FGFR Fusions in Cancer: From Diagnostic Approaches to Therapeutic Intervention

 , ,
, ,
Abstract
:1. Introduction
2. The FGFR/FGF System
3. Genetic Alterations of FGFRs in Human Cancers
3.1. FGFR Amplifications and Mutations
3.2. FGFR Family Gene Fusions
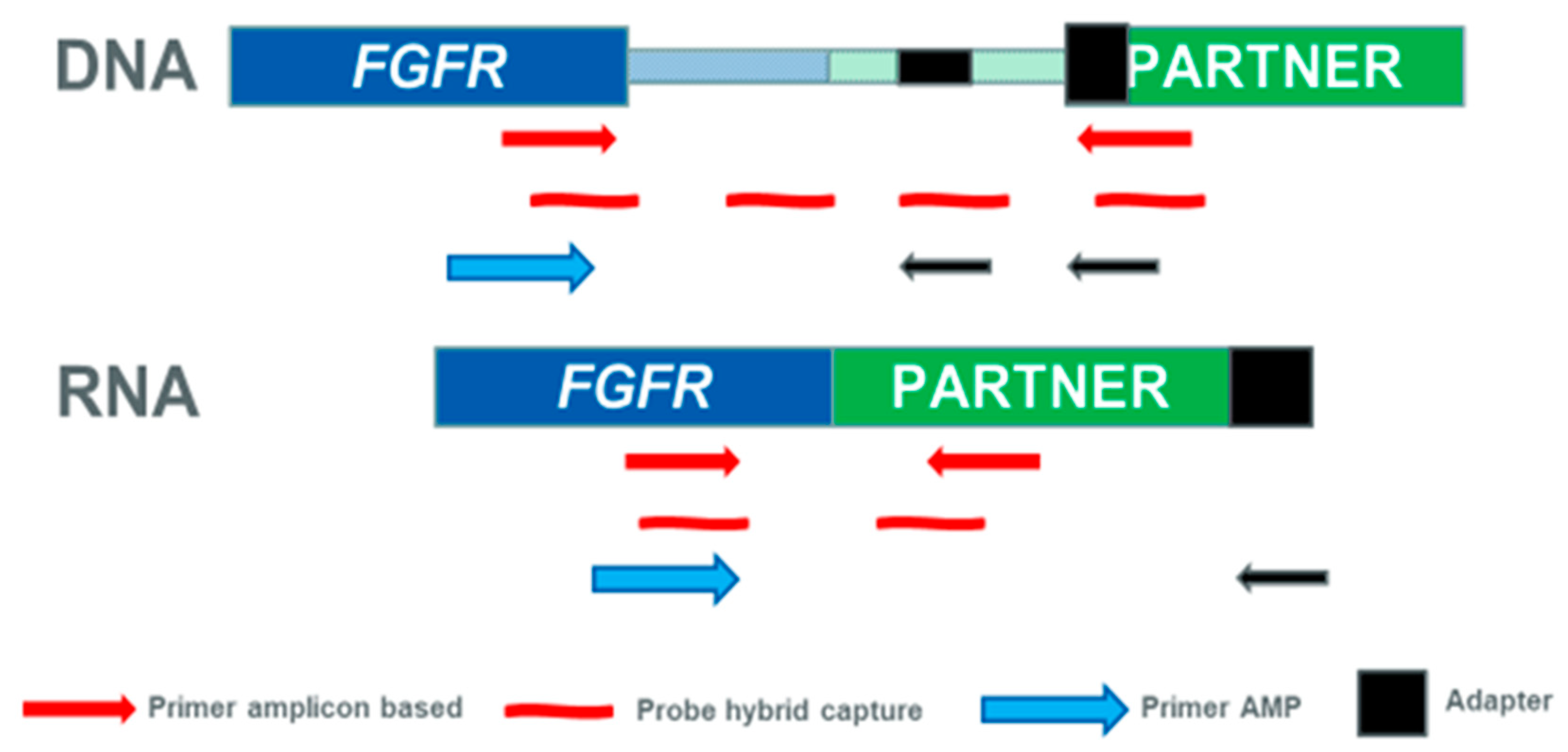
4. Approaches to Detect FGFR Fusions in Clinical Diagnostics
5. Prognostic Significance of FGFR Fusions
6. FGFR Fusions as Therapeutic Target for Solid Tumors
7. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Babina, I.S.; Turner, N.C. Advances and challenges in targeting FGFR signalling in cancer. Nat. Rev. Cancer 2017, 17, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Goetz, R.; Mohammadi, M. Exploring mechanisms of FGF signalling through the lens of structural biology. Nat. Rev. Mol. Cell Biol 2013, 14, 166–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, M. Fibroblast growth factor receptors as treatment targets in clinical oncology. Nat. Rev. Clin. Oncol. 2019, 16, 105–122. [Google Scholar] [CrossRef] [PubMed]
- Mertens, F.; Johansson, B.; Fioretos, T.; Mitelman, F. The emerging complexity of gene fusions in cancer. Nat. Rev. Cancer 2015, 15, 371–381. [Google Scholar] [CrossRef]
- Schram, A.M.; Chang, M.T.; Jonsson, P.; Drilon, A. Fusions in solid tumours: Diagnostic strategies, targeted therapy, and acquired resistance. Nat. Rev. Clin. Oncol. 2017, 14, 735–748. [Google Scholar] [CrossRef]
- Beenken, A.; Mohammadi, M. The FGF family: Biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 2009, 8, 235–253. [Google Scholar] [CrossRef] [Green Version]
- Kalinina, J.; Dutta, K.; Ilghari, D.; Beenken, A.; Goetz, R.; Eliseenkova, A.V.; Cowburn, D.; Mohammadi, M. The alternatively spliced acid box region plays a key role in FGF receptor autoinhibition. Structure 2012, 20, 77–88. [Google Scholar] [CrossRef] [Green Version]
- Sleeman, M.; Fraser, J.; McDonald, M.; Yuan, S.; White, D.; Grandison, P.; Kumble, K.; Watson, J.D.; Murison, J.G. Identification of a new fibroblast growth factor receptor, FGFR5. Gene 2001, 271, 171–182. [Google Scholar] [CrossRef]
- Trueb, B. Biology of FGFRL1, the fifth fibroblast growth factor receptor. Cell. Mol. Life Sci. 2011, 68, 951–964. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, L.; Steinberg, F.; Trueb, B. Receptor FGFRL1 acts as a tumor suppressor in nude mice when overexpressed in HEK 293 Tet-On cells. Oncol. Lett. 2016, 12, 4524–4530. [Google Scholar] [CrossRef] [Green Version]
- Holzmann, K.; Grunt, T.; Heinzle, C.; Sampl, S.; Steinhoff, H.; Reichmann, N.; Kleiter, M.; Hauck, M.; Marian, B. Alternative Splicing of Fibroblast Growth Factor Receptor IgIII Loops in Cancer. J. Nucleic Acids 2012, 2012, 950508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takaishi, S.; Sawada, M.; Morita, Y.; Seno, H.; Fukuzawa, H.; Chiba, T. Identification of a novel alternative splicing of human FGF receptor 4: Soluble-form splice variant expressed in human gastrointestinal epithelial cells. Biochem. Biophys. Res. Commun. 2000, 267, 658–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ezzat, S.; Zheng, L.; Yu, S.; Asa, S.L. A soluble dominant negative fibroblast growth factor receptor 4 isoform in human MCF-7 breast cancer cells. Biochem. Biophys. Res. Commun. 2001, 287, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Olsen, S.K.; Garbi, M.; Zampieri, N.; Eliseenkova, A.V.; Ornitz, D.M.; Goldfarb, M.; Mohammadi, M. Fibroblast growth factor (FGF) homologous factors share structural but not functional homology with FGFs. J. Biol. Chem. 2003, 278, 34226–34236. [Google Scholar] [CrossRef] [Green Version]
- Goetz, R.; Beenken, A.; Ibrahimi, O.A.; Kalinina, J.; Olsen, S.K.; Eliseenkova, A.V.; Xu, C.; Neubert, T.A.; Zhang, F.; Linhardt, R.J.; et al. Molecular insights into the klotho-dependent, endocrine mode of action of fibroblast growth factor 19 subfamily members. Mol. Cell Biol. 2007, 27, 3417–3428. [Google Scholar] [CrossRef] [Green Version]
- Gallo, L.H.; Nelson, K.N.; Meyer, A.N.; Donoghue, D.J. Functions of Fibroblast Growth Factor Receptors in cancer defined by novel translocations and mutations. Cytokine Growth Factor Rev. 2015, 26, 425–449. [Google Scholar] [CrossRef] [Green Version]
- Helsten, T.; Elkin, S.; Arthur, E.; Tomson, B.N.; Carter, J.; Kurzrock, R. The FGFR Landscape in Cancer: Analysis of 4,853 Tumors by Next-Generation Sequencing. Clin. Cancer Res. 2016, 22, 259–267. [Google Scholar] [CrossRef] [Green Version]
- The AACR Project GENIE Consortium. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef] [Green Version]
- Lew, E.D.; Furdui, C.M.; Anderson, K.S.; Schlessinger, J. The precise sequence of FGF receptor autophosphorylation is kinetically driven and is disrupted by oncogenic mutations. Sci. Signal. 2009, 2, ra6. [Google Scholar] [CrossRef] [Green Version]
- Tomlinson, D.C.; Hurst, C.D.; Knowles, M.A. Knockdown by shRNA identifies S249C mutant FGFR3 as a potential therapeutic target in bladder cancer. Oncogene 2007, 26, 5889–5899. [Google Scholar] [CrossRef] [Green Version]
- Knowles, M.A. Role of FGFR3 in urothelial cell carcinoma: Biomarker and potential therapeutic target. World J. Urol. 2007, 25, 581–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, J.G.t.; Cheuk, A.T.; Tsang, P.S.; Chung, J.Y.; Song, Y.K.; Desai, K.; Yu, Y.; Chen, Q.R.; Shah, K.; Youngblood, V.; et al. Identification of FGFR4-activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. J. Clin. Investig. 2009, 119, 3395–3407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Futami, T.; Kawase, T.; Mori, K.; Asaumi, M.; Kihara, R.; Shindoh, N.; Kuromitsu, S. Identification of a novel oncogenic mutation of FGFR4 in gastric cancer. Sci. Rep. 2019, 9, 14627. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.C.; Engels, M.; Annala, M.; Zhang, W. Emergence of FGFR family gene fusions as therapeutic targets in a wide spectrum of solid tumours. J. Pathol. 2014, 232, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Shi, E.; Chmielecki, J.; Tang, C.M.; Wang, K.; Heinrich, M.C.; Kang, G.; Corless, C.L.; Hong, D.; Fero, K.E.; Murphy, J.D.; et al. FGFR1 and NTRK3 actionable alterations in “Wild-Type” gastrointestinal stromal tumors. J. Transl. Med. 2016, 14, 339. [Google Scholar] [CrossRef] [Green Version]
- Di Stefano, A.L.; Fucci, A.; Frattini, V.; Labussiere, M.; Mokhtari, K.; Zoppoli, P.; Marie, Y.; Bruno, A.; Boisselier, B.; Giry, M.; et al. Detection, Characterization, and Inhibition of FGFR-TACC Fusions in IDH Wild-type Glioma. Clin. Cancer Res. 2015, 21, 3307–3317. [Google Scholar] [CrossRef] [Green Version]
- Singh, D.; Chan, J.M.; Zoppoli, P.; Niola, F.; Sullivan, R.; Castano, A.; Liu, E.M.; Reichel, J.; Porrati, P.; Pellegatta, S.; et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science 2012, 337, 1231–1235. [Google Scholar] [CrossRef] [Green Version]
- Stephens, P.J.; McBride, D.J.; Lin, M.L.; Varela, I.; Pleasance, E.D.; Simpson, J.T.; Stebbings, L.A.; Leroy, C.; Edkins, S.; Mudie, L.J.; et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature 2009, 462, 1005–1010. [Google Scholar] [CrossRef] [Green Version]
- Ross, J.S.; Wang, K.; Khaira, D.; Ali, S.M.; Fisher, H.A.; Mian, B.; Nazeer, T.; Elvin, J.A.; Palma, N.; Yelensky, R.; et al. Comprehensive genomic profiling of 295 cases of clinically advanced urothelial carcinoma of the urinary bladder reveals a high frequency of clinically relevant genomic alterations. Cancer 2016, 122, 702–711. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Wang, L.; Li, Y.; Hu, H.; Shen, L.; Shen, X.; Pan, Y.; Ye, T.; Zhang, Y.; Luo, X.; et al. FGFR1/3 tyrosine kinase fusions define a unique molecular subtype of non-small cell lung cancer. Clin. Cancer Res. 2014, 20, 4107–4114. [Google Scholar] [CrossRef] [Green Version]
- Qin, A.; Johnson, A.; Ross, J.S.; Miller, V.A.; Ali, S.M.; Schrock, A.B.; Gadgeel, S.M. Detection of Known and Novel FGFR Fusions in Non-Small Cell Lung Cancer by Comprehensive Genomic Profiling. J. Thorac. Oncol. 2019, 14, 54–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, Y.; Totoki, Y.; Hosoda, F.; Shirota, T.; Hama, N.; Nakamura, H.; Ojima, H.; Furuta, K.; Shimada, K.; Okusaka, T.; et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology 2014, 59, 1427–1434. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.S.; Wang, K.; Gay, L.; Al-Rohil, R.; Rand, J.V.; Jones, D.M.; Lee, H.J.; Sheehan, C.E.; Otto, G.A.; Palmer, G.; et al. New routes to targeted therapy of intrahepatic cholangiocarcinomas revealed by next-generation sequencing. Oncologist 2014, 19, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowery, M.A.; Ptashkin, R.; Jordan, E.; Berger, M.F.; Zehir, A.; Capanu, M.; Kemeny, N.E.; O’Reilly, E.M.; El-Dika, I.; Jarnagin, W.R.; et al. Comprehensive Molecular Profiling of Intrahepatic and Extrahepatic Cholangiocarcinomas: Potential Targets for Intervention. Clin. Cancer Res. 2018, 24, 4154–4161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, A.; Borad, M.J.; Kelley, R.K.; Wang, Y.; Abdel-Wahab, R.; Meric-Bernstam, F.; Baggerly, K.A.; Kaseb, A.O.; Al-shamsi, H.O.; Ahn, D.H.; et al. Cholangiocarcinoma With FGFR Genetic Aberrations: A Unique Clinical Phenotype. JCO Precis. Oncol. 2018, 1–12. [Google Scholar] [CrossRef]
- Borad, M.J.; Champion, M.D.; Egan, J.B.; Liang, W.S.; Fonseca, R.; Bryce, A.H.; McCullough, A.E.; Barrett, M.T.; Hunt, K.; Patel, M.D.; et al. Integrated genomic characterization reveals novel, therapeutically relevant drug targets in FGFR and EGFR pathways in sporadic intrahepatic cholangiocarcinoma. PLoS ONE Genet. 2014, 10, e1004135. [Google Scholar] [CrossRef]
- Sia, D.; Losic, B.; Moeini, A.; Cabellos, L.; Hao, K.; Revill, K.; Bonal, D.; Miltiadous, O.; Zhang, Z.; Hoshida, Y.; et al. Massive parallel sequencing uncovers actionable FGFR2-PPHLN1 fusion and ARAF mutations in intrahepatic cholangiocarcinoma. Nat. Commun. 2015, 6, 6087. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.M.; Su, F.; Kalyana-Sundaram, S.; Khazanov, N.; Ateeq, B.; Cao, X.; Lonigro, R.J.; Vats, P.; Wang, R.; Lin, S.F.; et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov. 2013, 3, 636–647. [Google Scholar] [CrossRef] [Green Version]
- Tanizaki, J.; Ercan, D.; Capelletti, M.; Dodge, M.; Xu, C.; Bahcall, M.; Tricker, E.M.; Butaney, M.; Calles, A.; Sholl, L.M.; et al. Identification of Oncogenic and Drug-Sensitizing Mutations in the Extracellular Domain of FGFR2. Cancer Res. 2015, 75, 3139–3146. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ding, X.; Wang, S.; Moser, C.D.; Shaleh, H.M.; Mohamed, E.A.; Chaiteerakij, R.; Allotey, L.K.; Chen, G.; Miyabe, K.; et al. Antitumor effect of FGFR inhibitors on a novel cholangiocarcinoma patient derived xenograft mouse model endogenously expressing an FGFR2-CCDC6 fusion protein. Cancer Lett. 2016, 380, 163–173. [Google Scholar] [CrossRef] [Green Version]
- Hood, F.E.; Royle, S.J. Pulling it together: The mitotic function of TACC3. Bioarchitecture 2011, 1, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.V.; Hurst, C.D.; Knowles, M.A. Oncogenic FGFR3 gene fusions in bladder cancer. Hum. Mol. Genet. 2013, 22, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, B.A.; Elvin, J.A.; Kamath, S.D.; Ali, S.M.; Paintal, A.S.; Restrepo, A.; Berry, E.; Giles, F.J.; Johnson, M.L. FGFR3-TACC3: A novel gene fusion in cervical cancer. Gynecol. Oncol. Rep. 2015, 13, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, Y.; Akiyama, N.; Tsukaguchi, T.; Fujii, T.; Satoh, Y.; Ishii, N.; Aoki, M. Mechanism of Oncogenic Signal Activation by the Novel Fusion Kinase FGFR3-BAIAP2L1. Mol. Cancer Ther. 2015, 14, 704–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar-Sinha, C.; Kalyana-Sundaram, S.; Chinnaiyan, A.M. Landscape of gene fusions in epithelial cancers: Seq and ye shall find. Genome Med. 2015, 7, 129. [Google Scholar] [CrossRef] [Green Version]
- Stam, K.; Heisterkamp, N.; Grosveld, G.; de Klein, A.; Verma, R.S.; Coleman, M.; Dosik, H.; Groffen, J. Evidence of a new chimeric bcr/c-abl mRNA in patients with chronic myelocytic leukemia and the Philadelphia chromosome. N. Engl. J. Med. 1985, 313, 1429–1433. [Google Scholar] [CrossRef]
- Speicher, M.R.; Carter, N.P. The new cytogenetics: Blurring the boundaries with molecular biology. Nat. Rev. Genet. 2005, 6, 782–792. [Google Scholar] [CrossRef]
- Cheng, L.; Zhang, S.; Wang, L.; MacLennan, G.T.; Davidson, D.D. Fluorescence in situ hybridization in surgical pathology: Principles and applications. J. Pathol. Clin. Res. 2017, 3, 73–99. [Google Scholar] [CrossRef] [Green Version]
- Kurobe, M.; Kojima, T.; Nishimura, K.; Kandori, S.; Kawahara, T.; Yoshino, T.; Ueno, S.; Iizumi, Y.; Mitsuzuka, K.; Arai, Y.; et al. Development of RNA-FISH Assay for Detection of Oncogenic FGFR3-TACC3 Fusion Genes in FFPE Samples. PLoS ONE 2016, 11, e0165109. [Google Scholar] [CrossRef]
- Yang, L.; Lee, M.S.; Lu, H.; Oh, D.Y.; Kim, Y.J.; Park, D.; Park, G.; Ren, X.; Bristow, C.A.; Haseley, P.S.; et al. Analyzing Somatic Genome Rearrangements in Human Cancers by Using Whole-Exome Sequencing. Am. J. Hum. Genet. 2016, 98, 843–856. [Google Scholar] [CrossRef] [Green Version]
- Yoshihara, K.; Wang, Q.; Torres-Garcia, W.; Zheng, S.; Vegesna, R.; Kim, H.; Verhaak, R.G. The landscape and therapeutic relevance of cancer-associated transcript fusions. Oncogene 2015, 34, 4845–4854. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Z.; Liebers, M.; Zhelyazkova, B.; Cao, Y.; Panditi, D.; Lynch, K.D.; Chen, J.; Robinson, H.E.; Shim, H.S.; Chmielecki, J.; et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat. Med. 2014, 20, 1479–1484. [Google Scholar] [CrossRef] [PubMed]
- Kongpetch, S.; Jusakul, A.; Lim, J.Q.; Ng, C.C.Y.; Chan, J.Y.; Rajasegaran, V.; Lim, T.H.; Lim, K.H.; Choo, S.P.; Dima, S.; et al. Lack of Targetable FGFR2 Fusions in Endemic Fluke-Associated Cholangiocarcinoma. JCO Glob. Oncol. 2020, 6, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.P.; Barr Fritcher, E.G.; Pestova, E.; Schulz, J.; Sitailo, L.A.; Vasmatzis, G.; Murphy, S.J.; McWilliams, R.R.; Hart, S.N.; Halling, K.C.; et al. Fibroblast growth factor receptor 2 translocations in intrahepatic cholangiocarcinoma. Hum. Pathol. 2014, 45, 1630–1638. [Google Scholar] [CrossRef] [PubMed]
- Churi, C.R.; Shroff, R.; Wang, Y.; Rashid, A.; Kang, H.C.; Weatherly, J.; Zuo, M.; Zinner, R.; Hong, D.; Meric-Bernstam, F.; et al. Mutation profiling in cholangiocarcinoma: Prognostic and therapeutic implications. PLoS ONE 2014, 9, e115383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, R.R.; Morganroth, J. Update on Cardiovascular Safety of Tyrosine Kinase Inhibitors: With a Special Focus on QT Interval, Left Ventricular Dysfunction and Overall Risk/Benefit. Drug Saf. 2015, 38, 693–710. [Google Scholar] [CrossRef]
- Konecny, G.E.; Finkler, N.; Garcia, A.A.; Lorusso, D.; Lee, P.S.; Rocconi, R.P.; Fong, P.C.; Squires, M.; Mishra, K.; Upalawanna, A.; et al. Second-line dovitinib (TKI258) in patients with FGFR2-mutated or FGFR2-non-mutated advanced or metastatic endometrial cancer: A non-randomised, open-label, two-group, two-stage, phase 2 study. Lancet Oncol. 2015, 16, 686–694. [Google Scholar] [CrossRef] [Green Version]
- Hahn, N.M.; Bivalacqua, T.J.; Ross, A.E.; Netto, G.J.; Baras, A.; Park, J.C.; Chapman, C.; Masterson, T.A.; Koch, M.O.; Bihrle, R.; et al. A Phase II Trial of Dovitinib in BCG-Unresponsive Urothelial Carcinoma with FGFR3 Mutations or Overexpression: Hoosier Cancer Research Network Trial HCRN 12-157. Clin. Cancer Res. 2017, 23, 3003–3011. [Google Scholar] [CrossRef] [Green Version]
- Mazzaferro, V.; El-Rayes, B.F.; Droz Dit Busset, M.; Cotsoglou, C.; Harris, W.P.; Damjanov, N.; Masi, G.; Rimassa, L.; Personeni, N.; Braiteh, F.; et al. Derazantinib (ARQ 087) in advanced or inoperable FGFR2 gene fusion-positive intrahepatic cholangiocarcinoma. Br. J. Cancer 2019, 120, 165–171. [Google Scholar] [CrossRef] [Green Version]
- Soria, J.C.; DeBraud, F.; Bahleda, R.; Adamo, B.; Andre, F.; Dientsmann, R.; Delmonte, A.; Cereda, R.; Isaacson, J.; Litten, J.; et al. Phase I/IIa study evaluating the safety, efficacy, pharmacokinetics, and pharmacodynamics of lucitanib in advanced solid tumors. Ann. Oncol. 2014, 25, 2244–2251. [Google Scholar] [CrossRef]
- Loriot, Y.; Necchi, A.; Park, S.H.; Garcia-Donas, J.; Huddart, R.; Burgess, E.; Fleming, M.; Rezazadeh, A.; Mellado, B.; Varlamov, S.; et al. Erdafitinib in Locally Advanced or Metastatic Urothelial Carcinoma. N. Engl. J. Med. 2019, 381, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Sahai, V.; Hollebecque, A.; Vaccaro, G.; Melisi, D.; Al-Rajabi, R.; Paulson, A.S.; Borad, M.J.; Gallinson, D.; Murphy, A.G.; et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: A multicentre, open-label, phase 2 study. Lancet Oncol. 2020, 21, 671–684. [Google Scholar] [CrossRef]
- Chae, Y.K.; Hong, F.; Vaklavas, C.; Cheng, H.H.; Hammerman, P.; Mitchell, E.P.; Zwiebel, J.A.; Ivy, S.P.; Gray, R.J.; Li, S.; et al. Phase II Study of AZD4547 in Patients With Tumors Harboring Aberrations in the FGFR Pathway: Results From the NCI-MATCH Trial (EAY131) Subprotocol W. J. Clin. Oncol. 2020, 38, 2407–2417. [Google Scholar] [CrossRef] [PubMed]
- Javle, M.; Lowery, M.; Shroff, R.T.; Weiss, K.H.; Springfeld, C.; Borad, M.J.; Ramanathan, R.K.; Goyal, L.; Sadeghi, S.; Macarulla, T.; et al. Phase II Study of BGJ398 in Patients With FGFR-Altered Advanced Cholangiocarcinoma. J. Clin. Oncol. 2018, 36, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Kalyukina, M.; Yosaatmadja, Y.; Middleditch, M.J.; Patterson, A.V.; Smaill, J.B.; Squire, C.J. TAS-120 Cancer Target Binding: Defining Reactivity and Revealing the First Fibroblast Growth Factor Receptor 1 (FGFR1) Irreversible Structure. ChemMedChem 2019, 14, 494–500. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Arkenau, H.; Tran, B.; Bahleda, R.; Kelley, R.; Hierro, C.; Ahn, D.; Zhu, A.; Javle, M.; Winkler, R.; et al. Efficacy of TAS-120, an irreversible fibroblast growth factor receptor (FGFR) inhibitor, in cholangiocarcinoma patients with FGFR pathway alterations who were previously treated with chemotherapy and other FGFR inhibitors. Ann. Oncol. 2018, 29, v100. [Google Scholar] [CrossRef]
- Lamarca, A.; Barriuso, J.; McNamara, M.G.; Valle, J.W. Molecular targeted therapies: Ready for “prime time” in biliary tract cancer. J. Hepatol. 2020, 73, 170–185. [Google Scholar] [CrossRef] [Green Version]
- Normanno, N.; Cervantes, A.; Ciardiello, F.; De Luca, A.; Pinto, C. The liquid biopsy in the management of colorectal cancer patients: Current applications and future scenarios. Cancer Treat. Rev. 2018, 70, 1–8. [Google Scholar] [CrossRef]
- Heitzer, E.; Haque, I.S.; Roberts, C.E.S.; Speicher, M.R. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet. 2019, 20, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Goyal, L.; Saha, S.K.; Liu, L.Y.; Siravegna, G.; Leshchiner, I.; Ahronian, L.G.; Lennerz, J.K.; Vu, P.; Deshpande, V.; Kambadakone, A.; et al. Polyclonal Secondary FGFR2 Mutations Drive Acquired Resistance to FGFR Inhibition in Patients with FGFR2 Fusion-Positive Cholangiocarcinoma. Cancer Discov. 2017, 7, 252–263. [Google Scholar] [CrossRef] [Green Version]
- Goyal, L.; Shi, L.; Liu, L.Y.; Fece de la Cruz, F.; Lennerz, J.K.; Raghavan, S.; Leschiner, I.; Elagina, L.; Siravegna, G.; Ng, R.W.S.; et al. TAS-120 Overcomes Resistance to ATP-Competitive FGFR Inhibitors in Patients with FGFR2 Fusion-Positive Intrahepatic Cholangiocarcinoma. Cancer Discov. 2019, 9, 1064–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Gene | 5′-Gene | 3′-Gene | Tumor Type | No. of Cases Reported (Ref.) |
|---|---|---|---|---|
| FGFR1 | FGFR1 | HOOK3 | GIST | 1/186 [25] |
| FGFR1 | TACC1 | GIST | 1/186 [25] | |
| Glioma | 1/795 [26] | |||
| Glioblastoma | 1/97 [27] | |||
| FGFR1 | ZNF703 | Breast cancer | 1/24 [28] | |
| FGFR1 | NTM | Bladder urothelial carcinoma | 1/295 [29] | |
| BAG4 | FGFR1 | Non-small cell lung cancer | 2/1328 [30]; 1/26,054 [31] | |
| FGFR2 | FGFR2 | AHCYL | Cholangiocarcinoma | 7/102 [32] |
| FGFR2 | BICC1 | Cholangiocarcinoma | 2/102 [32]; 6/195 [34]; 8/377 [35]; 40/107 [37]; | |
| Colorectal cancer | 1/149 [32]; | |||
| Hepatocarcinoma | 1/96 [32] | |||
| FGFR2 | PPHLN1 | Cholangiocarcinoma | 16/107 [37] | |
| FGFR2 | TACC3 | Cholangiocarcinoma | 1/6 [36] | |
| FGFR2 | CCDC6 | Cholangiocarcinoma | 3/377 [35] | |
| FGFR2 | KIAA1598 | Non-small cell lung cancer | 2/26054 [31] | |
| FGFR3 | FGFR3 | TACC3 | Glioblastoma | 2/97 [27] |
| Glioma | 20/795 [26] | |||
| Non-small cell lung cancer | 15/1328 [30]; 37/26,054 [31] | |||
| Bladder cancer | 3/2375 [38] | |||
| Head and neck squamous cancer | 2/2375 [38] | |||
| Lung squamous cell carcinoma | 4/2375 [38] | |||
| FGFR3 | BAIAP2L1 | Bladder cancer | 1/2375 [38]; 2/46 [44] | |
| Lung cancer | 2/83 [44] |
| Technology | Kit | Sample | Nucleic Acid | Input | No. of Genes | No. of Fusion Genes |
|---|---|---|---|---|---|---|
| Hybrid Capture-based | FoundationOneCDx (Foundation Medicine) | FFPE | DNA | Moderate (≥50 ng FFPE RNA) | 324 | 36, including FGFR1–3 |
| TruSight Tumor 170 (Illumina) | FFPE | DNA/RNA | Moderate (≥40 ng FFPE DNA/RNA) | 170 | 55, including FGFR1–4 | |
| TruSight Oncology 500 (Illumina) | FFPE | DNA/RNA | Moderate (≥40 ng FFPE DNA/RNA) | 523 | 55, including FGFR1–4 | |
| Amplicon-based | Oncomine comprehensive assay (Thermofisher) | FFPE | DNA/RNA | Low (≥10 ng FFPE DNA/RNA) | 161 | 51, including FGFR1–3 |
| Oncomine Focus Assay (Thermofisher) | FFPE | DNA/RNA | Low (≥10 ng FFPE DNA/RNA) | 52 | 23, including FGFR1–3 | |
| Anchored multiplex PCR-based | FusionPlex Oncology Research(ArcherDX) | Fresh, frozen, and FFPE | RNA | Moderate (≥50 ng § FFPE RNA) | 75 | 75¥, including FGFR1–3 |
| FusionPlex Solid Tumor (ArcherDX) | Fresh, frozen, and FFPE | RNA | Moderate (≥50 ng § FFPE RNA) | 53 | 53 ¥, including FGFR1–3 | |
| FusionPlex Comprehensive Thyroid and Lung (CTL) (ArcherDX) | Fresh, frozen, and FFPE | RNA | Moderate (≥50 ng § FFPE RNA) | 36 | 16 ¥, including FGFR1–3 | |
| FusionPlex Lung (ArcherDX) | Fresh, frozen, and FFPE | RNA | Moderate (≥50 ng § FFPE RNA) | 14 | 13 ¥, including FGFR1–3 |
| Compound | Target | Eligibility on the Basis of FGFR Alterations | Tumor Type | Phase | ClinicalTrial Identifier |
|---|---|---|---|---|---|
| Dovitinib | FGFR1–2–3; VEGFR1–2–3; PDGFRβ | FGFR3 mutation/over-expression | BCG refractory urothelial carcinoma | II | NCT01732107 |
| FGFR mutation/ amplification/ translocation | Solid and hematologic tumors | II | NCT01831726 | ||
| FGFR2 amplification | Metastatic or unresectable gastric cancer | II | NCT01719549 | ||
| FGFR2 mutation or FGFR2 wild type | Advanced and/or metastatic endometrial cancer | II | NCT01379534 | ||
| Lucitanib | FGFR1–2–3; VEGFR 1–2–3; PDGFRα-β; CSF1R | FGFR 1–3 gene fusion/activating mutation | Advanced/metastatic lung cancer | II | NCT02109016 |
| FGFR aberrations | Advanced cancers | II | NCT02747797 | ||
| FGFR1 amplification or FGFR1 wild type | Estrogen receptor-positive metastatic breast cancer | II | NCT02053636 | ||
| Nintedanib | FGFR1–2–3; VEGFR 1–2–3; PDGFRα-β | FGFR 1–3 alterations | Advanced non-small cell lung cancer | II | NCT02299141 |
| FGFR3 mutation/ overexpression or FGFR3 wild type | Advanced urothelial carcinoma | II | NCT02278978 | ||
| Ponatinib | FGFR1, VEGFR2; BCR–ABL, SRC; KIT; PDGFRα | FGFR mutation/ fusion/amplification | Advanced cancers | II | NCT02272998 |
| FGFR2 fusion | Advanced biliary cancer | II | NCT02265341 | ||
| Derazantinib | FGFR1–3, CSF1R, RET; KIT; PDGFRβ | FGFR aberrations | Advanced urothelial cancer | I/II | NCT04045613 |
| FGFR genetic alterations | Advanced solid tumors | I/II | NCT01752920 | ||
| FGFR2 fusion/ mutation/amplification | Inoperable or advanced intrahepatic cholangiocarcinoma | II | NCT03230318 |
| Drug | Target | Tumor Type | Phase | Status | Clinical Trial Identifier |
|---|---|---|---|---|---|
| Reversible FGFR inhibitors | |||||
| Erdafitinib (JNJ-42756493) | FGFR1–4 | FGFRaberrant advanced refractory solid tumors, lymphomas, or multiple myeloma | II | recruiting | NCT02465060 |
| FGFR-aberrant urothelial cancer | II | active | NCT02365597 | ||
| FGFR-aberrant advanced squamous non-small-cell lung carcinoma | II | recruiting | NCT03827850 | ||
| FGFR-aberrant urothelial cancer | III | recruiting | NCT03390504 | ||
| ER+/HER2-/FGFR-amplified metastatic breast cancer | I | recruiting | NCT03238196 | ||
| FGFR-aberrant advanced non-small-cell lung cancer, urothelial cancer, esophageal cancer, or cholangiocarcinoma | II | recruiting | NCT02699606 | ||
| Advanced solid tumor with FGFR mutation or gene fusion | II | recruiting | NCT04083976 | ||
| Pemigatinib (INCB054828) | FGFR1–3 | FGFR-aberrant advanced solid malignancies | I/II | recruiting | NCT02393248 |
| FGFR3 mutant or rearranged metastatic or unresectable urothelial carcinoma | II | recruiting | NCT04003610 | ||
| FGFR2 rearranged unresectable or metastatic cholangiocarcinoma | III | recruiting | NCT03656536 | ||
| FGFR-aberrant unresectable advanced, relapsed, or metastatic solid tumors | I | not yet recruiting | NCT04258527 | ||
| FGFR-aberrant metastatic or unresectable colorectal cancer | II | not yet recruiting | NCT04096417 | ||
| FGFR-aberrant metastatic or surgically unresectable urothelial carcinoma | II | recruiting | NCT02872714 | ||
| Locally advanced/metastatic or surgically unresectable solid tumor malignancies with activating FGFR mutations or translocations | II | recruiting | NCT03822117 | ||
| High-risk patients with urothelial carcinoma with activating FGFR mutations or translocations | II | not yet recruiting | NCT04294277 | ||
| Infigratinib (BGJ398) | FGFR1–3 | Advanced or metastatic cholangiocarcinoma with FGFR2 gene fusions or translocations or other FGFR genetic alterations | II | recruiting | NCT02150967 |
| Invasive urothelial carcinoma and FGFR3 genetic alterations | III | recruiting | NCT04197986 | ||
| Advanced or metastatic solid tumors with FGFR1–3 gene fusions or other FGFR genetic alterations | II | Recruiting | NCT04233567 | ||
| Unresectable locally advanced or metastatic cholangiocarcinoma with FGFR2 gene fusions/translocations | III | recruiting | NCT03773302 | ||
| Recurrent high-grade glioma with FGFR1 K656E or FGFR3 K650E mutation or FGFR3–TACC3 translocation | I | recruiting | NCT04424966 | ||
| Rogaratinib (BAY1163877) | FGFR1–4 | Recurrent or metastatic squamous cell carcinoma of the head and neck with FGFR1/2/3 mRNA overexpression | II | recruiting | NCT03088059 |
| Metastatic gastric cancer with FGFR mutation/fusion | II | not yet recruiting | NCT04077255 | ||
| FGFR-positive locally advanced or metastatic urothelial carcinoma | II/III | active, not recruiting | NCT03410693 | ||
| FGFR1–3-positive advanced squamous-cell non-small cell lung cancer | II | recruiting | NCT03762122 | ||
| AZD4547 | FGFR1–3 | FGFR-aberrant advanced refractory solid tumors, lymphomas, or multiple myeloma | II | recruiting | NCT02465060 |
| Muscle-invasive bladder cancer with FGFR mutations/fusions | I | active not recruiting | NCT02546661 | ||
| ER+ breast cancer patients with FGFR1 polysomy (FISH4/5) or gene amplification | I/II | completed | NCT01202591 | ||
| FGFR-aberrant squamous cell lung cancer | II/III | active not recruiting | NCT02965378 | ||
| FGFR1-amplified squamous non-small-cell lung cancer | I/II | completed | NCT01824901 | ||
| Recurrent malignant glioma expressing FGFR–TACC Gene Fusion | I/II | completed | NCT02824133 | ||
| Advanced refractory cancers/lymphomas/multiple myeloma | II | active not recruiting | NCT04439240 | ||
| DEBIO 1347 (CH5183284) | FGFR1–3 | Solid tumors harboring a fusion of FGFR1, FGFR2, or FGFR3 | II | active not recruiting | NCT03834220 |
| FGFR-amplified metastatic breast cancer | I/II | recruiting | NCT03344536 | ||
| FGFR-aberrant advanced solid tumors | I | active not recruiting | NCT01948297 | ||
| Irreversible FGFR inhibitors | |||||
| Futibatinib (TAS-120) | FGFR1–4 | FGFR-aberrant advanced or metastatic solid tumor, advanced or metastatic gastric or gastroesophageal cancer, myeloid or lymphoid neoplasms | II | not yet recruiting | NCT04189445 |
| Advanced cholangiocarcinoma harboring FGFR2 gene rearrangements | III | not yet recruiting | NCT04093362 | ||
| FGF/FGFR aberrant advanced solid tumors | I/II | active, not recruiting | NCT02052778 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Luca, A.; Esposito Abate, R.; Rachiglio, A.M.; Maiello, M.R.; Esposito, C.; Schettino, C.; Izzo, F.; Nasti, G.; Normanno, N. FGFR Fusions in Cancer: From Diagnostic Approaches to Therapeutic Intervention. Int. J. Mol. Sci. 2020, 21, 6856. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186856
De Luca A, Esposito Abate R, Rachiglio AM, Maiello MR, Esposito C, Schettino C, Izzo F, Nasti G, Normanno N. FGFR Fusions in Cancer: From Diagnostic Approaches to Therapeutic Intervention. International Journal of Molecular Sciences. 2020; 21(18):6856. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186856
Chicago/Turabian StyleDe Luca, Antonella, Riziero Esposito Abate, Anna Maria Rachiglio, Monica Rosaria Maiello, Claudia Esposito, Clorinda Schettino, Francesco Izzo, Guglielmo Nasti, and Nicola Normanno. 2020. "FGFR Fusions in Cancer: From Diagnostic Approaches to Therapeutic Intervention" International Journal of Molecular Sciences 21, no. 18: 6856. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186856