Transcriptomic Analysis of Human Astrocytes In Vitro Reveals Hypoxia-Induced Mitochondrial Dysfunction, Modulation of Metabolism, and Dysregulation of the Immune Response

 , ,
, ,
Abstract
:1. Introduction
2. Results
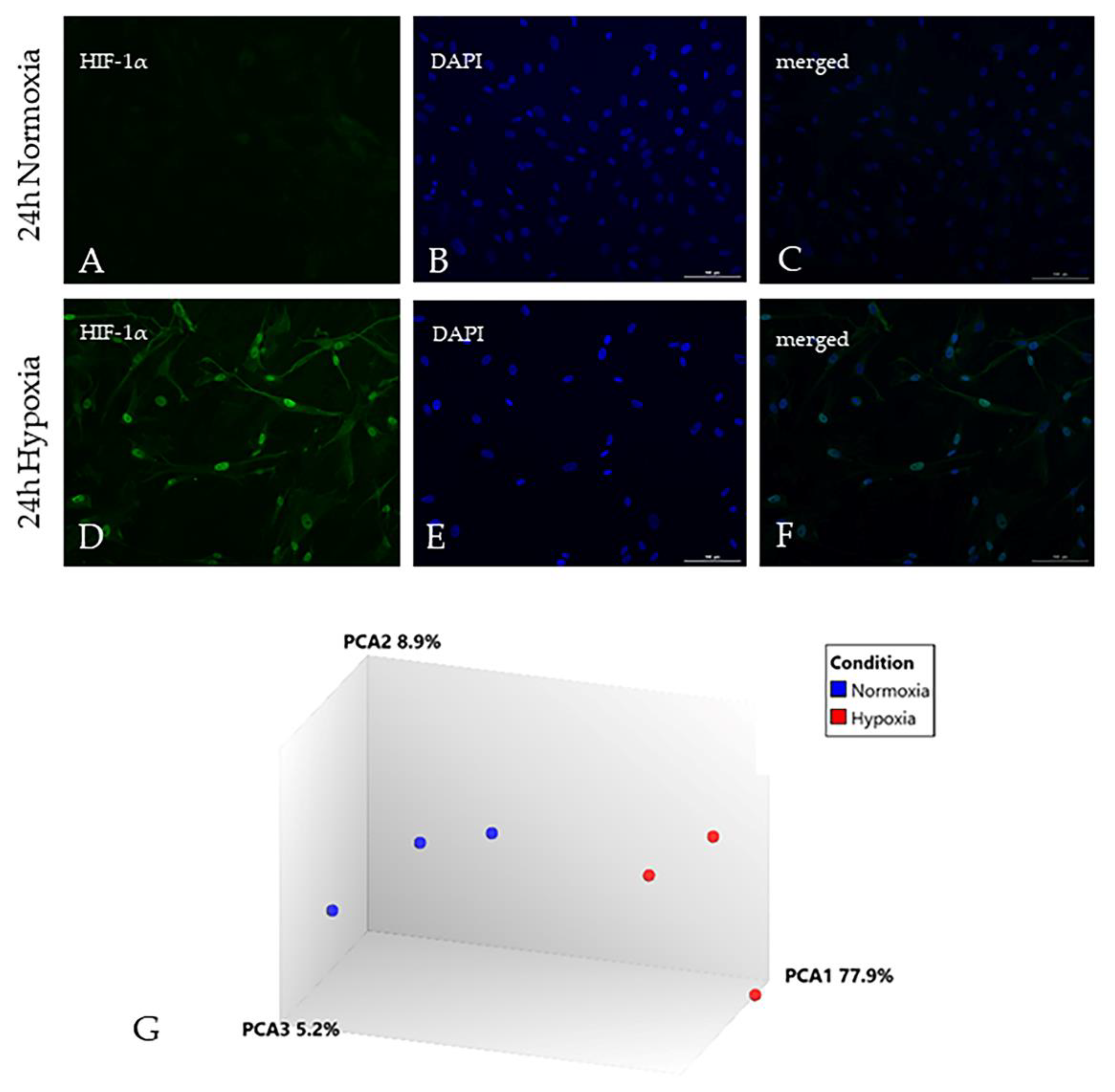
2.1. Hypoxia Significantly Alters the Astrocyte Transcriptome
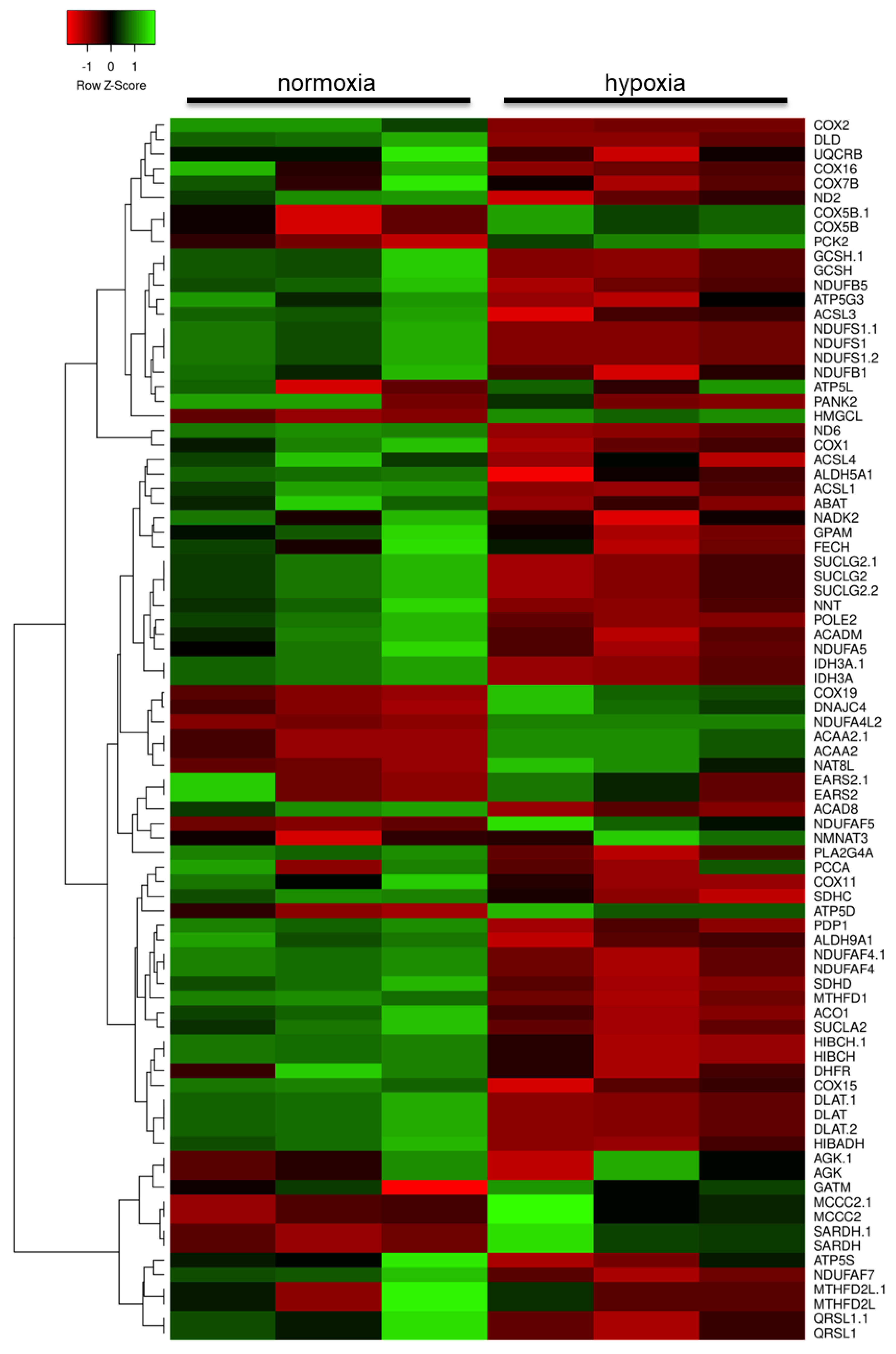
2.2. Hypoxia Modulates the Metabolic Profile of Astrocytes
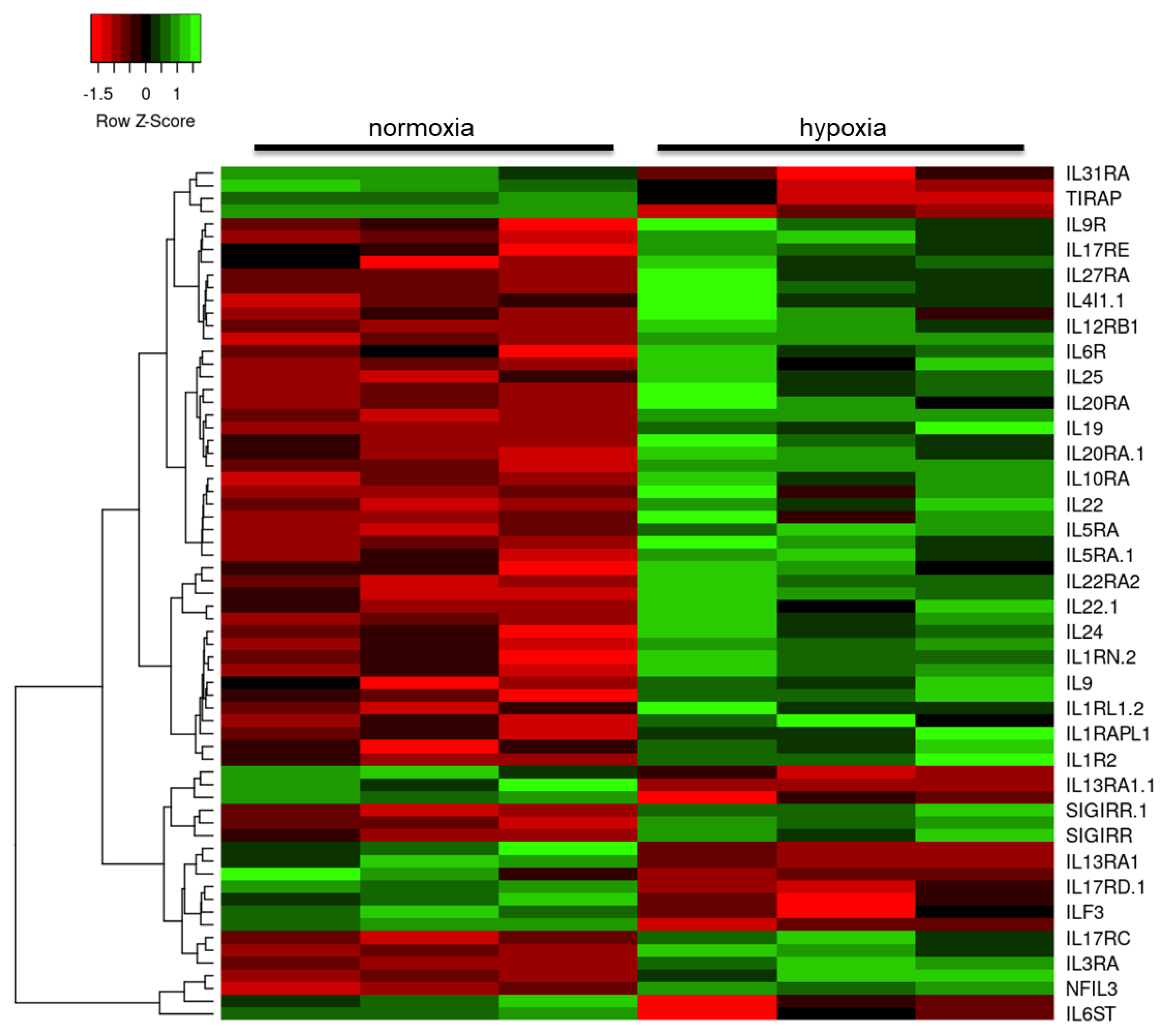
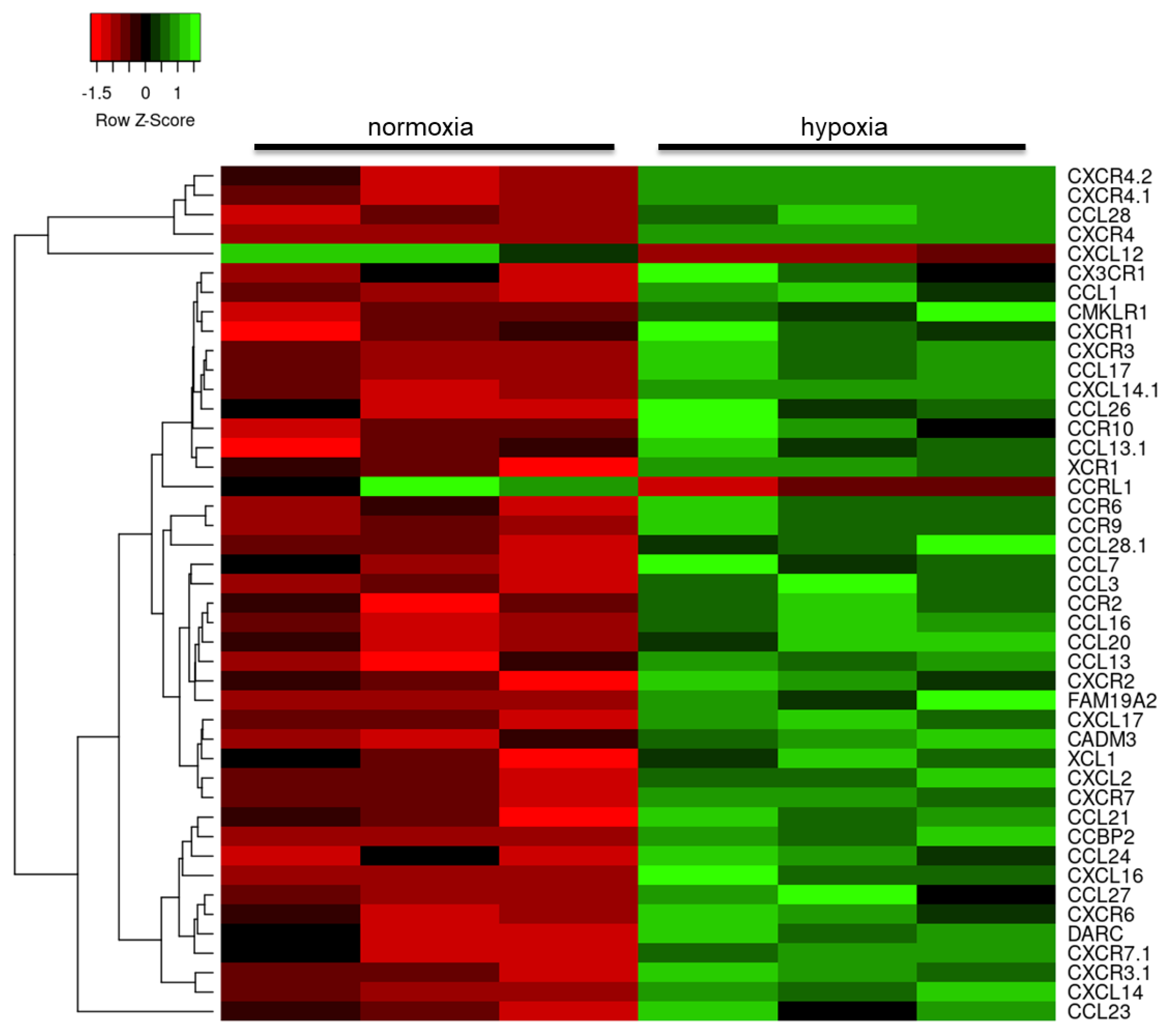
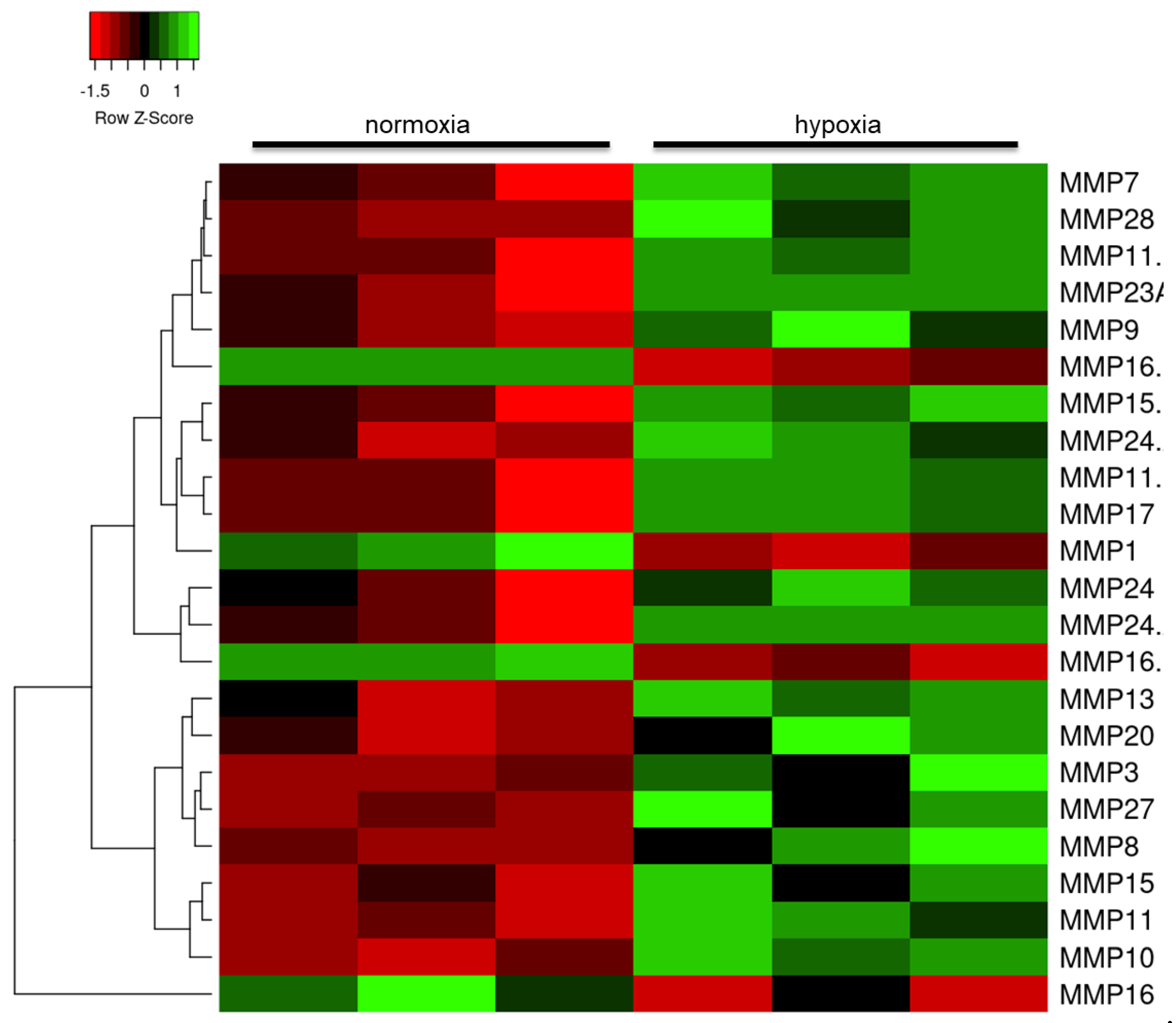
2.3. Hypoxia Modulates the Astrocyte Immune Response
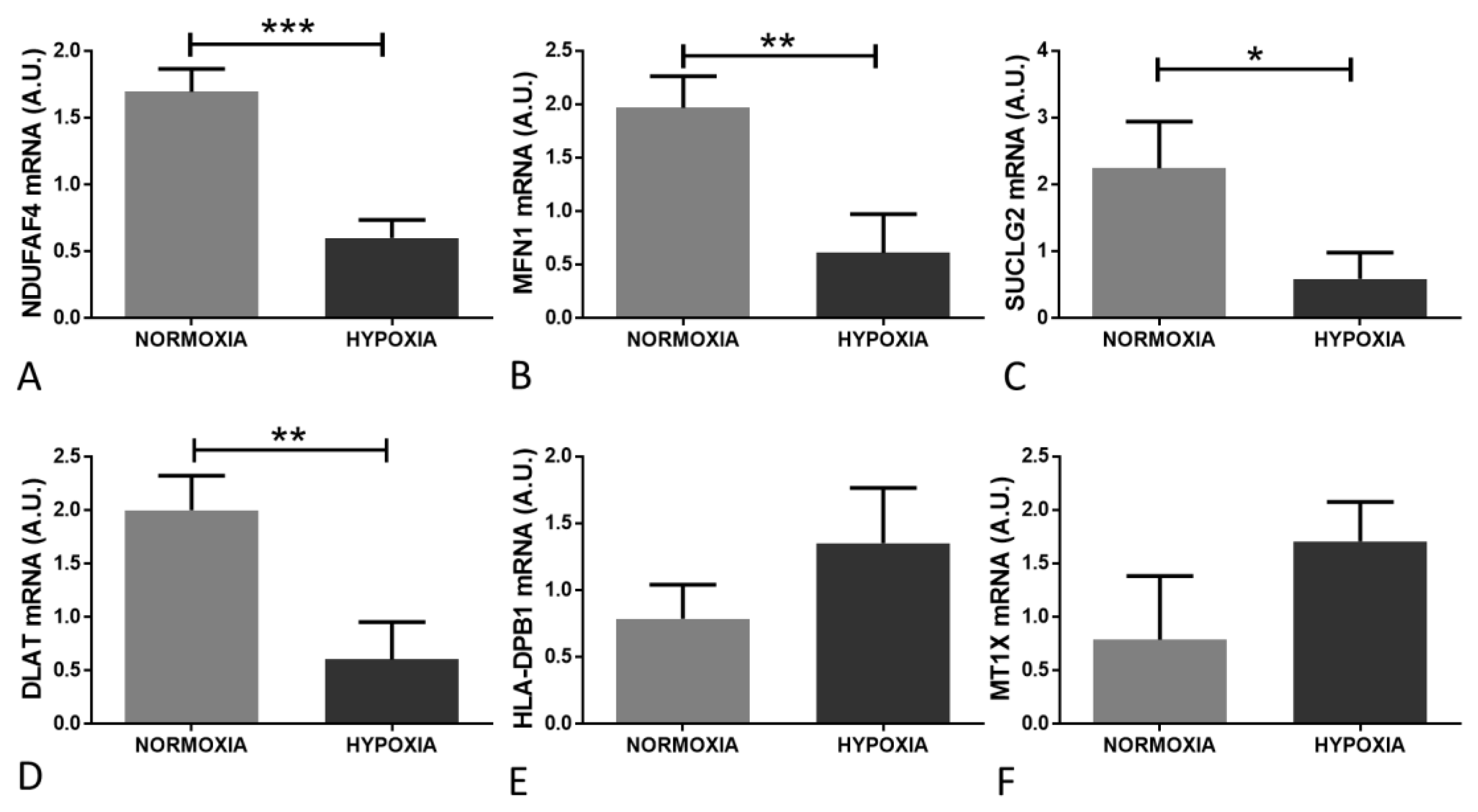
2.4. Validation of Candidate Gene Expression Changes
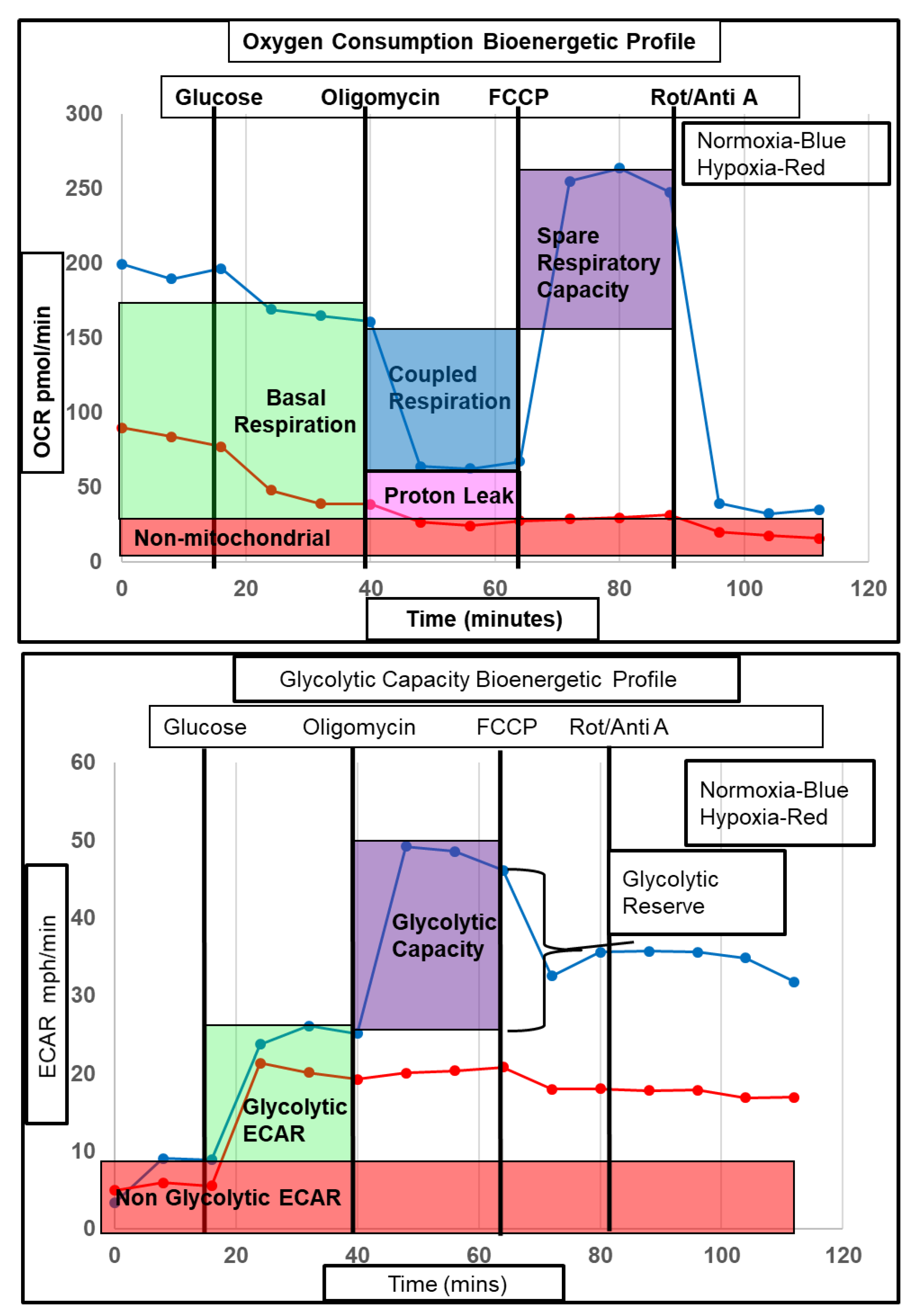
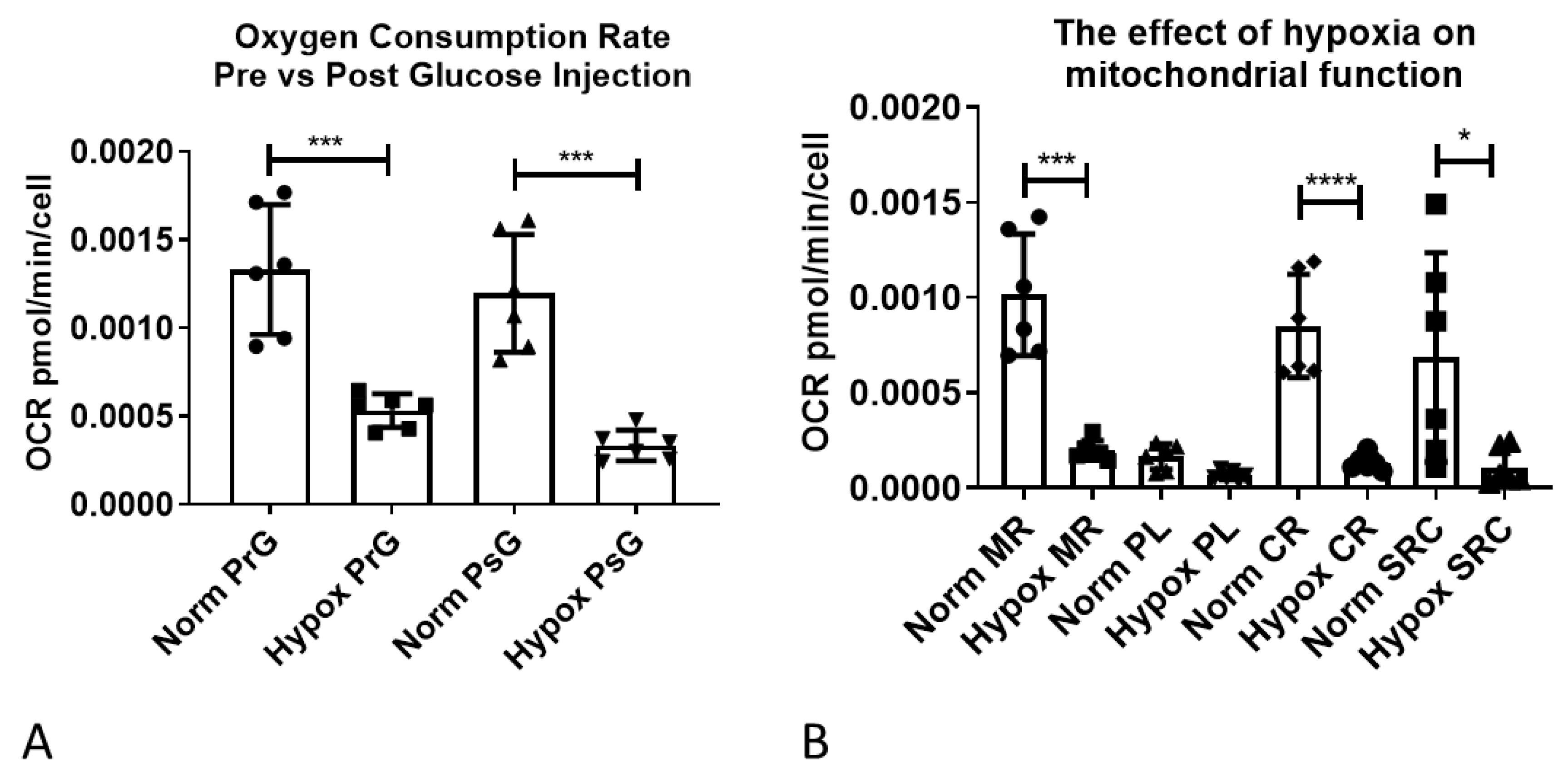
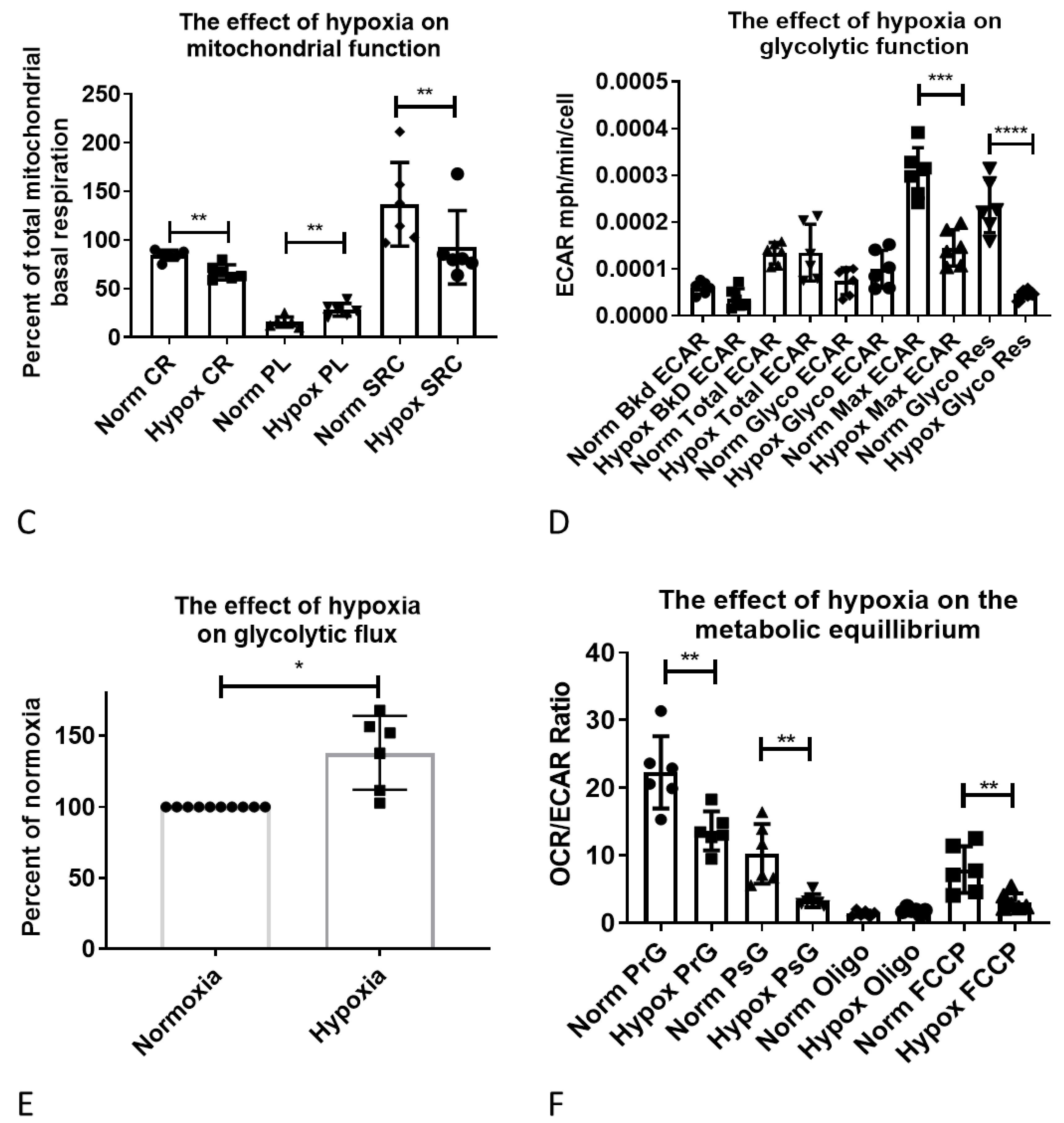
2.5. Functional Validation of Hypoxia-Induced Changes in Astrocyte Metabolism
3. Discussion
4. Materials and Methods
4.1. Primary Human Astrocytes
4.2. Microarray Analysis of Hypoxia-Induced Gene Expression Changes
4.3. Validation of Microarray Data: Quantitative Real-Time Polymerase Chain Reaction (qPCR)
4.4. XF Bioanalyser Metabolic Flux Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ADAM | a disintegrin and metalloproteinase |
| ALS | amyotrophic lateral sclerosis |
| ATP | adenosine tri-phosphate |
| CNS | central nervous system |
| CR | coupled to respiration ATP-linked flux |
| DAVID | Database for Annotation Visualisation and Integrated Discovery |
| ECAR | extracellular acidification rate |
| ETC | electron transport chain |
| FC | fold change |
| FCCP | carbonyl cyanide p-trifluoromethoxy phenylhydrazone |
| HIF-1 | hypoxia inducible factor-1 |
| HREs | hypoxia responsive elements |
| IGF | insulin-like growth factor |
| IGFR | insulin-like growth factor receptors |
| IL | interleukin |
| IR | insulin receptor |
| IRS | receptor substrates |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| MMP | matrix metalloproteinases |
| MR | mitochondrial respiratory flux |
| NGF | non-glycolytic flux |
| OCR | oxygen consumption rate |
| PD | Parkinson’s disease |
| PL | proton leak linked flux |
| PrG | pre-glucose |
| PsG | post-glucose |
| QPCR | quantitative polymerase chain reaction |
| RIN | RNA Integrity Number |
| SRC | spare respiratory capacity |
| TCA | tri-carboxylic acid cycle |
| TIMP | tissue inhibitor of metalloproteinase |
References
- Pluta, R.; Furmaga-Jablonska, W.; Maciejewski, R.; Ulamek-Koziol, M.; Jablonski, M. Brain ischemia activates beta-and gamma-secretase cleavage of amyloid precursor protein: Significance in sporadic Alzheimer’s disease. Mol. Neurobiol. 2013, 47, 425–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.; Tian, S.; Gao, H.; Xu, Y. Hypoxia increases Abeta-induced tau phosphorylation by calpain and promotes behavioral consequences in AD transgenic mice. J. Mol. Neurosci. 2013, 51, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Hypoxia/ischemia activate processing of Amyloid Precursor Protein: Impact of vascular dysfunction in the pathogenesis of Alzheimer’s disease. J. Neurochem. 2017, 140, 536–549. [Google Scholar] [CrossRef] [Green Version]
- Oosthuyse, B.; Moons, L.; Storkebaum, E.; Beck, H.; Nuyens, D.; Brusselmans, K.; Van Dorpe, J.; Hellings, P.; Gorselink, M.; Heymans, S.; et al. Deletion of the hypoxia-response element in the vascular endothelial growth factor promoter causes motor neuron degeneration. Nat. Genet. 2001, 28, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Morimoto, N.; Kurata, T.; Mimoto, T.; Miyazaki, K.; Ikeda, Y.; Abe, K. Impaired response of hypoxic sensor protein HIF-1alpha and its downstream proteins in the spinal motor neurons of ALS model mice. Brain Res. 2012, 1473, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Nagara, Y.; Tateishi, T.; Yamasaki, R.; Hayashi, S.; Kawamura, M.; Kikuchi, H.; Iinuma, K.M.; Tanaka, M.; Iwaki, T.; Matsushita, T.; et al. Impaired cytoplasmic-nuclear transport of hypoxia-inducible factor-1alpha in amyotrophic lateral sclerosis. Brain Pathol. 2013, 23, 534–546. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, J.; Li, H.; Wang, X.; Zhu, L.; Fan, M.; Wang, X. Hypoxia promotes dopaminergic differentiation of mesenchymal stem cells and shows benefits for transplantation in a rat model of Parkinson’s disease. PLoS ONE 2013, 8, e54296. [Google Scholar] [CrossRef]
- Kandil, E.A.; Sayed, R.H.; Ahmed, L.A.; Abd El Fattah, M.A.; El-Sayeh, B.M. Hypoxia-inducible factor 1 alpha and nuclear-related receptor 1 as targets for neuroprotection by albendazole in a rat rotenone model of Parkinson’s disease. Clin. Exp. Pharmacol. Physiol. 2019, 46, 1141–1150. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yan, J.; Chang, Y.; ShiDu Yan, S.; Shi, H. Hypoxia inducible factor-1 as a target for neurodegenerative diseases. Curr. Med. Chem. 2011, 18, 4335–4343. [Google Scholar] [CrossRef] [Green Version]
- Love, S.; Miners, J.S. Cerebral Hypoperfusion and the Energy Deficit in Alzheimer’s Disease. Brain Pathol. 2016, 26, 607–617. [Google Scholar] [CrossRef] [Green Version]
- Garwood, C.J.; Ratcliffe, L.E.; Simpson, J.E.; Heath, P.R.; Ince, P.G.; Wharton, S.B. Review: Astrocytes in Alzheimer’s disease and other age-associated dementias: A supporting player with a central role. Neuropathol. Appl. Neurobiol. 2017, 43, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Marina, N.; Kasymov, V.; Ackland, G.L.; Kasparov, S.; Gourine, A.V. Astrocytes and Brain Hypoxia. Adv. Exp. Med. Biol. 2016, 903, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Samy, Z.A.; Al-Abdullah, L.; Turcani, M.; Craik, J.; Redzic, Z. Rat astrocytes during anoxia: Secretome profile of cytokines and chemokines. Brain Behav. 2018, 8, e01013. [Google Scholar] [CrossRef] [PubMed]
- Ranjbar Taklimie, F.; Gasterich, N.; Scheld, M.; Weiskirchen, R.; Beyer, C.; Clarner, T.; Zendedel, A. Hypoxia Induces Astrocyte-Derived Lipocalin-2 in Ischemic Stroke. Int. J. Mol. Sci. 2019, 20, 1271. [Google Scholar] [CrossRef] [Green Version]
- LFS, E.S.; Brito, M.D.; Yuzawa, J.M.C.; Rosenstock, T.R. Mitochondrial Dysfunction and Changes in High-Energy Compounds in Different Cellular Models Associated to Hypoxia: Implication to Schizophrenia. Sci. Rep. 2019, 9, 18049. [Google Scholar] [CrossRef] [Green Version]
- Correia, S.C.; Moreira, P.I. Hypoxia-inducible factor 1: A new hope to counteract neurodegeneration? J. Neurochem. 2010, 112, 1–12. [Google Scholar] [CrossRef]
- Liu, W.; Shen, S.M.; Zhao, X.Y.; Chen, G.Q. Targeted genes and interacting proteins of hypoxia inducible factor-1. Int. J. Biochem Mol. Biol. 2012, 3, 165–178. [Google Scholar]
- Hasselblatt, M.; Paulus, W. Astrocytic gene expression profiling upon hypoxia. Neuroreport 2006, 17, 51–54. [Google Scholar] [CrossRef]
- Chadwick, W.; Boyle, J.P.; Zhou, Y.; Wang, L.; Park, S.S.; Martin, B.; Wang, R.; Becker, K.G.; Wood, W.H., 3rd; Zhang, Y.; et al. Multiple oxygen tension environments reveal diverse patterns of transcriptional regulation in primary astrocytes. PLoS ONE 2011, 6, e21638. [Google Scholar] [CrossRef]
- Salman, M.M.; Kitchen, P.; Woodroofe, M.N.; Bill, R.M.; Conner, A.C.; Heath, P.R.; Conner, M.T. Transcriptome Analysis of Gene Expression Provides New Insights into the Effect of Mild Therapeutic Hypothermia on Primary Human Cortical Astrocytes Cultured under Hypoxia. Front. Cell Neurosci. 2017, 11, 386. [Google Scholar] [CrossRef] [Green Version]
- Mense, S.M.; Sengupta, A.; Zhou, M.; Lan, C.; Bentsman, G.; Volsky, D.J.; Zhang, L. Gene expression profiling reveals the profound upregulation of hypoxia-responsive genes in primary human astrocytes. Physiol. Genom. 2006, 25, 435–449. [Google Scholar] [CrossRef]
- Dengler, V.L.; Galbraith, M.; Espinosa, J.M. Transcriptional regulation by hypoxia inducible factors. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Salazar, C.; Yanez, O.; Elorza, A.A.; Cortes, N.; Garcia-Beltran, O.; Tiznado, W.; Ruiz, L.M. Biosystem Analysis of the Hypoxia Inducible Domain Family Member 2A: Implications in Cancer Biology. Genes 2020, 11, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Mijn, J.C.; Fu, L.; Khani, F.; Zhang, T.; Molina, A.M.; Barbieri, C.E.; Chen, Q.; Gross, S.S.; Gudas, L.J.; Nanus, D.M. Combined Metabolomics and Genome-Wide Transcriptomics Analyses Show Multiple HIF1alpha-Induced Changes in Lipid Metabolism in Early Stage Clear Cell Renal Cell Carcinoma. Transl. Oncol. 2020, 13, 177–185. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, C.; Kiely, A.P.; Coakley, M.F.; Manning, S.; Long-Smith, C.M. Insulin and IGF-1 signalling: Longevity, protein homoeostasis and Alzheimer’s disease. Biochem. Soc. Trans. 2012, 40, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Bernard-Marissal, N.; Chrast, R.; Schneider, B.L. Endoplasmic reticulum and mitochondria in diseases of motor and sensory neurons: A broken relationship? Cell Death Dis. 2018, 9, 333. [Google Scholar] [CrossRef]
- Rivera, S.; Garcia-Gonzalez, L.; Khrestchatisky, M.; Baranger, K. Metalloproteinases and their tissue inhibitors in Alzheimer’s disease and other neurodegenerative disorders. Cell Mol. Life Sci. 2019, 76, 3167–3191. [Google Scholar] [CrossRef]
- Allen, S.P.; Hall, B.; Woof, R.; Francis, L.; Gatto, N.; Shaw, A.C.; Myszczynska, M.; Hemingway, J.; Coldicott, I.; Willcock, A.; et al. C9orf72 expansion within astrocytes reduces metabolic flexibility in amyotrophic lateral sclerosis. Brain 2019, 142, 3771–3790. [Google Scholar] [CrossRef]
- Miyazaki, K.; Masamoto, K.; Morimoto, N.; Kurata, T.; Mimoto, T.; Obata, T.; Kanno, I.; Abe, K. Early and progressive impairment of spinal blood flow-glucose metabolism coupling in motor neuron degeneration of ALS model mice. J. Cereb. Blood Flow Metab. 2012, 32, 456–467. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.M.; Kim, H.; Lee, J.S.; Park, K.S.; Jeon, G.S.; Shon, J.; Ahn, S.W.; Kim, S.H.; Lee, K.M.; Sung, J.J.; et al. Intermittent hypoxia can aggravate motor neuronal loss and cognitive dysfunction in ALS mice. PLoS ONE 2013, 8, e81808. [Google Scholar] [CrossRef] [Green Version]
- Shiota, S.; Takekawa, H.; Matsumoto, S.E.; Takeda, K.; Nurwidya, F.; Yoshioka, Y.; Takahashi, F.; Hattori, N.; Tabira, T.; Mochizuki, H.; et al. Chronic intermittent hypoxia/reoxygenation facilitate amyloid-beta generation in mice. J. Alzheimers Dis. 2013, 37, 325–333. [Google Scholar] [CrossRef] [Green Version]
- Lowe, R.; Shirley, N.; Bleackley, M.; Dolan, S.; Shafee, T. Transcriptomics technologies. PLoS Comput. Biol. 2017, 13, e1005457. [Google Scholar] [CrossRef] [Green Version]
- Verkhratsky, A.; Parpura, V.; Vardjan, N.; Zorec, R. Physiology of Astroglia. Adv. Exp. Med. Biol 2019, 1175, 45–91. [Google Scholar] [CrossRef]
- Hyder, F.; Rothman, D.L.; Bennett, M.R. Cortical energy demands of signaling and nonsignaling components in brain are conserved across mammalian species and activity levels. Proc. Natl. Acad. Sci. USA 2013, 110, 3549–3554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magistretti, P.J.; Allaman, I. A cellular perspective on brain energy metabolism and functional imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAvoy, K.; Kawamata, H. Glial mitochondrial function and dysfunction in health and neurodegeneration. Mol. Cell Neurosci. 2019, 101, 103417. [Google Scholar] [CrossRef]
- Allen, S.P.; Hall, B.; Castelli, L.M.; Francis, L.; Woof, R.; Siskos, A.P.; Kouloura, E.; Gray, E.; Thompson, A.G.; Talbot, K.; et al. Astrocyte adenosine deaminase loss increases motor neuron toxicity in amyotrophic lateral sclerosis. Brain 2019, 142, 586–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfleger, J.; He, M.; Abdellatif, M. Mitochondrial complex II is a source of the reserve respiratory capacity that is regulated by metabolic sensors and promotes cell survival. Cell Death Dis. 2015, 6, e1835. [Google Scholar] [CrossRef] [Green Version]
- Porter, L.; Toepfner, N.; Bashant, K.R.; Guck, J.; Ashcroft, M.; Farahi, N.; Chilvers, E.R. Metabolic Profiling of Human Eosinophils. Front. Immunol. 2018, 9, 1404. [Google Scholar] [CrossRef] [PubMed]
- Dimeloe, S.; Mehling, M.; Frick, C.; Loeliger, J.; Bantug, G.R.; Sauder, U.; Fischer, M.; Belle, R.; Develioglu, L.; Tay, S.; et al. The Immune-Metabolic Basis of Effector Memory CD4+ T Cell Function under Hypoxic Conditions. J. Immunol. 2016, 196, 106–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, H.; Board, M.; Pellagatti, A.; Turley, H.; Boultwood, J.; Callaghan, R. The Effects of Severe Hypoxia on Glycolytic Flux and Enzyme Activity in a Model of Solid Tumors. J. Cell Biochem. 2016, 117, 1890–1901. [Google Scholar] [CrossRef] [PubMed]
- Vlaski, M.; Negroni, L.; Kovacevic-Filipovic, M.; Guibert, C.; Brunet de la Grange, P.; Rossignol, R.; Chevaleyre, J.; Duchez, P.; Lafarge, X.; Praloran, V.; et al. Hypoxia/hypercapnia-induced adaptation maintains functional capacity of cord blood stem and progenitor cells at 4 degrees C. J. Cell Physiol. 2014, 229, 2153–2165. [Google Scholar] [CrossRef]
- Simmen, T.; Herrera-Cruz, M.S. Plastic mitochondria-endoplasmic reticulum (ER) contacts use chaperones and tethers to mould their structure and signaling. Curr. Opin. Cell Biol. 2018, 53, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Erpapazoglou, Z.; Mouton-Liger, F.; Corti, O. From dysfunctional endoplasmic reticulum-mitochondria coupling to neurodegeneration. Neurochem. Int. 2017, 109, 171–183. [Google Scholar] [CrossRef] [Green Version]
- Krols, M.; van Isterdael, G.; Asselbergh, B.; Kremer, A.; Lippens, S.; Timmerman, V.; Janssens, S. Mitochondria-associated membranes as hubs for neurodegeneration. Acta. Neuropathol. 2016, 131, 505–523. [Google Scholar] [CrossRef] [Green Version]
- Frank-Cannon, T.C.; Alto, L.T.; McAlpine, F.E.; Tansey, M.G. Does neuroinflammation fan the flame in neurodegenerative diseases? Mol. Neurodegener 2009, 4, 47. [Google Scholar] [CrossRef] [Green Version]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimers Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef]
- Chiot, A.; Lobsiger, C.S.; Boillee, S. New insights on the disease contribution of neuroinflammation in amyotrophic lateral sclerosis. Curr. Opin. Neurol. 2019, 32, 764–770. [Google Scholar] [CrossRef]
- Lee, Y.; Lee, S.; Chang, S.C.; Lee, J. Significant roles of neuroinflammation in Parkinson’s disease: Therapeutic targets for PD prevention. Arch. Pharm Res. 2019, 42, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.W.; Zhang, X.; Huang, W.J. Role of neuroinflammation in neurodegenerative diseases (Review). Mol. Med. Rep. 2016, 13, 3391–3396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kempuraj, D.; Thangavel, R.; Natteru, P.A.; Selvakumar, G.P.; Saeed, D.; Zahoor, H.; Zaheer, S.; Iyer, S.S.; Zaheer, A. Neuroinflammation Induces Neurodegeneration. J. Neurol. Neurosurg. Spine 2016, 1, 1003–1017. [Google Scholar] [PubMed]
- Qi, G.; Mi, Y.; Yin, F. Cellular Specificity and Inter-cellular Coordination in the Brain Bioenergetic System: Implications for Aging and Neurodegeneration. Front. Physiol. 2019, 10, 1531. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V. Multiple roles for astrocytes as effectors of cytokines and inflammatory mediators. Neuroscientist 2014, 20, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.H.; Fatima, M.; Mondal, A.C. Influence of microglia and astrocyte activation in the neuroinflammatory pathogenesis of Alzheimer’s disease: Rational insights for the therapeutic approaches. J. Clin. Neurosci. 2019, 59, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Guyon, A. CXCL12 chemokine and its receptors as major players in the interactions between immune and nervous systems. Front. Cell Neurosci. 2014, 8, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bezzi, P.; Domercq, M.; Brambilla, L.; Galli, R.; Schols, D.; De Clercq, E.; Vescovi, A.; Bagetta, G.; Kollias, G.; Meldolesi, J.; et al. CXCR4-activated astrocyte glutamate release via TNFalpha: Amplification by microglia triggers neurotoxicity. Nat. Neurosci. 2001, 4, 702–710. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Sobrino, T.; Orbe, J.; Purroy, A.; Martinez-Vila, E.; Castillo, J.; Paramo, J.A. proMetalloproteinase-10 is associated with brain damage and clinical outcome in acute ischemic stroke. J. Thromb. Haemost. 2013, 11, 1464–1473. [Google Scholar] [CrossRef]
- Galimberti, D.; Scarpini, E. Pioglitazone for the treatment of Alzheimer’s disease. Expert Opin. Investig. Drugs 2017, 26, 97–101. [Google Scholar] [CrossRef]
- Int’ Veld, B.A.; Ruitenberg, A.; Hofman, A.; Launer, L.J.; van Duijn, C.M.; Stijnen, T.; Breteler, M.M.; Stricker, B.H. Nonsteroidal antiinflammatory drugs and the risk of Alzheimer’s disease. N. Engl. J. Med. 2001, 345, 1515–1521. [Google Scholar] [CrossRef] [Green Version]
- Meyer, P.F.; Tremblay-Mercier, J.; Leoutsakos, J.; Madjar, C.; Lafaille-Maignan, M.E.; Savard, M.; Rosa-Neto, P.; Poirier, J.; Etienne, P.; Breitner, J.; et al. INTREPAD: A randomized trial of naproxen to slow progress of presymptomatic Alzheimer disease. Neurology 2019, 92, e2070–e2080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soucek, T.; Cumming, R.; Dargusch, R.; Maher, P.; Schubert, D. The regulation of glucose metabolism by HIF-1 mediates a neuroprotective response to amyloid beta peptide. Neuron 2003, 39, 43–56. [Google Scholar] [CrossRef] [Green Version]
- Greijer, A.E.; van der Wall, E. The role of hypoxia inducible factor 1 (HIF-1) in hypoxia induced apoptosis. J. Clin. Pathol. 2004, 57, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Nomura, E.; Ohta, Y.; Tadokoro, K.; Shang, J.; Feng, T.; Liu, X.; Shi, X.; Matsumoto, N.; Sasaki, R.; Tsunoda, K.; et al. Imaging Hypoxic Stress and the Treatment of Amyotrophic Lateral Sclerosis with Dimethyloxalylglycine in a Mice Model. Neuroscience 2019, 415, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, M.; Jain, I.H.; Goldberger, O.; Rezoagli, E.; Thoonen, R.; Cheng, K.H.; Sosnovik, D.E.; Scherrer-Crosbie, M.; Mootha, V.K.; Zapol, W.M. Hypoxia treatment reverses neurodegenerative disease in a mouse model of Leigh syndrome. Proc. Nat.l Acad. Sci. USA 2017, 114, E4241–E4250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutisya, E.M.; Bowling, A.C.; Beal, M.F. Cortical cytochrome oxidase activity is reduced in Alzheimer’s disease. J. Neurochem 1994, 63, 2179–2184. [Google Scholar] [CrossRef]
- Bonham, L.W.; Karch, C.M.; Fan, C.C.; Tan, C.; Geier, E.G.; Wang, Y.; Wen, N.; Broce, I.J.; Li, Y.; Barkovich, M.J.; et al. CXCR4 involvement in neurodegenerative diseases. Transl. Psychiatry 2018, 8, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, N.; Fang, B.; Li, Z.; Tian, A. Exogenous activation of cannabinoid-2 receptor modulates TLR4/MMP9 expression in a spinal cord ischemia reperfusion rat model. J. Neuroinflamm. 2020, 17, 101. [Google Scholar] [CrossRef] [Green Version]
- Ratcliffe, L.E.; Vazquez Villasenor, I.; Jennings, L.; Heath, P.R.; Mortiboys, H.; Schwartzentruber, A.; Karyka, E.; Simpson, J.E.; Ince, P.G.; Garwood, C.J.; et al. Loss of IGF1R in Human Astrocytes Alters Complex I Activity and Support for Neurons. Neuroscience 2018, 390, 46–59. [Google Scholar] [CrossRef]
- Babicki, S.; Arndt, D.; Marcu, A.; Liang, Y.; Grant, J.R.; Maciejewski, A.; Wishart, D.S. Heatmapper: Web-enabled heat mapping for all. Nucleic Acids Res. 2016, 44, W147–W153. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transcript | Gene Symbol | Gene Name | FC | p-Value |
|---|---|---|---|---|
| 202912_at | ADM | adrenomedullin | 3.4 | 0.011 |
| 217254_s_at | EPO | erythropoietin | 1.9 | 0.039 |
| 209328_x_at | HIGD2A | HIG1 hypoxia inducible domain family, member 2A | 1.3 | 0.042 |
| 209329_x_at | HIGD2A | HIG1 hypoxia inducible domain family, member 2A | 1.6 | 0.040 |
| 218507_at | HILPDA | hypoxia inducible lipid droplet-associated | 6.1 | 0.018 |
| 1554452_a_at | HILPDA | hypoxia inducible lipid droplet-associated | 4.4 | 0.018 |
| 207092_at | LEP | leptin | 1.6 | 0.030 |
| 229093_at | NOS3 | nitric oxide synthase 3 (endothelial cell) | 6.4 | 0.028 |
| 205581_s_at | NOS3 | nitric oxide synthase 3 (endothelial cell) | 1.8 | 0.035 |
| 217112_at | PDGFB | platelet-derived growth factor beta | 2.3 | 0.048 |
| 216061_x_at | PDGFB | platelet-derived growth factor beta | 1.8 | 0.028 |
| 203400_s_at | TF | transferrin | 1.8 | 0.031 |
| 212171_x_at | VEGFA | vascular endothelial growth factor A | 4.2 | 0.020 |
| 211527_x_at | VEGFA | vascular endothelial growth factor A | 2.7 | 0.054 |
| 210513_s_at | VEGFA | vascular endothelial growth factor A | 2.7 | 0.030 |
| 203683_s_at | VEGFB | vascular endothelial growth factor B | 2.0 | 0.052 |
| 209946_at | VEGFC | vascular endothelial growth factor C | 1.3 | 0.051 |
| KEGG Pathway | p-Value |
|---|---|
| Hypoxia inducible factor-1 (HIF-1) signalling pathway | 0.002 |
| Phosphatidylinositol 3-kinase and protein kinase B (PI3K-Akt) signalling pathway | 0.005 |
| Renal cell carcinoma | 0.006 |
| Glycolysis/Gluconeogenesis | 0.006 |
| Staphylococcus aureus infection | 0.007 |
| Oxytocin signalling pathway | 0.010 |
| Pathways in cancer | 0.013 |
| Insulin resistance | 0.014 |
| Human T- cell leukemia virus, type 1 (HTLV-I) infection | 0.019 |
| Biosynthesis of amino acids | 0.030 |
| adenosine monophosphate-activated protein kinase (AMPK) signalling pathway | 0.032 |
| Cell adhesion molecules (CAMs) | 0.035 |
| Circadian rhythm | 0.038 |
| Fructose and mannose metabolism | 0.043 |
| Intestinal immune network for IgA production | 0.045 |
| Graft-versus-host disease | 0.047 |
| KEGG Pathway | p-Value |
|---|---|
| Cell cycle | 6.1 × 10−9 |
| RNA transport | 1.9 × 10−7 |
| Ubiquitin mediated proteolysis | 4.5 × 10−7 |
| Oocyte meiosis | 1.9 × 10−5 |
| Protein processing in endoplasmic reticulum | 5.6 × 10−5 |
| RNA degradation | 1.6 × 10−4 |
| Spliceosome | 3.6 × 10−4 |
| Wnt signalling | 0.002 |
| Endocytosis | 0.006 |
| Pyramidine metabolism | 0.008 |
| Progesterone-mediated oocyte maturation | 0.012 |
| Peroxisome | 0.015 |
| Tricarboxylic acid cycle (TCA cycle) | 0.016 |
| p53 signalling | 0.016 |
| DNA replication | 0.019 |
| Pathways in cancer | 0.02 |
| Purine metabolism | 0.02 |
| Valine, leucine and isoleucine degradation | 0.02 |
| RNA polymerase | 0.02 |
| Fatty acid metabolism | 0.03 |
| Protein export | 0.03 |
| Chronic myeloid leukemia | 0.03 |
| mRNA surveillance pathway | 0.04 |
| HTLV-I infection | 0.04 |
| Gene | Sequence | |
|---|---|---|
| DLAT | Probe | 56-FAM/CGCTGTGCA/ZEN/ATAACCCGACGAATG/3IABkFQ |
| Primer 1 | CCAGTTCCTACAGGTGTCTTC | |
| Primer 2 | TGAGGTATGGTTTGCTTTGATTG | |
| HLADPB1 | Probe | 56-FAM/CCCACTCCA/ZEN/CAGATGATGAGCCC/3IABkFQ |
| Primer 1 | GCTCCTCCTGTGCATGAAG | |
| Primer 2 | CAAGTGGAGCACACCAG | |
| MFN1 | Probe | 56-FAM/AGCTTCTAC/ZEN/TCCCACTGCTCCTACC/3IABkFQ |
| Primer 1 | GAAATGCTCAAAGGGTGCTC | |
| Primer 2 | GTGATGCATTATCTGGCGTTG | |
| MT1X | Probe | 56-FAM/AGCTCGCCA/ZEN/TGGATCCCAACT/3IABkFQ |
| Primer 1 | GCAACCTGTCCCGACTCTA | |
| Primer 2 | AGCTTTTCTTGCAGGAGGTG | |
| NDUFAF4 | Probe | 56-FAM/CTGTGTCTT/ZEN/CCTTGCAGGTAAAAGCTG/3IABkFQ |
| Primer 1 | CCAGAAGTTAAAGGAGAGATTGCT | |
| Primer 2 | GAATTCCTTCGGCTCTTGAC | |
| SUCLG2 | Probe | 56-FAM/CACAGCTGA/ZEN/TCCTAAGGTTGAAGCCA/3IABkFQ |
| Primer 1 | TTGGAGGTGGTGTAAAGGAAG | |
| Primer 2 | GGCAATGATGGCACAGTTG | |
| ACTB | Probe | 56-FAM/CTGCCTCCA/ZEN/CCCACTCCCA/3IABkFQ |
| Primer 1 | GTCCCCCAACTTGAGATGTATG | |
| Primer 2 | AAGTCAGTGTACAGGTAAGCC | |
| GAPDH | Probe | 56-FAM/AAGGTCGGA/ZEN/GTCAACGGATTTGGTC/3IABkFQ |
| Primer 1 | ACATCGCTCAGACACCATG | |
| Primer 2 | TGTAGTTGAGGTCAATGAAGGG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allen, S.P.; Seehra, R.S.; Heath, P.R.; Hall, B.P.C.; Bates, J.; Garwood, C.J.; Matuszyk, M.M.; Wharton, S.B.; Simpson, J.E. Transcriptomic Analysis of Human Astrocytes In Vitro Reveals Hypoxia-Induced Mitochondrial Dysfunction, Modulation of Metabolism, and Dysregulation of the Immune Response. Int. J. Mol. Sci. 2020, 21, 8028. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218028
Allen SP, Seehra RS, Heath PR, Hall BPC, Bates J, Garwood CJ, Matuszyk MM, Wharton SB, Simpson JE. Transcriptomic Analysis of Human Astrocytes In Vitro Reveals Hypoxia-Induced Mitochondrial Dysfunction, Modulation of Metabolism, and Dysregulation of the Immune Response. International Journal of Molecular Sciences. 2020; 21(21):8028. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218028
Chicago/Turabian StyleAllen, Scott P., Rajpinder Singh Seehra, Paul R. Heath, Benjamin P. C. Hall, Jessica Bates, Claire J. Garwood, Martyna M. Matuszyk, Stephen B. Wharton, and Julie E. Simpson. 2020. "Transcriptomic Analysis of Human Astrocytes In Vitro Reveals Hypoxia-Induced Mitochondrial Dysfunction, Modulation of Metabolism, and Dysregulation of the Immune Response" International Journal of Molecular Sciences 21, no. 21: 8028. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218028