Priming with HDAC Inhibitors Sensitizes Ovarian Cancer Cells to Treatment with Cisplatin and HSP90 Inhibitors


 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. HDACi and HSP90i Mediated Biological Effects in Ovarian Cancer Cells
2.2. Treatment Order of HDACi and HSP90i Affects Increase in Cytotoxic Activity and Apoptosis Induction in A2780 and A2780CisR
2.3. Effects of HDACi and HSP90i on Cisplatin Induced Cytotoxicity and Apoptosis
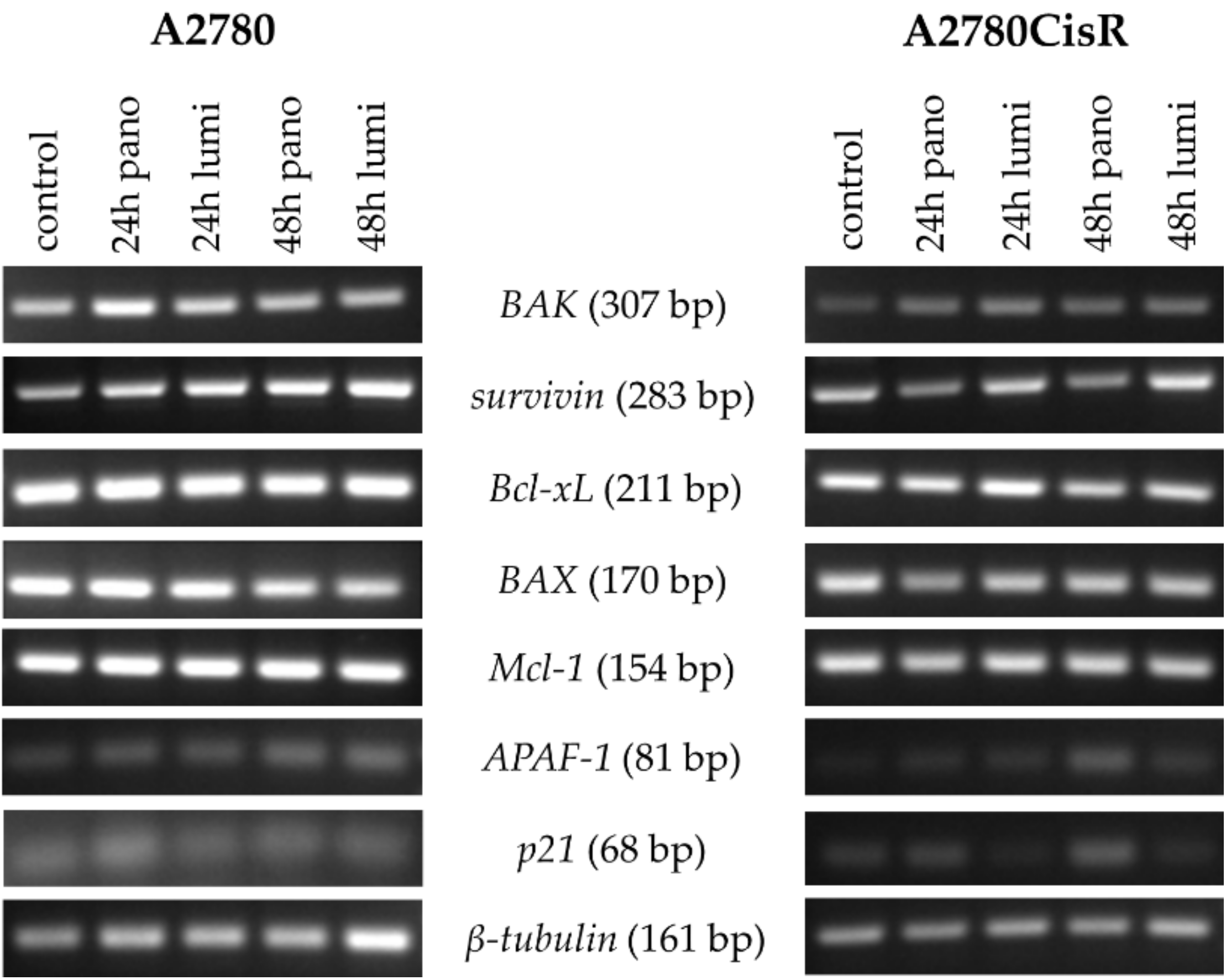
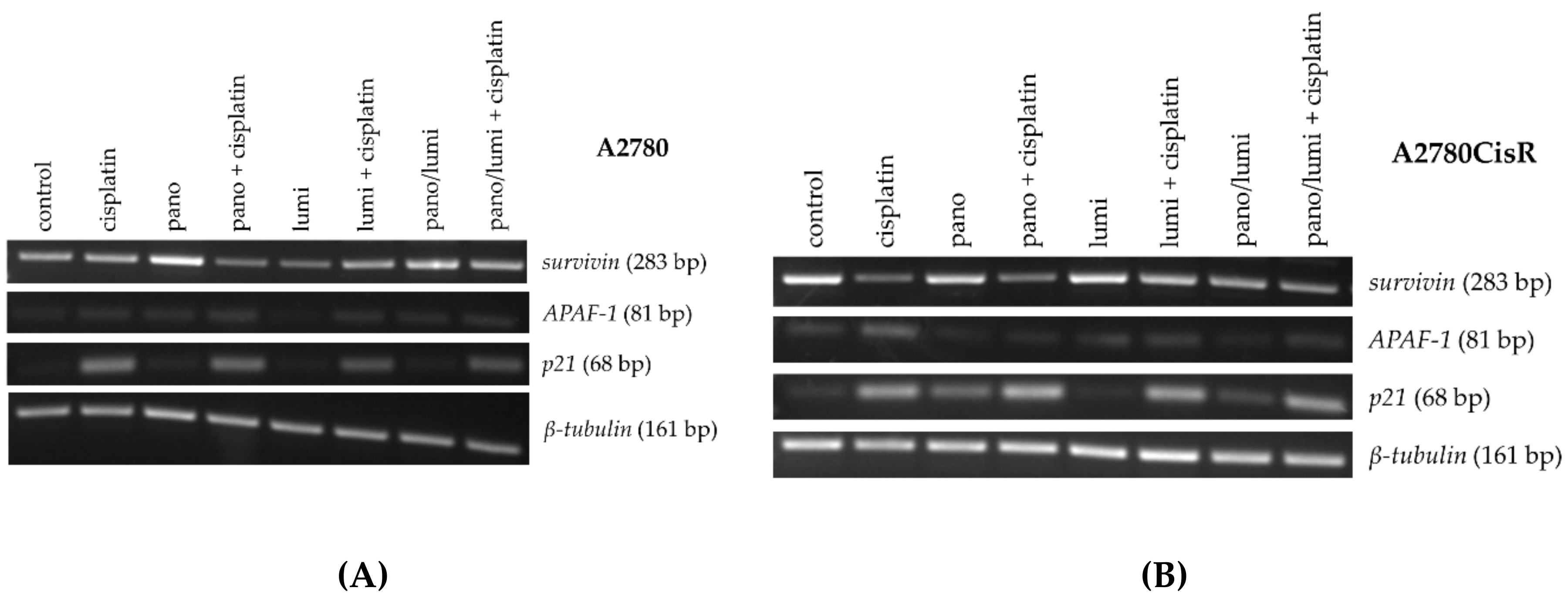
2.4. Effects of HDACi and/or HSP90i Plus Cisplatin Incubation on mRNA and Protein Expression of Pro-/Antiapoptotic Key Genes
2.5. Effects of Panobinostat or HSP990 Plus Cisplatin on Cell Viability in High Grade Serous Ovarian Cancer Cell Lines CaOV3 and OVCAR3 and Their Cisplatin-Resistant Sub-Cell Lines
2.6. Effects of HDACi or HSP90i and Cisplatin on the Non-Cancer Cell Line HEK293
2.7. Long Term Treatment with Low-Dose HDACi or HSP90i can Overcome and Prevent the Development of Cisplatin Resistance in A2780 Ovarian Cancer Cells.
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Lines
4.3. Cell Viability Assay
4.4. Doubling Time
4.5. Analysis of Apoptosis Induction
4.6. Immunoblotting
4.7. RNA Extraction and PCR
4.8. Caspase 3/7 Activation Assay
4.9. Statistical Methods
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| (p)Akt/AKT | (phosphorylated) protein kinase B |
| APAF-1 | apoptotic protease activating factor 1 |
| BAK | Bcl-2 homologous antagonist killer |
| BAX | Bcl-2-associated X protein |
| Bcl-xL | B-cell lymphoma-extra large |
| cDDP | cis-diamminedichloroplatinum(II) (cisplatin) |
| coinc | coincubation |
| HDAC | histone deacetylase |
| HDACi | histone deacetylase inhibitor |
| HSP90 | heat shock protein 90 |
| HSP90i | heat shock protein 90 inhibitor |
| HSP990 | NVP-HSP990 |
| lumi | luminespib; NVP-AUY922 |
| pano | panobinostat |
| PCR | polymerase chain reaction |
| preinc | preincubation |
References
- Ward, E.M.; Sherman, R.L.; Henley, S.J.; Jemal, A.; Siegel, D.A.; Feuer, E.J.; Firth, A.U.; Kohler, B.A.; Scott, S.; Ma, J.; et al. Annual Report to the Nation on the Status of Cancer, Featuring Cancer in Men and Women Age 20–49 Years. J. Natl Cancer Inst. 2019, 111, 1279–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer of the Ovary—Cancer Stat Facts. Available online: https://seer.cancer.gov/statfacts/html/ovary.html (accessed on 1 March 2020).
- Krebs—Datenbankabfrage—RKI. Available online: https://www.krebsdaten.de/Krebs/SiteGlobals/Forms/Datenbankabfrage/datenbankabfrage_stufe2_form.html (accessed on 1 March 2020).
- Buttmann-Schweiger, N.; Kraywinkel, K. Epidemiologie von Eierstockkrebs in Deutschland. Onkologe 2019, 25, 92–98. [Google Scholar] [CrossRef]
- Home | American Cancer Society—Cancer Facts & Statistics. Available online: https://cancerstatisticscenter.cancer.org/#!/ (accessed on 18 October 2020).
- Wagner, U.; Harter, P.; Hilpert, F.; Mahner, S.; Reuß, A.; du Bois, A.; Petru, E.; Meier, W.; Ortner, P.; König, K.; et al. S3-Guideline on Diagnostics, Therapy and Follow-up of Malignant Ovarian Tumours: Short version 1.0—AWMF registration number: 032/035OL, June 2013. Geburtshilfe Frauenheilkd 2013, 73, 874–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pchejetski, D.; Alfraidi, A.; Sacco, K.; Alshaker, H.; Muhammad, A.; Monzon, L. Histone deacetylases as new therapy targets for platinum-resistant epithelial ovarian cancer. J. Cancer Res. Clin. Oncol 2016, 142, 1659–1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenhauer, E.A. Real-world evidence in the treatment of ovarian cancer. Ann. Oncol. 2017, 28, viii61–viii65. [Google Scholar] [CrossRef] [PubMed]
- Kehoe, S.; Hook, J.; Nankivell, M.; Jayson, G.C.; Kitchener, H.; Lopes, T.; Luesley, D.; Perren, T.; Bannoo, S.; Mascarenhas, M.; et al. Primary chemotherapy versus primary surgery for newly diagnosed advanced ovarian cancer (CHORUS): An open-label, randomised, controlled, non-inferiority trial. Lancet 2015, 386, 249–257. [Google Scholar] [CrossRef]
- Vergote, I.; Tropé, C.G.; Amant, F.; Kristensen, G.B.; Ehlen, T.; Johnson, N.; Verheijen, R.H.M.; van der Burg, M.E.L.; Lacave, A.J.; Panici, P.B.; et al. Neoadjuvant Chemotherapy or Primary Surgery in Stage IIIC or IV Ovarian Cancer. New Engl. J. Med. 2010, 363, 943–953. [Google Scholar] [CrossRef] [Green Version]
- Balch, C.; Huang, T.H.-M.; Brown, R.; Nephew, K.P. The epigenetics of ovarian cancer drug resistance and resensitization. Am. J. Obstet. Gynecol. 2004, 191, 1552–1572. [Google Scholar] [CrossRef]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khabele, D. The Therapeutic Potential of Class I Selective Histone Deacetylase Inhibitors in Ovarian Cancer. Front. Oncol. 2014, 4. [Google Scholar] [CrossRef] [Green Version]
- Eckl, J.M.; Richter, K. Functions of the Hsp90 chaperone system: Lifting client proteins to new heights. Int J. Biochem Mol. Biol. 2013, 4, 157–165. [Google Scholar]
- Prodromou, C. Mechanisms of Hsp90 regulation. Biochem J. 2016, 473, 2439–2452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Xiao, F.; Serebriiskii, I.G.; O’Brien, S.W.; Maglaty, M.A.; Astsaturov, I.; Litwin, S.; Martin, L.P.; Proia, D.A.; Golemis, E.A.; et al. Network analysis identifies an HSP90-central hub susceptible in ovarian cancer. Clin. Cancer Res. 2013, 19, 5053–5067. [Google Scholar] [CrossRef] [Green Version]
- Maloney, A.; Clarke, P.A.; Naaby-Hansen, S.; Stein, R.; Koopman, J.-O.; Akpan, A.; Yang, A.; Zvelebil, M.; Cramer, R.; Stimson, L.; et al. Gene and protein expression profiling of human ovarian cancer cells treated with the heat shock protein 90 inhibitor 17-allylamino-17-demethoxygeldanamycin. Cancer Res. 2007, 67, 3239–3253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, K.; Hendershot, L.M.; Freeman, B.C. The cellular world according to Hsp90. Nat. Struct. Mol. Biol. 2007, 14, 90–94. [Google Scholar] [CrossRef]
- Mielczarek-Lewandowska, A.; Hartman, M.L.; Czyz, M. Inhibitors of HSP90 in melanoma. Apoptosis 2020, 25, 12–28. [Google Scholar] [CrossRef] [Green Version]
- Vasilevskaya, I.A.; Rakitina, T.V.; O’Dwyer, P.J. Quantitative effects on c-Jun N-terminal protein kinase signaling determine synergistic interaction of cisplatin and 17-allylamino-17-demethoxygeldanamycin in colon cancer cell lines. Mol. Pharmacol. 2004, 65, 235–243. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Xie, Z.; Sun, G.; Yang, P.; Li, J.; Yang, H.; Xiao, S.; Liu, Y.; Qiu, H.; Qin, L.; et al. Reversing drug resistance of cisplatin by hsp90 inhibitors in human ovarian cancer cells. Int J. Clin. Exp. Med. 2015, 8, 6687–6701. [Google Scholar]
- Hoter, A.; Rizk, S.; Naim, H.Y. The Multiple Roles and Therapeutic Potential of Molecular Chaperones in Prostate Cancer. Cancers (Basel) 2019, 11, 1194. [Google Scholar] [CrossRef] [Green Version]
- Kryeziu, K.; Bruun, J.; Guren, T.K.; Sveen, A.; Lothe, R.A. Combination therapies with HSP90 inhibitors against colorectal cancer. Biochim Biophys Acta Rev. Cancer 2019, 1871, 240–247. [Google Scholar] [CrossRef]
- Dutta Gupta, S.; Bommaka, M.K.; Banerjee, A. Inhibiting protein-protein interactions of Hsp90 as a novel approach for targeting cancer. Eur J. Med. Chem 2019, 178, 48–63. [Google Scholar] [CrossRef]
- Eckstein, N.; Servan, K.; Hildebrandt, B.; Pölitz, A.; von Jonquières, G.; Wolf-Kümmeth, S.; Napierski, I.; Hamacher, A.; Kassack, M.U.; Budczies, J.; et al. Hyperactivation of the insulin-like growth factor receptor I signaling pathway is an essential event for cisplatin resistance of ovarian cancer cells. Cancer Res. 2009, 69, 2996–3003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelke, L.H.; Hamacher, A.; Proksch, P.; Kassack, M.U. Ellagic Acid and Resveratrol Prevent the Development of Cisplatin Resistance in the Epithelial Ovarian Cancer Cell Line A2780. J. Cancer 2016, 7, 353–363. [Google Scholar] [CrossRef] [Green Version]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int J. Mol. Sci 2017, 18, 1414. [Google Scholar] [CrossRef]
- Kim, H.-J.; Bae, S.-C. Histone deacetylase inhibitors: Molecular mechanisms of action and clinical trials as anti-cancer drugs. Am. J. Transl Res. 2011, 3, 166–179. [Google Scholar]
- Kaiser, M.; Lamottke, B.; Mieth, M.; Jensen, M.R.; Quadt, C.; Garcia-Echeverria, C.; Atadja, P.; Heider, U.; von Metzler, I.; Türkmen, S.; et al. Synergistic action of the novel HSP90 inhibitor NVP-AUY922 with histone deacetylase inhibitors, melphalan, or doxorubicin in multiple myeloma. Eur. J. Haematol. 2010, 84, 337–344. [Google Scholar] [CrossRef]
- Khabele, D.; Son, D.-S.; Parl, A.K.; Goldberg, G.L.; Augenlicht, L.H.; Mariadason, J.M.; Rice, V.M. Drug-induced inactivation or gene silencing of class I histone deacetylases suppresses ovarian cancer cell growth: Implications for therapy. Cancer Biol. Ther. 2007, 6, 795–801. [Google Scholar] [CrossRef] [Green Version]
- Ozaki, K.; Kishikawa, F.; Tanaka, M.; Sakamoto, T.; Tanimura, S.; Kohno, M. Histone deacetylase inhibitors enhance the chemosensitivity of tumor cells with cross-resistance to a wide range of DNA-damaging drugs. Cancer Sci. 2008, 99, 376–384. [Google Scholar] [CrossRef]
- Marek, L.; Hamacher, A.; Hansen, F.K.; Kuna, K.; Gohlke, H.; Kassack, M.U.; Kurz, T. Histone deacetylase (HDAC) inhibitors with a novel connecting unit linker region reveal a selectivity profile for HDAC4 and HDAC5 with improved activity against chemoresistant cancer cells. J. Med. Chem. 2013, 56, 427–436. [Google Scholar] [CrossRef]
- Bandolik, J.J.; Hamacher, A.; Schrenk, C.; Weishaupt, R.; Kassack, M.U. Class I-Histone Deacetylase (HDAC) Inhibition is Superior to pan-HDAC Inhibition in Modulating Cisplatin Potency in High Grade Serous Ovarian Cancer Cell Lines. IJMS 2019, 20, 3052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.H.; Kang, J.G.; Kim, C.S.; Ihm, S.-H.; Choi, M.G.; Yoo, H.J.; Lee, S.J. Novel Heat Shock Protein 90 Inhibitor NVP-AUY922 Synergizes With the Histone Deacetylase Inhibitor PXD101 in Induction of Death of Anaplastic Thyroid Carcinoma Cells. J. Clin. Endocrinol. Metab. 2015, 100, E253–E261. [Google Scholar] [CrossRef] [Green Version]
- New, M.; Olzscha, H.; La Thangue, N.B. HDAC inhibitor-based therapies: Can we interpret the code? Mol. Oncol 2012, 6, 637–656. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Kang, J.G.; Kim, C.S.; Ihm, S.-H.; Choi, M.G.; Yoo, H.J.; Lee, S.J. The heat shock protein 90 inhibitor SNX5422 has a synergistic activity with histone deacetylase inhibitors in induction of death of anaplastic thyroid carcinoma cells. Endocrine 2016, 51, 274–282. [Google Scholar] [CrossRef]
- Lamottke, B.; Kaiser, M.; Mieth, M.; Heider, U.; Gao, Z.; Nikolova, Z.; Jensen, M.R.; Sterz, J.; von Metzler, I.; Sezer, O. The novel, orally bioavailable HSP90 inhibitor NVP-HSP990 induces cell cycle arrest and apoptosis in multiple myeloma cells and acts synergistically with melphalan by increased cleavage of caspases. Eur. J. Haematol. 2012, 88, 406–415. [Google Scholar] [CrossRef] [PubMed]
- Gohr, K.; Hamacher, A.; Engelke, L.H.; Kassack, M.U. Inhibition of PI3K/Akt/mTOR overcomes cisplatin resistance in the triple negative breast cancer cell line HCC38. Bmc Cancer 2017, 17, 711. [Google Scholar] [CrossRef] [Green Version]
- Kaletsch, A.; Pinkerneil, M.; Hoffmann, M.J.; Jaguva Vasudevan, A.A.; Wang, C.; Hansen, F.K.; Wiek, C.; Hanenberg, H.; Gertzen, C.; Gohlke, H.; et al. Effects of novel HDAC inhibitors on urothelial carcinoma cells. Clin. Epigenet 2018, 10, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solár, P.; Horváth, V.; Kleban, J.; Koval’, J.; Solárová, Z.; Kozubík, A.; Fedorocko, P. Hsp90 inhibitor geldanamycin increases the sensitivity of resistant ovarian adenocarcinoma cell line A2780cis to cisplatin. Neoplasma 2007, 54, 127–130. [Google Scholar]
- Diedrich, D.; Hamacher, A.; Gertzen, C.G.W.; Alves Avelar, L.A.; Reiss, G.J.; Kurz, T.; Gohlke, H.; Kassack, M.U.; Hansen, F.K. Rational design and diversity-oriented synthesis of peptoid-based selective HDAC6 inhibitors. Chem. Commun. (Camb.) 2016, 52, 3219–3222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pflieger, M.; Hamacher, A.; Öz, T.; Horstick-Muche, N.; Boesen, B.; Schrenk, C.; Kassack, M.U.; Kurz, T. Novel α,β-unsaturated hydroxamic acid derivatives overcome cisplatin resistance. Bioorganic Med. Chem. 2019, 27, 115036. [Google Scholar] [CrossRef]
- Weberpals, J.I.; O’Brien, A.M.; Niknejad, N.; Garbuio, K.D.; Clark-Knowles, K.V.; Dimitroulakos, J. The effect of the histone deacetylase inhibitor M344 on BRCA1 expression in breast and ovarian cancer cells. Cancer Cell Int 2011, 11, 29. [Google Scholar] [CrossRef] [Green Version]
- Steele, N.; Finn, P.; Brown, R.; Plumb, J.A. Combined inhibition of DNA methylation and histone acetylation enhances gene re-expression and drug sensitivity in vivo. Br. J. Cancer 2009, 100, 758–763. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.-Y.; Liao, W.S.-L.; Lu, Z.; Bornmann, W.G.; Hennessey, V.; Washington, M.N.; Rosner, G.L.; Yu, Y.; Ahmed, A.A.; Bast, R.C. Decitabine and suberoylanilide hydroxamic acid (SAHA) inhibit growth of ovarian cancer cell lines and xenografts while inducing expression of imprinted tumor suppressor genes, apoptosis, G2/M arrest, and autophagy. Cancer 2011, 117, 4424–4438. [Google Scholar] [CrossRef] [Green Version]
- Zitzmann, K.; Ailer, G.; Vlotides, G.; Spoettl, G.; Maurer, J.; Göke, B.; Beuschlein, F.; Auernhammer, C.J. Potent antitumor activity of the novel HSP90 inhibitors AUY922 and HSP990 in neuroendocrine carcinoid cells. Int. J. Oncol. 2013, 43, 1824–1832. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.; Curry, E.; Magnani, L.; Wilhelm-Benartzi, C.S.; Borley, J. Poised epigenetic states and acquired drug resistance in cancer. Nat. Rev. Cancer 2014, 14, 747–753. [Google Scholar] [CrossRef]
- Chou, T.-C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Ong, P.-S.; Wang, X.-Q.; Lin, H.-S.; Chan, S.-Y.; Ho, P.C. Synergistic effects of suberoylanilide hydroxamic acid combined with cisplatin causing cell cycle arrest independent apoptosis in platinum-resistant ovarian cancer cells. Int. J. Oncol. 2012, 40, 1705–1713. [Google Scholar] [CrossRef] [Green Version]
- Sidera, K.; Patsavoudi, E. HSP90 inhibitors: Current development and potential in cancer therapy. Recent Pat. Anticancer Drug Discov 2014, 9, 1–20. [Google Scholar] [CrossRef]
- Johnson, M.L.; Yu, H.A.; Hart, E.M.; Weitner, B.B.; Rademaker, A.W.; Patel, J.D.; Kris, M.G.; Riely, G.J. Phase I/II Study of HSP90 Inhibitor AUY922 and Erlotinib for EGFR-Mutant Lung Cancer With Acquired Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors. J. Clin. Oncol. 2015, 33, 1666–1673. [Google Scholar] [CrossRef] [Green Version]
- Spreafico, A.; Delord, J.-P.; De Mattos-Arruda, L.; Berge, Y.; Rodon, J.; Cottura, E.; Bedard, P.L.; Akimov, M.; Lu, H.; Pain, S.; et al. A first-in-human phase I, dose-escalation, multicentre study of HSP990 administered orally in adult patients with advanced solid malignancies. Br. J. Cancer 2015, 112, 650–659. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Beck, J.; Mita, M.; Paul, S.; Woo, M.M.; Squier, M.; Gadbaw, B.; Prince, H.M. A phase I dose-escalation study of intravenous panobinostat in patients with lymphoma and solid tumors. Invest. New Drugs 2013, 31, 974–985. [Google Scholar] [CrossRef]
- Kovacs, J.J.; Murphy, P.J.M.; Gaillard, S.; Zhao, X.; Wu, J.-T.; Nicchitta, C.V.; Yoshida, M.; Toft, D.O.; Pratt, W.B.; Yao, T.-P. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol. Cell 2005, 18, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Whitesell, L.; Lindquist, S.L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 2005, 5, 761–772. [Google Scholar] [CrossRef]
- Gosepath, E.M.; Eckstein, N.; Hamacher, A.; Servan, K.; von Jonquieres, G.; Lage, H.; Györffy, B.; Royer, H.D.; Kassack, M.U. Acquired cisplatin resistance in the head-neck cancer cell line Cal27 is associated with decreased DKK1 expression and can partially be reversed by overexpression of DKK1. Int. J. Cancer 2008, 123, 2013–2019. [Google Scholar] [CrossRef] [PubMed]
- Takai, N.; Narahara, H. Human Endometrial and Ovarian Cancer Cells: Histone Deacetylase Inhibitors Exhibit Antiproliferative Activity, Potently Induce Cell Cycle Arrest, and Stimulate Apoptosis. Available online: http://www.eurekaselect.com/59959/article (accessed on 14 May 2019).
- Rong, B.; Yang, S. Molecular mechanism and targeted therapy of Hsp90 involved in lung cancer: New discoveries and developments (Review). Int. J. Oncol. 2018, 52, 321–336. [Google Scholar] [CrossRef] [PubMed]
- Buurman, R.; Sandbothe, M.; Schlegelberger, B.; Skawran, B. HDAC inhibition activates the apoptosome via Apaf1 upregulation in hepatocellular carcinoma. Eur. J. Med. Res. 2016, 21, 26. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, P.K.; Goel, A.; Mittal, R.D. Survivin: A molecular biomarker in cancer. Indian J. Med. Res. 2015, 141, 389–397. [Google Scholar] [CrossRef]
- Rose, S.L.; Goodheart, M.J.; DeYoung, B.R.; Smith, B.J.; Buller, R.E. p21 expression predicts outcome in p53-null ovarian carcinoma. Clin. Cancer Res. 2003, 9, 1028–1032. [Google Scholar]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. Bmc Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | HDACi | HSP90i | ||||||
|---|---|---|---|---|---|---|---|---|
| Panobinostat | LMK235 | Luminespib | HSP990 | |||||
| IC50 [nM] | pIC50 ± SEM | IC50 [nM] | pIC50 ± SEM | IC50 [nM] | pIC50 ± SEM | IC50 [nM] | pIC50 ± SEM | |
| A2780 | 28.0 | 7.55 ± 0.02 | 847 | 6.07 ± 0.03 | 11.2 | 7.95 ± 0.06 | 20.1 | 7.70 ± 0.06 |
| A2780CisR | 28.2 | 7.55 ± 0.03 | 644 | 6.19 ± 0.02 | 16.4 | 7.78 ± 0.04 | 29.8 | 7.53 ± 0.02 |
| A – Dual Combination (inhibitor + cDDP) | |||||||||
| cell line | cDDP IC50 [µM] | ||||||||
| Control (cDDP only) | HSP90i | HDACi | |||||||
| Luminespib | HSP990 | Panobinostat | LMK235 | ||||||
| Coinc | preinc | coinc | preinc | coinc | preinc | coinc | preinc | ||
| A2780 | 3.34 | 2.41 | 0.71 | 2.22 | 0.50 | 1.63 | 0.57 | 0.69 | 0.57 |
| A2780CisR | 19.7 | 24.9 | 13.2 | 25.7 | 11.8 | 16.6 | 6.53 | 11.0 | 7.05 |
| B – Triple Combination I (HSP90i prior to HSP90i + HDACi + cDDP) | |||||||||
| A2780 cDDP IC50 [µM] | A2780CisR cDDP IC50 [µM] | ||||||||
| cDDP | HDACi/cDDP | cDDP | HDACi/cDDP | ||||||
| pano | LMK235 | pano | LMK235 | ||||||
| HSP90i | lumi | 0.71 | 0.65 | 0.63 | 13.2 | 12.3 | 9.69 | ||
| HSP990 | 0.50 | 0.70 | 0.95 | 11.8 | 7.78 | 7.07 | |||
| C – Triple Combination II (HDACi prior to HDACi + HSP90i + cDDP) | |||||||||
| A2780 cDDP IC50 [µM] | A2780CisR cDDP IC50 [µM] | ||||||||
| cDDP | HSP90i/cDDP | cDDP | HSP90i/cDDP | ||||||
| lumi | HSP990 | lumi | HSP990 | ||||||
| HDACi | pano | 0.57 | 0.91 | 0.84 | 6.53 | 6.07 | 5.91 | ||
| LMK235 | 0.57 | 0.70 | 0.84 | 7.05 | 9.23 | 9.26 | |||
| Cell Line | Panobinostat | HSP990 | ||
|---|---|---|---|---|
| IC50 [nM] | pIC50 ± SEM | IC50 [nM] | pIC50 ± SEM | |
| CaOV3 | 15.9 | 7.80 ± 0.02 | 40.9 | 7.39 ± 0.03 |
| CaOV3CisR | 16.9 | 7.77 ± 0.02 | 55.6 | 7.26 ± 0.02 |
| OVCAR3 | 41.3 | 7.38 ± 0.02 | 27.6 | 7.56 ± 0.03 |
| OVCAR3CisR | 30.5 | 7.52 ± 0.01 | 25.0 | 7.60 ± 0.02 |
| Cell Line | Control (Cisplatin Only) | + 48h Pretreatment | |||
|---|---|---|---|---|---|
| HSP990 | Panobinostat | ||||
| Cisplatin IC50 [µM] | SF | Cisplatin IC50 [µM] | SF | ||
| CaOV3 | 1.92 | 0.79 | 2.4 *** | 1.04 | 1.8 *** |
| CaOV3CisR | 4.80 | 3.20 | 1.5 ** | 1.38 | 3.5 *** |
| OVCAR3 | 3.94 | 2.77 | 1.4 ns | 1.50 | 2.6 ** |
| OVCAR3CisR | 37.7 | 12.6 | 3.0 * | 8.03 | 4.7 *** |
| Gene. | Primer Forward | Primer Reverse | Size [bp] |
|---|---|---|---|
| BAK | GAACAGGAGGCTGAAGGGGT | TCAGGCCATGCTGGTAGACG | 307 |
| survivin | CGAGGCTGGCTTCATCCACT | ACGGCGCACTTTCTTCGCA | 283 |
| Bcl-xL | CTGAATCGGAGATGGAGACC | TGGGATGTCAGGTCACTGAA | 211 |
| BAX | GATGCGTCCACCAAGAAGCT | CGGCCCCAGTTGAAGTTG | 170 |
| β-tubulin (TUBB) | TCTACCTCCCTCACTCAGCT | CCAGAGTCAGGGGTGTTCAT | 161 |
| Mcl-1 | GGACATCAAAAACGAAGACG | GCAGCTTTCTTGGTTTATGG | 154 |
| APAF-1 | ACAATGCTCTACTACATGAAGGATATAAAGA | CACTGGAAGAAGAGACAACAGGAA | 81 |
| p21 | CCTAATCCGCCCACAGGAA | AAGATGTAGAGCGGGCCTTTG | 68 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodrigues Moita, A.J.; Bandolik, J.J.; Hansen, F.K.; Kurz, T.; Hamacher, A.; Kassack, M.U. Priming with HDAC Inhibitors Sensitizes Ovarian Cancer Cells to Treatment with Cisplatin and HSP90 Inhibitors. Int. J. Mol. Sci. 2020, 21, 8300. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218300
Rodrigues Moita AJ, Bandolik JJ, Hansen FK, Kurz T, Hamacher A, Kassack MU. Priming with HDAC Inhibitors Sensitizes Ovarian Cancer Cells to Treatment with Cisplatin and HSP90 Inhibitors. International Journal of Molecular Sciences. 2020; 21(21):8300. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218300
Chicago/Turabian StyleRodrigues Moita, Ana J., Jan J. Bandolik, Finn K. Hansen, Thomas Kurz, Alexandra Hamacher, and Matthias U. Kassack. 2020. "Priming with HDAC Inhibitors Sensitizes Ovarian Cancer Cells to Treatment with Cisplatin and HSP90 Inhibitors" International Journal of Molecular Sciences 21, no. 21: 8300. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218300