Effects of Rationally Designed Physico-Chemical Variants of the Peptide PuroA on Biocidal Activity towards Bacterial and Mammalian Cells

 , and
, and
Abstract
:1. Introduction
2. Results
2.1. Effects of the Altered Physico-Chemical Parameters on the Antimicrobial Activity and Stability of the Designed Peptides
2.2. Effects on Biofilm Formation of the Clinical MRSA Isolates
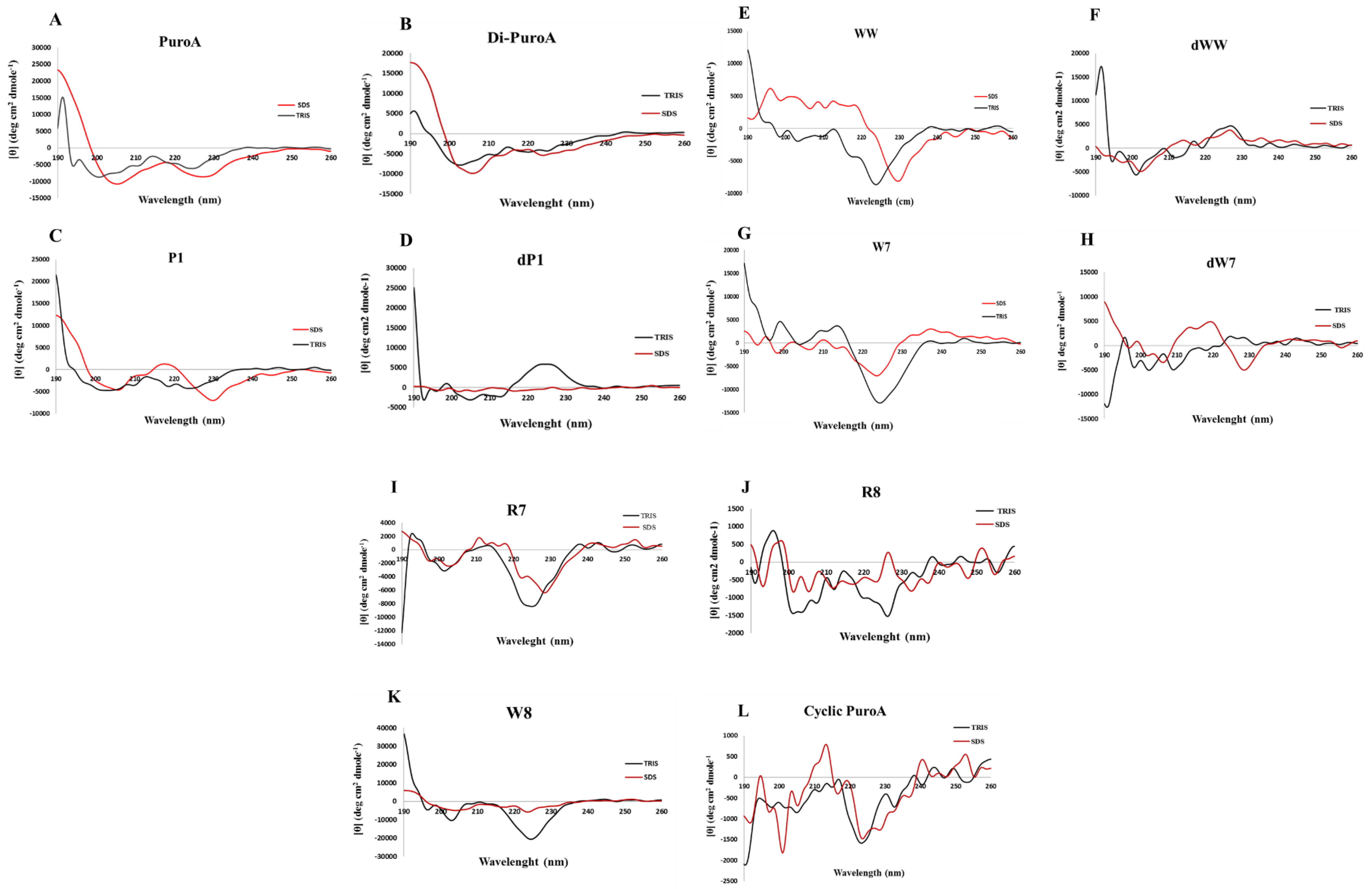
2.3. Prediction of 3D Structures and Determination of Peptide Conformations
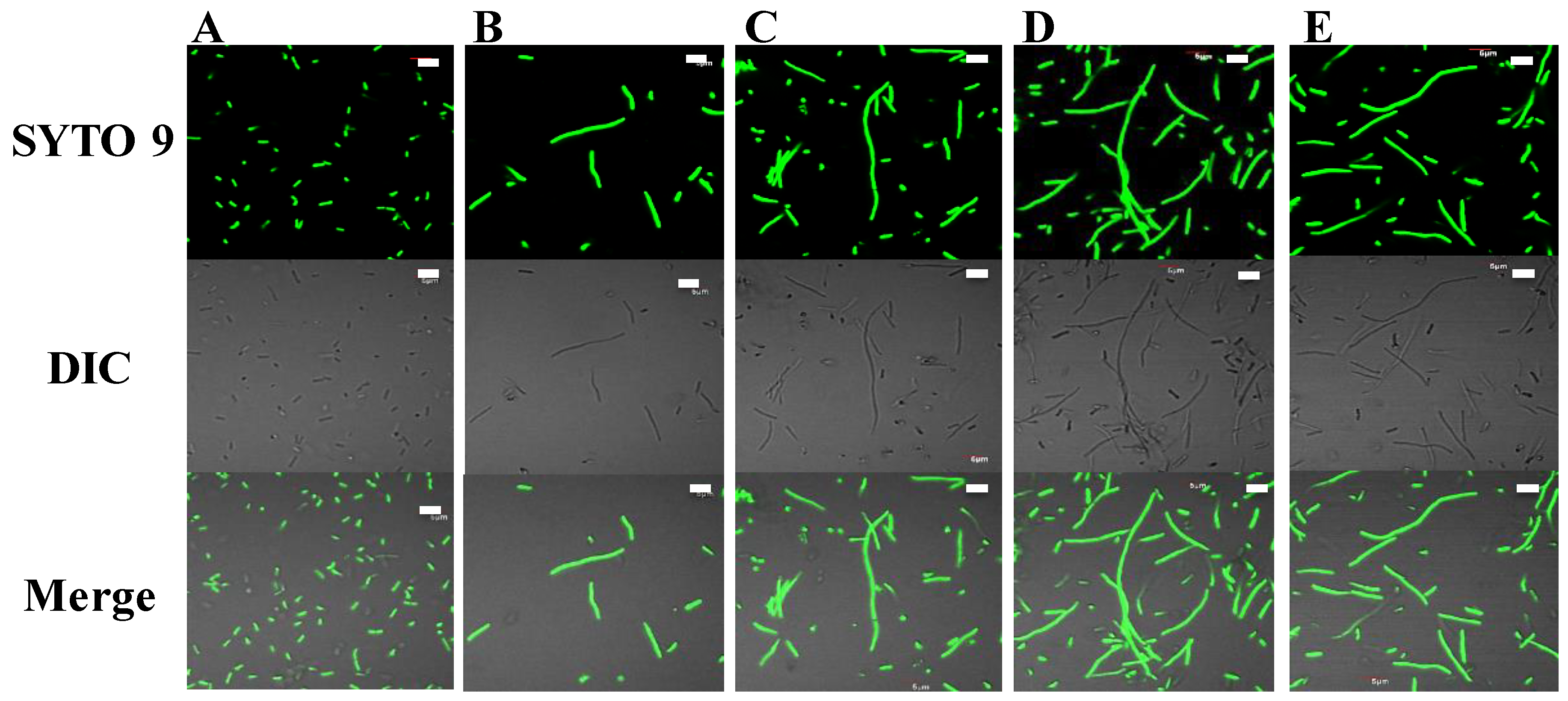
2.4. Potential DNA-Binding Ability and Intracellular Action
2.5. Haemolytic Activity and Cytotoxicity Against Mammalian Cells
3. Discussion
4. Materials and Methods
4.1. Peptide Design
4.2. Prediction of the Physicochemical Properties of Peptides
4.3. Antibacterial Activity and Salt Stability Assays
4.4. Anti-Candida Activity Assay
4.5. Proteases Stability Assay
4.6. Three Dimensional Structure Prediction
4.7. Circular Dichroism Spectroscopy
4.8. In Vitro DNA-Binding Assay
4.9. E. coli Filamentation Assay
4.10. Testing the Activity of Peptides on Biofilms of Clinical MRSA Isolate M173525
4.11. Biofilm Biomass Assay (Crystal Violet Assay)
4.12. Biofilm Metabolic Activity Assay (MTT Assay)
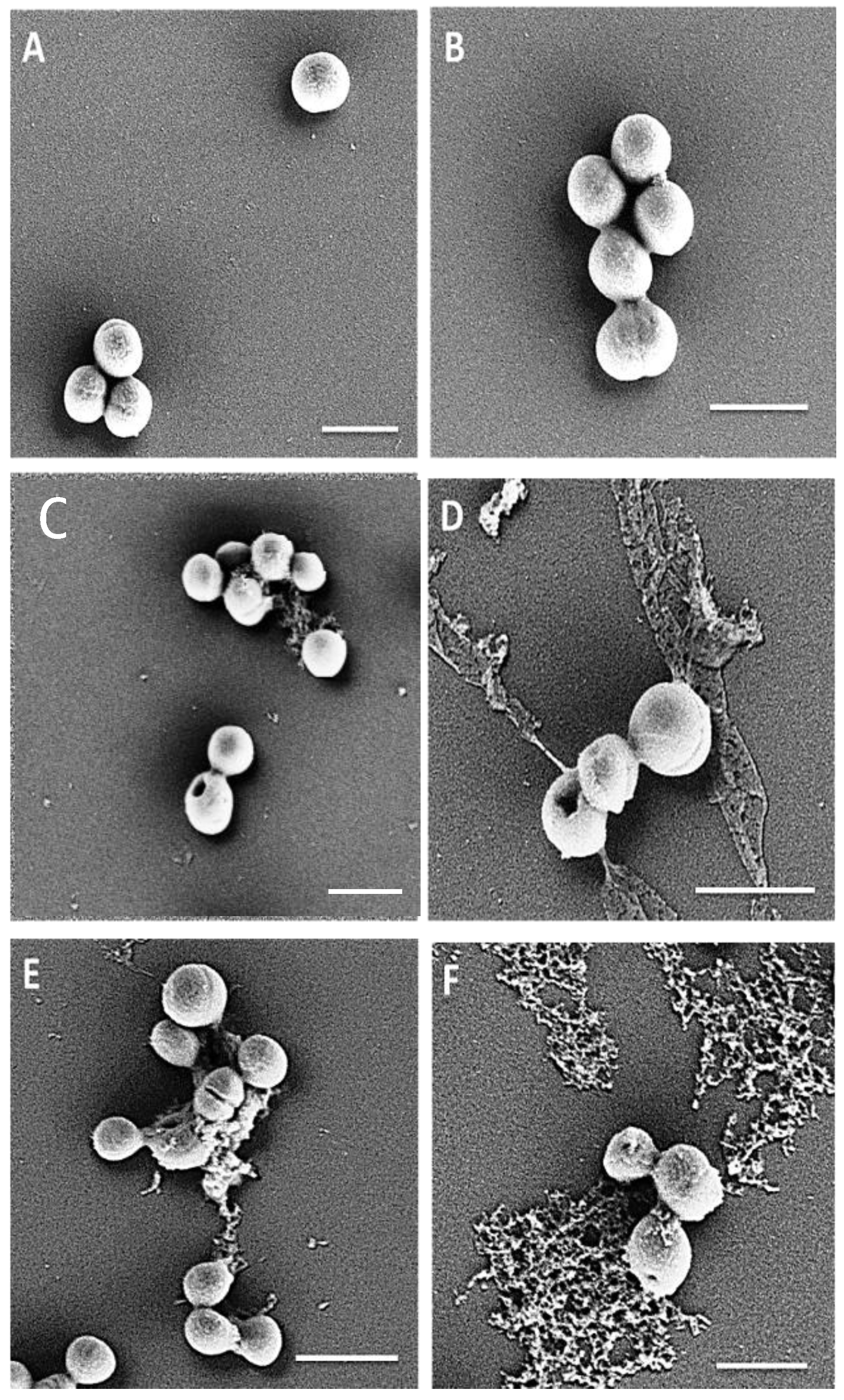
4.13. Visualization of the Effect of Peptides on Initial Adhesion of MRSA M173525
4.14. Haemolytic Activity
4.15. Cytotoxicity Against Mammalian Cells
4.16. Scanning Electron Microscopy
4.17. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AMP | antimicrobial peptide(s) |
| CD | circular dichroism |
| SEM | scanning electron microscopy |
| pI | isoelectric point |
| ChD | charge density |
| H | hydrophobicity |
| HR | hydrophobic ratio |
| µH | hydrophobic moment |
| MIC | Minimum Inhibitory Concentration |
| CV | crystal violet |
| MTT | methylthiazolyldiphenyl-tetrazolium |
References
- Hancock, R.E.W.; Sahl, H.G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Pushpanathan, M.; Gunasekaran, P.; Rajendhran, J. Antimicrobial peptides: Versatile biological properties. Int. J. Pept. 2013, 2013, 675391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nat. Cell Biol. 2002, 415, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Yeaman, M.R.; Yount, N.Y. Mechanisms of antimicrobial peptide action and resistance. Pharmacol. Rev. 2003, 55, 27–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bechinger, B.; Gorr, S.U. Antimicrobial Peptides: Mechanisms of Action and Resistance. J. Dent. Res. 2017, 96, 254–260. [Google Scholar] [CrossRef] [Green Version]
- Chan, D.I.; Prenner, E.J.; Vogel, H.J. Trytophan- and arginine-rich antimicrobial peptides: Structures and mechanisms of action. Biochim. Biophys. Acta 2006, 1758, 1184–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasupuleti, M.; Schmidtchen, A.; Malmsten, M. Antimicrobial peptides: Key components of the innate immune system. Crit. Rev. Biotechnol. 2012, 32, 143–171. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, V.; Feio, M.J.; Bastos, M. Role of lipids in the interaction of antimicrobial peptides with membranes. Prog. Lipid Res. 2012, 51, 149–177. [Google Scholar] [CrossRef]
- Shagaghi, N.; Palombo, E.A.; Clayton, A.H.A.; Bhave, M. Archetypal tryptophan-rich antimicrobial peptides: Properties and applications. World J. Microbiol. Biotechnol. 2016, 32, 31. [Google Scholar] [CrossRef]
- Schibli, D.J.; Epand, R.F.; Vogel, H.J.; Epand, R.M. Tryptophan-rich antimicrobial peptides: Comparative properties and membrane interactions. Biochem. Cell Biol. 2002, 80, 667–677. [Google Scholar] [CrossRef]
- Gautier, M.-F.; Aleman, M.-E.; Guirao, A.; Marion, D.; Joudrier, P. Triticum aestivum puroindolines, two basic cystine-rich seed proteins: cDNA sequence analysis and developmental gene expression. Plant Mol. Biol. 1994, 25, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Bhave, M.; Morris, C.F. Molecular genetics of puroindolines and related genes: Regulation of expression, membrane binding properties and applications. Plant Mol. Biol. 2008, 66, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Jing, W.; Demcoe, A.R.; Vogel, H.J. Conformation of a bactericidal domain of puroindoline a: Structure and mechanism of action of a 13-residue antimicrobial peptide. J. Bacteriol. 2003, 185, 4938–4947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, R.L.; Palombo, E.A.; Panozzo, J.F.; Bhave, M. Puroindolines, Pin alleles, hordoindolines and grain softness proteins are sources of bactericidal and fungicidal peptides. J. Cereal Sci. 2011, 53, 112–117. [Google Scholar] [CrossRef]
- Alfred, R.L.; Palombo, E.A.; Panozzo, J.F.; Bariana, H.; Bhave, M. Stability of puroindoline peptides and effects on wheat rust. World J. Microbiol. Biotechnol. 2013, 29, 1409–1419. [Google Scholar] [CrossRef]
- Vogel, H.J.; Schibli, D.J.; Jing, W.; Lohmeier-Vogel, E.M.; Epand, R.F.; Epand, R.M. Towards a structure-function analysis of bovine lactoferricin and related tryptophan- and arginine-containing peptides. Biochem. Cell Biol. 2002, 80, 49–63. [Google Scholar] [CrossRef]
- Staubitz, P.; Peschel, A.; Nieuwenhuizen, W.F.; Otto, M.; Götz, F.; Jung, G.; Jack, R.W. Structure-function relationships in the tryptophan-rich, antimicrobial peptide indolicidin. J. Pept. Sci. 2001, 7, 552–564. [Google Scholar] [CrossRef]
- Shagaghi, N.; Bhave, M.; Palombo, E.A.; Clayton, A.H.A. Revealing the sequence of interactions of PuroA peptide with Candida albicans cells by live-cell imaging. Sci. Rep. 2017, 7, 43542. [Google Scholar] [CrossRef]
- Papo, N.; Oren, Z.; Pag, U.; Sahl, H.G.; Shai, Y. The consequence of sequence alteration of an amphipathic α-helical antimicrobial peptide and its diastereomers. J. Biol. Chem. 2002, 277, 33913–33921. [Google Scholar] [CrossRef] [Green Version]
- Eisenberg, D.; Weiss, R.M.; Terwilliger, T.C. The helical hydrophobic moment: A measure of the amphiphilicity of a helix. Nat. Cell Biol. 1982, 299, 371–374. [Google Scholar] [CrossRef]
- Schiffer, M.; Edmundson, A.B. Use of helical wheels to represent the structures of proteins and to identify segments with helical potential. Biophys. J. 1967, 7, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Wiegand, I.; Hilpert, K.; Hancock, R.E.W. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 2008, 3, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Nan, Y.H.; Shin, S.Y. Effect of disulphide bond position on salt resistance and LPS-neutralizing activity of α-helical homo-dimeric model antimicrobial peptides. BMB Rep. 2011, 44, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Keil, B. Specificity of Proteolysis, 1st ed.; Springer: Berlin, Germany, 1992. [Google Scholar]
- Falla, T.J.; Karunaratne, D.N.; Hancock, R.E.W. Mode of action of the antimicrobial peptide indolicidin. J. Biol. Chem. 1996, 271, 19298–19303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schibli, D.J.; Hwang, P.M.; Vogel, H.J. Structure of the antimicrobial peptide tritrpticin bound to micelles: A distinct membrane-bound peptide fold. Biochemistry 1999, 38, 16749–16755. [Google Scholar] [CrossRef] [PubMed]
- Subbalakshmi, C.; Sitaram, N. Mechanism of antimicrobial action of indolocidin. FEMS Microbiol. Lett. 1998, 160, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Lutkenhaus, J. Regulation of cell division in E. coli. Trends Genet. 1990, 6, 22–25. [Google Scholar] [CrossRef]
- Botta, G.A.; Park, J.T. Evidence for involvement of penicillin-binding protein 3 in murein synthesis during septation but not during cell elongation. J. Bacteriol. 1981, 145, 333–340. [Google Scholar] [CrossRef] [Green Version]
- Di Somma, A.; Avitabile, C.; Cirillo, A.; Moretta, A.; Merlino, A.; Paduano, L.; Duilio, A.; Romanelli, A. The antimicrobial peptide Temporin L impairs E. coli cell division by interacting with FtsZ and the divisome complex. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129606. [Google Scholar] [CrossRef]
- Yin, C.M.; Wong, J.H.; Xia, J.; Ng, T.B. Studies on anticancer activities of lactoferrin and lactoferricin. Curr. Protein Pept. Sci. 2013, 14, 492–503. [Google Scholar] [CrossRef]
- Haug, B.E.; Camilio, K.A.; Eliassen, L.T.; Stensen, W.; Svendsen, J.S.; Berg, K.; Mortensen, B.; Serin, G.; Mirjolet, J.F.; Bichat, F.; et al. Discovery of a 9-mer cationic peptide (LTX-315) as a potential first in class oncolytic peptide. J. Med. Chem. 2016, 59, 2918–2927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Mant, C.T.; Farmer, S.W.; Hancock, R.E.W.; Vasil, M.L.; Hodges, R.S. Rational design of α-helical antimicrobial peptides with enhanced activities and specificity/therapeutic index. J. Biol. Chem. 2005, 280, 12316–12329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.B.; Wang, X.F.; Wang, H.Y.; Liu, Y.; Chen, Y. Studies on mechanism of action of anticancer peptides by modulation of hydrophobicity within a defined structural framework. Mol. Cancer Ther. 2011, 10, 416–426. [Google Scholar] [CrossRef] [Green Version]
- Dennison, S.R.; Whittaker, M.; Harris, F.; Phoenix, D.A. Anticancer α-helical peptides and structure/function relationships underpinning their interactions with tumour cell membranes. Curr. Protein Pept. Sci. 2006, 7, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Strandberg, E.; Morein, S.; Rijkers, D.T.S.; Liskamp, R.M.J.; Van Der Wel, P.C.A.; Antoinette Killian, J. Lipid dependence of membrane anchoring properties and snorkeling behavior of aromatic and charged residues in transmembrane peptides. Biochemistry 2002, 41, 7190–7198. [Google Scholar] [CrossRef] [PubMed]
- Dathe, M.; Nikolenko, H.; Meyer, J.; Beyermann, M.; Bienert, M. Optimization of the antimicrobial activity of magainin peptides by modification of charge. FEBS Lett. 2001, 501, 146–150. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Saravanan, R.; Kwak, S.K.; Leong, S.S.J. Biomolecular engineering of a human beta defensin model for increased salt resistance. Chem. Eng. Sci. 2013, 95, 128–137. [Google Scholar] [CrossRef]
- Andreu, D.; Rivas, L. Animal antimicrobial peptides: An overview. Biopolym. Pept. Sci. Sect. 1998, 47, 415–433. [Google Scholar] [CrossRef]
- Dennison, S.R.; Harris, F.; Bhatt, T.; Singh, J.; Phoenix, D.A. The effect of c-terminal amidation on the efficacy and selectivity of antimicrobial and anticancer peptides. Mol. Cell. Biochem. 2009, 332, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Bahar, A.A.; Ren, D. Antimicrobial peptides. Pharmaceuticals 2013, 6, 1543–1575. [Google Scholar] [CrossRef] [Green Version]
- Subbalakshmi, C.; Nagaraj, R.; Sitaram, N. Biological activities of C-terminal 15-residue synthetic fragment of melittin: Design of an analog with improved antibacterial activity. FEBS Lett. 1999, 448, 62–66. [Google Scholar] [CrossRef] [Green Version]
- Westerhoff, H.V.; Juretic, D.; Hendler, R.W.; Zasloff, M. Magainins and the disruption of membrane-linked free-energy transduction. Proc. Natl. Acad. Sci. USA 1989, 86, 6597–6601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, F.; Dennison, S.R.; Singh, J.; Phoenix, D.A. On the selectivity and efficacy of defense peptides with respect to cancer cells. Med. Res. Rev. 2013, 33, 190–234. [Google Scholar] [CrossRef] [PubMed]
- Strandberg, E.; Zerweck, J.; Horn, D.; Pritz, G.; Berditsch, M.; Bürck, J.; Wadhwani, P.; Ulrich, A.S. Influence of hydrophobic residues on the activity of the antimicrobial peptide magainin 2 and its synergy with PGLa. J. Pept. Sci. 2015, 21, 436–445. [Google Scholar] [CrossRef]
- Seelig, J. Thermodynamics of lipid-peptide interactions. Biochim. Biophys. Acta Biomembr. 2004, 1666, 40–50. [Google Scholar] [CrossRef] [Green Version]
- Hwang, P.M.; Zhou, N.; Shan, X.; Arrowsmith, C.H.; Vogel, H.J. Three-dimensional solution structure of lactoferricin B, an antimicrobial peptide derived from bovine lactoferrin. Biochemistry 1998, 37, 4288–4298. [Google Scholar] [CrossRef]
- Rozek, A.; Friedrich, C.L.; Hancock, R.E.W. Structure of the bovine antimicrobial peptide indolicidin bound to dodecylphosphocholine and sodium dodecyl sulfate micelles. Biochemistry 2000, 39, 15765–15774. [Google Scholar] [CrossRef]
- Rozek, A.; Powers, J.P.S.; Friedrich, C.L.; Hancock, R.E.W. Structure-Based Design of an Indolicidin Peptide Analogue with Increased Protease Stability. Biochemistry 2003, 42, 14130–14138. [Google Scholar] [CrossRef]
- Carmona, G.; Rodriguez, A.; Juarez, D.; Corzo, G.; Villegas, E. Improved protease stability of the antimicrobial peptide pin2 substituted with d-Amino acids. Protein. J. 2013, 32, 456–466. [Google Scholar] [CrossRef]
- Shai, Y.; Oren, Z. Diastereomers of cytolysins, a novel class of potent antibacterial peptides. J. Biol. Chem. 1996, 271, 7305–7308. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; He, L.; Li, G.; Zhai, N.; Jiang, H.; Chen, Y. Role of helicity of α-helical antimicrobial peptides to improve specificity. Protein Cell 2014, 5, 631–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.T.; Chau, J.K.; Zaat, S.A.J.; Vogel, H.J. Cyclic Tritrpticin Analogs with Distinct Biological Activities. Probiotics Antimicrob. Proteins 2011, 3, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Rosengren, K.J.; Clark, R.J.; DalY, N.L.; Göransson, U.; Jones, A.; Craik, D.J. Microcin J25 has a threaded sidechain-to-backbone ring structure and not a head-to-tail cyclized backbone. J. Am. Chem. Soc. 2003, 125, 12464–12474. [Google Scholar] [CrossRef] [PubMed]
- Melo, M.N.; Ferre, R.; Castanho, M.A.R.B. Antimicrobial peptides: Linking partition, activity and high membrane-bound concentrations. Nat. Rev. Microbiol. 2009, 7, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.L.; Shin, S.Y. Effects of dimerization of the cell-penetrating peptide Tat analog on antimicrobial activity and mechanism of bactericidal action. J. Pept. Sci. 2009, 15, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Krajewski, K.; Marchand, C.; Long, Y.-Q.; Pommier, Y.; Roller, P.P. Synthesis and HIV-1 integrase inhibitory activity of dimeric and tetrameric analogs of indolicidin. Bioorg Med. Chem. Lett. 2004, 14, 5595–5598. [Google Scholar] [CrossRef] [PubMed]
- Lorenzón, E.N.; Sanches, P.R.S.; Nogueira, L.G.; Bauab, T.M.; Cilli, E.M. Dimerization of aurein 1.2: Effects in structure, antimicrobial activity and aggregation of Cândida albicans cells. Amino Acids 2013, 44, 1521–1528. [Google Scholar] [CrossRef]
- Bucki, R.; Janmey, P.A. Interaction of the gelsolin-derived antibacterial PBP 10 peptide with lipid bilayers and cell membranes. Antimicrob. Agents Chemother. 2006, 50, 2932–2940. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Kim, I.W.; Kim, S.H.; Yun, E.Y.; Nam, S.H.; Ahn, M.Y.; Kang, D.C.; Hwang, J.S. Anticancer activity of CopA3 dimer peptide in human gastric cancer cells. BMB Rep. 2015, 48, 324–329. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.H.; Chen, C.; Jou, M.L.; Yueh-Luen Lee, A.; Lin, Y.C.; Yu, Y.P.; Huang, W.T.; Wu, S.H. Structural and DNA-binding studies on the bovine antimicrobial peptide, indolicidin: Evidence for multiple conformations involved in binding to membranes and DNA. Nucleic Acids Res. 2005, 33, 4053–4064. [Google Scholar] [CrossRef] [Green Version]
- Nan, Y.H.; Park, K.H.; Park, Y.; Jeon, Y.J.; Kim, Y.; Park, I.S.; Hahm, K.S.; Shin, S.Y. Investigating the effects of positive charge and hydrophobicity on the cell selectivity, mechanism of action and anti-inflammatory activity of a Trp-rich antimicrobial peptide indolicidin. FEMS Microbiol. Lett. 2009, 292, 134–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, A.; Kar, R.K.; Jana, J.; Saha, A.; Jana, B.; Krishnamoorthy, J.; Kumar, D.; Ghosh, S.; Chatterjee, S.; Bhunia, A. Indolicidin targets duplex DNA: Structural and mechanistic insight through a combination of spectroscopy and microscopy. ChemMedChem 2014, 9, 2052–2058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulvatne, H.; Samuelsen, Ø.; Haukland, H.H.; Krämer, M.; Vorland, L.H. Lactoferricin B inhibits bacterial macromolecular synthesis in Escherichia coli and Bacillus subtilis. FEMS Microbiol. Lett. 2004, 237, 377–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfred, R.L.; Palombo, E.A.; Panozzo, J.F.; Bhave, M. The antimicrobial domains of wheat puroindolines are cell-penetrating peptides with possible intracellular mechanisms of action. PLoS ONE 2013, 8, e75488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenberg, D.; Schwarz, E.; Komaromy, M.; Wall, R. Analysis of membrane and surface protein sequences with the hydrophobic moment plot. J. Mol. Biol. 1984, 179, 125–142. [Google Scholar] [CrossRef]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef] [Green Version]
- Jadhav, S.; Shah, R.; Bhave, M.; Palombo, E.A. Inhibitory activity of yarrow essential oil on Listeria planktonic cells and biofilms. Food Control 2013, 29, 125–130. [Google Scholar] [CrossRef]
- Kouidhi, B.; Zmantar, T.; Bakhrouf, A. Anti-cariogenic and anti-biofilms activity of Tunisian propolis extract and its potential protective effect against cancer cells proliferation. Anaerobe 2010, 16, 566–571. [Google Scholar] [CrossRef]
- Schillaci, D.; Arizza, V.; Dayton, T.; Camarda, L.; Di Stefano, V. In vitro anti-biofilm activity of Boswellia spp. oleogum resin essential oils. Lett. Appl. Microbiol. 2008, 47, 433–438. [Google Scholar] [CrossRef]
- Anunthawan, T.; De La Fuente-Núñez, C.; Hancock, R.E.W.; Klaynongsruang, S. Cationic amphipathic peptides KT2 and RT2 are taken up into bacterial cells and kill planktonic and biofilm bacteria. Biochim. Biophys. Acta Biomembr. 2015, 1848, 1352–1358. [Google Scholar] [CrossRef] [Green Version]
- Dathe, M.; Schümann, M.; Wieprecht, T.; Winkler, A.; Beyermann, M.; Krause, E.; Matsuzaki, K.; Murase, O.; Bienert, M. Peptide helicity and membrane surface charge modulate the balance of electrostatic and hydrophobic interactions with lipid bilayers and biological membranes. Biochemistry 1996, 35, 12612–12622. [Google Scholar] [CrossRef] [PubMed]
- Scudiero, D.A.; Shoemaker, R.H.; Paull, K.D.; Monks, A.; Tierney, S.; Nofziger, T.H.; Currens, M.J.; Seniff, D.; Boyd, M.R. Evaluation of a Soluble Tetrazolium/Formazan Assay for Cell Growth and Drug Sensitivity in Culture Using Human and Other Tumor Cell Lines. Cancer Res. 1988, 48, 4827–4833. [Google Scholar] [PubMed]
- Tripathi, A.; Kumari, T.; Tandon, A.; Sayeed, M.; Afshan, T.; Kathuria, M.; Shukla, P.K.; Mitra, K.; Ghosh, J.K. Selective phenylalanine to proline substitution for improved antimicrobial and anticancer activities of peptides designed on phenylalanine heptad repeat. Acta Biomater. 2017, 57, 170–186. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide ID | Sequences * | Length | Mw | Number of Trp | Net Charge | Isoelectic Point (pI) | Charge Density (ChD) | Hydrophobicity (H) | Hydropobic Ratio (HR) | GRAVY | Hydrophobic Moment (µH) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| PuroA | FPVTWRWWKWWKG-NH2 | 13 | 1863 | 5 | +3 | 11.17 | 0.23 | −0.08 | 61.5 | −0.962 | 5.21 |
| Cyclic PuroA | FPVTWRWWKWWKG | 13 | 1845 | 5 | +3 | 11.17 | 0.23 | −0.08 | 61.5 | −0.962 | 5.21 |
| Di-PuroA | (FPVTWRWWKWWKG)2k-NH2 | 27 | 3836 | 10 | +7 | 12.04 | 0.25 | −0.11 | 59 | −1.070 | 3.56 |
| PuroA-OH | FPVTWRWWKWWKG | 13 | 1863 | 5 | +3 | 11.17 | 0.23 | −0.08 | 61.5 | −0.962 | 5.21 |
| R8 | RRRRWRWWRWWRR-NH2 | 13 | 2198 | 5 | +8 | 12.85 | 0.61 | −0.94 | 38 | −3.115 | 5.01 |
| P1 | RKRWWRWWKWWKR-NH2 | 14 | 2144 | 6 | +7 | 12.48 | 0.5 | −0.58 | 43 | −2.700 | 7.88 |
| dP1 | RKRWWRWWKWWKR-NH2 | 14 | 2144 | 6 | +7 | 12.48 | 0.5 | ND | 43 | ND | ND |
| R6 | RWWKWW-NH2 | 6 | 1047 | 4 | +2 | 11.00 | 0.33 | −0.23 | 66 | −2.000 | 5.56 |
| R7 | RWWKWWK-NH2 | 7 | 1175 | 4 | +3 | 11.17 | 0.42 | −0.35 | 57 | −2.271 | 6.01 |
| W7 | WRWWKWW-NH2 | 7 | 1233 | 5 | +2 | 11.00 | 0.28 | −0.14 | 71 | −1.843 | 4.51 |
| dW7 | WRWWKWW-NH2 | 7 | 1233 | 5 | +2 | 11.00 | 0.28 | ND | 71 | ND | ND |
| W8 | WRWWKWWK-NH2 | 8 | 1361 | 5 | +3 | 11.17 | 0.37 | −0.26 | 62.5 | −2.100 | 4.71 |
| WW | WWRWWKWW-NH2 | 8 | 1419 | 6 | +2 | 11.00 | 0.25 | −0.08 | 75 | −1.725 | 6.32 |
| dWW | WWRWWKWW-NH2 | 8 | 1419 | 6 | +2 | 11.00 | 0.25 | ND | 75 | ND | ND |
| Peptide ID | Sequence | MIC (μg/mL) * | |||||
|---|---|---|---|---|---|---|---|
| E. coli ATCC25922 | S. aureus ATCC25923 | P. aeruginosa ATCC9027 | C. albicans FRR 5580 | MRSA ** M173525 | MRSA ** M180920 | ||
| PuroA | FPVTWRWWKWWKG-NH2 | 16 | 16 | 64 | 125 | 16 | 16 |
| Cyclic PuroA | FPVTWRWWKWWKG | 250 | 125 | >250 | >250 | N/T | N/T |
| Di-PuroA | (FPVTWRWWKWWKG)2k-NH2 | 250 | 250 | >250 | 125 | N/T | N/T |
| PuroA-OH | FPVTWRWWKWWKG | 32 | 64 | 125 | 250 | N/T | N/T |
| R8 | RRRRWRWWRWWRR-NH2 | 64 | 64 | 64 | 250 | N/T | N/T |
| P1 | RKRWWRWWKWWKR-NH2 | 16 | 16 | 16 | 64 | 16 | 8 |
| dP1 | RKRWWRWWKWWKR-NH2 | 16 | 16 | 16 | 64 | N/T | N/T |
| R6 | RWWKWW-NH2 | 32 | 16 | 32 | 64 | N/T | N/T |
| R7 | RWWKWWK-NH2 | 8 | 16 | 32 | 64 | N/T | N/T |
| W7 | WRWWKWW-NH2 | 4 | 8 | 32 | 64 | 8 | 8 |
| dW7 | WRWWKWW-NH2 | 8 | 8 | 16 | 64 | N/T | N/T |
| W8 | WRWWKWWK-NH2 | 4 | 8 | 32 | 64 | N/T | N/T |
| WW | WWRWWKWW-NH2 | 8 | 8 | 32 | 64 | 4 | 4 |
| dWW | WWRWWKWW-NH2 | 8 | 8 | 32 | 64 | N/T | N/T |
| Peptide ID | Salt Tolerance * | Protease Stability ** | Binding to DNA in vitro (µg/mL) |
|---|---|---|---|
| PuroA | Up to 50mM | Unstable | 32 |
| Cyclic PuroA | Intolerant | Unstable | 64 |
| Di-PuroA | Intolerant | Unstable | 8 |
| PuroA-OH | Intolerant | Unstable | 32 |
| R8 | Up to 150 mM | Unstable | 64 |
| P1 | Up to 150 mM | Unstable | 8 |
| dP1 | Up to 150 mM | Stable | 8 |
| R6 | Intolerant | Unstable | 250 |
| R7 | Intolerant | Unstable | 250 |
| W7 | Up to 150 mM | Unstable | 16 |
| dw7 | Up to 150 mM | Stable | 32 |
| W8 | Up to 150 mM | Unstable | 16 |
| WW | Up to 150mM | Unstable | 32 |
| dWW | Up to 150 mM | Stable | 32 |
| Peptide ID | Peptide Concentration | Percentage of Inhibition of Initial MRSA Cells Attachment | % Inhibition of Preformed 6 h MRSA Biofilm after 1 h Incubation | ||
|---|---|---|---|---|---|
| CV | MTT | CV | MTT | ||
| PuroA | 0.5 × MIC | 70 ± 5.6 | 92 ± 1.5 | 68 ± 9.2 | 90 ± 2.5 |
| 1 × MIC | 75 ± 4.0 | 95 ± 1.1 | 75 ± 7.5 | 92 ± 4.0 | |
| 2 × MIC | 79 ± 4.5 | 95 ± 0.5 | 77 ± 7.0 | 95 ± 2.6 | |
| P1 | 0.5 × MIC | 72 ± 3.1 | 92 ± 3.0 | 70 ± 6.9 | 93 ± 5.0 |
| 1 × MIC | 75 ± 1.9 | 95 ± 2.3 | 74 ± 5.8 | 95 ± 3.8 | |
| 2 × MIC | 78 ± 6.1 | 95 ± 2.5 | 78 ± 3.1 | 95 ± 4.0 | |
| W7 | 0.5 × MIC | 69 ± 3.6 | 90 ± 2.9 | 67 ± 6.0 | 88 ± 2.5 |
| 1 × MIC | 75 ± 5.0 | 95 ± 4.0 | 75.5 ± 3.0 | 90 ± 4.7 | |
| 2 × MIC | 76 ± 4.8 | 96 ± 1.5 | 75 ± 5.5 | 94 ± 0.5 | |
| WW | 0.5 × MIC | 71.5 ± 3.5 | 90 ± 2.0 | 69 ± 7.8 | 91 ± 3.0 |
| 1 × MIC | 73 ± 6.2 | 95 ± 3.1 | 71 ± 4.5 | 92 ± 3.5 | |
| 2 × MIC | 79 ± 7.5 | 96 ± 0.5 | 74 ± 2.0 | 95 ± 1.7 | |
| Peptide ID | * IC50 (μg/mL) | ** Therapeutic Index (TI) | |
|---|---|---|---|
| NIH-3T3 | HeLa | ||
| PuroA | 250 | >250 | <1 |
| Cyclic PuroA | >250 | >250 | ND |
| Di-PuroA | 250 | 32 | 7.8 |
| PuroA-OH | >250 | >250 | ND |
| R8 | >250 | 250 | >1 |
| P1 | 250 | 32 | 7.8 |
| dP1 | 250 | 125 | 2 |
| R6 | 250 | >250 | <1 |
| R7 | 250 | >250 | <1 |
| W7 | 250 | 250 | 2 |
| dW7 | >250 | 125 | >3 |
| W8 | >250 | 250 | >2 |
| WW | >250 | 250 | >2 |
| dWW | 250 | 125 | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shagaghi, N.; Clayton, A.H.A.; Aguilar, M.-I.; Lee, T.-H.; Palombo, E.A.; Bhave, M. Effects of Rationally Designed Physico-Chemical Variants of the Peptide PuroA on Biocidal Activity towards Bacterial and Mammalian Cells. Int. J. Mol. Sci. 2020, 21, 8624. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228624
Shagaghi N, Clayton AHA, Aguilar M-I, Lee T-H, Palombo EA, Bhave M. Effects of Rationally Designed Physico-Chemical Variants of the Peptide PuroA on Biocidal Activity towards Bacterial and Mammalian Cells. International Journal of Molecular Sciences. 2020; 21(22):8624. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228624
Chicago/Turabian StyleShagaghi, Nadin, Andrew H. A. Clayton, Marie-Isabel Aguilar, Tzong-Hsien Lee, Enzo A. Palombo, and Mrinal Bhave. 2020. "Effects of Rationally Designed Physico-Chemical Variants of the Peptide PuroA on Biocidal Activity towards Bacterial and Mammalian Cells" International Journal of Molecular Sciences 21, no. 22: 8624. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228624