1. Introduction
Gene expression is controlled by various stimuli in order to coordinate cellular metabolism and achieve cell-specific responses that ensure cell adaptation to the changed environment [
1]. New technologies and approaches, developed over the past 25 years, have led to a more detailed understanding of the mechanisms, which provide different patterns of gene expression, evoked by the multiple stimuli in different types of cells [
2]. The transcriptional response is usually very complex and often affects the activity of many hundreds of genes (the so-called transcription program). The response to environmental stimuli typically involves activation of immediate early response genes (the so-called “first wave” of expression) which encode transcription factors that affect activity of other genes (“second wave”). However, it is still difficult to discern the effect of a single stimulus on gene expression, because when applied to a cell the stimulus usually triggers several different signaling pathways that produce multiple messengers, which also induce gene expression.
The electrochemical gradient of Na
+ and K
+, existing across the plasma membrane in all animal cells, is maintained by Na,K-ATPase (Na-pump). This gradient is used by animal cells to support numerous physiological processes: generation of action membrane potentials (spikes), cell volume regulation, and secondary transport of ions (calcium, proton, phosphate, chloride) and nutrients (amino acids, nucleotides, sugars) [
3]. An imbalance of intracellular Na
+i and K
+i concentrations, in response to different stimuli (e.g., hypoxia, inflammation, osmolality changes of extracellular fluids), can affect transcription and translation processes, thus contributing to the development of various diseases. Indeed, it has recently been demonstrated that an increase in [Na
+]
i/[K
+]
i-ratio in various types of cells results in differential expression of immediate early response genes, including
FOS [
4], as well as cell-specific genes, in particular, Na,K-ATPase α1-, α2-, and α3-subunits [
5], tumor growth factor-β, skeletal muscle actin, myosin light chains, atrial natriuretic factor [
6], and mortalin [
7]. It was initially suggested that the alteration of gene expression caused by an increase in [Na
+]
i/[K
+]
i-ratio is associated with a change in the intracellular Ca
2+ concentration, which is mediated by Na
+/Ca
2+ exchanger [
8] and/or voltage dependent Ca
2+-channels [
9]. However, we have recently shown that ouabain, Na,K-ATPase inhibitor, triggers similar gene expression alterations in the presence of Ca
2+ channel blockers and extra- and intracellular Ca
2+ chelators in rat vascular smooth muscle cells (RVSM), human adenocarcinoma cell line (HeLa), and human umbilical vein endothelial cells (HUVEC) [
10].
The data suggest that an increase in [Na
+]
i/[K
+]
i-ratio affects gene transcription through Ca
2+i-independent mechanism(s), and that alteration of this parameter may be considered as a trigger of gene expression. Dissipation of the Na
+i/K
+i gradient in endothelial cells can lead to their dysfunction, and thus contribute to the development of cardiovascular diseases. The object of this study was human umbilical vein endothelial cells. These cells are a convenient model for the investigations of pathophysiological aspects that contribute to the development of cardiovascular diseases [
11]. Using Affymetrix technology, we tried to identify the first gene targets (upstream genes), whose expression change was induced by the elevation of [Na
+]
i/[K
+]
i-ratio. We performed a comparative analysis of time-dependent modulation of the [Na
+]
i/[K
+]
i-ratio and transcriptomic changes in HUVEC triggered by ouabain and K
+-free medium, i.e., two independent approaches to Na,K-ATPase inhibition, with a final goal of identifying intermediates of the upstream signaling pathway. It is known that ouabain not only inhibits Na,K-ATPase, but, being bound to the pump molecule, also triggers some signaling pathways through protein–protein interactions [
12]. This signaling affects gene expression too. We considered the differentially expressed genes common to both stimuli as Na
+i/K
+i sensitive genes. Our results revealed upstream [Na
+]
i/[K
+]
i sensitive gene targets, and allowed us to suggest how they might be regulated by the change in [Na
+]
i/[K
+]
i-ratio.
3. Discussions
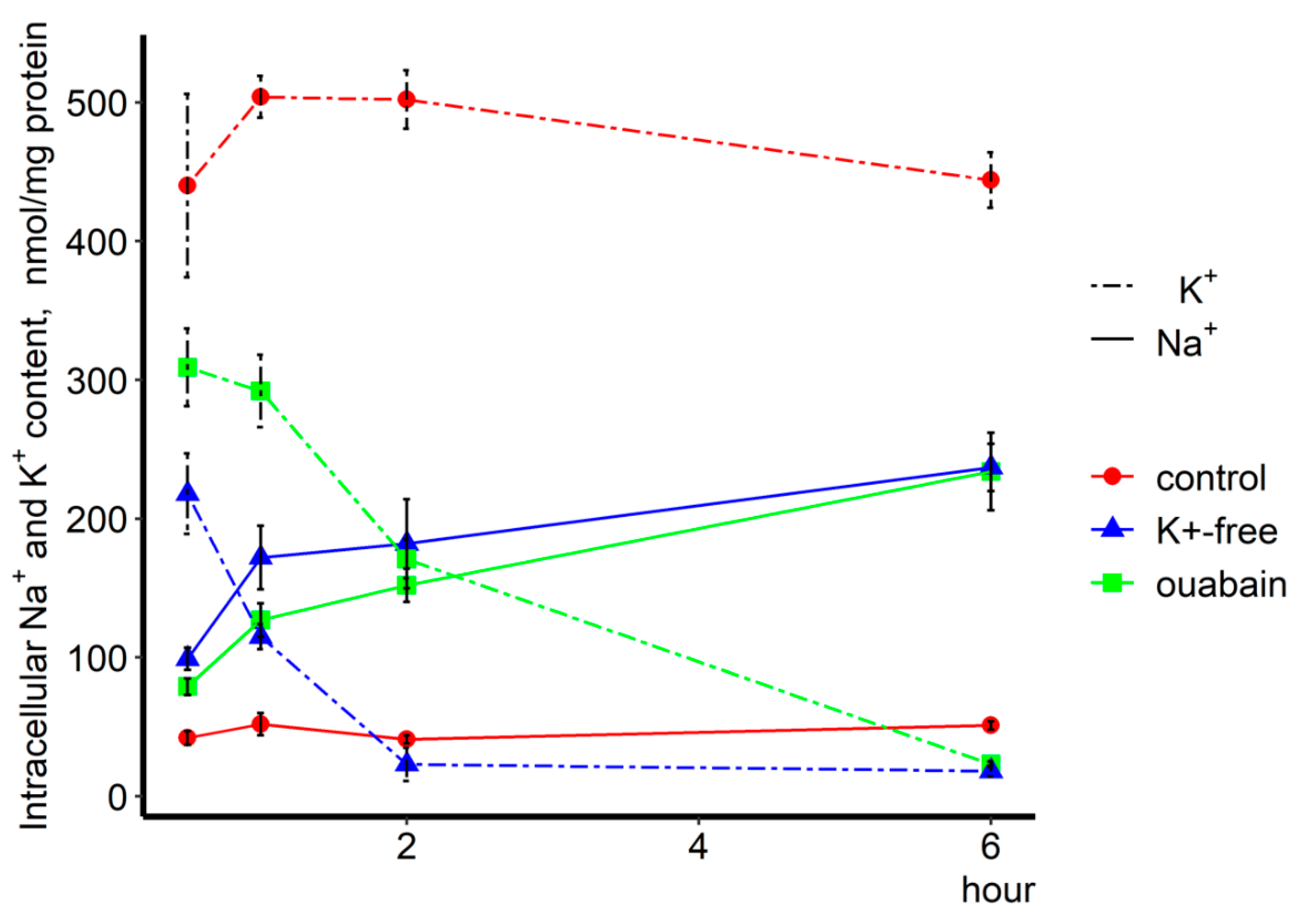
Numerous studies have shown that inhibition of Na,K-ATPase, resulting in the elevation of [Na
+]
i/[K
+]
i-ratio, leads to the alteration of gene expression in various types of cells. Among them were found RVSM, HUVEC, HeLa [
10], cardiomyocytes [
6], renal epithelial cells [
32], hepatocytes [
33], and neuronal cells [
34]. Treatment of these cells by ouabain at concentrations that completely inhibit Na,K-ATPase induces expression of immediate early response genes, in particular,
FOS. In this study we confirmed that 0.5–6 h incubation of HUVEC with ouabain or K
+-free medium, i.e., two independent approaches to Na,K-ATPase suppression, affects gene expression. These transcriptomic changes are accompanied by the gain of Na
+i and loss of K
+i, thus suggesting a key role of [Na
+]
i/[K
+]
i-mediated, rather than [Na
+]
i/[K
+]
i-independent, signaling.
According to the currently accepted paradigm, inhibition of Na,K-ATPase by ouabain and subsequent increase of [Na
+]
i/[K
+]
i-ratio results in the elevation of intracellular Ca
2+ concentration, through the activation of Na
+/Ca
2+ exchanger [
8], or conductivity of L-type voltage-gated Ca
2+-channels [
9]. Both types of these transporters were found in endothelial cells [
35]. It is known that increase of Ca
2+i, in turn, controls gene expression, mainly by CaMKI (II, III)/NFκB, calcineurine/NFAT, and CaMKII (IV)/CREB signaling pathways [
36]. However, we previously found that Ca
2+ depletion, triggered by addition of extra-and intracellular Ca
2+ chelators, increased rather than decreased the number of ubiquitous and cell-type specific Na
+i/K
+i sensitive genes in RVSM, HeLa, and HUVEC. These results as well as early reports of Ca
2+-independent
c-Fos expression in RVSMC [
4] and HeLa cells [
26] allowed us to conclude that, along with the canonical Ca
2+i-mediated signaling, a sustained increase in [Na
+]
i/[K
+]
i-ratio affects gene transcription via unknown Ca
2+i-independent mechanism(s).
Another method of gene expression activation by ouabain was disclosed by Xie and Askari for cardiomyocytes [
12]. They demonstrated that after ouabain binding to Na,K-ATPase, the enzyme interacts with membrane protein-partners that, in turn, trigger signal cascades. The signaling pathways start from the interaction of Na,K-ATPase with Src kinase, phosphorylation of the epidermal growth factor receptor, and consequent activation of Ras and p42/44 mitogen-activated protein kinases.
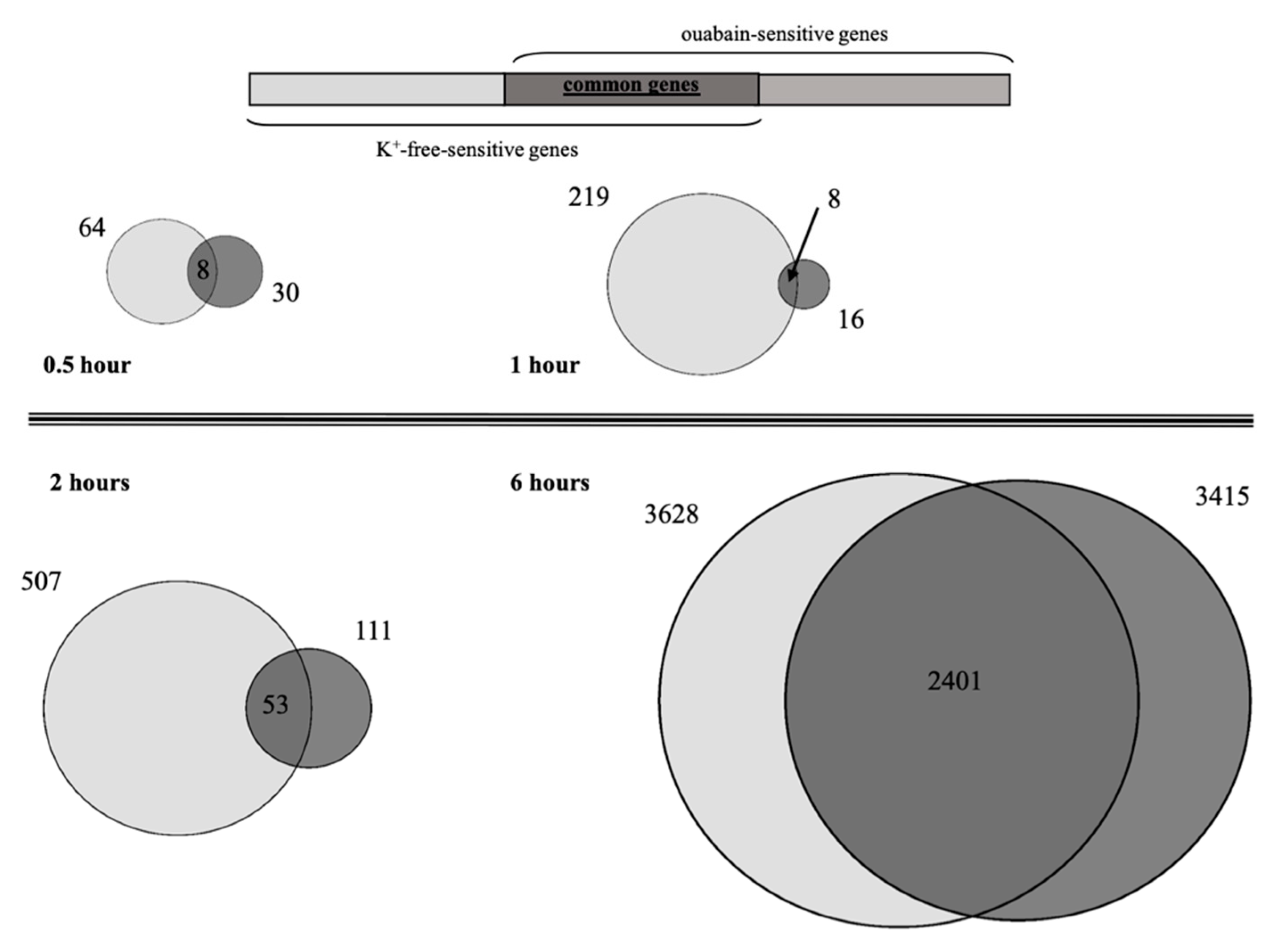
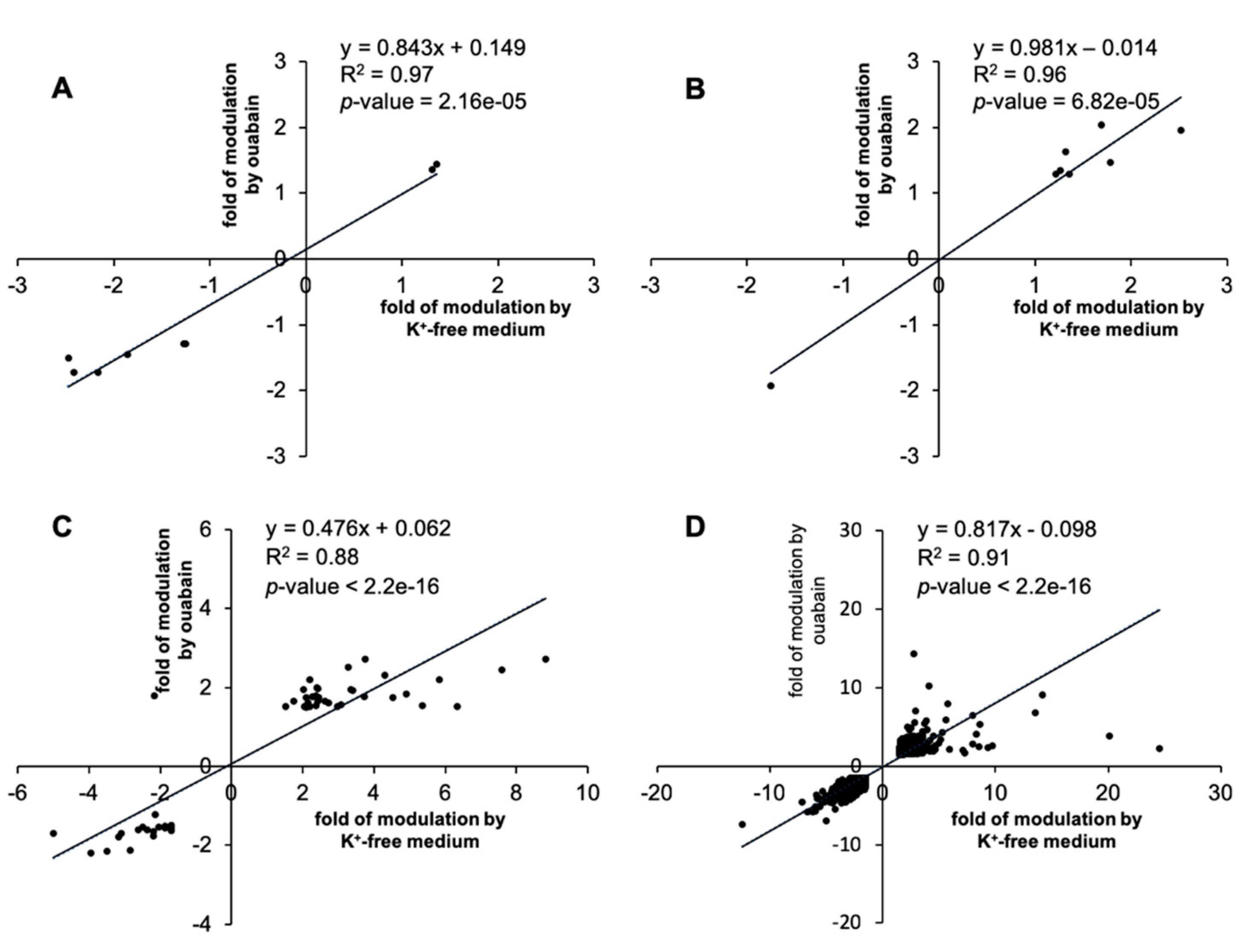
Taking into account that ouabain itself can evoke signaling pathways that affect gene expression, we compared transcriptomic changes triggered by ouabain and K
+-free medium. Thus, we were able to identify common genes, whose expression is caused by both stimuli and mainly depends on changes in [Na
+]
i/[K
+]
i-ratio (see
Figure 3). This suggests that in both cases, there was basically a common (identical) trigger for the transcriptomic changes.
The main goal of this study was to reveal upstream Na
+i/K
+i sensitive genes, namely, the genes that are the first targets of a trigger that induces the alteration of gene expression. We found that eight common, differentially expressed, genes were the same for both stimuli after 0.5 h of Na,K-ATPase inhibition by ouabain or in K
+-free medium (
Table 3). Furthermore, all of them have a similar expression pattern (activation or suppression). After 1 h of Na,K-ATPase inhibition, we also detected only eight common genes with a similar expression pattern for both stimuli. However, the set of common genes at this time point was altered compared to the set found after 0.5 h of Na,K-ATPase inhibition. The expression of seven of these genes was increased, while the expression of only one of them was decreased (
Table 3). The sets and expression patterns of the eight revealed common genes, triggered after 0.5 and 1 h of Na,K-ATPase inhibition, were the same, and did not depend on whether inhibition was produced by ouabain or K
+-free medium. Thus, we can conclude that these transcriptomic changes are triggered by the common (identical) starting stimulus, namely, the alteration of [Na
+]
i/[K
+]
i-ratio.
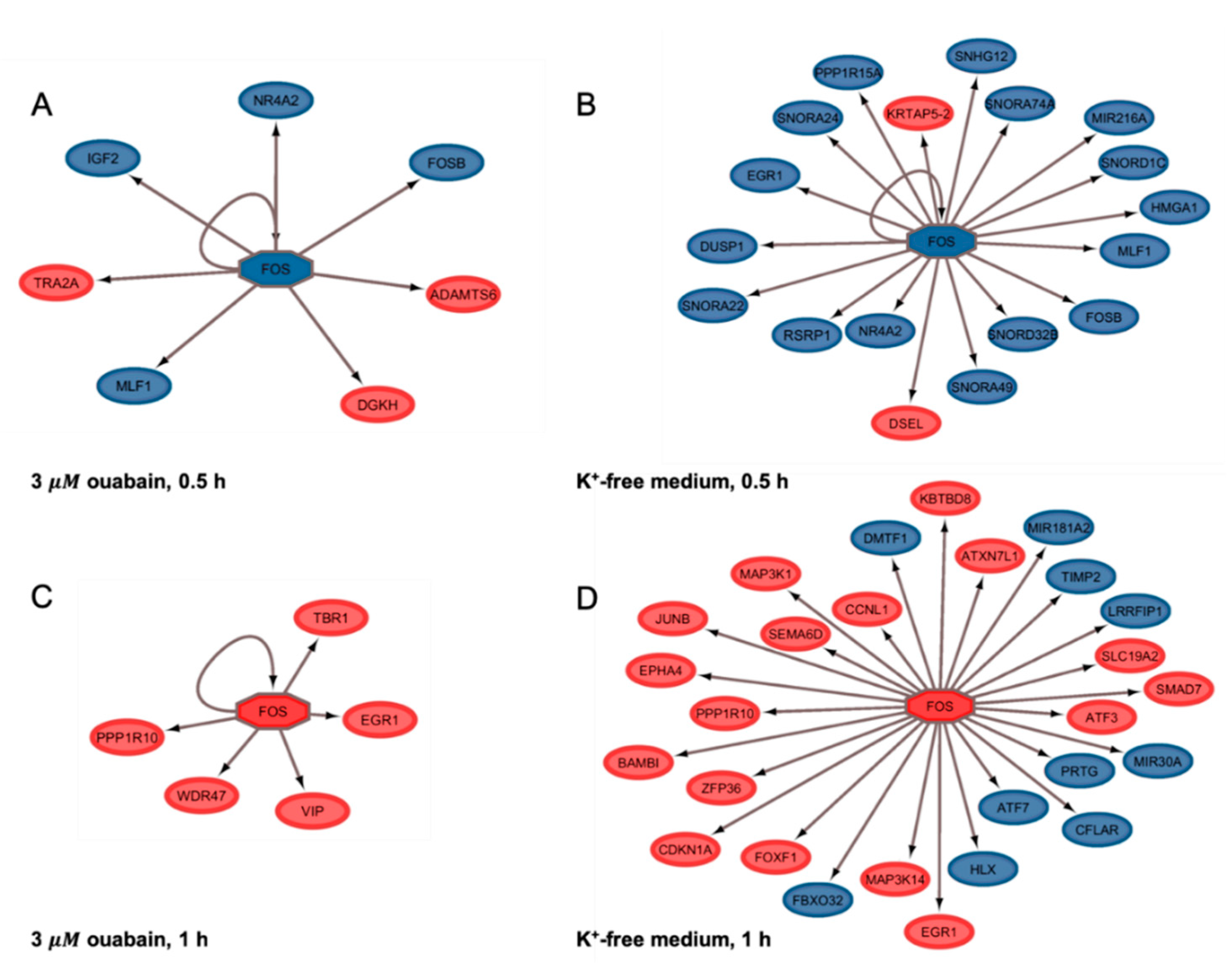
All genes whose expression changed after 0.5 and 1 h of Na,K-ATPase inhibition are “first wave” genes that are necessary for the alteration of the expression of other genes. Among their products we can see RNAs required for splicing, small nucleolar RNA, microRNAs which are involved in the regulation processes at the post-transcriptional level, and proteins which are transcriptional regulation factors. The data demonstrating that expression of some of them (e.g., FOS and FOSB) was downregulated after 0.5 h and then upregulated later, apparently demonstrates that during the first hour after Na,K-ATPase inhibition we observed the “reorganization” of the expression pattern of cells.
Special attention was focused on the
FOS gene. This gene encodes one of the immediately expressed transcription factors, which induce a “second wave” of gene transcription. The
FOS gene product (c-Fos protein) is a member of the leucine zipper proteins family, which, as homo- or heterodimers, bind to a specific region of double-stranded DNA, and which leads to varying effects on transcription. It is believed that c-Fos acts by combining short-term extracellular signals with long-term changes, through coordinated changes in the expression of target genes. Curiously, c-Fos is currently considered as a marker of neuronal activity [
37]. It is known that neuronal activity is coupled with a significant change of [Na
+]
i/[K
+]
i-ratio. Activation of
FOS expression was also observed in contracting C2C12 myotubes, where it also correlates with an increase in this parameter [
38]. We have previously shown that
FOS gene expression occurs via Ca
2+-independent mechanism(s) [
4,
25]. It should be noted that this phenomenon was also noticed earlier. For example, Nakagawa et al., in the experiments with deletion within
FOS gene promoter, have demonstrated that expression of this gene is mediated by ouabain via Na,K-ATPase inhibition, regardless of the cAMP/Ca
2+ response element ability [
39]. In another study, these authors noted that ouabain-induced
FOS gene expression is due to intracellular monovalent cations perturbations [
40].
The c-Fos proteins form heterodimers with Jun family proteins, and thus regulate transcription of many cellular genes through mitogen-activated protein kinases (MAPKs). It is known that Akt modulates
FOS gene transcription [
41]. It has also been shown that
FOS gene promoters contain a serum response element (SRE) and a Ca
2+/cAMP response element (CRE), which are activated by [Ca
2+]
i increments in the cytoplasm and nucleus, respectively [
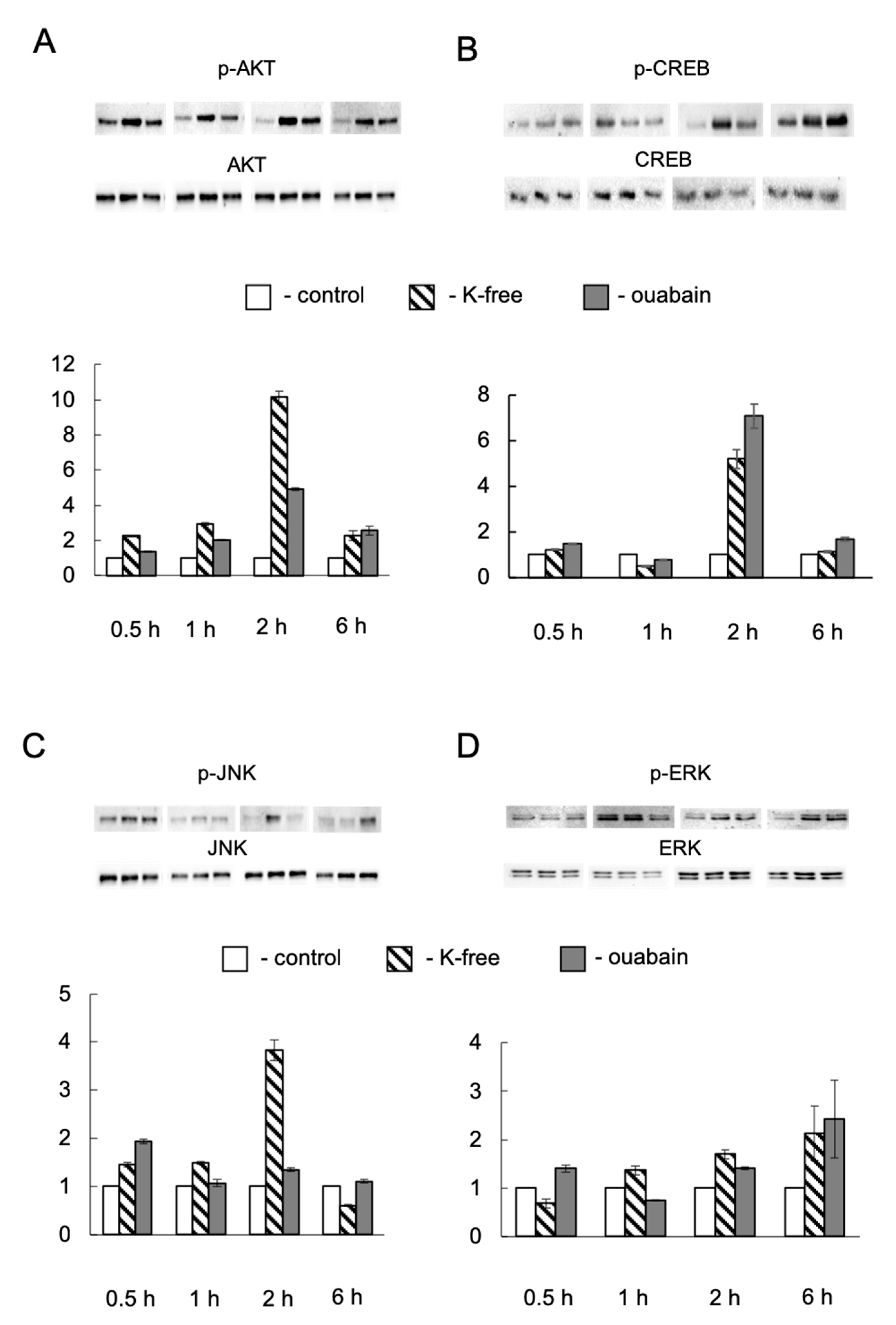
42], as well as protein kinase B (Akt) and its target, the transcription factor CREB. Our data demonstrated CREB activation only in 2 h of Na,K-ATPase inhibition. Thus, we can assume that in the early stages of Na
+i/K
+i gradient dissipation, the coordination of the transcription factors network is likely to occur, which, probably, is not mediated primarily by Ca
2+ signaling.
It is well known that monovalent ions intracellular concentrations change temporarily in response to various stimuli. Orlov and Hamet have formulated a hypothesis which proposes monovalent ions as secondary messengers [
43]. Data on monovalent cation intracellular sensors has been reviewed in recent publication [
44]. In our opinion, G-quadruplexes are the most suitable candidate for this role [
45]. These structures are abundant in the human genome and formed within guanine-rich regions of nucleic acids, DNA, and RNA. It was also shown that their structure is stabilized by monovalent cations [
23]. Telomeres and the promoter region of genes are the main locations of DNA G-quadruplexes [
46]. The formation, or the reorganization, of these structures within gene promoters can affect transcription processes sterically. We speculate that Na
+i/K
+i imbalance mediates transcriptomic changes directly, via change of DNA conformation G-quadruplexes. Sen and Gilbert were the first to show that the ratio of Na
+ and K
+ ions is critical for the DNA G-quadruplexes’ formation, the so-called “sodium–potassium switch” [
47]. These non-canonical structures occur in promoters of oncogenes, such as
MYC,
MYB,
FOS, and
ABL [
48]. In addition, experimental data show that G-quadruplexes are more frequent in cancer cells in comparison with normal cells [
49]. In this regard, these structures are currently considered as molecular anti-cancer targets. It is worth mentioning that viruses also have G-quadruplexes in the regulatory regions of the genome, which are the anti-viral targets [
50]. These observations are consistent with the data demonstrated by the application of cardiotonic steroids (CTS) in anti-cancer [
51,
52,
53] and anti-viral therapy [
54,
55,
56,
57]. It is well documented that different members of the CTS family have different affinities to the Na,K-ATPase and, as a consequence, different inhibitory properties [
58,
59]. It is likely that the various therapeutic effects of these compounds are due to these parameters.
4. Materials and Methods
4.1. Cell Culture
The human umbilical vein endothelial cells (HUVEC) were purchased from Lonza (Maryland, Walkersville, MD, USA) and passaged up to 6 times. Cells were cultured in complete endothelial cell growth medium-2 (EGM-2 BulletKit, CC3162, Lonza, Maryland, Walkersville, MD, USA) containing 10% fetal bovine serum (FBS), and maintained in a humidified atmosphere with 5% CO2/balance air at 37 °C. To establish quiescence, cells were incubated for 24 h in media in which concentration of FBS was reduced to 0.2%.
4.2. Intracellular Na+ and K+ Content
Intracellular content of exchangeable Na
+ and K
+ was measured as the steady-state distribution of extra- and intracellular
86Rb and
22Na, respectively. To establish isotope equilibrium, cells growing in 12-well plates were preincubated for 3 h in control medium containing 0.5 µCi/mL
86RbCl or 3 µCi/mL
22NaCl, and then with or without 3 µM ouabain, and in K
+-free medium for 0.5, 1, 2, and 6 h. The cells were transferred onto ice, washed 4 times with 2 mL of ice-cold medium W containing 100 mM MgCl
2 and 10 mM HEPES-tris buffer (pH 7.4). The medium was aspirated and cells were lysed with 1% SDS and 4 mM EDTA solution. Radioactivity of incubation media and cell lysates was quantified, and intracellular cation content was calculated as A/a × m, where A was the radioactivity of the samples (cpm), a was the specific radioactivity of
86Rb (K
+ congener) and
22Na in the medium (cpm/nmol), and m was the protein content. For more details, see [
10].
4.3. RNA Isolation
Total RNA was extracted from cells grown in 6-well plates, using TRIzol® reagent (Invitrogen, California, Carlsbad, CA, USA) and purified with a RNeasy® MinElute cleanup kit (Qiagen, California, Valencia, CA, USA), following the manufacturers’ protocols. Only the RNA samples that had a more than 7.0 RNA integrity number (RIN) and no detectable genomic DNA contamination were used for the subsequent gene array analyses. RNA quality was assessed by 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA). Microarray experiments were performed with GeneChip® Human Gene 1.0 ST array (which detects 28,869 gene products). In this case, each gene was represented by approximately 26 probes along the entire length of the transcript (Affymetrix, California, Santa Clara, CA, USA). 100 ng of total RNA for each sample was processed with Ambion® WT Expression Kit (Invitrogen). This kit uses a reverse transcription priming method that specifically primes non-ribosomal RNA, including both poly(A) and non-poly(A) mRNA, and generates sense-strand cDNA as the final product. 5.5 µg of the single-stranded cDNA was fragmented and labeled using a Affymetrix GeneChip® WT Terminal Labeling Kit, and 2.0 µg of the resulting cDNA was hybridized on the chip.
4.4. Gene Chip Expression Analysis
The whole hybridization procedure was conducted with the Affymetrix GeneChip
® system, according to the protocol recommended by the manufacturer. The hybridization results were evaluated with Affymetrix GeneChip
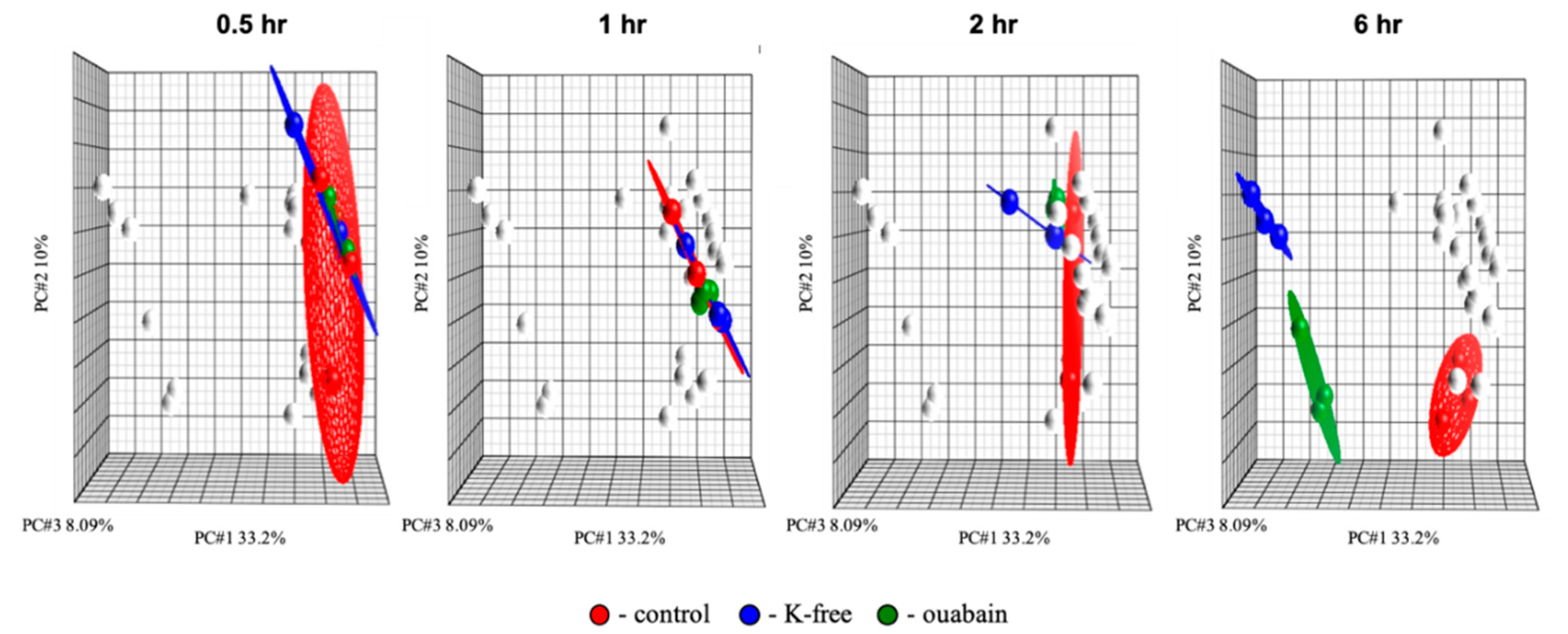
® Command Console software (AGCC, version 0.0.0.676). Quality of the chips was determined using Affymetrix Expression Console. Data analysis was performed within Partek Genomics Suite (Partek, Missouri, St. Louis, MO, USA). The data were initially normalized by the robust multichip average (RMA) algorithm, which uses background adjustment, quantile normalization, and summarization. Then, normalized data were analyzed by principal component analysis (PCA) [
60] to identify patterns in the dataset, and highlight similarities and differences among the samples. Major sources of variability identified within the dataset by PCA were used as grouping variabilities for analysis of variance (ANOVA), with
n = 3 for each group of samples. The ensuing data were filtered to identify transcripts with statistically significant variation of expression among the groups that are modulated by at least 20%, with multiple testing correction by the false discovery rate (
p-VALUE).
4.5. GO and KEGG Pathways Analysis
Active subnetwork search and enrichment analysis was provided by the pathfindR package in R [
61]. Biogrid and KEGG sets were used for the identification of protein–protein interaction network (PIN) and necessary gene sets to obtain for enrichment analysis, respectively. A greedy search (GR) algorithm was used to perform an active subnetwork search. Network of the set of co-regulated genes was predicted by using iRegulon in Cytoscope software [
62]. The Ggplot2 package was used for data visualization [
63].
4.6. G-Quadruplex-Forming Region Analysis
The search for quadruplexes within the DNA sequences of early response genes (0.5 and 1 h) was performed using the G4Catchall online tool [
64], NCBI Nucleotide, and Gene databases. Values with a score greater than 0 were considered reliable.
4.7. Western Blot Analysis
The harvested lysates of cells were prepared by solubilization in RIPA buffer, containing cocktails of proteases and phosphatases inhibitors (Thermo Fisher Scientific, Massachusetts, Waltham, MA, USA). Protein concentrations were determined using the Lowry protein assay [
65]. SDS-PAGE was performed in accordance with the Laemmli method [
66], with 6% stacking gel and 10% running gel. Proteins were transferred from gel to nitrocellulose membrane (BioRad, California, Hercules, CA, USA), followed by blocking with 5% fat-free milk (Valio, Helsinki, Finland) in PBST 1X (PBS supplemented with Tween 20 0.1%) for 1 h at room temperature. Akt, CREB, and their phosphorylated forms were detected by overnight incubation with the corresponding antibodies (#9272S, #4060S, #4820S, #9198S, #9252S, #4668S, #4695S #4377S, Cell Signaling, Massachusetts, Danvers, MA, USA) at 4 °C in 5% milk powder in PBST. These samples were then incubated with HRP-conjugated secondary antibodies monoclonal anti-rabbit IgG (a1949, Sigma-Aldrich, Missouri, St. Louis, MO, USA) for 1 h at room temperature in 5% milk powder in PBST. GAPDH (#2118S, Cell Signaling, Massachusetts, Danvers, MA, USA) was chosen as the house-keeping protein. Antigen-antibody complexes were visualized using an ECL kit and ChemiDoc XRS+ Molecular Imager (BioRad, California, Hercules, CA, USA).
4.8. Chemicals
22NaCl and 86RbCl were obtained from PerkinElmer (Massachusetts, Waltham, MA, USA) and Isotope (St-Petersburg, Russia). The remaining chemicals were supplied by Gibco BRL (Missouri, Gaithersburg, MO, USA), Calbiochem (California, La Jolla, CA, USA), Sigma-Aldrich (Missouri, St. Louis, MO, USA), and Anachemia Canada Inc. (Quebec, Montreal, QC, Canada).

 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}