GOLPH3 Regulates EGFR in T98G Glioblastoma Cells by Modulating Its Glycosylation and Ubiquitylation


Abstract
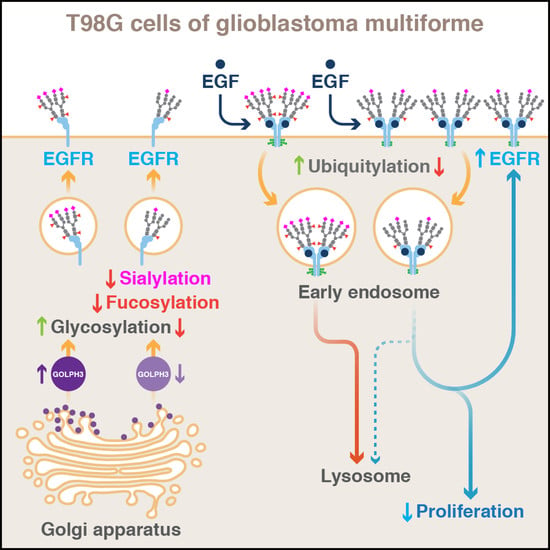
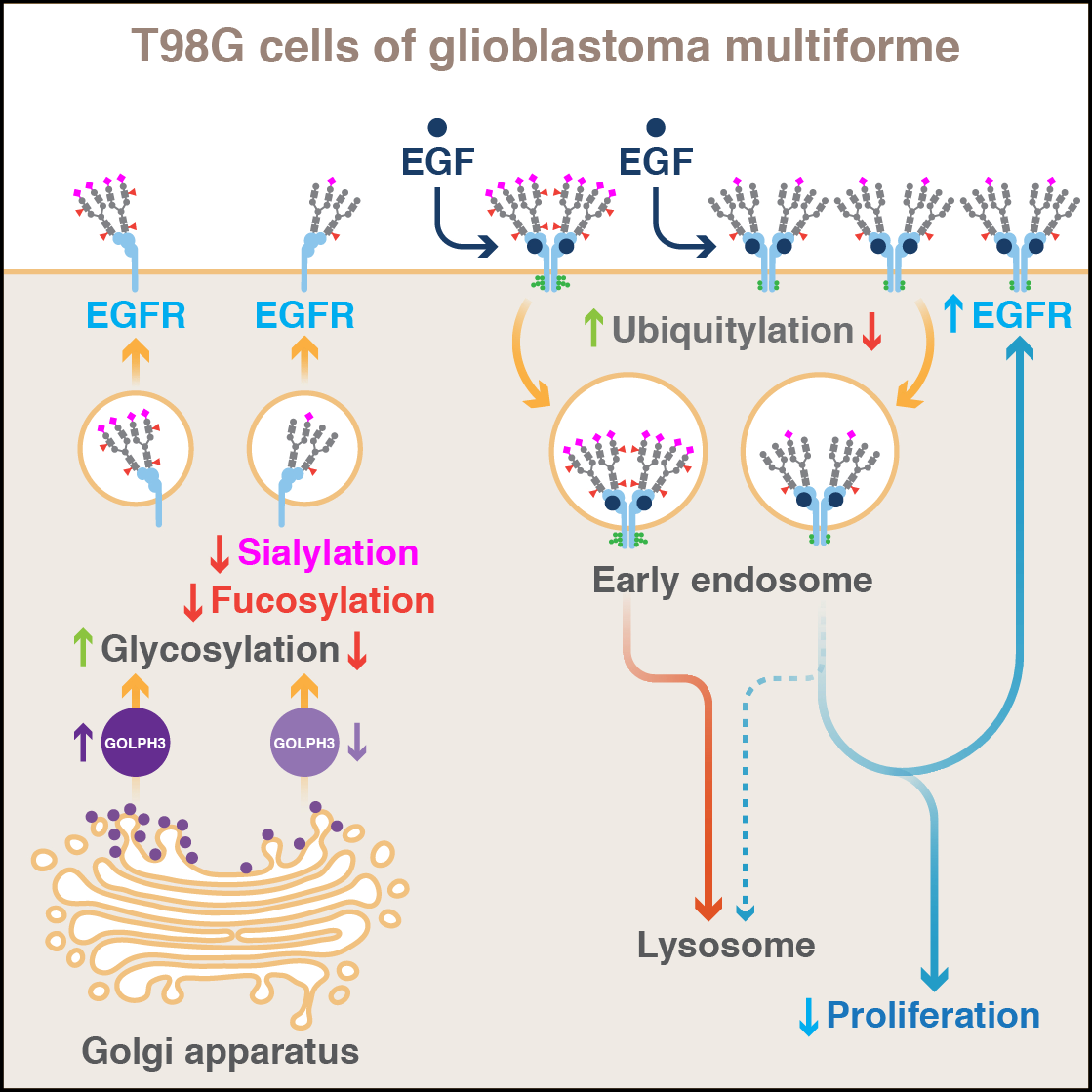
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
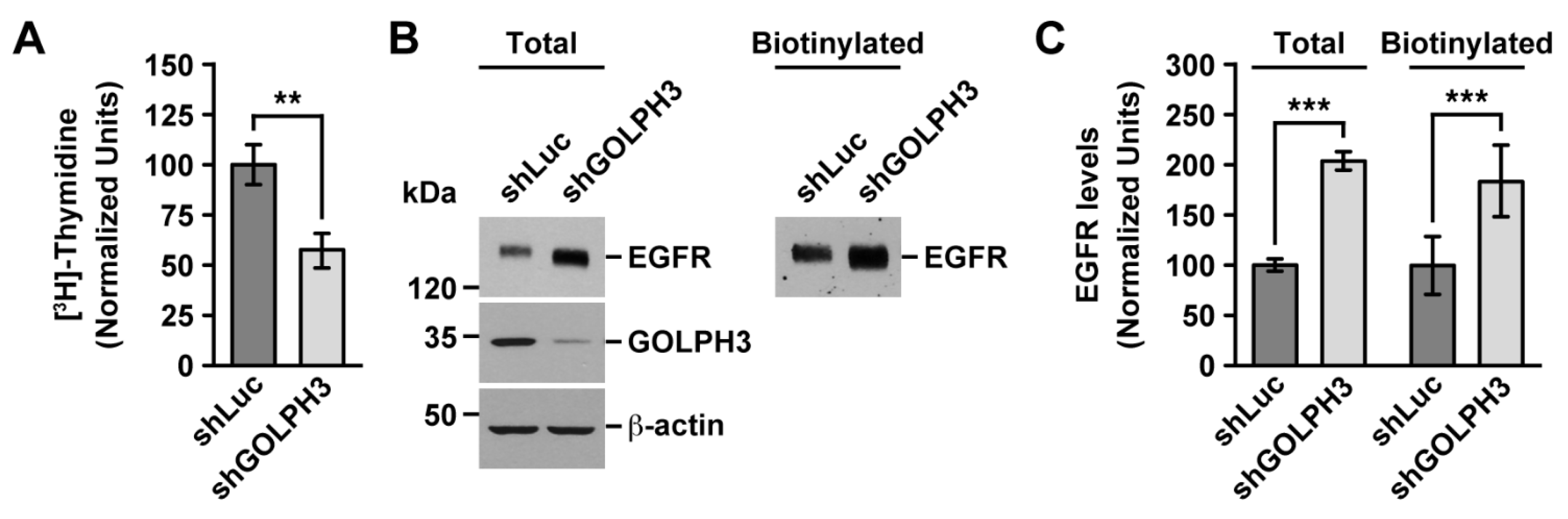
2.1. The Knockdown of GOLPH3 in T98G Cells Decreases Cell Proliferation, but Increases the Levels of EGFR
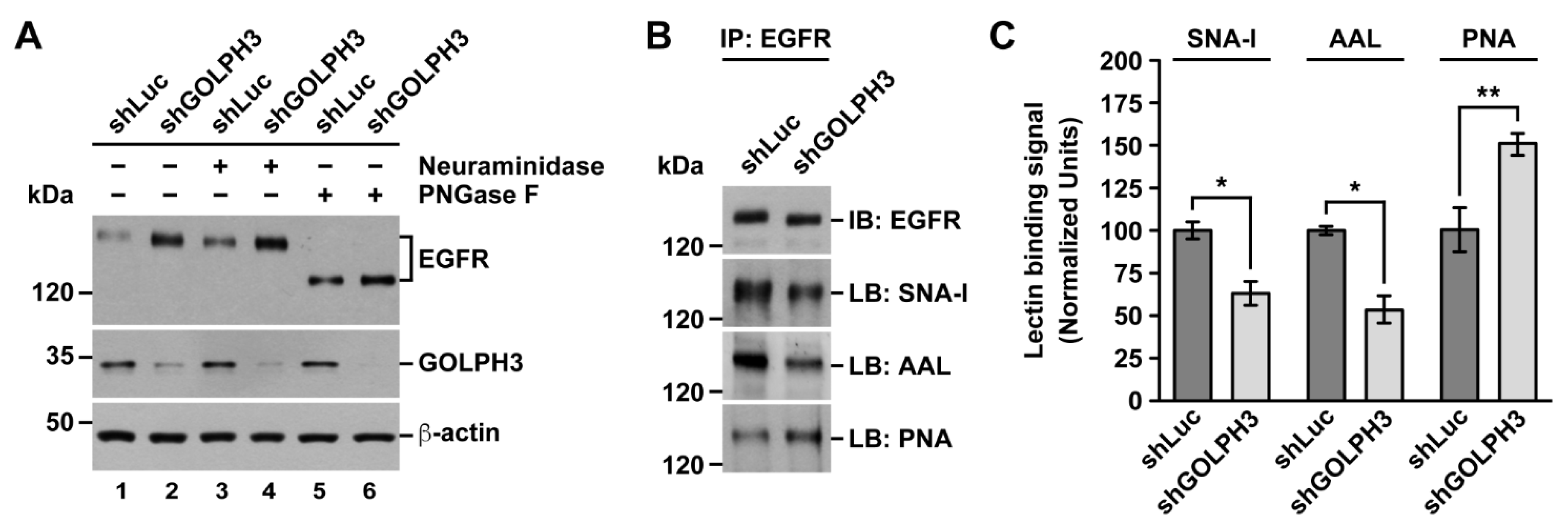
2.2. The Knockdown of GOLPH3 in T98G Cells Perturbs EGFR Glycosylation
2.3. The Knockdown of GOLPH3 in T98G Cells Does Not Affect the Kinetics of EGFR Trafficking from the Endoplasmic Reticulum to the Cell Surface
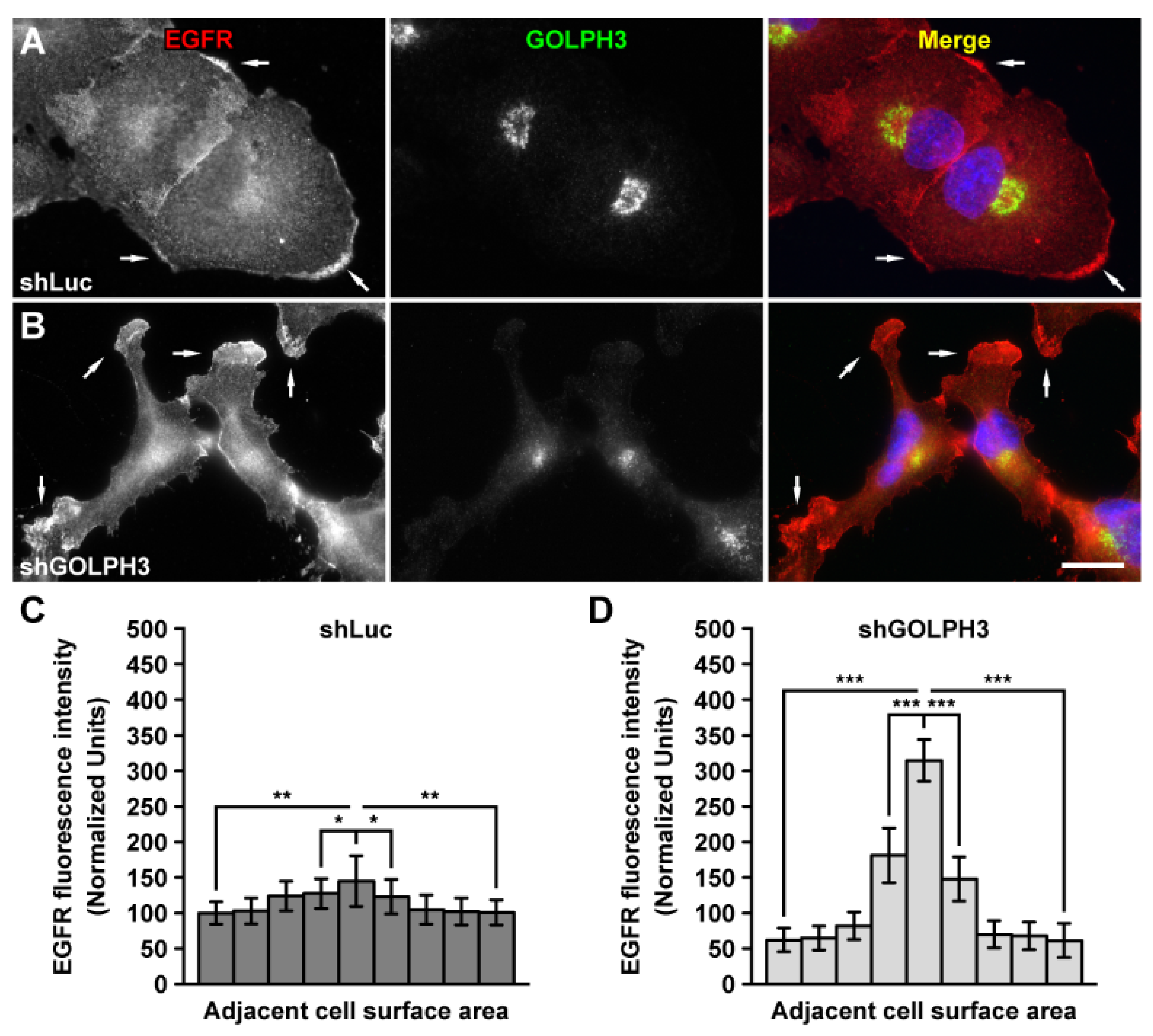
2.4. The Knockdown of GOLPH3 Increases the Recycling of EGFR at the Cell Surface in T98G Cells
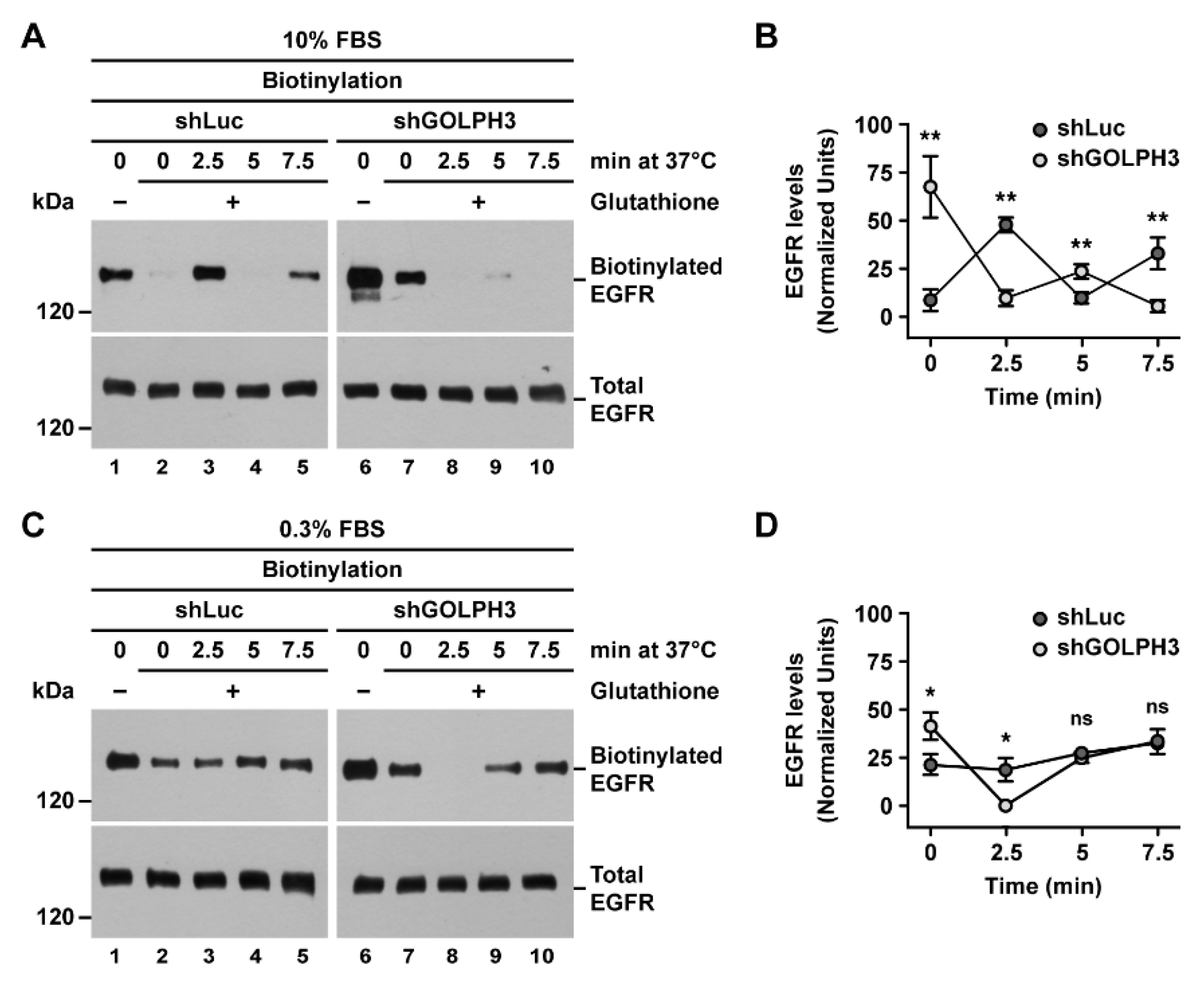
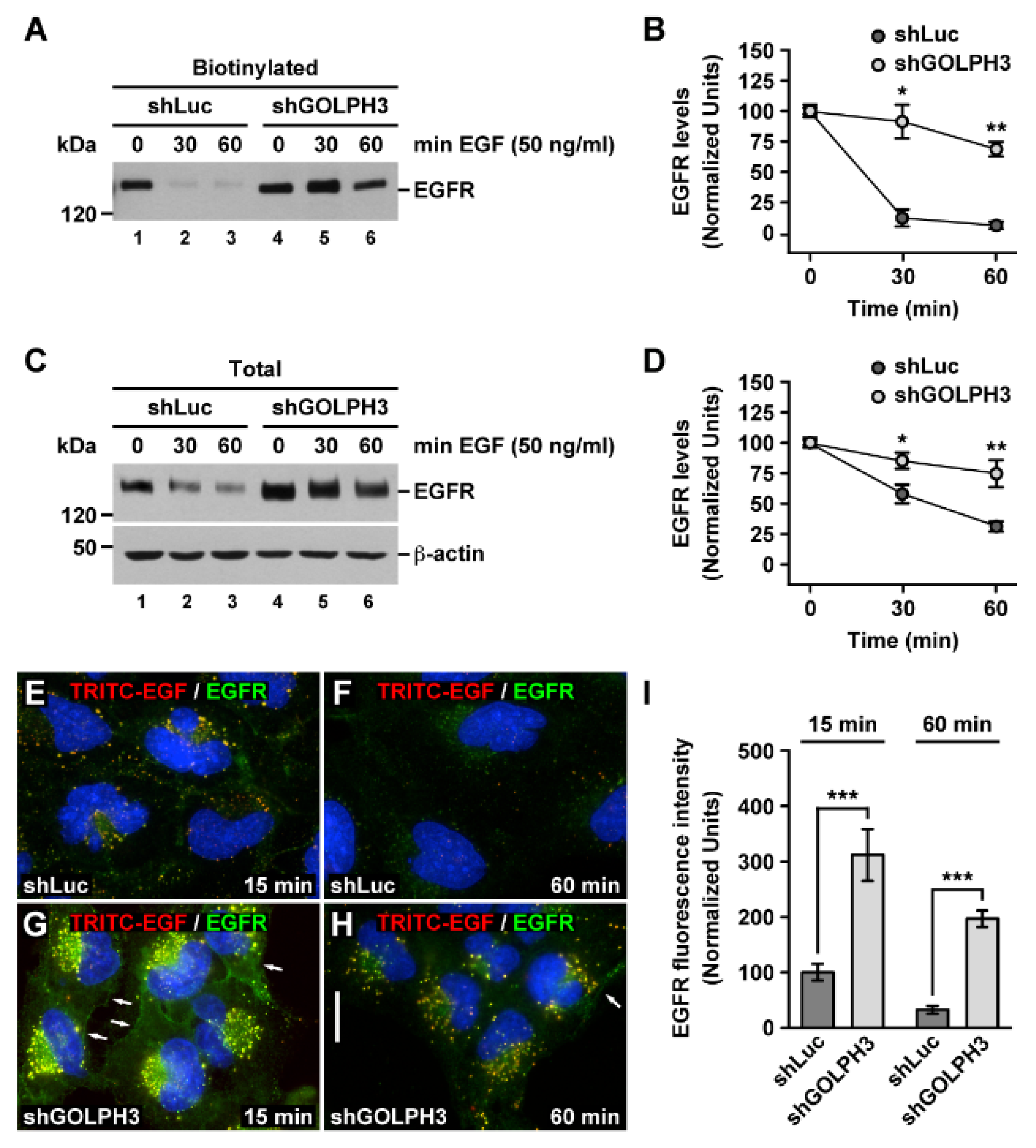
2.5. The Knockdown of GOLPH3 in T98G Cells Affects the Internalization and Degradation of EGFR
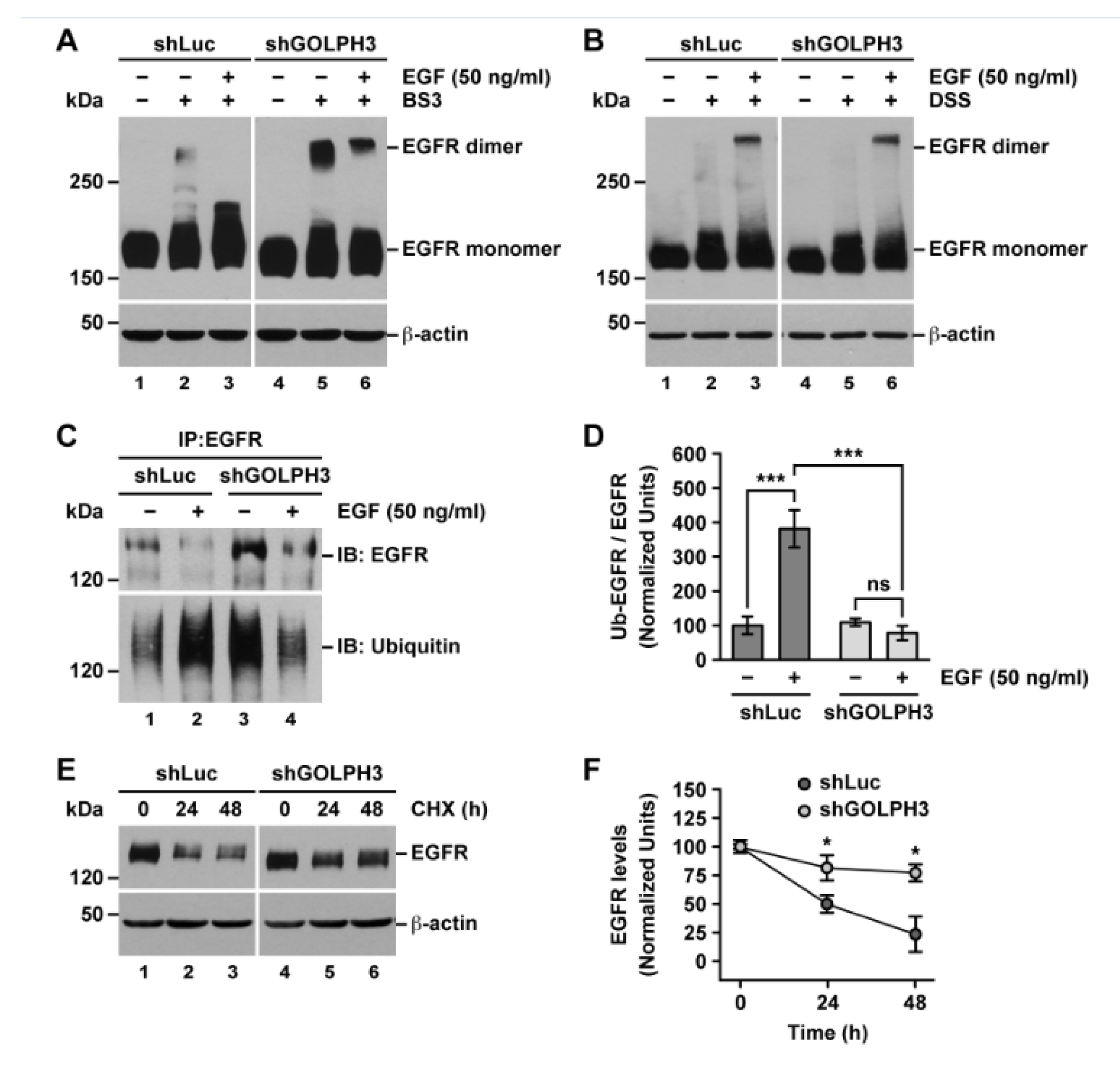
2.6. The Knockdown of GOLPH3 in T98G Cells Does Not Affect EGFR Dimerization but Affects EGFR Ubiquitylation
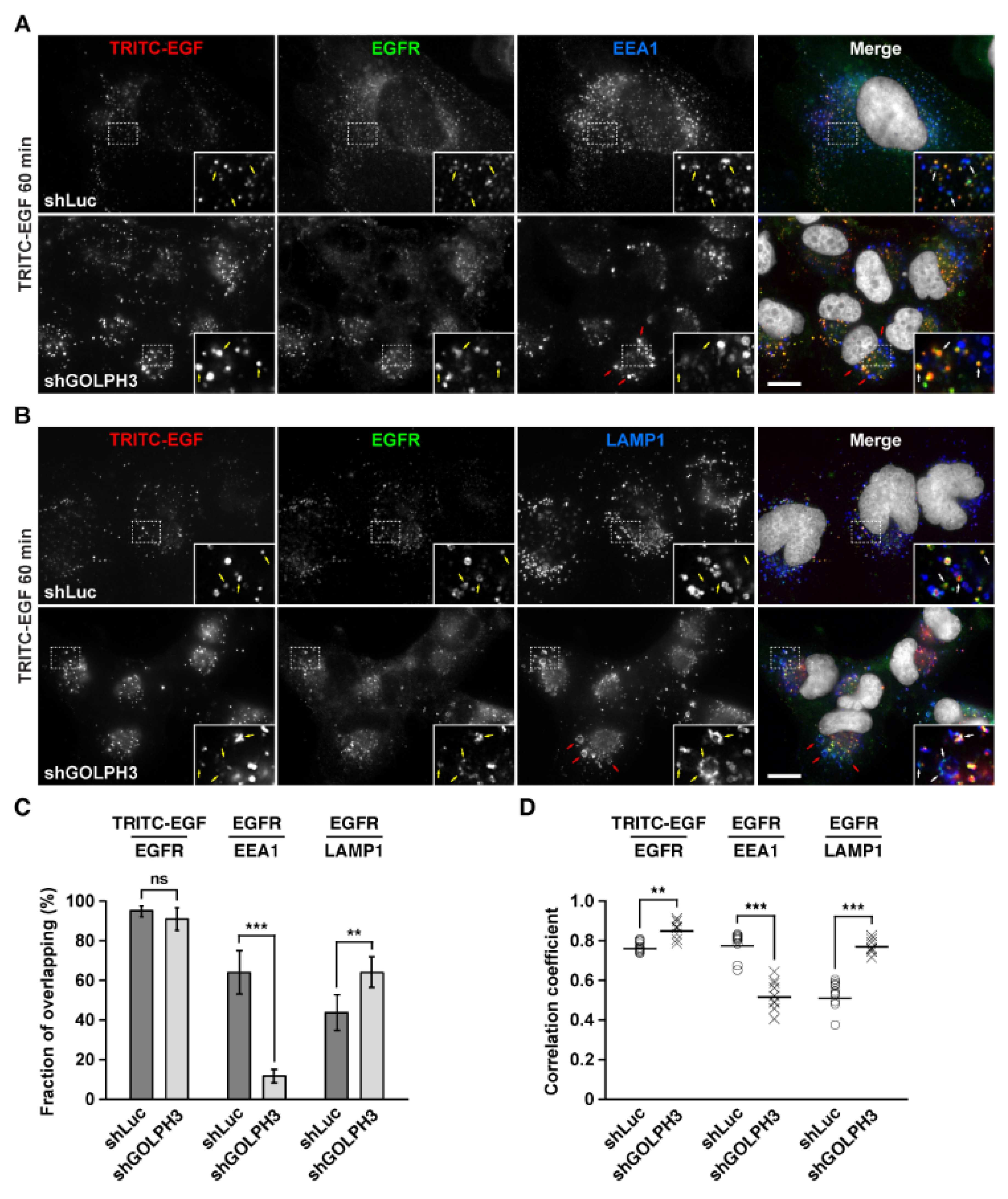
2.7. The Knockdown of GOLPH3 Promotes the Accumulation in Endo-lysosomal Compartments of EGF-Stimulated Internalized EGFR
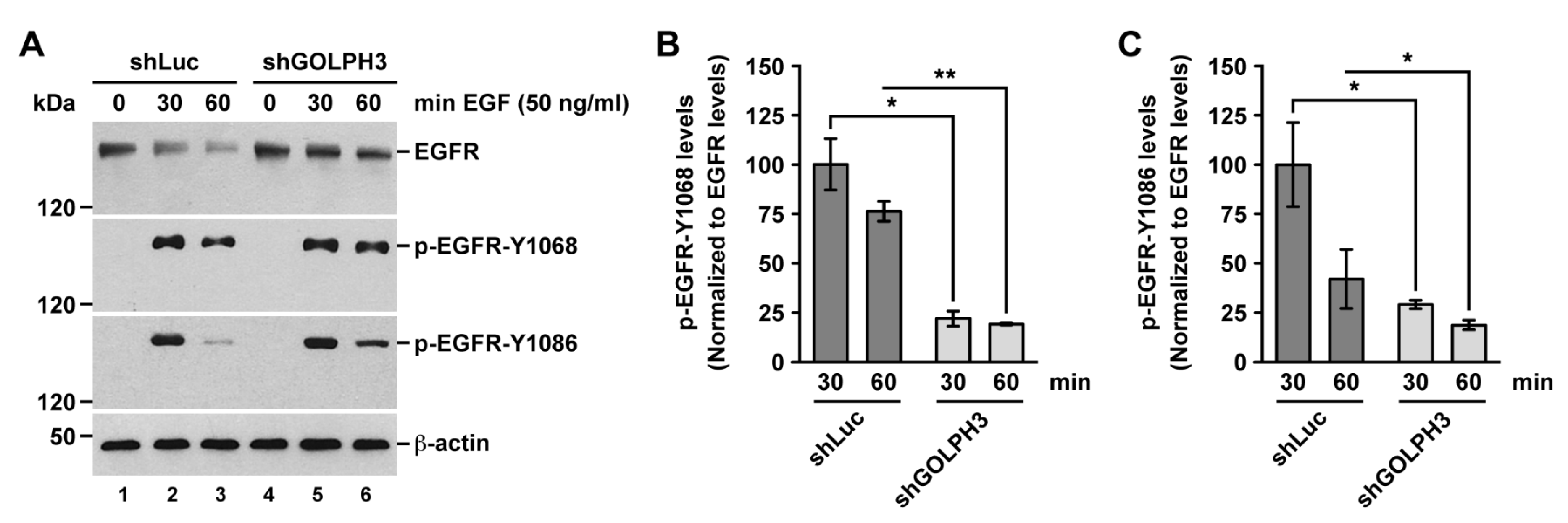
2.8. The Knockdown of GOLPH3 Negatively Affects the Activation of EGFR
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Generation of Cell Lines
4.2. Antibodies and Cell and Lectin Blotting Reagents
4.3. Cell Proliferation Assay, Immunoblotting and Densitometry Quantification
4.4. Cell Surface Biotinylation
4.5. Enzymatic Deglycosylation, Immunoprecipitation and Lectin Blotting
4.6. DNA Constructs, Cell Transfection, Fluorescence Microscopy, and Image Analysis
4.7. Crosslinking, Internalization, Degradation and Turnover Assays
4.8. Recycling Assay
4.9. Statistical Analysis
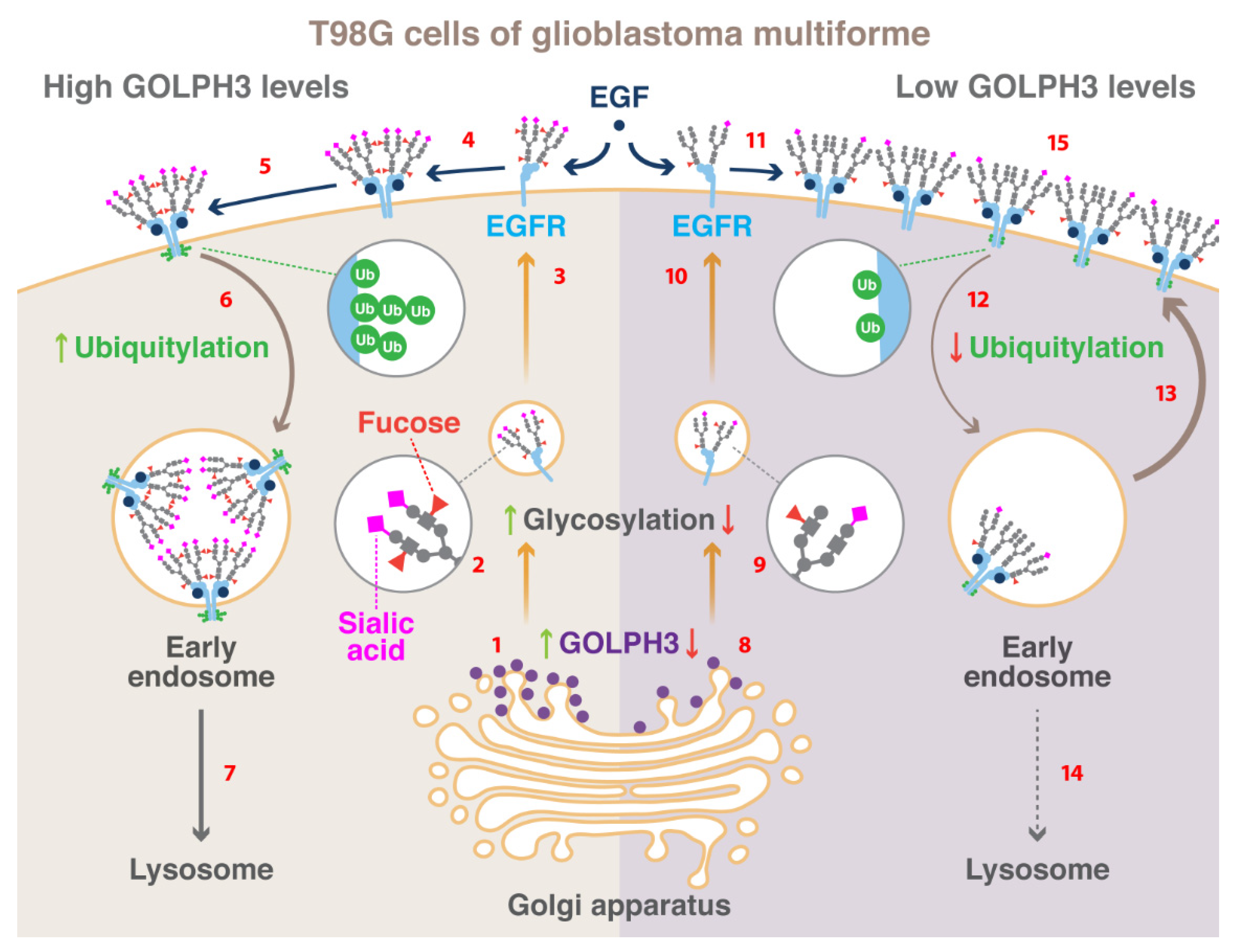
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kulkarni-Gosavi, P.; Makhoul, C.; Gleeson, P.A. Form and function of the Golgi apparatus: Scaffolds, cytoskeleton and signalling. FEBS Lett. 2019, 593, 2289–2305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donizy, P.; Marczuk, J. Selected Golgi-Localized Proteins and Carcinogenesis: What Do We Know? Results Probl. Cell Differ. 2019, 67, 487–529. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, R.; Parashuraman, S.; D’Angelo, G.; Luini, A. GOLPH3 and oncogenesis: What is the molecular link? Tissue Cell 2017, 49, 170–174. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Su, Y.; Zhao, Y.; Pan, C.; Chen, L. Golgi phosphoprotein3 overexpression is associated with poor survival in patients with solid tumors: A meta-analysis. Int. J. Clin. Exp. Pathol. 2015, 8, 10615–10624. [Google Scholar]
- Sechi, S.; Frappaolo, A.; Belloni, G.; Colotti, G.; Giansanti, M.G. The multiple cellular functions of the oncoprotein Golgi phosphoprotein 3. Oncotarget 2015, 6, 3493–3506. [Google Scholar] [CrossRef] [Green Version]
- Tenorio, M.J.; Ross, B.H.; Luchsinger, C.; Rivera-Dictter, A.; Arriagada, C.; Acuña, D.; Aguilar, M.; Cavieres, V.; Burgos, P.V.; Ehrenfeld, P.; et al. Distinct Biochemical Pools of Golgi Phosphoprotein 3 in the Human Breast Cancer Cell Lines MCF7 and MDA-MB-231. PLoS ONE 2016, 11, e0154719. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.C.; Taylor, R.S.; Lane, D.R.; Ladinsky, M.S.; Weisz, J.A.; Howell, K.E. GMx33: A novel family of trans-Golgi proteins identified by proteomics. Traffic 2000, 1, 963–975. [Google Scholar] [CrossRef]
- Bell, A.W.; Ward, M.A.; Blackstock, W.P.; Freeman, H.N.; Choudhary, J.S.; Lewis, A.P.; Chotai, D.; Fazel, A.; Gushue, J.N.; Paiement, J.; et al. Proteomics characterization of abundant Golgi membrane proteins. J. Biol. Chem. 2001, 276, 5152–5165. [Google Scholar] [CrossRef] [Green Version]
- Dippold, H.C.; Ng, M.M.; Farber-Katz, S.E.; Lee, S.K.; Kerr, M.L.; Peterman, M.C.; Sim, R.; Wiharto, P.A.; Galbraith, K.A.; Madhavarapu, S.; et al. GOLPH3 bridges phosphatidylinositol-4- phosphate and actomyosin to stretch and shape the Golgi to promote budding. Cell 2009, 139, 337–351. [Google Scholar] [CrossRef] [Green Version]
- Wood, C.S.; Schmitz, K.R.; Bessman, N.J.; Setty, T.G.; Ferguson, K.M.; Burd, C.G. PtdIns4P recognition by Vps74/GOLPH3 links PtdIns 4-kinase signaling to retrograde Golgi trafficking. J. Cell Biol. 2009, 187, 967–975. [Google Scholar] [CrossRef] [Green Version]
- Snyder, C.M.; Mardones, G.A.; Ladinsky, M.S.; Howell, K.E. GMx33 associates with the trans-Golgi matrix in a dynamic manner and sorts within tubules exiting the Golgi. Mol. Biol. Cell 2006, 17, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Rahajeng, J.; Kuna, R.S.; Makowski, S.L.; Tran, T.T.T.; Buschman, M.D.; Li, S.; Cheng, N.; Ng, M.M.; Field, S.J. Efficient Golgi Forward Trafficking Requires GOLPH3-Driven, PI4P-Dependent Membrane Curvature. Dev. Cell 2019, 50, 573–585.e575. [Google Scholar] [CrossRef] [PubMed]
- Bishe, B.; Syed, G.H.; Field, S.J.; Siddiqui, A. Role of phosphatidylinositol 4-phosphate (PI4P) and its binding protein GOLPH3 in hepatitis C virus secretion. J. Biol. Chem. 2012, 287, 27637–27647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, K.L.; Kabbarah, O.; Liang, M.C.; Ivanova, E.; Anagnostou, V.; Wu, J.; Dhakal, S.; Wu, M.; Chen, S.; Feinberg, T.; et al. GOLPH3 modulates mTOR signalling and rapamycin sensitivity in cancer. Nature 2009, 459, 1085–1090. [Google Scholar] [CrossRef] [Green Version]
- Bugarcic, A.; Zhe, Y.; Kerr, M.C.; Griffin, J.; Collins, B.M.; Teasdale, R.D. Vps26A and Vps26B subunits define distinct retromer complexes. Traffic 2011, 12, 1759–1773. [Google Scholar] [CrossRef]
- Tu, L.; Chen, L.; Banfield, D.K. A conserved N-terminal arginine-motif in GOLPH3-family proteins mediates binding to coatomer. Traffic 2012, 13, 1496–1507. [Google Scholar] [CrossRef]
- Eckert, E.S.; Reckmann, I.; Hellwig, A.; Rohling, S.; El-Battari, A.; Wieland, F.T.; Popoff, V. Golgi phosphoprotein 3 triggers signal-mediated incorporation of glycosyltransferases into coatomer-coated (COPI) vesicles. J. Biol. Chem. 2014, 289, 31319–31329. [Google Scholar] [CrossRef] [Green Version]
- Isaji, T.; Im, S.; Gu, W.; Wang, Y.; Hang, Q.; Lu, J.; Fukuda, T.; Hashii, N.; Takakura, D.; Kawasaki, N.; et al. An oncogenic protein Golgi phosphoprotein 3 up-regulates cell migration via sialylation. J. Biol. Chem. 2014, 289, 20694–20705. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.F.; Chachadi, V.B.; Petrosyan, A.; Cheng, P.W. Golgi phosphoprotein 3 determines cell binding properties under dynamic flow by controlling Golgi localization of core 2 N-acetylglucosaminyltransferase 1. J. Biol. Chem. 2012, 287, 39564–39577. [Google Scholar] [CrossRef] [Green Version]
- Pereira, N.A.; Pu, H.X.; Goh, H.; Song, Z. Golgi phosphoprotein 3 mediates the Golgi localization and function of protein O-linked mannose beta-1,2-N-acetlyglucosaminyltransferase 1. J. Biol. Chem. 2014, 289, 14762–14770. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Ding, Z.; Mo, J.; Sang, B.; Shi, Q.; Hu, J.; Xie, S.; Zhan, W.; Lu, D.; Yang, M.; et al. GOLPH3 promotes glioblastoma cell migration and invasion via the mTOR-YB1 pathway in vitro. Mol. Carcinog. 2015, 54, 1252–1263. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.H.; Yuan, L.J.; Liang, R.X.; Liu, Z.G.; Li, B.H.; Wen, Z.S.; Huang, S.T.; Zheng, M. GOLPH3 promotes cell proliferation and tumorigenicity in esophageal squamous cell carcinoma via mTOR and Wnt/betacatenin signal activation. Mol. Med. Rep. 2017, 16, 7138–7144. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Wang, X.; Feng, B.; Tang, L.; Li, W.; Zheng, X.; Liu, Y.; Peng, Y.; Zheng, G.; He, Q. Golgi phosphoprotein 3 (GOLPH3) promotes hepatocellular carcinoma progression by activating mTOR signaling pathway. BMC Cancer 2018, 18, 661. [Google Scholar] [CrossRef]
- Liu, J.; Wei, H.; Lai, L.; Wang, Y.; Han, X.; Zhang, Z. Golgi phosphoprotein-3 promotes invasiveness of gastric cancer cells through the mTOR signalling pathway. Clin. Investig. Med. 2019, 42, E38–E47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A. Ligand-induced ErbB receptor dimerization. Exp. Cell Res. 2009, 315, 638–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorkin, A.; Goh, L.K. Endocytosis and intracellular trafficking of ErbBs. Exp. Cell Res. 2009, 315, 683–696. [Google Scholar] [CrossRef] [Green Version]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Cai, X.; Sughrue, M.E. Glioblastoma: New therapeutic strategies to address cellular and genomic complexity. Oncotarget 2018, 9, 9540–9554. [Google Scholar] [CrossRef] [Green Version]
- Heimberger, A.B.; Hlatky, R.; Suki, D.; Yang, D.; Weinberg, J.; Gilbert, M.; Sawaya, R.; Aldape, K. Prognostic effect of epidermal growth factor receptor and EGFRvIII in glioblastoma multiforme patients. Clin. Cancer Res. 2005, 11, 1462–1466. [Google Scholar] [CrossRef] [Green Version]
- Eskilsson, E.; Rosland, G.V.; Solecki, G.; Wang, Q.; Harter, P.N.; Graziani, G.; Verhaak, R.G.W.; Winkler, F.; Bjerkvig, R.; Miletic, H. EGFR heterogeneity and implications for therapeutic intervention in glioblastoma. Neuro-Oncology 2018, 20, 743–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Xie, S.; Wu, S.; Qi, Y.; Wang, Z.; Zhang, H.; Lu, D.; Wang, X.; Dong, Y.; Liu, G.; et al. Golgi phosphoprotein 3 promotes glioma progression via inhibiting Rab5-mediated endocytosis and degradation of epidermal growth factor receptor. Neuro-Oncology 2017, 19, 1628–1639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Antonyak, M.A.; Singh, G.; Cerione, R.A. A mechanism for the upregulation of EGF receptor levels in glioblastomas. Cell Rep. 2013, 3, 2008–2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.Y.; Liu, W.; Chen, S.F.; Zhang, L.Q.; Li, X.G.; Wang, L.X. Expression of the Golgi phosphoprotein-3 gene in human gliomas: A pilot study. J. Neuro-Oncol. 2011, 105, 159–163. [Google Scholar] [CrossRef]
- Zhou, J.; Xu, T.; Qin, R.; Yan, Y.; Chen, C.; Chen, Y.; Yu, H.; Xia, C.; Lu, Y.; Ding, X.; et al. Overexpression of Golgi phosphoprotein-3 (GOLPH3) in glioblastoma multiforme is associated with worse prognosis. J. Neuro-Oncol. 2012, 110, 195–203. [Google Scholar] [CrossRef]
- Wu, S.; Fu, J.; Dong, Y.; Yi, Q.; Lu, D.; Wang, W.; Qi, Y.; Yu, R.; Zhou, X. GOLPH3 promotes glioma progression via facilitating JAK2-STAT3 pathway activation. J. Neuro-Oncol. 2018, 139, 269–279. [Google Scholar] [CrossRef]
- Arriagada, C.; Luchsinger, C.; González, A.E.; Schwenke, T.; Arriagada, G.; Folch, H.; Ehrenfeld, P.; Burgos, P.V.; Mardones, G.A. The knocking down of the oncoprotein Golgi phosphoprotein 3 in T98G cells of glioblastoma multiforme disrupts cell migration by affecting focal adhesion dynamics in a focal adhesion kinase-dependent manner. PLoS ONE 2019, 14, e0212321. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Zhen, Y.; Caprioli, R.M.; Staros, J.V. Characterization of glycosylation sites of the epidermal growth factor receptor. Biochemistry 2003, 42, 5478–5492. [Google Scholar] [CrossRef] [Green Version]
- Maley, F.; Trimble, R.B.; Tarentino, A.L.; Plummer, T.H., Jr. Characterization of glycoproteins and their associated oligosaccharides through the use of endoglycosidases. Anal. Biochem. 1989, 180, 195–204. [Google Scholar] [CrossRef]
- Britain, C.M.; Holdbrooks, A.T.; Anderson, J.C.; Willey, C.D.; Bellis, S.L. Sialylation of EGFR by the ST6Gal-I sialyltransferase promotes EGFR activation and resistance to gefitinib-mediated cell death. J. Ovarian Res. 2018, 11, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roggentin, P.; Berg, W.; Schauer, R. Purification and Characterization of Sialidase from Clostridium sordellii G12. Glycoconj. J. 1987, 4, 349–359. [Google Scholar] [CrossRef]
- Shibuya, N.; Goldstein, I.J.; Broekaert, W.F.; Nsimba-Lubaki, M.; Peeters, B.; Peumans, W.J. The elderberry (Sambucus nigra L.) bark lectin recognizes the Neu5Ac(alpha 2-6)Gal/GalNAc sequence. J. Biol. Chem. 1987, 262, 1596–1601. [Google Scholar] [PubMed]
- Christiansen, M.N.; Chik, J.; Lee, L.; Anugraham, M.; Abrahams, J.L.; Packer, N.H. Cell surface protein glycosylation in cancer. Proteomics 2014, 14, 525–546. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Yokote, H.; Arao, T.; Maegawa, M.; Tanaka, K.; Fujita, Y.; Shimizu, C.; Hanafusa, T.; Fujiwara, Y.; Nishio, K. N-Glycan fucosylation of epidermal growth factor receptor modulates receptor activity and sensitivity to epidermal growth factor receptor tyrosine kinase inhibitor. Cancer Sci. 2008, 99, 1611–1617. [Google Scholar] [CrossRef]
- Kochibe, N.; Furukawa, K. Purification and properties of a novel fucose-specific hemagglutinin of Aleuria aurantia. Biochemistry 1980, 19, 2841–2846. [Google Scholar] [CrossRef]
- Lotan, R.; Skutelsky, E.; Danon, D.; Sharon, N. The purification, composition, and specificity of the anti-T lectin from peanut (Arachis hypogaea). J. Biol. Chem. 1975, 250, 8518–8523. [Google Scholar]
- Al-Akhrass, H.; Naves, T.; Vincent, F.; Magnaudeix, A.; Durand, K.; Bertin, F.; Melloni, B.; Jauberteau, M.O.; Lalloue, F. Sortilin limits EGFR signaling by promoting its internalization in lung cancer. Nat. Commun. 2017, 8, 1182. [Google Scholar] [CrossRef] [Green Version]
- Waters, C.M.; Oberg, K.C.; Carpenter, G.; Overholser, K.A. Rate constants for binding, dissociation, and internalization of EGF: Effect of receptor occupancy and ligand concentration. Biochemistry 1990, 29, 3563–3569. [Google Scholar] [CrossRef]
- Herbst, J.J.; Opresko, L.K.; Walsh, B.J.; Lauffenburger, D.A.; Wiley, H.S. Regulation of postendocytic trafficking of the epidermal growth factor receptor through endosomal retention. J. Biol. Chem. 1994, 269, 12865–12873. [Google Scholar]
- Roepstorff, K.; Grandal, M.V.; Henriksen, L.; Knudsen, S.L.; Lerdrup, M.; Grovdal, L.; Willumsen, B.M.; van Deurs, B. Differential effects of EGFR ligands on endocytic sorting of the receptor. Traffic 2009, 10, 1115–1127. [Google Scholar] [CrossRef] [PubMed]
- Scharaw, S.; Iskar, M.; Ori, A.; Boncompain, G.; Laketa, V.; Poser, I.; Lundberg, E.; Perez, F.; Beck, M.; Bork, P.; et al. The endosomal transcriptional regulator RNF11 integrates degradation and transport of EGFR. J. Cell Biol. 2016, 215, 543–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boncompain, G.; Divoux, S.; Gareil, N.; de Forges, H.; Lescure, A.; Latreche, L.; Mercanti, V.; Jollivet, F.; Raposo, G.; Perez, F. Synchronization of secretory protein traffic in populations of cells. Nat. Methods 2012, 9, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Wiley, H.S.; Herbst, J.J.; Walsh, B.J.; Lauffenburger, D.A.; Rosenfeld, M.G.; Gill, G.N. The role of tyrosine kinase activity in endocytosis, compartmentation, and down-regulation of the epidermal growth factor receptor. J. Biol. Chem. 1991, 266, 11083–11094. [Google Scholar]
- Hanover, J.A.; Willingham, M.C.; Pastan, I. Kinetics of transit of transferrin and epidermal growth factor through clathrin-coated membranes. Cell 1984, 39, 283–293. [Google Scholar] [CrossRef]
- Bakker, J.; Spits, M.; Neefjes, J.; Berlin, I. The EGFR odyssey—From activation to destruction in space and time. J. Cell Sci. 2017, 130, 4087–4096. [Google Scholar] [CrossRef] [Green Version]
- Fanger, B.O.; Stephens, J.E.; Staros, J.V. High-yield trapping of EGF-induced receptor dimers by chemical cross-linking. FASEB J. 1989, 3, 71–75. [Google Scholar] [CrossRef]
- Eden, E.R.; Huang, F.; Sorkin, A.; Futter, C.E. The role of EGF receptor ubiquitination in regulating its intracellular traffic. Traffic 2012, 13, 329–337. [Google Scholar] [CrossRef]
- Mu, F.T.; Callaghan, J.M.; Steele-Mortimer, O.; Stenmark, H.; Parton, R.G.; Campbell, P.L.; McCluskey, J.; Yeo, J.P.; Tock, E.P.; Toh, B.H. EEA1, an early endosome-associated protein. EEA1 is a conserved alpha-helical peripheral membrane protein flanked by cysteine “fingers” and contains a calmodulin-binding IQ motif. J. Biol. Chem. 1995, 270, 13503–13511. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, G.; Hoflack, B.; Simons, K.; Mellman, I.; Kornfeld, S. The mannose 6-phosphate receptor and the biogenesis of lysosomes. Cell 1988, 52, 329–341. [Google Scholar] [CrossRef]
- Batzer, A.G.; Rotin, D.; Urena, J.M.; Skolnik, E.Y.; Schlessinger, J. Hierarchy of binding sites for Grb2 and Shc on the epidermal growth factor receptor. Mol. Cell. Biol. 1994, 14, 5192–5201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Xue, P.; Yang, M.; Shi, H.; Lu, D.; Wang, Z.; Shi, Q.; Hu, J.; Xie, S.; Zhan, W.; et al. Protein kinase D2 promotes the proliferation of glioma cells by regulating Golgi phosphoprotein 3. Cancer Lett. 2014, 355, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zhan, W.; Bian, W.; Hua, L.; Shi, Q.; Xie, S.; Yang, D.; Li, Y.; Zhang, X.; Liu, G.; et al. GOLPH3 regulates the migration and invasion of glioma cells though RhoA. Biochem. Biophys. Res. Commun. 2013, 433, 338–344. [Google Scholar] [CrossRef]
- Dai, T.; Zhang, D.; Cai, M.; Wang, C.; Wu, Z.; Ying, Z.; Wu, J.; Li, M.; Xie, D.; Li, J.; et al. Golgi phosphoprotein 3 (GOLPH3) promotes hepatocellular carcinoma cell aggressiveness by activating the NF-kappaB pathway. J. Pathol. 2015, 235, 490–501. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Yang, X.; Zhang, R.; Liu, S.; Gan, X.; Xi, X.; Zhang, Z.; Feng, Y.; Sun, Y. GOLPH3 induces epithelial-mesenchymal transition via Wnt/beta-catenin signaling pathway in epithelial ovarian cancer. Cancer Med. 2017, 6, 834–844. [Google Scholar] [CrossRef]
- Wang, M.Z.; Qiu, C.Z.; Yu, W.S.; Guo, Y.T.; Wang, C.X.; Chen, Z.X. GOLPH3 expression promotes the resistance of HT29 cells to 5fluorouracil by activating multiple signaling pathways. Mol. Med. Rep. 2018, 17, 542–548. [Google Scholar] [CrossRef] [Green Version]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef] [Green Version]
- Pinho, S.S.; Reis, C.A. Glycosylation in cancer: Mechanisms and clinical implications. Nat. Rev. Cancer 2015, 15, 540–555. [Google Scholar] [CrossRef]
- Sato, C.; Kim, J.H.; Abe, Y.; Saito, K.; Yokoyama, S.; Kohda, D. Characterization of the N-oligosaccharides attached to the atypical Asn-X-Cys sequence of recombinant human epidermal growth factor receptor. J. Biochem. 2000, 127, 65–72. [Google Scholar] [CrossRef]
- Liu, Y.C.; Yen, H.Y.; Chen, C.Y.; Chen, C.H.; Cheng, P.F.; Juan, Y.H.; Chen, C.H.; Khoo, K.H.; Yu, C.J.; Yang, P.C.; et al. Sialylation and fucosylation of epidermal growth factor receptor suppress its dimerization and activation in lung cancer cells. Proc. Natl. Acad. Sci. USA 2011, 108, 11332–11337. [Google Scholar] [CrossRef] [Green Version]
- Park, J.J.; Yi, J.Y.; Jin, Y.B.; Lee, Y.J.; Lee, J.S.; Lee, Y.S.; Ko, Y.G.; Lee, M. Sialylation of epidermal growth factor receptor regulates receptor activity and chemosensitivity to gefitinib in colon cancer cells. Biochem. Pharmacol. 2012, 83, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Gu, J.; Ihara, H.; Miyoshi, E.; Honke, K.; Taniguchi, N. Core fucosylation regulates epidermal growth factor receptor-mediated intracellular signaling. J. Biol. Chem. 2006, 281, 2572–2577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contessa, J.N.; Bhojani, M.S.; Freeze, H.H.; Rehemtulla, A.; Lawrence, T.S. Inhibition of N-linked glycosylation disrupts receptor tyrosine kinase signaling in tumor cells. Cancer Res. 2008, 68, 3803–3809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, C.; Pan, B.; Xu, H.; Zhao, Z.; Shen, J.; Lu, J.; Yu, R.; Liu, H. Co-delivery of GOLPH3 siRNA and gefitinib by cationic lipid-PLGA nanoparticles improves EGFR-targeted therapy for glioma. J. Mol. Med. 2019, 97, 1575–1588. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, Z.; Zhang, Y.; Wang, Y.; Zhang, H.; Xie, S.; Xie, P.; Yu, R.; Zhou, X. Golgi phosphoprotein 3 sensitizes the tumour suppression effect of gefitinib on gliomas. Cell Prolif. 2019, 52, e12636. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Kizuka, Y.; Ohtsubo, K.; Gu, J.; Taniguchi, N. Disease-associated glycans on cell surface proteins. Mol. Asp. Med. 2016, 51, 56–70. [Google Scholar] [CrossRef]
- Bonangelino, C.J.; Chavez, E.M.; Bonifacino, J.S. Genomic screen for vacuolar protein sorting genes in Saccharomyces cerevisiae. Mol. Biol. Cell 2002, 13, 2486–2501. [Google Scholar] [CrossRef] [Green Version]
- Sechi, S.; Colotti, G.; Belloni, G.; Mattei, V.; Frappaolo, A.; Raffa, G.D.; Fuller, M.T.; Giansanti, M.G. GOLPH3 is essential for contractile ring formation and Rab11 localization to the cleavage site during cytokinesis in Drosophila melanogaster. PLoS Genet. 2014, 10, e1004305. [Google Scholar] [CrossRef] [Green Version]
- Saravanan, C.; Liu, F.T.; Gipson, I.K.; Panjwani, N. Galectin-3 promotes lamellipodia formation in epithelial cells by interacting with complex N-glycans on alpha3beta1 integrin. J. Cell Sci. 2009, 122, 3684–3693. [Google Scholar] [CrossRef] [Green Version]
- Przybylo, M.; Pochec, E.; Link-Lenczowski, P.; Litynska, A. Beta1-6 branching of cell surface glycoproteins may contribute to uveal melanoma progression by up-regulating cell motility. Mol. Vis. 2008, 14, 625–636. [Google Scholar]
- Harms, B.D.; Bassi, G.M.; Horwitz, A.R.; Lauffenburger, D.A. Directional persistence of EGF-induced cell migration is associated with stabilization of lamellipodial protrusions. Biophys. J. 2005, 88, 1479–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gagliardi, P.A.; di Blasio, L.; Puliafito, A.; Seano, G.; Sessa, R.; Chianale, F.; Leung, T.; Bussolino, F.; Primo, L. PDK1-mediated activation of MRCKalpha regulates directional cell migration and lamellipodia retraction. J. Cell Biol. 2014, 206, 415–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, V.W.; Bai, M.; Li, J. Getting active: Protein sorting in endocytic recycling. Nat. Rev. Mol. Cell Biol. 2012, 13, 323–328. [Google Scholar] [CrossRef]
- Sigismund, S.; Woelk, T.; Puri, C.; Maspero, E.; Tacchetti, C.; Transidico, P.; Di Fiore, P.P.; Polo, S. Clathrin-independent endocytosis of ubiquitinated cargos. Proc. Natl. Acad. Sci. USA 2005, 102, 2760–2765. [Google Scholar] [CrossRef] [Green Version]
- Johannes, L.; Parton, R.G.; Bassereau, P.; Mayor, S. Building endocytic pits without clathrin. Nat. Rev. Mol. Cell Biol. 2015, 16, 311–321. [Google Scholar] [CrossRef]
- Dunn, W.A.; Connolly, T.P.; Hubbard, A.L. Receptor-mediated endocytosis of epidermal growth factor by rat hepatocytes: Receptor pathway. J. Cell Biol. 1986, 102, 24–36. [Google Scholar] [CrossRef] [Green Version]
- Henne, W.M.; Buchkovich, N.J.; Emr, S.D. The ESCRT pathway. Dev. Cell 2011, 21, 77–91. [Google Scholar] [CrossRef] [Green Version]
- Ye, Q.H.; Zhu, W.W.; Zhang, J.B.; Qin, Y.; Lu, M.; Lin, G.L.; Guo, L.; Zhang, B.; Lin, Z.H.; Roessler, S.; et al. GOLM1 Modulates EGFR/RTK Cell-Surface Recycling to Drive Hepatocellular Carcinoma Metastasis. Cancer Cell 2016, 30, 444–458. [Google Scholar] [CrossRef] [Green Version]
- Chung, I.; Akita, R.; Vandlen, R.; Toomre, D.; Schlessinger, J.; Mellman, I. Spatial control of EGF receptor activation by reversible dimerization on living cells. Nature 2010, 464, 783–787. [Google Scholar] [CrossRef]
- Gamou, S.; Shimizu, N. Glycosylation of the epidermal growth factor receptor and its relationship to membrane transport and ligand binding. J. Biochem. 1988, 104, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, T.; Ikeda, Y.; Taniguchi, N. The Asn-420-linked sugar chain in human epidermal growth factor receptor suppresses ligand-independent spontaneous oligomerization. Possible role of a specific sugar chain in controllable receptor activation. J. Biol. Chem. 2000, 275, 21988–21994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, M.; Yokoe, S.; Asahi, M.; Lee, S.H.; Li, W.; Osumi, D.; Miyoshi, E.; Taniguchi, N. N-glycan of ErbB family plays a crucial role in dimer formation and tumor promotion. Biochim. Biophys. Acta 2008, 1780, 520–524. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.B.; Johnson, H.; Randolph, M.; Lee, I.; Pierce, M. Knockdown of GnT-Va expression inhibits ligand-induced downregulation of the epidermal growth factor receptor and intracellular signaling by inhibiting receptor endocytosis. Glycobiology 2009, 19, 547–559. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.L.; Lin, Y.S.; Shi, G.Y.; Chang, C.F.; Wu, H.L. Lewisy promotes migration of oral cancer cells by glycosylation of epidermal growth factor receptor. PLoS ONE 2015, 10, e0120162. [Google Scholar] [CrossRef]
- Mathew, M.P.; Tan, E.; Saeui, C.T.; Bovonratwet, P.; Sklar, S.; Bhattacharya, R.; Yarema, K.J. Metabolic flux-driven sialylation alters internalization, recycling, and drug sensitivity of the epidermal growth factor receptor (EGFR) in SW1990 pancreatic cancer cells. Oncotarget 2016, 7, 66491–66511. [Google Scholar] [CrossRef]
- Raiborg, C.; Stenmark, H. The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature 2009, 458, 445–452. [Google Scholar] [CrossRef]
- Henne, W.M.; Stenmark, H.; Emr, S.D. Molecular mechanisms of the membrane sculpting ESCRT pathway. Cold Spring Harb. Perspect. Biol. 2013, 5, a016766. [Google Scholar] [CrossRef] [Green Version]
- Clague, M.J.; Liu, H.; Urbe, S. Governance of endocytic trafficking and signaling by reversible ubiquitylation. Dev. Cell 2012, 23, 457–467. [Google Scholar] [CrossRef] [Green Version]
- Umebayashi, K.; Stenmark, H.; Yoshimori, T. Ubc4/5 and c-Cbl continue to ubiquitinate EGF receptor after internalization to facilitate polyubiquitination and degradation. Mol. Biol. Cell 2008, 19, 3454–3462. [Google Scholar] [CrossRef] [Green Version]
- Luchsinger, C.; Aguilar, M.; Burgos, P.V.; Ehrenfeld, P.; Mardones, G.A. Functional disruption of the Golgi apparatus protein ARF1 sensitizes MDA-MB-231 breast cancer cells to the antitumor drugs Actinomycin D and Vinblastine through ERK and AKT signaling. PLoS ONE 2018, 13, e0195401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, B.H.; Lin, Y.; Corales, E.A.; Burgos, P.V.; Mardones, G.A. Structural and functional characterization of cargo-binding sites on the mu4-subunit of adaptor protein complex 4. PLoS ONE 2014, 9, e88147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, H.A.; Rivera-Dictter, A.; Cavieres, V.A.; Muñoz, V.C.; González, A.; Lin, Y.; Mardones, G.A.; Burgos, P.V. Turnover of C99 is controlled by a crosstalk between ERAD and ubiquitin-independent lysosomal degradation in human neuroglioma cells. PLoS ONE 2013, 8, e83096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tenorio, M.J.; Luchsinger, C.; Mardones, G.A. Protein Kinase A Activity Is Necessary for Fission and Fusion of Golgi to Endoplasmic Reticulum Retrograde Tubules. PLoS ONE 2015, 10, e0135260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González, A.E.; Muñoz, V.C.; Cavieres, V.A.; Bustamante, H.A.; Cornejo, V.H.; Januário, Y.C.; González, I.; Hetz, C.; daSilva, L.L.; Rojas-Fernández, A.; et al. Autophagosomes cooperate in the degradation of intracellular C-terminal fragments of the amyloid precursor protein via the MVB/lysosomal pathway. FASEB J. 2017, 31, 2446–2459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crupi, M.J.F.; Richardson, D.S.; Mulligan, L.M. Cell surface biotinylation of receptor tyrosine kinases to investigate intracellular trafficking. Methods Mol. Biol. 2015, 1233, 91–102. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arriagada, C.; Cavieres, V.A.; Luchsinger, C.; González, A.E.; Muñoz, V.C.; Cancino, J.; Burgos, P.V.; Mardones, G.A. GOLPH3 Regulates EGFR in T98G Glioblastoma Cells by Modulating Its Glycosylation and Ubiquitylation. Int. J. Mol. Sci. 2020, 21, 8880. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228880
Arriagada C, Cavieres VA, Luchsinger C, González AE, Muñoz VC, Cancino J, Burgos PV, Mardones GA. GOLPH3 Regulates EGFR in T98G Glioblastoma Cells by Modulating Its Glycosylation and Ubiquitylation. International Journal of Molecular Sciences. 2020; 21(22):8880. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228880
Chicago/Turabian StyleArriagada, Cecilia, Viviana A. Cavieres, Charlotte Luchsinger, Alexis E. González, Vanessa C. Muñoz, Jorge Cancino, Patricia V. Burgos, and Gonzalo A. Mardones. 2020. "GOLPH3 Regulates EGFR in T98G Glioblastoma Cells by Modulating Its Glycosylation and Ubiquitylation" International Journal of Molecular Sciences 21, no. 22: 8880. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228880