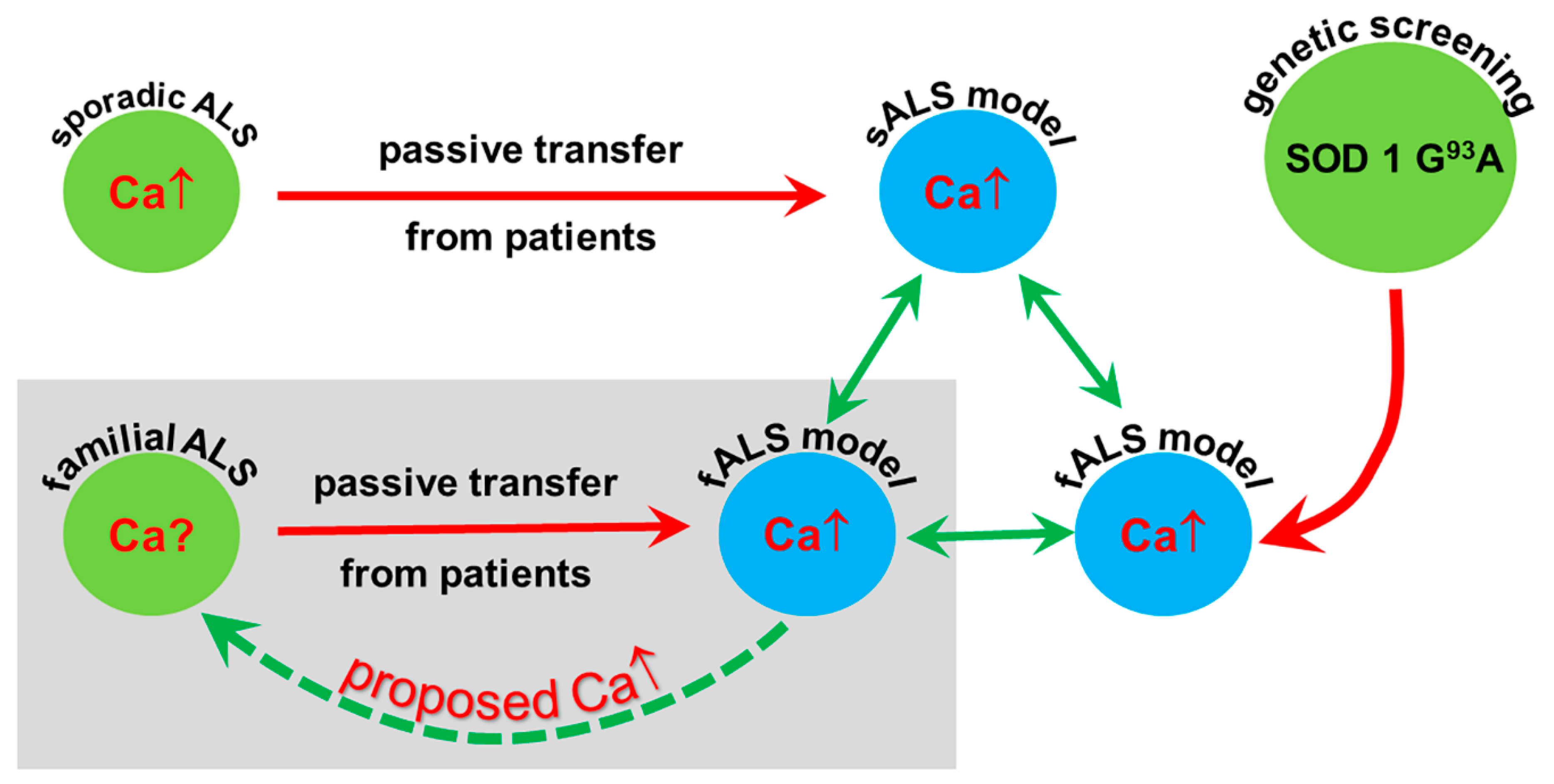
:
1. Introduction
2. Results
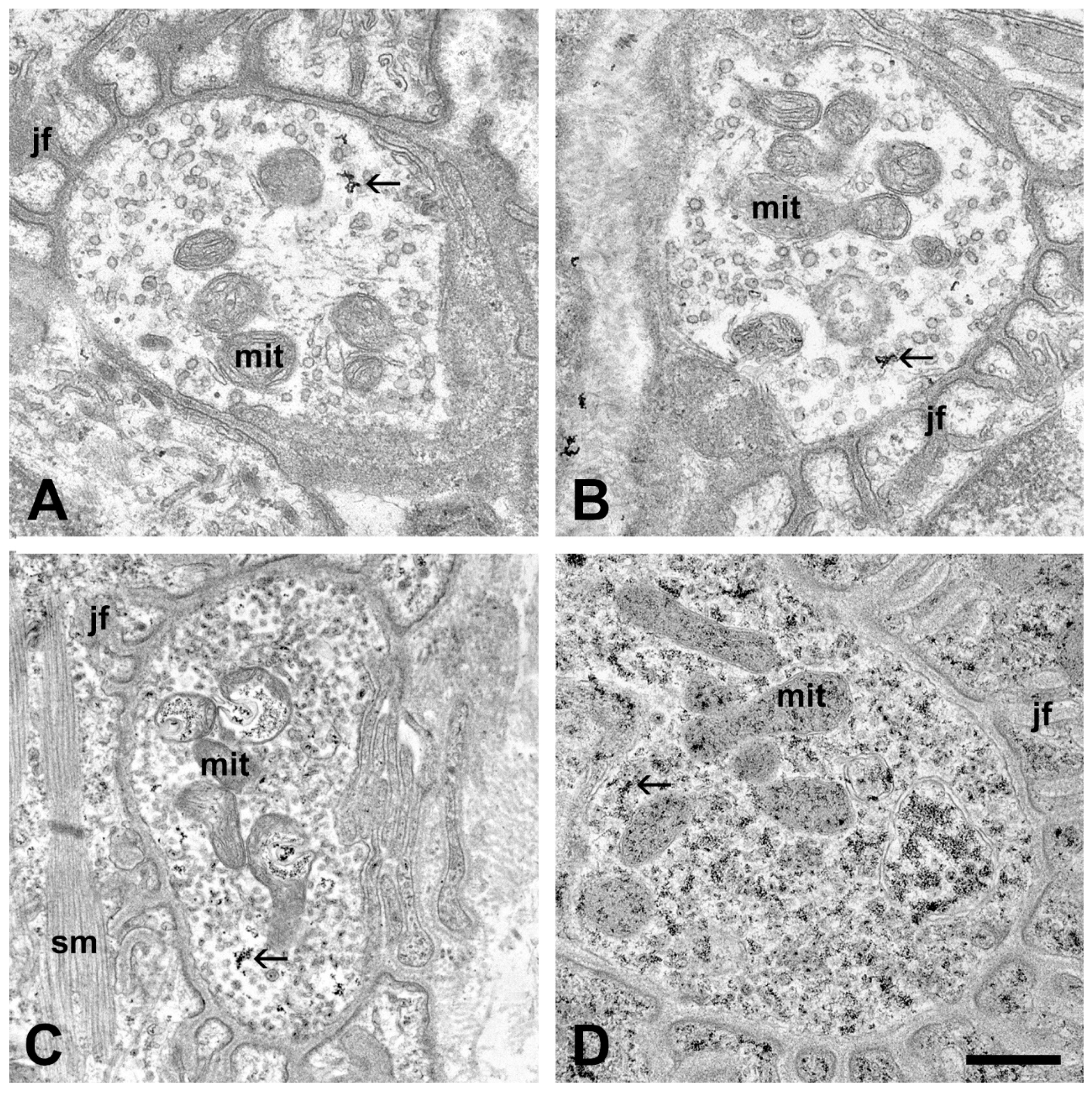
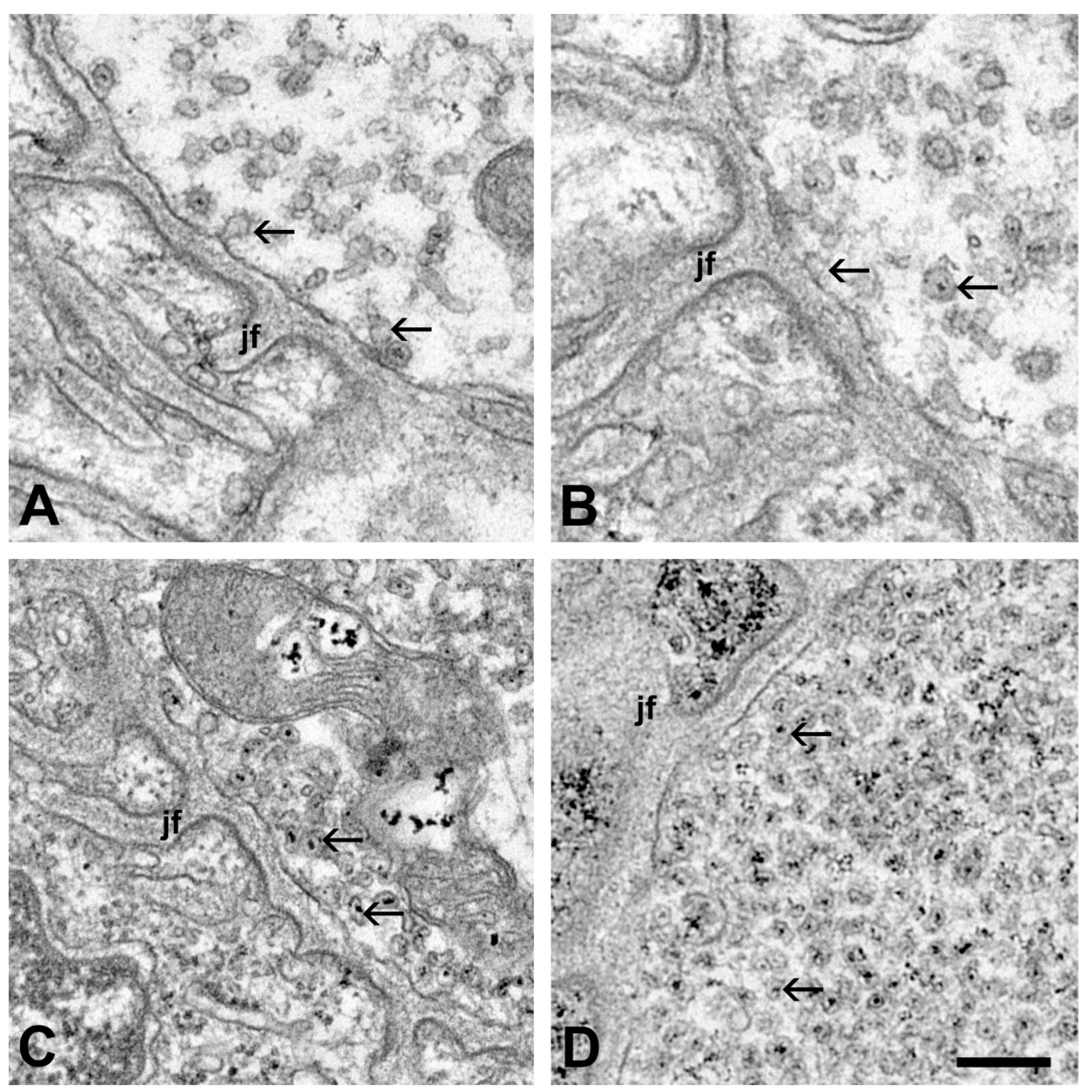
2.1. Ultrastructural Alterations of the Motor Axon Terminals after Inoculation with ALS Sera
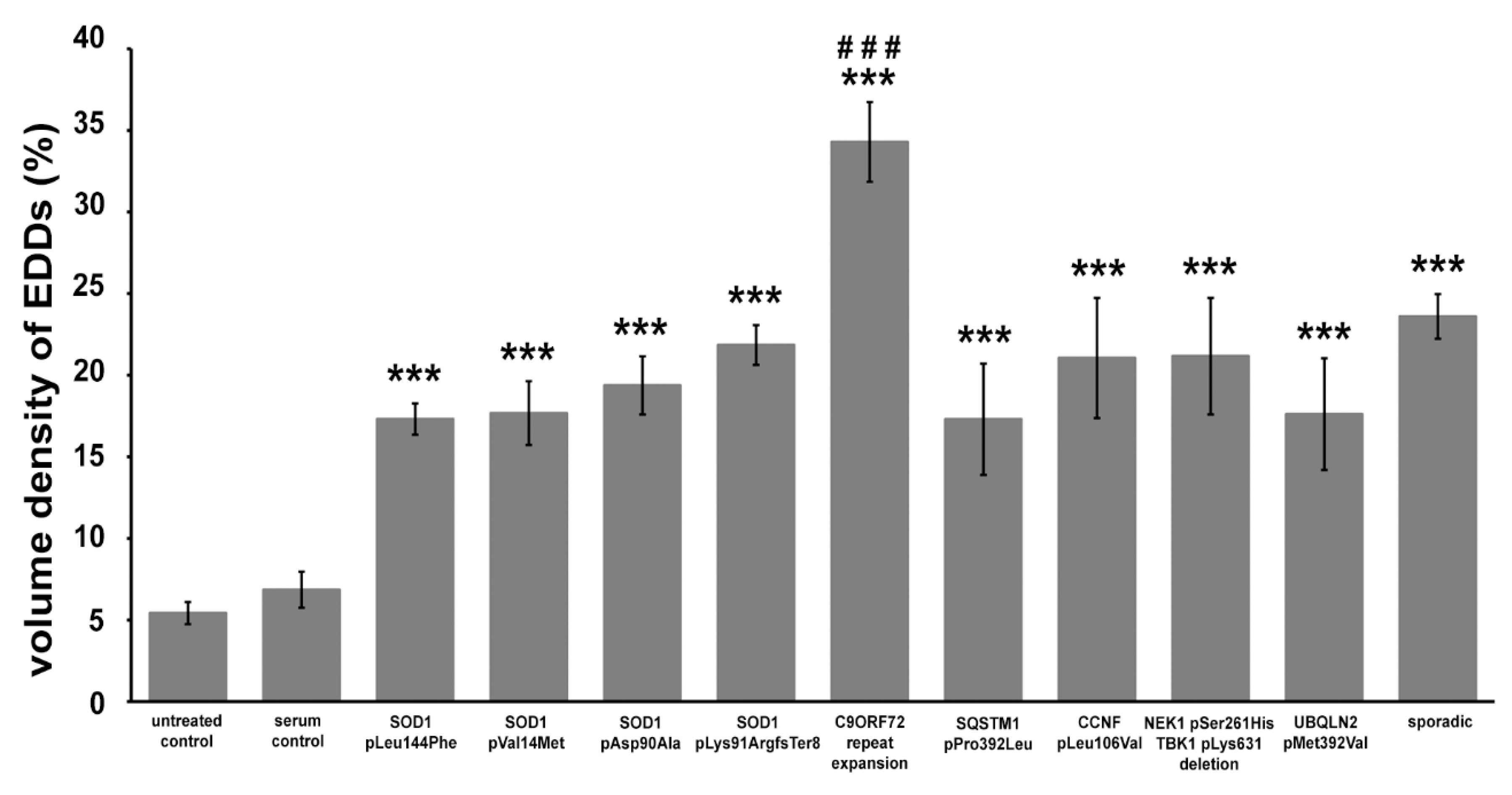
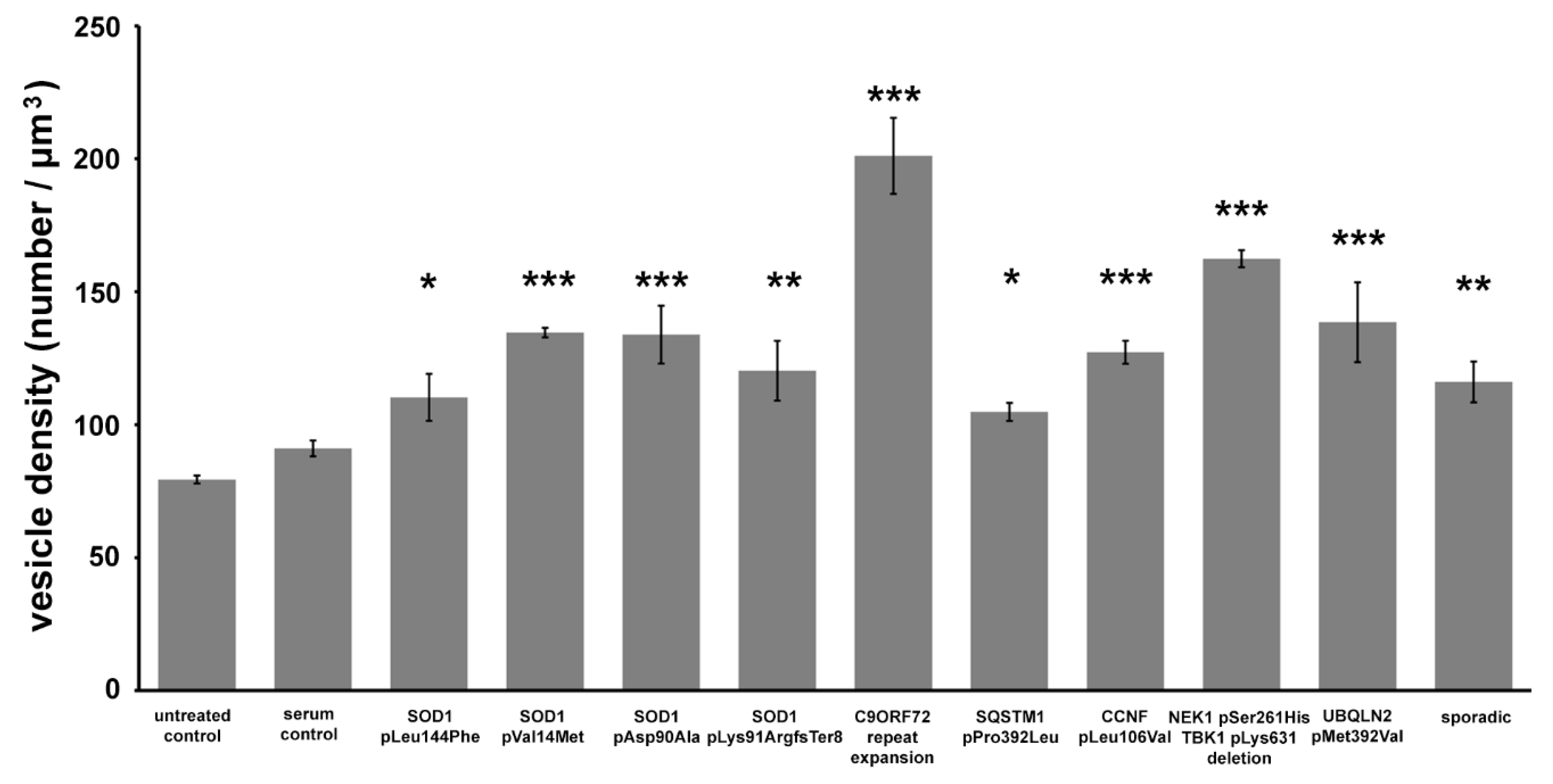
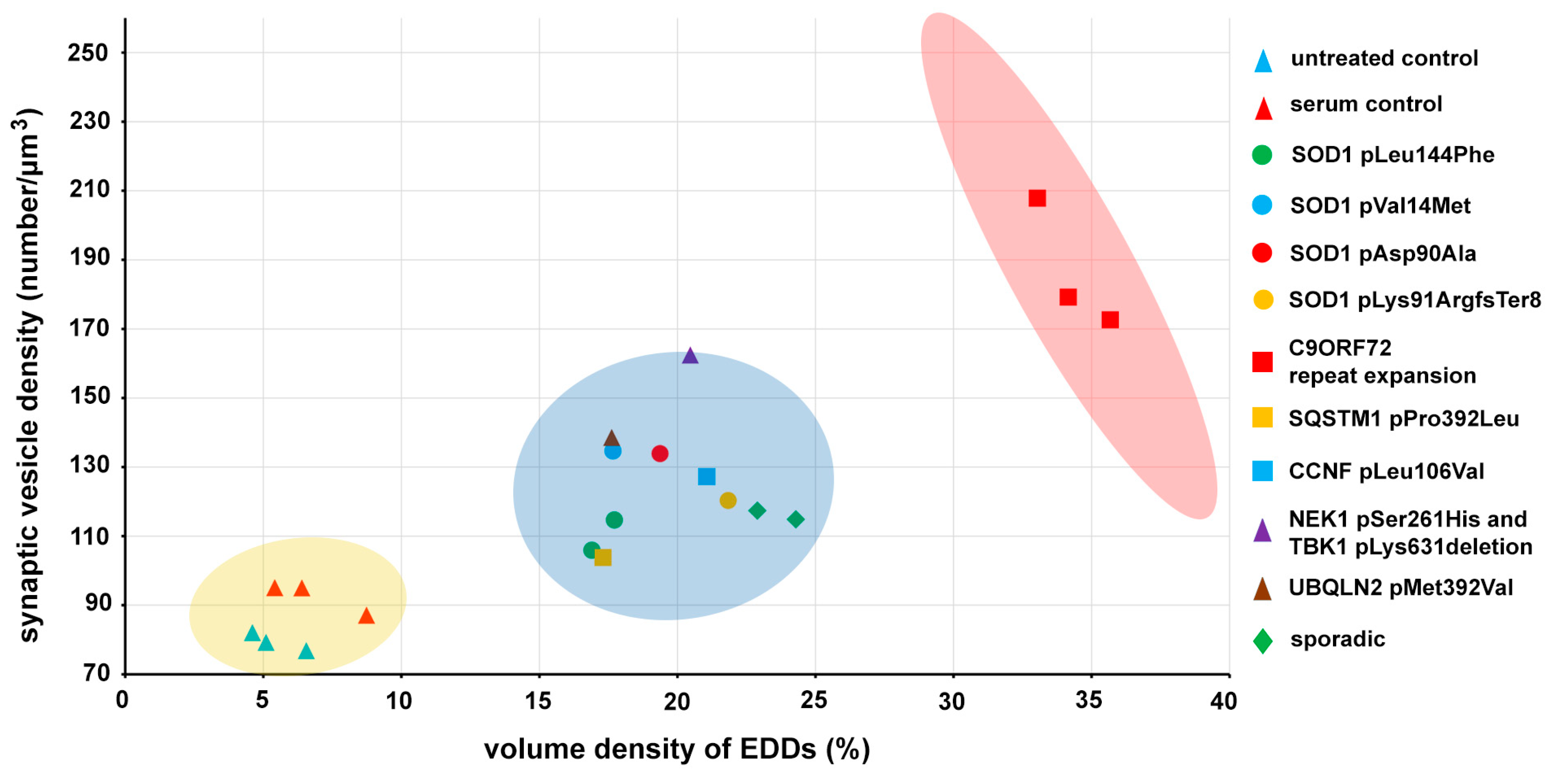
2.2. Quantitative Analysis of the Change in the Number of Synaptic Vesicles and Increase in Intracellular Calcium in Motor Axon Terminals after an Inoculation with ALS Sera
3. Discussion
4. Materials and Methods
4.1. Ethics Approval and Consent to Participate
4.2. Patients
4.3. Passive Transfer with Human Sera and Tissue Preparation
4.4. Quantification of the Intracellular Calcium Levels in the Motor Axon Terminals
4.5. Quantification of the Density of Synaptic Vesicles
4.6. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALS | Amyotrophic lateral sclerosis |
| ALSFRS-R | Revised amyotrophic lateral sclerosis functional rating scale |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid |
| ANG | Angiogenin |
| ANOVA | Analysis of variance |
| B | Bulbar |
| C9ORF72 | Chromosome 9 open reading frame 72 |
| CCNF | G2/mitotic-specific cyclin F |
| EDDs | Electron-dense deposits |
| IgG | Immunoglobulin G |
| LMN | Lower motor neuron |
| mALS | Amyotrophic lateral sclerosis with an identified mutation |
| mfALS | Familial amyotrophic lateral sclerosis with an identified mutation |
| MMSE | Mini-Mental State Examination |
| NEK1 | NIMA-related kinase 1 |
| PB | Pseudobulbar |
| RNA | Ribonucleic acid |
| sALS | Sporadic amyotrophic lateral sclerosis |
| s.e.m. | Standard error of the mean |
| SOD1 | Superoxide dismutase 1 |
| SQSTM1 | Sequestosome 1 |
| TBK1 | TANK-binding kinase 1 |
| UBQLN2 | Ubiquilin 2 |
| UMN | Upper motor neuron |
References
- Rowland, L.P.; Shneider, N.A. Amyotrophic lateral sclerosis. N. Eng. J. Med. 2001, 344, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Barber, S.C.; Shaw, P.J. Oxidative stress in ALS: Key role in motor neuron injury and therapeutic target. Free Radic. Biol. Med. 2010, 48, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Kurland, L.T.; Mulder, D.W. Epidemiologic investigations of amyotrophic lateral sclerosis. 2. Familial aggregations indicative of dominant inheritance. II. Neurology 1955, 5, 249–268. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef] [Green Version]
- Turner, M.R.; Hardiman, O.; Benatar, M.; Brooks, B.R.; Chio, A.; de Carvalho, M.; Ince, P.G.; Lin, C.; Miller, R.G.; Mitsumoto, H.; et al. Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol. 2013, 12, 310–322. [Google Scholar] [CrossRef] [Green Version]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Swinnen, B.; Robberecht, W. The phenotypic variability of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2014, 10, 661–670. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Dawson, T.M.; Dawson, V.L. Cell Death Mechanisms of Neurodegeneration. Adv. Neurobiol. 2017, 15, 403–425. [Google Scholar] [CrossRef]
- Malik, A.R.; Willnow, T.E. Excitatory Amino Acid Transporters in Physiology and Disorders of the Central Nervous System. Int. J. Mol. Sci. 2019, 20, 5671. [Google Scholar] [CrossRef] [Green Version]
- Bano, D.; Ankarcrona, M. Beyond the critical point: An overview of excitotoxicity, calcium overload and the downstream consequences. Neurosci. Lett. 2018, 663, 79–85. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, E.; Devasahayam, G. Neurodegeneration by oxidative stress: A review on prospective use of small molecules for neuroprotection. Mol. Biol. Rep. 2020, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Yaribeygi, H.; Panahi, Y.; Javadi, B.; Sahebkar, A. The Underlying Role of Oxidative Stress in Neurodegeneration: A Mechanistic Review. CNS Neurol. Disord. Drug Targets 2018, 17, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.L.; Soeters, M.R.; Wüst, R.; Houtkooper, R.H. Metabolic Flexibility as an Adaptation to Energy Resources and Requirements in Health and Disease. Endocr. Rev. 2018, 39, 489–517. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Zhao, D.; Shah, S.; Zhang, X.; Lai, M.; Yang, D.; Wu, X.; Guan, Z.; Li, J.; Zhao, H.; et al. OPA1 overexpression ameliorates mitochondrial cristae remodeling, mitochondrial dysfunction, and neuronal apoptosis in prion diseases. Cell Death Dis. 2019, 10, 710. [Google Scholar] [CrossRef]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef]
- Obál, I.; Nógrádi, B.; Meszlényi, V.; Patai, R.; Ricken, G.; Kovacs, G.G.; Tripolszki, K.; Széll, M.; Siklós, L.; Engelhardt, J.I. Experimental Motor Neuron Disease Induced in Mice with Long-Term Repeated Intraperitoneal Injections of Serum from ALS Patients. Int. J. Mol. Sci. 2019, 20, 2573. [Google Scholar] [CrossRef] [Green Version]
- Voet, S.; Srinivasan, S.; Lamkanfi, M.; van Loo, G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol. Med. 2019, 11, e10248. [Google Scholar] [CrossRef]
- Bezprozvanny, I. Calcium signaling and neurodegenerative diseases. Trends Mol. Med. 2009, 15, 89–100. [Google Scholar] [CrossRef] [Green Version]
- Enders, M.; Heider, T.; Ludwig, A.; Kuerten, S. Strategies for Neuroprotection in Multiple Sclerosis and the Role of Calcium. Int. J. Mol. Sci. 2020, 21, 1663. [Google Scholar] [CrossRef] [Green Version]
- Glaser, T.; Arnaud Sampaio, V.F.; Lameu, C.; Ulrich, H. Calcium signalling: A common target in neurological disorders and neurogenesis. Semin. Cell Dev. Biol. 2019, 95, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Pchitskaya, E.; Popugaeva, E.; Bezprozvanny, I. Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium 2018, 70, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Ureshino, R.P.; Erustes, A.G.; Bassani, T.B.; Wachilewski, P.; Guarache, G.C.; Nascimento, A.C.; Costa, A.J.; Smaili, S.S.; Pereira, G. The Interplay between Ca2+ Signaling Pathways and Neurodegeneration. Int. J. Mol. Sci. 2019, 20, 6004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belsh, J.M.; Schiffman, P.L. The amyotrophic lateral sclerosis (ALS) patient perspective on misdiagnosis and its repercussions. J. Neurol. Sci. 1996, 139, 110–116. [Google Scholar] [CrossRef]
- Patai, R.; Nógrádi, B.; Engelhardt, J.I.; Siklós, L. Calcium in the pathomechanism of amyotrophic lateral sclerosis—Taking center stage? Biochem. Biophys. Res. Commun. 2017, 483, 1031–1039. [Google Scholar] [CrossRef] [PubMed]
- Siklós, L.; Engelhardt, J.; Harati, Y.; Smith, R.G.; Joó, F.; Appel, S.H. Ultrastructural evidence for altered calcium in motor nerve terminals in amyotropic lateral sclerosis. Ann. Neurol. 1996, 39, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Longinetti, E.; Fang, F. Epidemiology of amyotrophic lateral sclerosis: An update of recent literature. Curr. Opin. Neurol. 2019, 32, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, J.I.; Siklós, L.; Kömüves, L.; Smith, R.G.; Appel, S.H. Antibodies to calcium channels from ALS patients passively transferred to mice selectively increase intracellular calcium and induce ultrastructural changes in motoneurons. Synapse 1995, 20, 185–199. [Google Scholar] [CrossRef]
- Sattler, R.; Tymianski, M. Molecular mechanisms of calcium-dependent excitotoxicity. J. Mol. Med. 2000, 78, 3–13. [Google Scholar] [CrossRef]
- Choi, D.W.; Maulucci-Gedde, M.; Kriegstein, A.R. Glutamate neurotoxicity in cortical cell culture. J. Neurosci. 1987, 7, 357–368. [Google Scholar] [CrossRef]
- Choi, D.W. Glutamate neurotoxicity and diseases of the nervous system. Neuron 1988, 1, 623–634. [Google Scholar] [CrossRef]
- Choi, D.W. Glutamate neurotoxicity in cortical cell culture is calcium dependent. Neurosci. Lett. 1985, 58, 293–297. [Google Scholar] [CrossRef]
- Arundine, M.; Tymianski, M. Molecular mechanisms of calcium-dependent neurodegeneration in excitotoxicity. Cell Calcium 2003, 34, 325–337. [Google Scholar] [CrossRef]
- Alvarez, J.; Alvarez-Illera, P.; García-Casas, P.; Fonteriz, R.I.; Montero, M. The Role of Ca2+ Signaling in Aging and Neurodegeneration: Insights from Caenorhabditis elegans Models. Cells 2020, 9, 204. [Google Scholar] [CrossRef] [Green Version]
- Berridge, M.J. Calcium hypothesis of Alzheimer’s disease. Pflugers Archiv 2010, 459, 441–449. [Google Scholar] [CrossRef]
- Betzer, C.; Jensen, P.H. Reduced Cytosolic Calcium as an Early Decisive Cellular State in Parkinson’s Disease and Synucleinopathies. Front. Neurosci. 2018, 12, 819. [Google Scholar] [CrossRef]
- Grosskreutz, J.; Van Den Bosch, L.; Keller, B.U. Calcium dysregulation in amyotrophic lateral sclerosis. Cell Calcium 2010, 47, 165–174. [Google Scholar] [CrossRef]
- Wang, X.; Zheng, W. Ca2+ homeostasis dysregulation in Alzheimer’s disease: A focus on plasma membrane and cell organelles. FASEB J. 2019, 33, 6697–6712. [Google Scholar] [CrossRef]
- von Lewinski, F.; Fuchs, J.; Vanselow, B.K.; Keller, B.U. Low Ca2+ buffering in hypoglossal motoneurons of mutant SOD1 (G93A) mice. Neurosci. Lett. 2008, 445, 224–228. [Google Scholar] [CrossRef]
- von Lewinski, F.; Keller, B.U. Ca2+, mitochondria and selective motoneuron vulnerability: Implications for ALS. Trends Neurosci. 2005, 28, 494–500. [Google Scholar] [CrossRef]
- May, C.; Nordhoff, E.; Casjens, S.; Turewicz, M.; Eisenacher, M.; Gold, R.; Brüning, T.; Pesch, B.; Stephan, C.; Woitalla, D.; et al. Highly immunoreactive IgG antibodies directed against a set of twenty human proteins in the sera of patients with amyotrophic lateral sclerosis identified by protein array. PLoS ONE 2014, 9, e89596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pullen, A.H.; Humphreys, P. Ultrastructural analysis of spinal motoneurones from mice treated with IgG from ALS patients, healthy individuals, or disease controls. J. Neurol. Sci. 2000, 180, 35–45. [Google Scholar] [CrossRef]
- Pullen, A.H.; Demestre, M.; Howard, R.S.; Orrell, R.W. Passive transfer of purified IgG from patients with amyotrophic lateral sclerosis to mice results in degeneration of motor neurons accompanied by Ca2+ enhancement. Acta Neuropathol. 2004, 107, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Demestre, M.; Pullen, A.; Orrell, R.W.; Orth, M. ALS-IgG-induced selective motor neurone apoptosis in rat mixed primary spinal cord cultures. J. Neurochem. 2005, 94, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.G.; Hamilton, S.; Hofmann, F.; Schneider, T.; Nastainczyk, W.; Birnbaumer, L.; Stefani, E.; Appel, S.H. Serum antibodies to L-type calcium channels in patients with amyotrophic lateral sclerosis. N. Engl. J. Med. 1992, 327, 1721–1728. [Google Scholar] [CrossRef] [PubMed]
- Delbono, O.; García, J.; Appel, S.H.; Stefani, E. IgG from amyotrophic lateral sclerosis affects tubular calcium channels of skeletal muscle. Am. J. Physiol. 1991, 260, C1347–C1351. [Google Scholar] [CrossRef]
- Magnelli, V.; Sawada, T.; Delbono, O.; Smith, R.G.; Appel, S.H.; Stefani, E. The action of amyotrophic lateral sclerosis immunoglobulins on mammalian single skeletal muscle Ca2+ channels. J. Physiol. 1993, 461, 103–118. [Google Scholar] [CrossRef]
- Mosier, D.R.; Baldelli, P.; Delbono, O.; Smith, R.G.; Alexianu, S.E.; Appel, S.H.; Stefani, E. Amyotrophic lateral sclerosis immunoglobulins increase Ca2+ currents in a motoneuron cell line. Ann. Neurol. 1995, 37, 102–109. [Google Scholar] [CrossRef]
- Llinás, R.; Sugimori, M.; Cherksey, B.D.; Smith, R.G.; Delbono, O.; Stefani, E.; Appel, S. IgG from amyotrophic lateral sclerosis patients increases current through P-type calcium channels in mammalian cerebellar Purkinje cells and in isolated channel protein in lipid bilayer. Proc. Natl. Acad. Sci. USA 1993, 90, 11743–11747. [Google Scholar] [CrossRef] [Green Version]
- Engelhardt, J.I.; Siklos, L.; Appel, S.H. Altered calcium homeostasis and ultrastructure in motoneurons of mice caused by passively transferred anti-motoneuronal IgG. J. Neuropathol. Exp. Neurol. 1997, 56, 21–39. [Google Scholar] [CrossRef] [Green Version]
- La Bella, V.; Goodman, J.C.; Appel, S.H. Increased CSF glutamate following injection of ALS immunoglobulins. Neurology 1997, 48, 1270–1272. [Google Scholar] [CrossRef] [PubMed]
- Obál, I.; Klausz, G.; Mándi, Y.; Deli, M.; Siklós, L.; Engelhardt, J.I. Intraperitoneally administered IgG from patients with amyotrophic lateral sclerosis or from an immune-mediated goat model increase the levels of TNF-α, IL-6, and IL-10 in the spinal cord and serum of mice. J. Neuroinflamm. 2016, 13, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milošević, M.; Milićević, K.; Božić, I.; Lavrnja, I.; Stevanović, I.; Bijelić, D.; Dubaić, M.; Zivković, I.; Stević, Z.; Giniatullin, R.; et al. Immunoglobulins G from Sera of Amyotrophic Lateral Sclerosis Patients Induce Oxidative Stress and Upregulation of Antioxidative System in BV-2 Microglial Cell Line. Front. Immunol. 2017, 8, 1619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milošević, M.; Stenovec, M.; Kreft, M.; Petrušić, V.; Stević, Z.; Trkov, S.; Andjus, P.R.; Zorec, R. Immunoglobulins G from patients with sporadic amyotrophic lateral sclerosis affects cytosolic Ca2+ homeostasis in cultured rat astrocytes. Cell Calcium 2013, 54, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Miller, R.G.; Swash, M.; Munsat, T.L.; World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2000, 1, 293–299. [Google Scholar] [CrossRef]
- Appel, S.H.; Engelhardt, J.I.; García, J.; Stefani, E. Immunoglobulins from animal models of motor neuron disease and from human amyotrophic lateral sclerosis patients passively transfer physiological abnormalities to the neuromuscular junction. Proc. Natl. Acad. Sci. USA 1991, 88, 647–651. [Google Scholar] [CrossRef] [Green Version]
- Mosier, D.R.; Siklós, L.; Appel, S.H. Resistance of extraocular motoneuron terminals to effects of amyotrophic lateral sclerosis sera. Neurology 2000, 54, 252–255. [Google Scholar] [CrossRef]
- Balendra, R.; Isaacs, A.M. C9orf72-mediated ALS and FTD: Multiple pathways to disease. Nat. Rev. Neurol. 2018, 14, 544–558. [Google Scholar] [CrossRef]
- Lall, D.; Baloh, R.H. Microglia and C9orf72 in neuroinflammation and ALS and frontotemporal dementia. J. Clin. Investig. 2017, 127, 3250–3258. [Google Scholar] [CrossRef]
- Sudria-Lopez, E.; Koppers, M.; de Wit, M.; van der Meer, C.; Westeneng, H.J.; Zundel, C.A.; Youssef, S.A.; Harkema, L.; de Bruin, A.; Veldink, J.H.; et al. Full ablation of C9orf72 in mice causes immune system-related pathology and neoplastic events but no motor neuron defects. Acta Neuropathol. 2016, 132, 145–147. [Google Scholar] [CrossRef] [Green Version]
- Siklós, L.; Engelhardt, J.I.; Alexianu, M.E.; Gurney, M.E.; Siddique, T.; Appel, S.H. Intracellular calcium parallels motoneuron degeneration in SOD-1 mutant mice. J. Neuropathol. Exp. Neurol. 1998, 57, 571–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lips, M.B.; Keller, B.U. Endogenous calcium buffering in motoneurones of the nucleus hypoglossus from mouse. J. Physiol. 1998, 511, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Palecek, J.; Lips, M.B.; Keller, B.U. Calcium dynamics and buffering in motoneurones of the mouse spinal cord. J. Physiol. 1999, 520, 485–502. [Google Scholar] [CrossRef] [PubMed]
- Vanselow, B.K.; Keller, B.U. Calcium dynamics and buffering in oculomotor neurones from mouse that are particularly resistant during amyotrophic lateral sclerosis (ALS)-related motoneurone disease. J. Physiol. 2000, 525, 433–445. [Google Scholar] [CrossRef] [PubMed]
- Beers, D.R.; Ho, B.K.; Siklós, L.; Alexianu, M.E.; Mosier, D.R.; Mohamed, A.H.; Otsuka, Y.; Kozovska, M.E.; McAlhany, R.E.; Smith, R.G.; et al. Parvalbumin overexpression alters immune-mediated increases in intracellular calcium, and delays disease onset in a transgenic model of familial amyotrophic lateral sclerosis. J. Neurochem. 2001, 79, 499–509. [Google Scholar] [CrossRef] [Green Version]
- Van Den Bosch, L.; Van Damme, P.; Bogaert, E.; Robberecht, W. The role of excitotoxicity in the pathogenesis of amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2006, 1762, 1068–1082. [Google Scholar] [CrossRef] [Green Version]
- Tortarolo, M.; Grignaschi, G.; Calvaresi, N.; Zennaro, E.; Spaltro, G.; Colovic, M.; Fracasso, C.; Guiso, G.; Elger, B.; Schneider, H.; et al. Glutamate AMPA receptors change in motor neurons of SOD1G93A transgenic mice and their inhibition by a noncompetitive antagonist ameliorates the progression of amytrophic lateral sclerosis-like disease. J. Neurosci. Res. 2006, 83, 134–146. [Google Scholar] [CrossRef]
- Paizs, M.; Tortarolo, M.; Bendotti, C.; Engelhardt, J.I.; Siklós, L. Talampanel reduces the level of motoneuronal calcium in transgenic mutant SOD1 mice only if applied presymptomatically. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2011, 12, 340–344. [Google Scholar] [CrossRef]
- Tripolszki, K.; Danis, J.; Padhi, A.K.; Gomes, J.; Bozó, R.; Nagy, Z.F.; Nagy, D.; Klivényi, P.; Engelhardt, J.I.; Széll, M. Angiogenin mutations in Hungarian patients with amyotrophic lateral sclerosis: Clinical, genetic, computational, and functional analyses. Brain Behav. 2019, 9, e01293. [Google Scholar] [CrossRef]
- Costa, J.; Swash, M.; de Carvalho, M. Awaji criteria for the diagnosis of amyotrophic lateral sclerosis: A systematic review. Arch. Neurol. 2012, 69, 1410–1416. [Google Scholar] [CrossRef]
- Cedarbaum, J.M.; Stambler, N.; Malta, E.; Fuller, C.; Hilt, D.; Thurmond, B.; Nakanishi, A. The ALSFRS-R: A revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III). J. Neurol. Sci. 1999, 169, 13–21. [Google Scholar] [CrossRef]
- Borgers, M.; De Brabander, M.; Van Reempts, J.; Awouters, F.; Jacob, W.A. Intranuclear microtubules in lung mast cells of guinea pigs in anaphylactic shock. Lab. Investig. 1977, 37, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Borgers, M. The role of calcium in the toxicity of the myocardium. Histochem. J. 1981, 13, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Adalbert, R.; Engelhardt, J.I.; Siklós, L. DL-Homocysteic acid application disrupts calcium homeostasis and induces degeneration of spinal motor neurons in vivo. Acta Neuropathol. 2002, 103, 428–436. [Google Scholar] [CrossRef]
- Obál, I.; Engelhardt, J.I.; Siklós, L. Axotomy induces contrasting changes in calcium and calcium-binding proteins in oculomotor and hypoglossal nuclei of Balb/c mice. J. Comp. Neurol. 2006, 499, 17–32. [Google Scholar] [CrossRef]
- Paizs, M.; Engelhardt, J.I.; Katarova, Z.; Siklós, L. Hypoglossal motor neurons display a reduced calcium increase after axotomy in mice with upregulated parvalbumin. J. Comp. Neurol. 2010, 518, 1946–1961. [Google Scholar] [CrossRef]
- Patai, R.; Paizs, M.; Tortarolo, M.; Bendotti, C.; Obál, I.; Engelhardt, J.I.; Siklós, L. Presymptomatically applied AMPA receptor antagonist prevents calcium increase in vulnerable type of motor axon terminals of mice modeling amyotrophic lateral sclerosis. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1739–1748. [Google Scholar] [CrossRef]
- Siklós, L.; Kuhnt, U.; Párducz, A.; Szerdahelyi, P. Intracellular calcium redistribution accompanies changes in total tissue Na+, K+ and water during the first two hours of in vitro incubation of hippocampal slices. Neuroscience 1997, 79, 1013–1022. [Google Scholar] [CrossRef]
- Maxwell, M.H. Two rapid and simple methods used for the removal of resins from 1.0 micron thick epoxy sections. J. Microsc. 1978, 112, 253–255. [Google Scholar] [CrossRef]
- Richardson, K.C.; Jarett, L.; Finke, E.H. Embedding in epoxy resins for ultrathin sectioning in electron microscopy. Stain Technol. 1960, 35, 313–323. [Google Scholar] [CrossRef]
- Cruz-Orive, L.M.; Weibel, E.R. Sampling designs for stereology. J. Microsc. 1981, 122, 235–257. [Google Scholar] [CrossRef] [PubMed]
- Hayat, M.A. Principles and Techniques of Electron Microscopy: Biological Applications, 4th ed.; Cambridge University Press: Cambridge, UK, 2000. [Google Scholar]
- Reynolds, E.S. The use of lead citrate at high pH as an electron-opaque stain in electron microscopy. J. Cell Biol. 1963, 17, 208–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayhew, T.M. A review of recent advances in stereology for quantifying neural structure. J. Neurocytol. 1992, 21, 313–328. [Google Scholar] [CrossRef] [PubMed]
- Weibel, E.R. Stereological Methods Vol. 1. Practical Methods for Biological Morphometry; Academic Press: London, UK; New York, NY, USA; Toronto, ON, USA; Sydney, Australia; San Francisco, CA, USA, 1979. [Google Scholar]
- Domoki, F.; Bari, F.; Nagy, K.; Busija, D.W.; Siklós, L. Diazoxide prevents mitochondrial swelling and Ca2+ accumulation in CA1 pyramidal cells after cerebral ischemia in newborn pigs. Brain Res. 2004, 1019, 97–104. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patients | Age at Onset (Years) | Duration of the Disease at the Study | Initial Symptoms | Clinical Signs | ALS FRS-R | MMSE | Genetic Alteration | Family History | Therapy | Other Disease |
|---|---|---|---|---|---|---|---|---|---|---|
| mALS 1 | 63 | 1 year | proximal bilateral lower limb weakness | LMN, UMN | 32/48 | 28/30 | SOD1 pVal14 Met ANG Met24Ile | negative | Riluzole, Perindopril, Aspirin, Piracetam, Vinpocetine, Nebivolol | atherosclerosis, hypertension |
| mALS 2 | 75 | 12 years | bilateral lower limb weakness | LMN, UMN, B | 19/48 | 29/30 | SOD1 pAsp90Ala | negative | Riluzole | cervical and lumbar spondylarthrosis, hyperlipidemia |
| mfALS 3 | 29 | 2 years | gait disturbance | LMN, UMN | 36/48 | 30/30 | SOD1 pLeu144Phe | grandmother (fraternal) | Riluzole | - |
| mfALS 4 | 49 | 3 years | distal weakness of lower limbs | LMN, UMN, B, PB | 25/48 | 28/30 | SOD1 pLeu144Phe | grandmother (maternal) | Riluzole, Citalopram | depression, lumbar discs’ herniation |
| mALS 5 | 67 | 6 months | four limbs weakness | B, PB, UMN, LMN | 39/48 | 30/30 | SOD1 pLys91Arg fs Ter8 | negative | Riluzole, Atorvastatin, Valsartan | breast cancer (irradiated 8 years ago), hypercholesterolemia, cervical and lumbar discs’ protrusion |
| mALS 6 | 68 | 6 months | bilateral peroneal palsy, dysarthria | LMN, B, UMN | 44/48 | 30/30 | C9ORF72 repeat expansion | negative | Alprazolam, Perindopril, Duloxetine | hyperparathyroidism (cured), generalized lipomatosis, osteoporosis, hypertension, depression |
| mALS 7 | 55 | 1 year | dysarthria, dysphagia | B, PB, LMN, UMN | 37/48 | 27/30 | C9ORF72 repeat expansion | negative | Riluzole, L-thyroxin | Hashimoto’s thyroiditis |
| mfALS 8 | 56 | 8 months | dysarthria, dysphagia | B, PB, LMN, UMN | 36/48 | 30/30 | C9ORF72 repeat expansion | mother with suspected ALS (not documented) | Riluzole, L-thyroxin | hypothyroidism |
| mALS 9 | 54 | 6 months | dyspnea | B, PB, LMN, UMN | 40/48 | 30/30 | SQSTM1 pPro392Leu | negative | Valsartan-HCT | hypertension |
| mALS 10 | 61 | 6 months | UMN, LMN lesions in the lower limbs | LMN, UMN, B | 42/48 | 30/30 | CCNF pLeu106Val | negative | Valsartan, Riluzole | hypertension, cervical and lumbar discs’ protrusion |
| mALS 11 | 65 | 6 months | four limbs weakness | LMN, UMN | 43/48 | 29/30 | UBQLN2 pMet392Val | negative | Riluzole | hypertension depression |
| mALS 12 | 37 | 6 months | four limbs weakness, dysarthria, cognitive deficit | UMN, LMN, B, PB | 39/48 | 23/30 | NEK1 pSer261His TBK1 pLys631 deletion | negative | Riluzole, Perindopril, Paroxetine | hypertension depression, frontotemporal dementia |
| sALS1 | 71 | 1 year | weakness of the right arm and leg (peroneal) | UMN, LMN | 41/48 | 28/30 | - | negative | Piracetam, Diclofenac, Aspirin, Perindopril, Isosorbide-mononitrate, Bisoprolol | hypertension, hypercholesterolemia, atherosclerosis, post zoster neuralgia |
| sALS2 | 74 | 9 months | dysarthria, dysphagia | B, UMN, LMN | 39/48 | 26/26 | - | negative | Amlodipine, Perindopril, Metoprolol, Atorvastatin, Riluzole | hypertension, hypercholesterolemia |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meszlényi, V.; Patai, R.; Polgár, T.F.; Nógrádi, B.; Körmöczy, L.; Kristóf, R.; Spisák, K.; Tripolszki, K.; Széll, M.; Obál, I.; et al. Passive Transfer of Sera from ALS Patients with Identified Mutations Evokes an Increased Synaptic Vesicle Number and Elevation of Calcium Levels in Motor Axon Terminals, Similar to Sera from Sporadic Patients. Int. J. Mol. Sci. 2020, 21, 5566. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155566
Meszlényi V, Patai R, Polgár TF, Nógrádi B, Körmöczy L, Kristóf R, Spisák K, Tripolszki K, Széll M, Obál I, et al. Passive Transfer of Sera from ALS Patients with Identified Mutations Evokes an Increased Synaptic Vesicle Number and Elevation of Calcium Levels in Motor Axon Terminals, Similar to Sera from Sporadic Patients. International Journal of Molecular Sciences. 2020; 21(15):5566. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155566
Chicago/Turabian StyleMeszlényi, Valéria, Roland Patai, Tamás F. Polgár, Bernát Nógrádi, Laura Körmöczy, Rebeka Kristóf, Krisztina Spisák, Kornélia Tripolszki, Márta Széll, Izabella Obál, and et al. 2020. "Passive Transfer of Sera from ALS Patients with Identified Mutations Evokes an Increased Synaptic Vesicle Number and Elevation of Calcium Levels in Motor Axon Terminals, Similar to Sera from Sporadic Patients" International Journal of Molecular Sciences 21, no. 15: 5566. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155566