Comparative Analysis of Proteomes of a Number of Nosocomial Pathogens by KEGG Modules and KEGG Pathways


Abstract
:1. Introduction
2. Results and Discussion
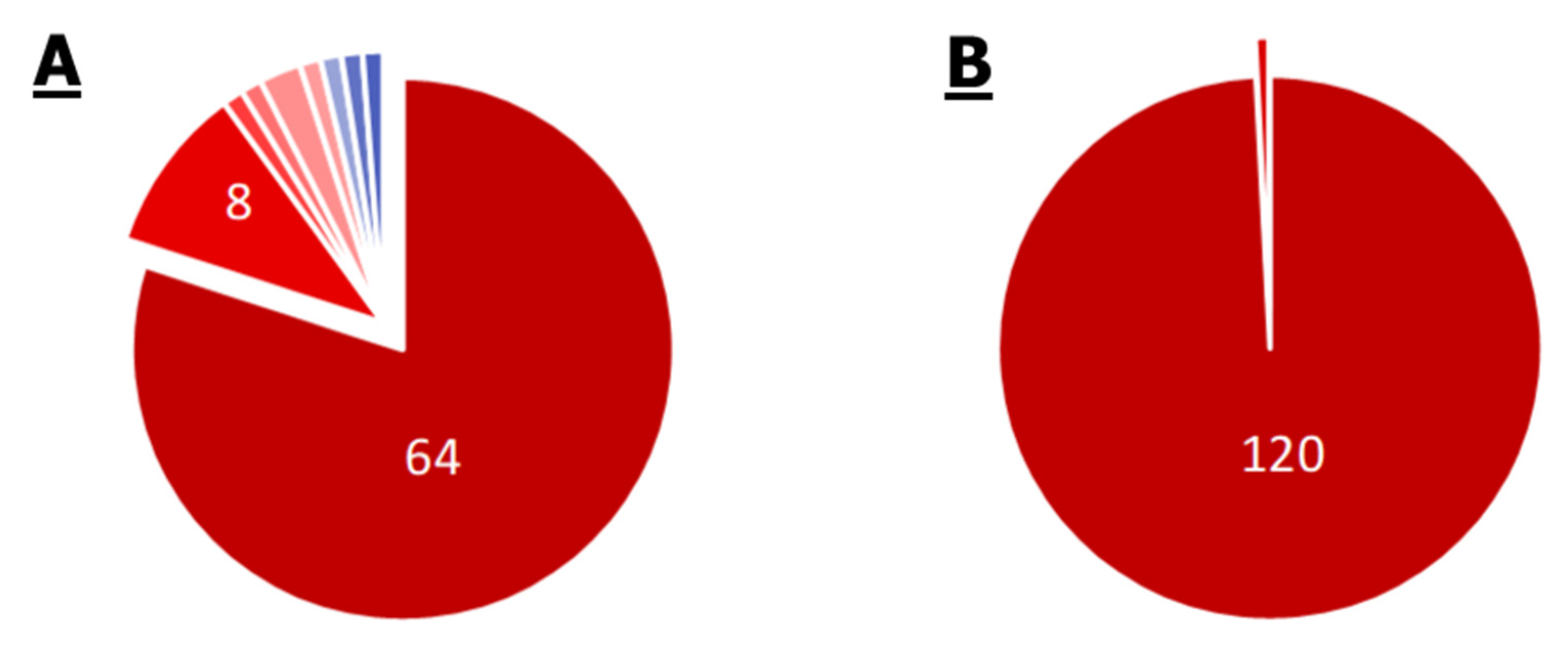
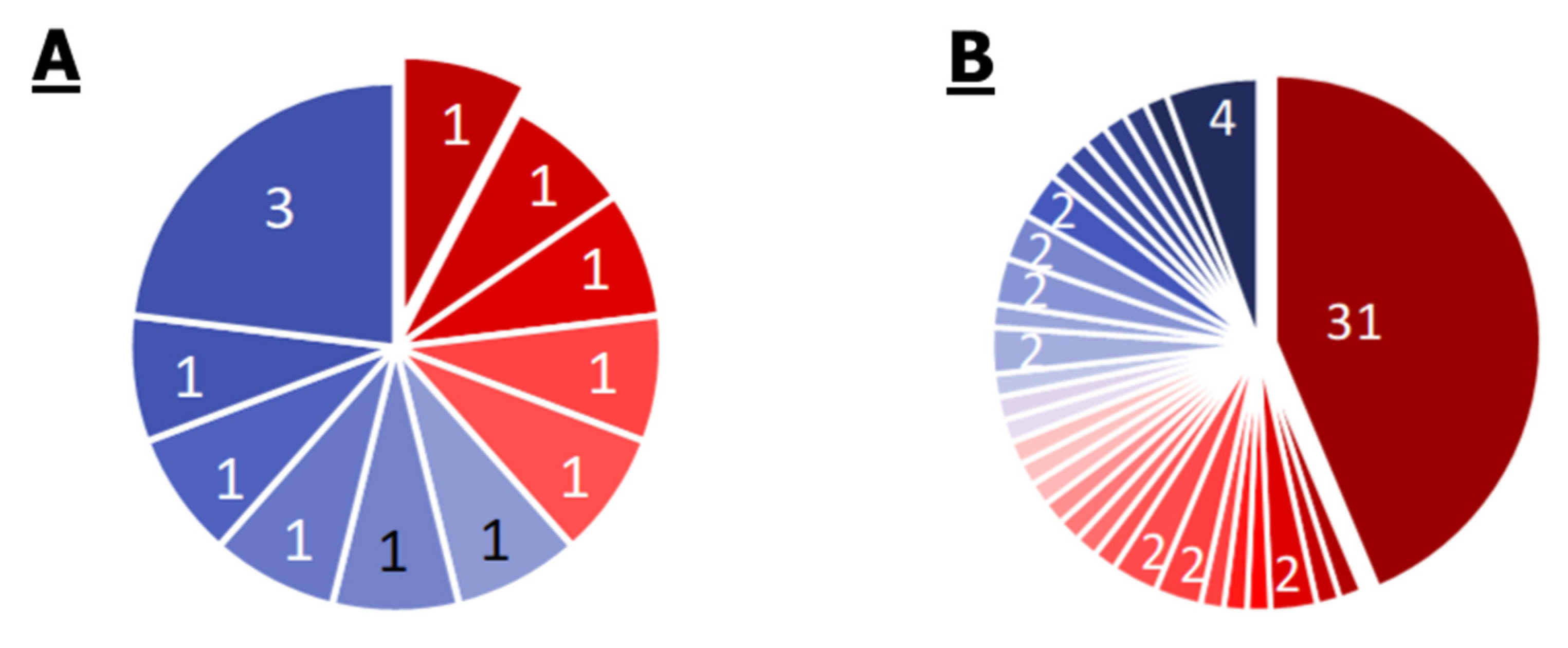
2.1. Comparison of Single Genomes of S. aureus, E. cloacae, P. aeruginosa, M. mobile
2.2. Analysis of Pangenomes and Core Genomes
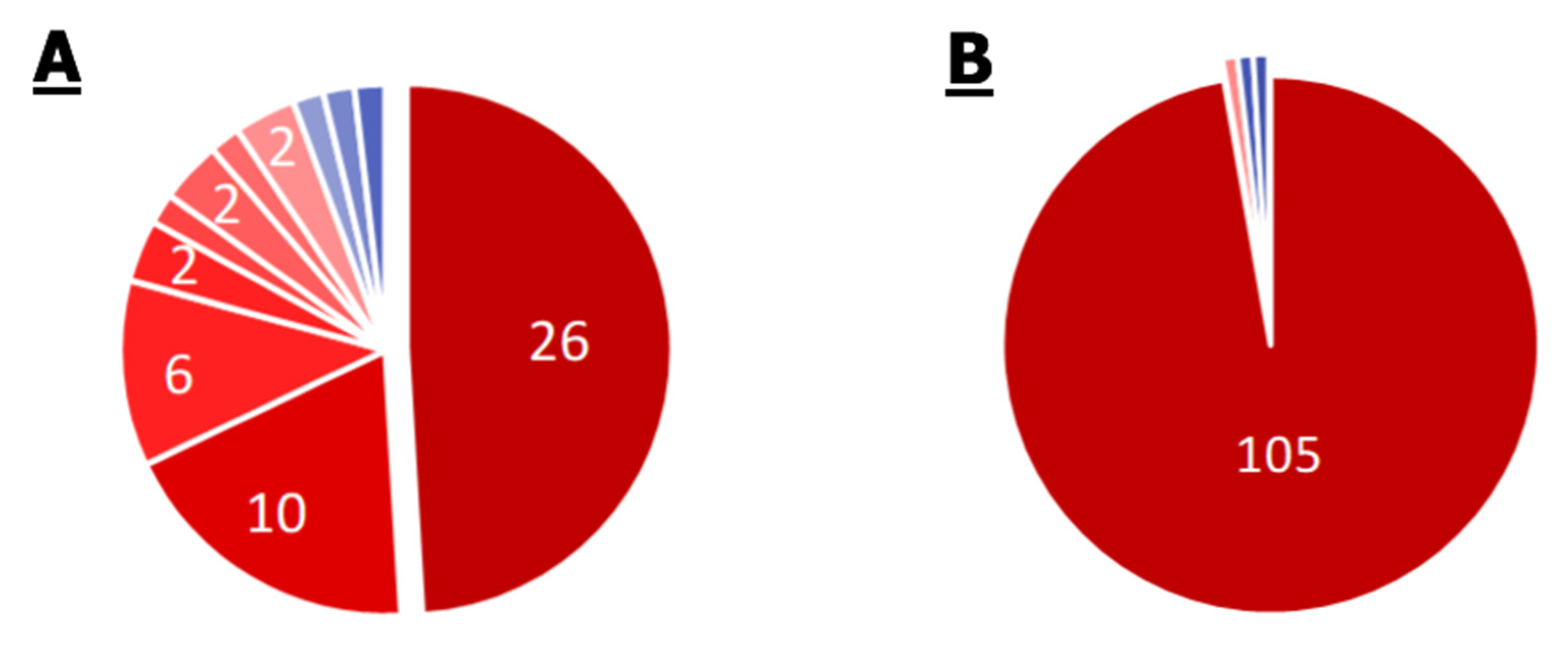
2.2.1. S. aureus
2.2.2. Enterobacter spp.
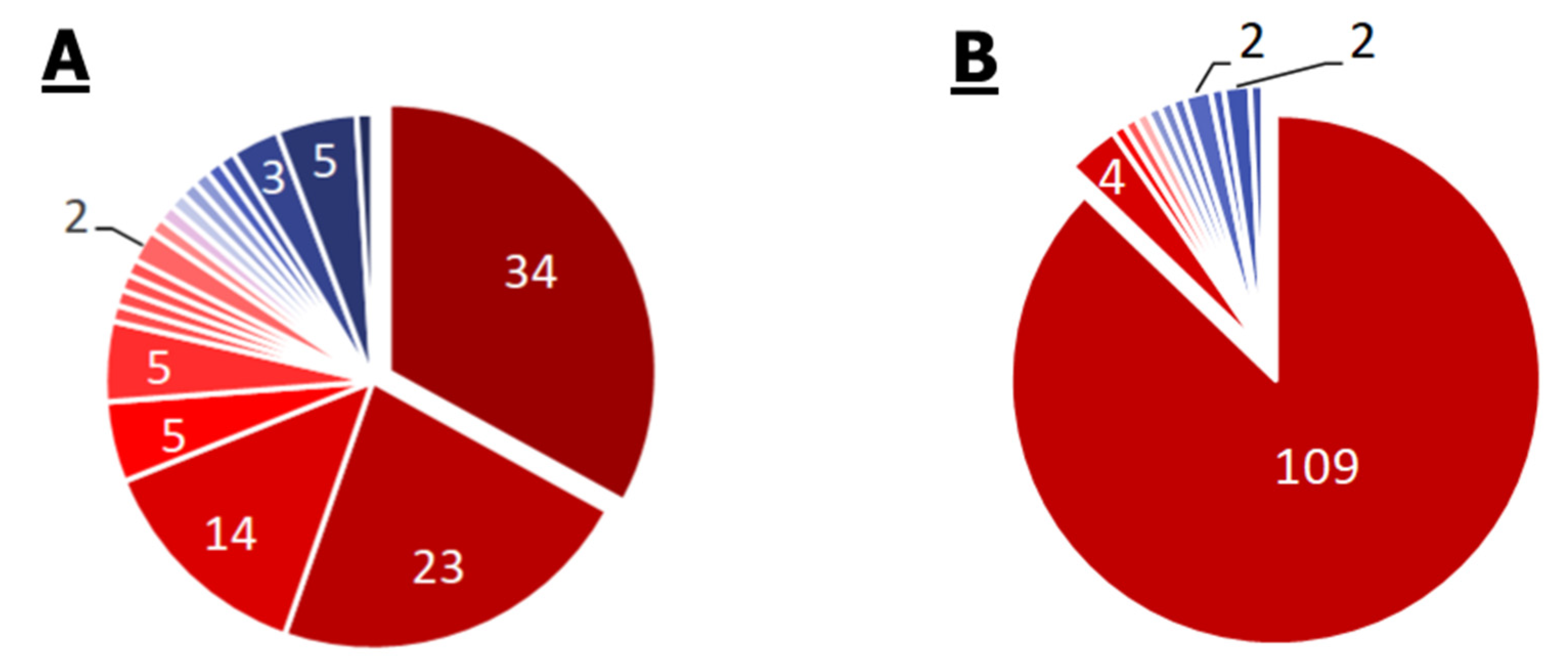
2.2.3. P. aeruginosa
2.2.4. Mycoplasma spp.
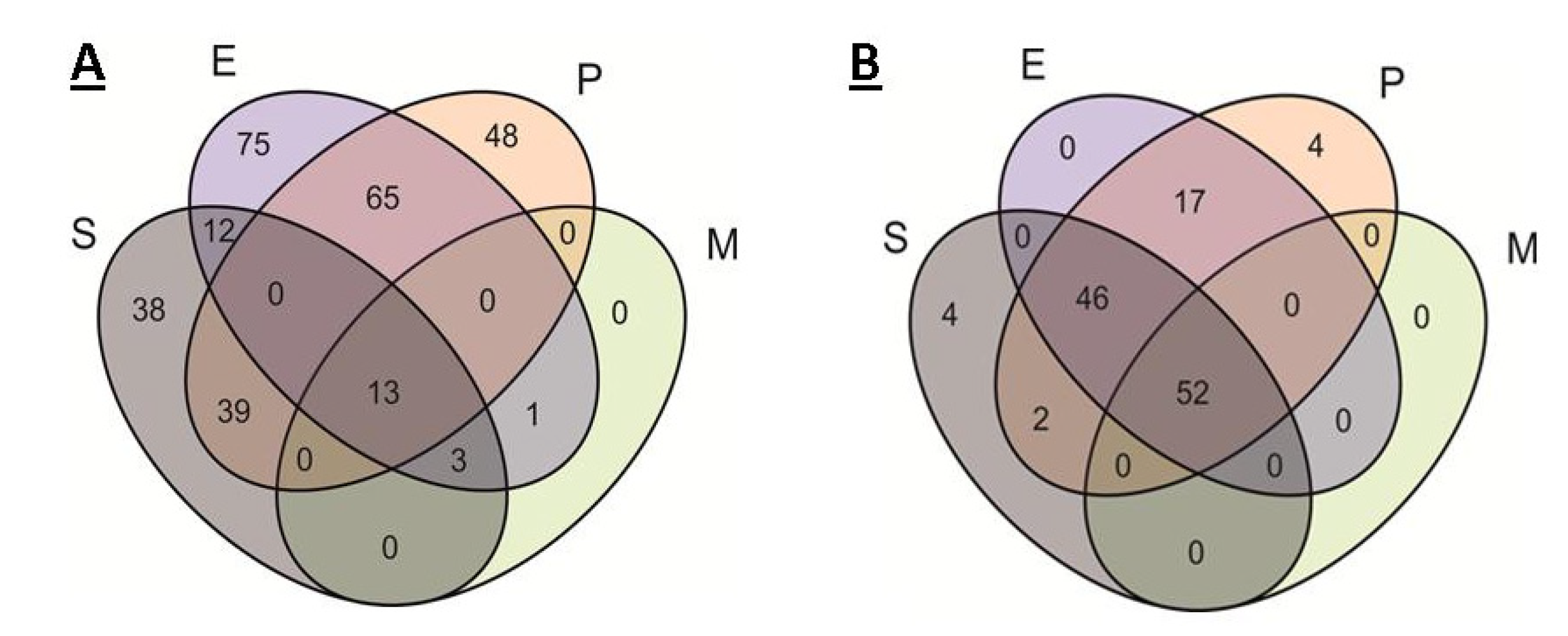
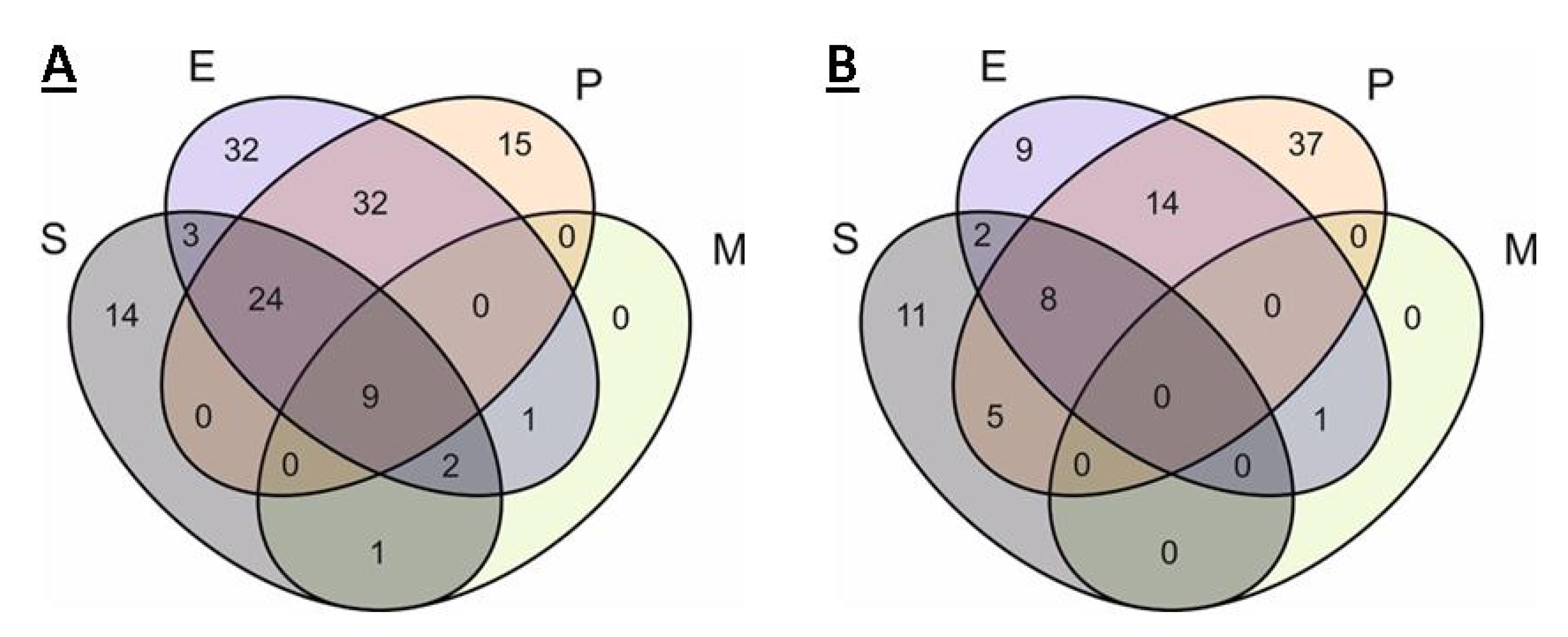
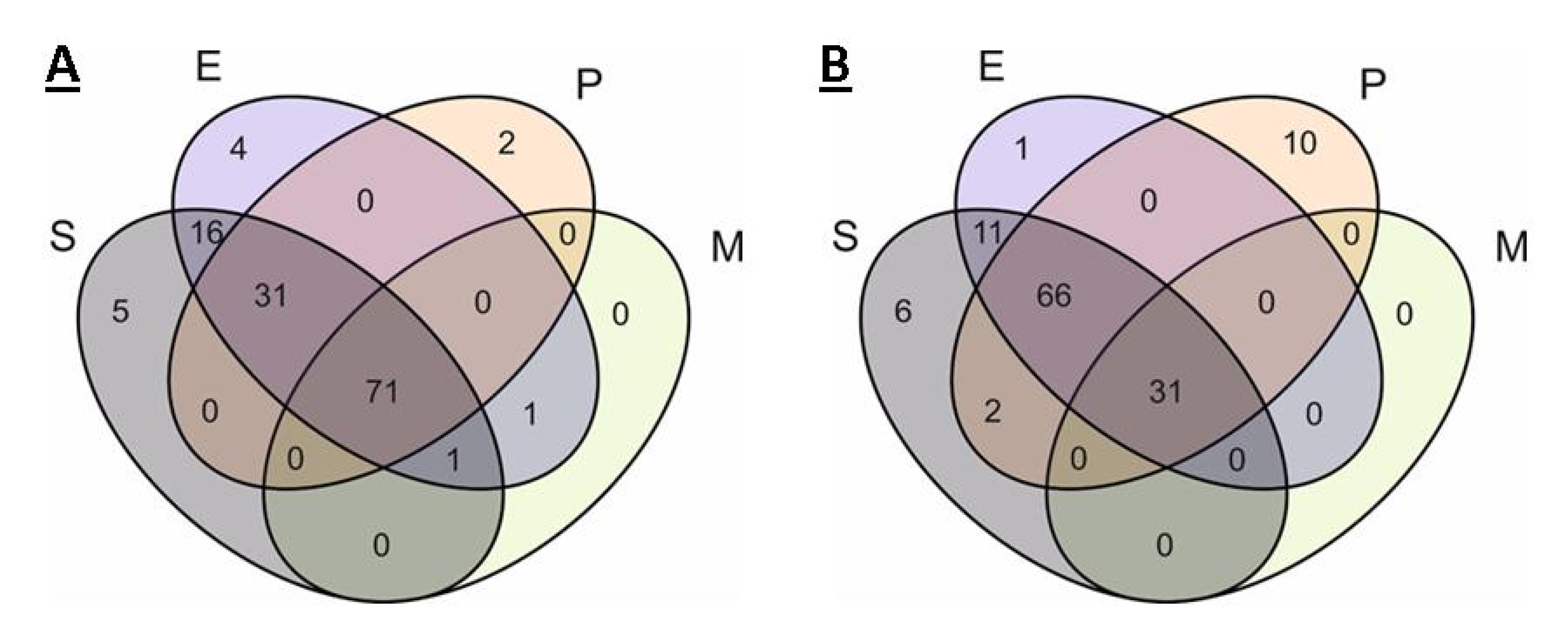
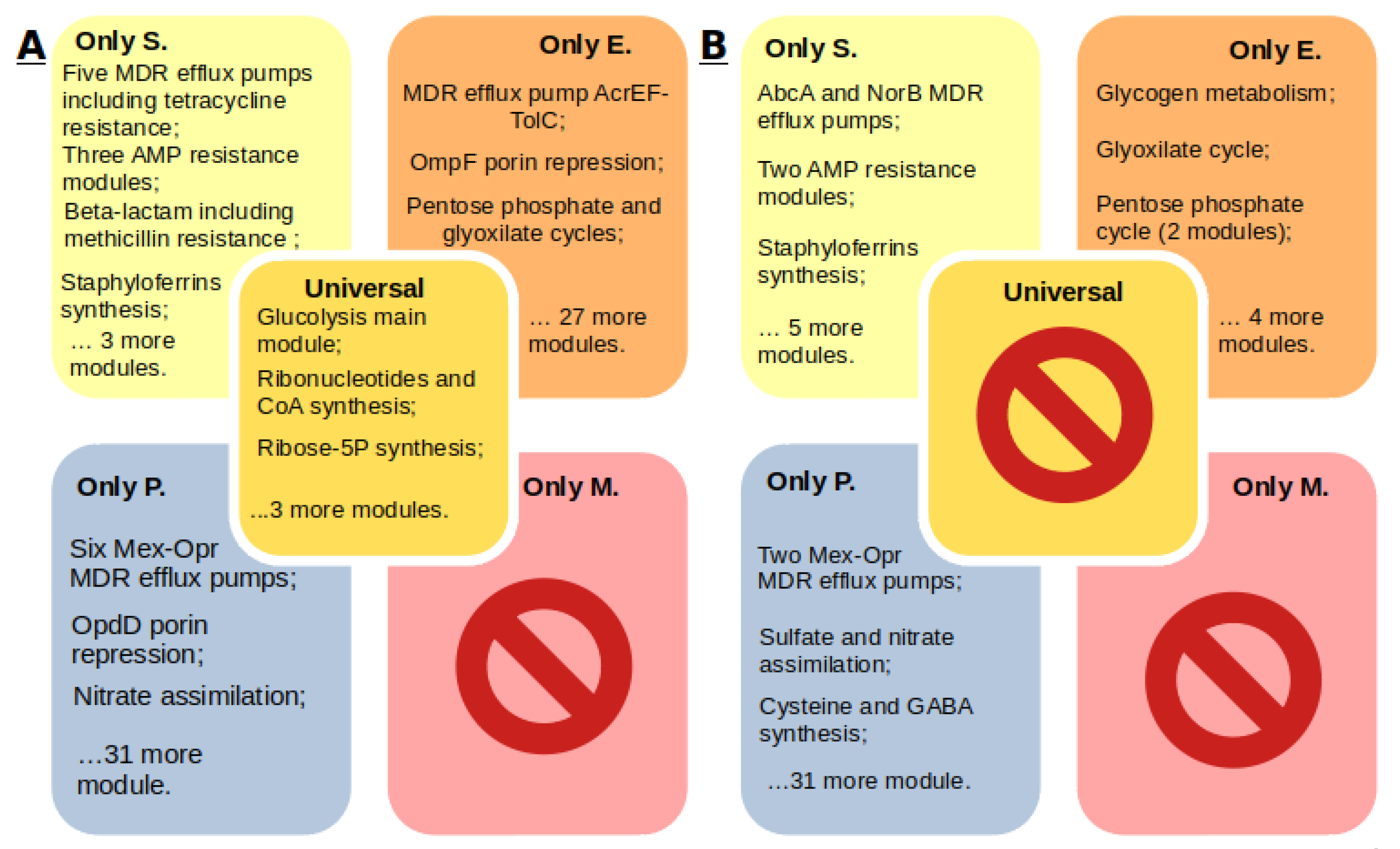
2.3. Comparison of Core Genomes and Pangenomes
3. Materials and Methods
3.1. KEGG Databases
3.2. Organisms of Interest
- Staphylococcus aureus. Reference strain NCTC 8325, 53 genomes of S. aureus were used.
- Enterobacter species. Reference strain EcWSU1, 35 genomes of Enterobacter spp. were used.
- Pseudomonas aeruginosa. Reference strain ATCC 15692, 21 genomes of P. aeruginosa were used.
- Mycoplasma species. Reference strain ATCC 43663, 92 genomes of Mycoplasma spp. were used.
3.3. Algorithm of Comparison
4. Conclusions
- (1)
- Each S. aureus strain has multidrug resistance systems, even if they are susceptible to methicillin. Twenty-five percent of the strains in the KEGG database are methicillin resistant, 85% of the strains are tetracycline resistant, 45% of the strains have the beta-lactam resistance module, and 21% of the genomes have the QacA efflux system multidrug resistance module.
- (2)
- Enterobacter spp. have a wide range of antibiotic resistance systems, however, there are strains without such systems at all. Seventy-six percent of the strains have at least one such system. Twenty-one percent of the Enterobacter spp. strains in the KEGG database are resistant to carbapenems. Seventy-four percent of the strains have the AcrEF-TolC efflux system module.
- (3)
- P. aeruginosa has a wide range of unique efflux systems. One hundred percent of the strains have at least two drug resistance systems, and 75% of the strains have seven. Resistance to carbapenems was found in 14% of the strains. The M00718 module of the MexAB-OprM efflux system, which confers multidrug resistance, and the M00745 module of the OprD repression system, conferring resistance to imipenem, were found in 20 of the 21 genomes.
- (4)
- Mycoplasma spp. do not have annotated antibiotic resistance or antimicrobial peptide resistance modules, although resistance has been proven for some Mycoplasma species. Determination of resistance mechanisms requires additional research.
- (5)
- Each of the organisms has a characteristic set of metabolic traits, whose contribution to drug resistance can be determined in future studies.
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AMP | Antimicrobial peptide |
| MDR | Multidrug resistance |
References
- Rosenthal, V.D.; Bijie, H.; Maki, D.G.; Mehta, Y.; Apisarnthanarak, A.; Medeiros, E.A.; Leblebicioglu, H.; Fisher, D.; Álvarez-Moreno, C.; Khader, I.A.; et al. International Nosocomial Infection Control Consortium (INICC) report, data summary of 36 countries, for 2004–2009. Am. J. Infect. Control 2012, 40, 396–407. [Google Scholar] [CrossRef] [PubMed]
- Boev, C.; Kiss, E. Hospital-Acquired Infections. Crit. Care Nurs. Clin. N. Am. 2017, 29, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Ventola, C.L. The antibiotic resistance crisis: Part 1: Causes and threats. Pharm. Ther. 2015, 40, 277–283. [Google Scholar]
- Wieler, L.H.; Ewers, C.; Guenther, S.; Walther, B.; Lübke-Becker, A. Methicillin-resistant staphylococci (MRS) and extended-spectrum beta-lactamases (ESBL)-producing Enterobacteriaceae in companion animals: Nosocomial infections as one reason for the rising prevalence of these potential zoonotic pathogens in clinical samples. Int. J. Med. Microbiol. 2011, 301, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Kourtis, A.P.; Hatfield, K.; Baggs, J.; Mu, Y.; See, I.; Epson, E.; Nadle, J.; Kainer, M.A.; Dumyati, G.; Petit, S.; et al. Vital Signs: Epidemiology and Recent Trends in Methicillin-Resistant and in Methicillin-Susceptible Staphylococcus aureus Bloodstream Infections—United States. MMWR Morb. Mortal. Wkly. Rep. 2019, 68, 214–219. [Google Scholar] [CrossRef] [Green Version]
- DeLeo, F.R.; Otto, M.; Kreiswirth, B.N.; Chambers, H.F. Community-associated meticillin-resistant Staphylococcus aureus. Lancet 2010, 375, 1557–1568. [Google Scholar] [CrossRef] [Green Version]
- Drougka, E.; Foka, A.; Liakopoulos, A.; Doudoulakakis, A.; Jelastopulu, E.; Chini, V.; Spiliopoulou, A.; Levidiotou, S.; Panagea, T.; Vogiatzi, A.; et al. A 12-year survey of methicillin-resistant Staphylococcus aureus infections in Greece: ST80-IV epidemic? Clin. Microbiol. Infect. 2014, 20, O796–O803. [Google Scholar] [CrossRef] [Green Version]
- Kaasch, A.J.; Barlow, G.; Edgeworth, J.D.; Fowler, V.G.; Hellmich, M.; Hopkins, S.; Kern, W.V.; Llewelyn, M.J.; Rieg, S.; Rodriguez-Baño, J.; et al. Staphylococcus aureus bloodstream infection: A pooled analysis of five prospective, observational studies. J. Infect. 2014, 68, 242–251. [Google Scholar] [CrossRef] [Green Version]
- Gellatly, S.L.; Hancock, R.E.W. Pseudomonas aeruginosa: New insights into pathogenesis and host defenses. Pathog. Dis. 2013, 67, 159–173. [Google Scholar] [CrossRef] [Green Version]
- Yayan, J.; Ghebremedhin, B.; Rasche, K. Antibiotic Resistance of Pseudomonas aeruginosa in Pneumonia at a Single University Hospital Center in Germany over a 10-Year Period. PLoS ONE 2015, 10, e0139836. [Google Scholar] [CrossRef] [Green Version]
- Treepong, P.; Kos, V.N.; Guyeux, C.; Blanc, D.S.; Bertrand, X.; Valot, B.; Hocquet, D. Global emergence of the widespread Pseudomonas aeruginosa ST235 clone. Clin. Microbiol. Infect. 2018, 24, 258–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabot, G.; López-Causapé, C.; Ocampo-Sosa, A.A.; Sommer, L.M.; Domínguez, M.Á.; Zamorano, L.; Juan, C.; Tubau, F.; Rodríguez, C.; Moyà, B.; et al. Deciphering the resistome of the widespread P. aeruginosa ST175 international high-risk clone through whole genome sequencing. Antimicrob. Agents Chemother. 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, J.W.; Floyd, R.V.; Fothergill, J.L. The contribution of Pseudomonas aeruginosa virulence factors and host factors in the establishment of urinary tract infections. FEMS Microbiol. Lett. 2017, 364. [Google Scholar] [CrossRef] [PubMed]
- Davin-Regli, A.; Pagès, J.-M. Enterobacter aerogenes and Enterobacter cloacae; versatile bacterial pathogens confronting antibiotic treatment. Front. Microbiol. 2015, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malek, A.; McGlynn, K.; Taffner, S.; Fine, L.; Tesini, B.; Wang, J.; Mostafa, H.; Petry, S.; Perkins, A.; Graman, P.; et al. Next-Generation-Sequencing-Based Hospital Outbreak Investigation Yields Insight into Klebsiella aerogenes Population Structure and Determinants of Carbapenem Resistance and Pathogenicity. Antimicrob. Agents Chemother. 2019, 63, e02577-18. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Simmonds, A.; Annavajhala, M.K.; Wang, Z.; Macesic, N.; Hu, Y.; Giddins, M.J.; O’Malley, A.; Toussaint, N.C.; Whittier, S.; Torres, V.J.; et al. Genomic and Geographic Context for the Evolution of High-Risk Carbapenem-Resistant Enterobacter cloacae Complex Clones ST171 and ST78. mBio 2018, 9, e00542-18. [Google Scholar] [CrossRef] [Green Version]
- Sader, H.S.; Biedenbach, D.J.; Jones, R.N. Global patterns of susceptibility for 21 commonly utilized antimicrobial agents tested against 48,440 Enterobacteriaceae in the SENTRY Antimicrobial Surveillance Program (1997–2001). Diagn. Microbiol. Infect. Dis. 2003, 47, 361–364. [Google Scholar] [CrossRef]
- Thiolas, A.; Bollet, C.; La Scola, B.; Raoult, D.; Pagès, J.-M. Successive Emergence of Enterobacter aerogenes Strains Resistant to Imipenem and Colistin in a Patient. Antimicrob. Agents Chemother. 2005, 49, 1354–1358. [Google Scholar] [CrossRef] [Green Version]
- Band, V.I.; Crispell, E.K.; Napier, B.A.; Herrera, C.M.; Tharp, G.K.; Vavikolanu, K.; Pohl, J.; Read, T.D.; Bosinger, S.E.; Trent, M.S.; et al. Antibiotic failure mediated by a resistant subpopulation in Enterobacter cloacae. Nat. Microbiol. 2016, 1, 16053. [Google Scholar] [CrossRef]
- Elias, S.; Banin, E. Multi-species biofilms: Living with friendly neighbors. FEMS Microbiol. Rev. 2012, 36, 990–1004. [Google Scholar] [CrossRef]
- Tettelin, H.; Riley, D.; Cattuto, C.; Medini, D. Comparative genomics: The bacterial pan-genome. Curr. Opin. Microbiol. 2008, 11, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Ochman, H.; Lerat, E.; Daubin, V. Examining bacterial species under the specter of gene transfer and exchange. Proc. Natl. Acad. Sci. USA 2005, 102, 6595–6599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vernikos, G.; Medini, D.; Riley, D.R.; Tettelin, H. Ten years of pan-genome analyses. Curr. Opin. Microbiol. 2015, 23, 148–154. [Google Scholar] [CrossRef]
- Mira, A. The bacterial pan-genome: A new paradigm in microbiology. Int. Microbiol. 2010, 45–57. [Google Scholar] [CrossRef]
- Mosquera-Rendón, J.; Rada-Bravo, A.M.; Cárdenas-Brito, S.; Corredor, M.; Restrepo-Pineda, E.; Benítez-Páez, A. Pangenome-wide and molecular evolution analyses of the Pseudomonas aeruginosa species. BMC Genom. 2016, 17, 45. [Google Scholar] [CrossRef] [Green Version]
- Hassan, A.; Naz, A.; Obaid, A.; Paracha, R.Z.; Naz, K.; Awan, F.M.; Muhmmad, S.A.; Janjua, H.A.; Ahmad, J.; Ali, A. Pangenome and immuno-proteomics analysis of Acinetobacter baumannii strains revealed the core peptide vaccine targets. BMC Genom. 2016, 17, 732. [Google Scholar] [CrossRef] [Green Version]
- Pohl, P.C.; Klafke, G.M.; Carvalho, D.D.; Martins, J.R.; Daffre, S.; da Silva Vaz, I.; Masuda, A. ABC transporter efflux pumps: A defense mechanism against ivermectin in Rhipicephalus (Boophilus) microplus. Int. J. Parasitol. 2011, 41, 1323–1333. [Google Scholar] [CrossRef]
- Strange, R.E. Bacterial “Glycogen” and Survival. Nature 1968, 220, 606–607. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Genes | RNA Genes | KEGG Modules | KEGG Pathways | |
|---|---|---|---|---|
| S. aureus | 2767 | 77 | 105 | 104 |
| E. cloacae | 4619 | 108 | 208 | 118 |
| P. aeruginosa | 5572 | 106 | 165 | 121 |
| M. mobile | 635 | 34 | 17 | 52 |
| Total | Drug Resistance | Metabolic | Transport | Regulation | AMP Resistance | |
|---|---|---|---|---|---|---|
| S | 38 | 7 | 6 | 12 | 10 | 3 |
| P | 48 | 10 | 14 | 11 | 13 | 0 |
| E | 75 | 5 | 27 | 30 | 9 | 4 |
| EP | 65 | 5 | 24 | 26 | 10 | 1 |
| SE | 12 | 0 | 3 | 9 | 0 | 0 |
| EM | 1 | 0 | 1 | 0 | 0 | 0 |
| SEP | 39 | 0 | 26 | 11 | 2 | 0 |
| SEM | 3 | 0 | 1 | 2 | 0 | 0 |
| SEPM | 13 | 0 | 9 | 4 | 0 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Slizen, M.V.; Galzitskaya, O.V. Comparative Analysis of Proteomes of a Number of Nosocomial Pathogens by KEGG Modules and KEGG Pathways. Int. J. Mol. Sci. 2020, 21, 7839. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21217839
Slizen MV, Galzitskaya OV. Comparative Analysis of Proteomes of a Number of Nosocomial Pathogens by KEGG Modules and KEGG Pathways. International Journal of Molecular Sciences. 2020; 21(21):7839. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21217839
Chicago/Turabian StyleSlizen, Mikhail V., and Oxana V. Galzitskaya. 2020. "Comparative Analysis of Proteomes of a Number of Nosocomial Pathogens by KEGG Modules and KEGG Pathways" International Journal of Molecular Sciences 21, no. 21: 7839. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21217839