Multifaceted Role of PRDM Proteins in Human Cancer

 ,
,  ,
,  , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Role of PRDM Genes in Cancer
2.1. PRDM1
2.2. PRDM2
2.3. MECOM/PRDM3
2.4. PRDM4
2.5. PRDM5
2.6. PRDM6
2.7. PRDM7
2.8. PRDM8
2.9. PRDM9
2.10. PRDM10
2.11. PRDM11
2.12. PRDM12
2.13. PRDM13
2.14. PRDM14
2.15. PRDM15
2.16. PRDM16
2.17. ZNF408/PRDM17
2.18. ZFPM1/FOG1
2.19. ZFPM2/FOG2
3. Clinical Value of PRDMs in Cancer and Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABC | Activated-B cell like |
| ACC | Adrenocortical carcinoma |
| ALL | Acute lymphoblastic leukemia |
| AML | Acute myeloid leukemia |
| ARHGAP30 | Rho GTPase-activating protein 30 |
| ATM | Ataxia telangiectasia mutated |
| CEBPβ | CCAAT/enhancer-binding protein beta-2 isoform |
| ceRNA | Competitive endogenous RNA |
| CIN | Chromosomal Instability |
| CML | Chronic myeloid leukemia |
| DKK | Dickkopf-1 LDL low-density lipoprotein |
| DLBCL | Diffuse large B cell lymphoma |
| DLC | Deleted in liver cancer |
| DSB | Double-strand break |
| EGF | Epidermal growth factor |
| EMT | Epithelial-to-mesenchymal transition |
| ER | Estrogen receptor |
| FOG | Friend of GATA |
| GCB | Germinal center B-cell |
| GRP78 | Glucose-regulated protein 78 |
| HDAC | Histone deacetylases |
| HMT | Histone methyltransferases |
| HNSCC | Head and neck squamous cell carcinoma |
| HPV | Human papillomavirus |
| IGF-1 | Insulin-like growth factor-1 |
| IMiDs | Immunomodulatory drugs |
| ITGB2 | Leukocyte-specific integrin β2 |
| KMT | Lysine methyltransferases |
| LL | Lymphoblastic leukemia |
| LUAD | Lung adenocarcinoma |
| MAGL | Monoacylglycerol lipase |
| MIN | Microsatellite Instability |
| MMP | Matrix Metalloproteinase |
| MUC4 | Mucin-4 |
| NAP1L1 | Nucleosome assembly protein 1-like 1 |
| PC | Prostate cancer |
| PRDM | PRD-BF1 and RIZ homology domain containing |
| RTK | Tyrosine kinase receptor |
| SERPIN | Serine protease inhibitor |
| TCGA | The Cancer Genome Atlas |
| TGF | Transforming growth factor |
| TIMP | Tissue inhibitor of metalloproteinases |
| TXNIP | Thioredoxin binding protein |
| VEGF | Vascular endothelial growth factor |
| YAP | Yes-associated protein |
References
- Di Zazzo, E.; De Rosa, C.; Abbondanza, C.; Moncharmont, B. PRDM Proteins: Molecular Mechanisms in Signal Transduction and Transcriptional Regulation. Biology 2013, 2, 107–141. [Google Scholar] [CrossRef] [Green Version]
- Mzoughi, S.; Tan, Y.X.; Low, D.; Guccione, E. The role of PRDMs in cancer: One family, two sides. Curr. Opin. Genet. Dev. 2016, 36, 83–91. [Google Scholar] [CrossRef]
- Sorrentino, A.; Rienzo, M.; Ciccodicola, A.; Casamassimi, A.; Abbondanza, C. Human PRDM2: Structure, function and pathophysiology. Biochim. Biophys. Acta Gene Regul. Mech. 2018, 1861, 657–671. [Google Scholar] [CrossRef]
- Clifton, M.K.; Westman, B.J.; Thong, S.Y.; O’Connell, M.R.; Webster, M.W.; Shepherd, N.E.; Quinlan, K.G.; Crossley, M.; Blobel, G.A.; Mackay, J.P. The identification and structure of an N-terminal PR domain show that FOG1 is a member of the PRDM family of proteins. PLoS ONE 2014, 9, e106011. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, K.; Yoshida, K.; Matsui, Y. A histone H3 methyltransferase controls epigenetic events required for meiotic prophase. Nature 2005, 438, 374–378. [Google Scholar] [CrossRef]
- Eram, M.S.; Bustos, S.P.; Lima-Fernandes, E.; Siarheyeva, A.; Senisterra, G.; Hajian, T.; Chau, I.; Duan, S.; Wu, H.; Dombrovski, L.; et al. Trimethylation of histone H3 lysine 36 by human methyltransferase PRDM9 protein. J. Biol. Chem. 2014, 289, 12177–12188. [Google Scholar] [CrossRef] [Green Version]
- Pinheiro, I.; Margueron, R.; Shukeir, N.; Eisold, M.; Fritzsch, C.; Richter, F.M.; Mittler, G.; Genoud, C.; Goyama, S.; Kurokawa, M.; et al. Prdm3 and Prdm16 are H3K9me1 methyltransferases required for mammalian heterochromatin integrity. Cell 2012, 150, 948–960. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Shao, G.; Liu, L. The PR domain of the Rb-binding zinc finger protein RIZ1 is a protein binding interface and is related to the SET domain functioning in chromatin-mediated gene expression. J. Biol. Chem. 1998, 273, 15933–15939. [Google Scholar] [CrossRef] [Green Version]
- Ren, B.; Chee, K.J.; Kim, T.H.; Maniatis, T. PRDI-BF1/Blimp-1 repression is mediated by corepressors of the Groucho family of proteins. Genes Dev. 1999, 13, 125–137. [Google Scholar] [CrossRef] [Green Version]
- Seale, P.; Bjork, B.; Yang, W.; Kajimura, S.; Chin, S.; Kuang, S.; Scimè, A.; Devarakonda, S.; Conroe, H.M.; Erdjument-Bromage, H.; et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature 2008, 454, 961–967. [Google Scholar] [CrossRef] [Green Version]
- Okashita, N.; Suwa, Y.; Nishimura, O.; Sakashita, N.; Kadota, M.; Nagamatsu, G.; Kawaguchi, M.; Kashida, H.; Nakajima, A.; Tachibana, M.; et al. PRDM14 Drives OCT3/4 Recruitment via Active Demethylation in the Transition from Primed to Naive Pluripotency. Stem Cell Rep. 2016, 7, 1072–1086. [Google Scholar] [CrossRef] [Green Version]
- Chi, J.; Cohen, P. The Multifaceted Roles of PRDM16: Adipose Biology and Beyond. Trends Endocrinol. Metab. 2016, 27, 11–23. [Google Scholar] [CrossRef]
- Keller, A.D.; Maniatis, T. Identification and characterization of a novel repressor of beta-interferon gene expression. Genes Dev. 1991, 5, 868–879. [Google Scholar] [CrossRef] [Green Version]
- Turner, C.A.; Mack, D.H.; Davis, M.M. Blimp-1, a novel zinc finger-containing protein that can drive the maturation of B lymphocytes into immunoglobulin-secreting cells. Cell 1994, 77, 297–306. [Google Scholar] [CrossRef]
- Martins, G.A.; Cimmino, L.; Shapiro-Shelef, M.; Szabolcs, M.; Herron, A.; Magnusdottir, E.; Calame, K. Transcriptional repressor Blimp-1 regulates T cell homeostasis and function. Nat. Immunol. 2006, 7, 457–465. [Google Scholar] [CrossRef]
- Kallies, A.; Hawkins, E.D.; Belz, G.T.; Metcalf, D.; Hommel, M.; Corcoran, L.M.; Hodgkin, P.D.; Nutt, S.L. Transcriptional repressor Blimp-1 is essential for T cell homeostasis and self-tolerance. Nat. Immunol. 2006, 7, 466–474. [Google Scholar] [CrossRef]
- De Mel, S.; Hue, S.S.; Jeyasekharan, A.D.; Chng, W.J.; Ng, S.B. Molecular pathogenic pathways in extranodal NK/T cell lymphoma. J. Hematol. Oncol. 2019, 12, 33. [Google Scholar] [CrossRef]
- Boi, M.; Zucca, E.; Inghirami, G.; Bertoni, F. PRDM1/BLIMP1: A tumor suppressor gene in B and T cell lymphomas. Leuk. Lymphoma 2015, 56, 1223–1228. [Google Scholar] [CrossRef]
- Pasqualucci, L.; Compagno, M.; Houldsworth, J.; Monti, S.; Grunn, A.; Nandula, S.V.; Aster, J.C.; Murty, V.V.; Shipp, M.A.; Dalla-Favera, R. Inactivation of the PRDM1/BLIMP1 gene in diffuse large B cell lymphoma. J. Exp. Med. 2006, 203, 311–317. [Google Scholar] [CrossRef] [Green Version]
- Tam, W.; Gomez, M.; Chadburn, A.; Lee, J.W.; Chan, W.C.; Knowles, D.M. Mutational analysis of PRDM1 indicates a tumor-suppressor role in diffuse large B-cell lymphomas. Blood 2006, 107, 4090–4100. [Google Scholar] [CrossRef]
- Iqbal, J.; Kucuk, C.; Deleeuw, R.J.; Srivastava, G.; Tam, W.; Geng, H.; Klinkebiel, D.; Christman, J.K.; Patel, K.; Cao, K.; et al. Genomic analyses reveal global functional alterations that promote tumor growth and novel tumor suppressor genes in natural killer-cell malignancies. Leukemia 2009, 23, 1139–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Küçük, C.; Iqbal, J.; Hu, X.; Gaulard, P.; De Leval, L.; Srivastava, G.; Au, W.Y.; McKeithan, T.W.; Chan, W.C. PRDM1 is a tumor suppressor gene in natural killer cell malignancies. Proc. Natl. Acad. Sci. USA 2011, 108, 20119–20124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boi, M.; Rinaldi, A.; Kwee, I.; Bonetti, P.; Todaro, M.; Tabbò, F.; Piva, R.; Rancoita, P.M.; Matolcsy, A.; Timar, B.; et al. PRDM1/BLIMP1 is commonly inactivated in anaplastic large T-cell lymphoma. Blood 2013, 122, 2683–2693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, K.; Gomez, M.; Landgraf, P.; Garcia, J.F.; Liu, Y.; Tan, L.H.; Chadburn, A.; Tuschl, T.; Knowles, D.M.; Tam, W. MicroRNA-mediated down-regulation of PRDM1/Blimp-1 in Hodgkin/Reed-Sternberg cells: A potential pathogenetic lesion in Hodgkin lymphomas. Am. J. Pathol. 2008, 173, 242–252. [Google Scholar] [CrossRef] [Green Version]
- Nie, K.; Zhang, T.; Allawi, H.; Gomez, M.; Liu, Y.; Chadburn, A.; Wang, Y.L.; Knowles, D.M.; Tam, W. Epigenetic down-regulation of the tumor suppressor gene PRDM1/Blimp-1 in diffuse large B cell lymphomas: A potential role of the microRNA let-7. Am. J. Pathol. 2010, 177, 1470–1479. [Google Scholar] [CrossRef]
- Xia, Y.; Xu-Monette, Z.Y.; Tzankov, A.; Li, X.; Manyam, G.C.; Murty, V.; Bhagat, G.; Zhang, S.; Pasqualucci, L.; Visco, C.; et al. Loss of PRDM1/BLIMP-1 function contributes to poor prognosis of activated B-cell-like diffuse large B-cell lymphoma. Leukemia 2017, 31, 625–636. [Google Scholar] [CrossRef]
- Mandelbaum, J.; Bhagat, G.; Tang, H.; Mo, T.; Brahmachary, M.; Shen, Q.; Chadburn, A.; Rajewsky, K.; Tarakhovsky, A.; Pasqualucci, L.; et al. BLIMP1 is a tumor suppressor gene frequently disrupted in activated B cell-like diffuse large B cell lymphoma. Cancer Cell 2010, 18, 568–579. [Google Scholar] [CrossRef] [Green Version]
- Wan, Z.; Lu, Y.; Rui, L.; Yu, X.; Li, Z. PRDM1 overexpression induce G0/G1 arrest in DF-1 cell line. Gene 2016, 592, 119–127. [Google Scholar] [CrossRef]
- Calado, D.P.; Zhang, B.; Srinivasan, L.; Sasaki, Y.; Seagal, J.; Unitt, C.; Rodig, S.; Kutok, J.; Tarakhovsky, A.; Schmidt-Supprian, M.; et al. Constitutive canonical NF-κB activation cooperates with disruption of BLIMP1 in the pathogenesis of activated B cell-like diffuse large cell lymphoma. Cancer Cell 2010, 18, 580–589. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Liang, L.; Li, D.; Nong, L.; Zheng, Y.; Huang, S.; Zhang, B.; Li, T. JAK3/STAT3 oncogenic pathway and PRDM1 expression stratify clinicopathologic features of extranodal NK/T-cell lymphoma, nasal type. Oncol. Rep. 2019, 41, 3219–3232. [Google Scholar] [CrossRef]
- Baytak, E.; Gong, Q.; Akman, B.; Yuan, H.; Chan, W.C.; Küçük, C. Whole transcriptome analysis reveals dysregulated oncogenic lncRNAs in natural killer/T-cell lymphoma and establishes MIR155HG as a target of PRDM1. Tumour Biol. 2017, 39, 1010428317701648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Liang, L.; Li, D.; Nong, L.; Liu, J.; Qu, L.; Qu, L.; Zheng, Y.; Zhang, B.; Li, T. Hypermethylation of PRDM1/Blimp-1 promoter in extranodal NK/T-cell lymphoma, nasal type: An evidence of predominant role in its downregulation. Hematol. Oncol. 2017, 35, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Zhang, Z.; Wang, Y.; Nong, L.; Zheng, Y.; Qu, L.; Zhang, B.; Li, T. The Genetic Deletion of 6q21 and PRDM1 and Clinical Implications in Extranodal NK/T Cell Lymphoma, Nasal Type. Biomed. Res. Int. 2015, 2015, 435423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Angelin-Duclos, C.; Greenwood, J.; Liao, J.; Calame, K. Transcriptional repression by blimp-1 (PRDI-BF1) involves recruitment of histone deacetylase. Mol. Cell Biol. 2000, 20, 2592–2603. [Google Scholar] [CrossRef] [Green Version]
- Gyory, I.; Wu, J.; Fejér, G.; Seto, E.; Wright, K.L. PRDI-BF1 recruits the histone H3 methyltransferase G9a in transcriptional silencing. Nat. Immunol. 2004, 5, 299–308. [Google Scholar] [CrossRef]
- Montes-Moreno, S.; Martinez-Magunacelaya, N.; Zecchini-Barrese, T.; Villambrosía, S.G.; Linares, E.; Ranchal, T.; Rodriguez-Pinilla, M.; Batlle, A.; Cereceda-Company, L.; Revert-Arce, J.B.; et al. Plasmablastic lymphoma phenotype is determined by genetic alterations in MYC and PRDM1. Mod. Pathol. 2017, 30, 85–94. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Belguise, K.; O’Neill, C.F.; Sánchez-Morgan, N.; Romagnoli, M.; Eddy, S.F.; Mineva, N.D.; Yu, Z.; Min, C.; Trinkaus-Randall, V.; et al. RelB NF-kappaB represses estrogen receptor alpha expression via induction of the zinc finger protein Blimp1. Mol. Cell Biol. 2009, 29, 3832–3844. [Google Scholar] [CrossRef] [Green Version]
- Romagnoli, M.; Belguise, K.; Yu, Z.; Wang, X.; Landesman-Bollag, E.; Seldin, D.C.; Chalbos, D.; Barillé-Nion, S.; Jézéquel, P.; Seldin, M.L.; et al. Epithelial-to-mesenchymal transition induced by TGF-β1 is mediated by Blimp-1-dependent repression of BMP-5. Cancer Res. 2012, 72, 6268–6278. [Google Scholar] [CrossRef] [Green Version]
- Sciortino, M.; Camacho-Leal, M.D.P.; Orso, F.; Grassi, E.; Costamagna, A.; Provero, P.; Tam, W.; Turco, E.; Defilippi, P.; Taverna, D.; et al. Dysregulation of Blimp1 transcriptional repressor unleashes p130Cas/ErbB2 breast cancer invasion. Sci. Rep. 2017, 7, 1145. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Jiang, J.; Lim, C.A.; Wu, Q.; Ng, H.H.; Chin, K.C. BLIMP1 regulates cell growth through repression of p53 transcription. Proc. Natl. Acad. Sci. USA 2007, 104, 1841–1846. [Google Scholar] [CrossRef] [Green Version]
- Györy, I.; Fejér, G.; Ghosh, N.; Seto, E.; Wright, K.L. Identification of a functionally impaired positive regulatory domain I binding factor 1 transcription repressor in myeloma cell lines. J. Immunol. 2003, 170, 3125–3133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Banister, C.E.; Weige, C.C.; Altomare, D.; Richardson, J.H.; Contreras, C.M.; Buckhaults, P.J. PRDM1 silences stem cell-related genes and inhibits proliferation of human colon tumor organoids. Proc. Natl. Acad. Sci. USA 2018, 115, E5066–E5075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Wang, K.; Han, L.; Zhang, A.; Shi, Z.; Zhang, K.; Zhang, H.; Yang, S.; Pu, P.; Shen, C.; et al. PRDM1 is directly targeted by miR-30a-5p and modulates the Wnt/β-catenin pathway in a Dkk1-dependent manner during glioma growth. Cancer Lett. 2013, 331, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Wang, H.; Wei, Y.; Meng, F.; Liu, Z.; Zhang, Z. Downregulation of PRDM1 promotes cellular invasion and lung cancer metastasis. Tumour Biol. 2017, 39, 1010428317695929. [Google Scholar] [CrossRef] [Green Version]
- Sorrentino, A.; Federico, A.; Rienzo, M.; Gazzerro, P.; Bifulco, M.; Ciccodicola, A.; Casamassimi, A.; Abbondanza, C. PR/SET Domain Family and Cancer: Novel Insights from the Cancer Genome Atlas. Int. J. Mol. Sci. 2018, 19, 3250. [Google Scholar] [CrossRef] [Green Version]
- Desmots, F.; Roussel, M.; Pangault, C.; Llamas-Gutierrez, F.; Pastoret, C.; Guiheneuf, E.; Le Priol, J.; Camara-Clayette, V.; Caron, G.; Henry, C.; et al. Pan-HDAC Inhibitors Restore PRDM1 Response to IL21 in CREBBP-Mutated Follicular Lymphoma. Clin. Cancer Res. 2019, 25, 735–746. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Shao, G.; Steele-Perkins, G.; Huang, S. The retinoblastoma interacting zinc finger gene RIZ produces a PR domain-lacking product through an internal promoter. J. Biol. Chem. 1997, 272, 2984–2991. [Google Scholar] [CrossRef] [Green Version]
- Huang, S. The retinoblastoma protein-interacting zinc finger gene RIZ in 1p36-linked cancers. Front. Biosci. 1999, 4, D528–D532. [Google Scholar] [CrossRef] [Green Version]
- Abbondanza, C.; De Rosa, C.; D’Arcangelo, A.; Pacifico, M.; Spizuoco, C.; Piluso, G.; Di Zazzo, E.; Gazzerro, P.; Medici, N.; Moncharmont, B.; et al. Identification of a functional estrogen-responsive enhancer element in the promoter 2 of PRDM2 gene in breast cancer cell lines. J. Cell. Physiol. 2012, 227, 964–975. [Google Scholar] [CrossRef]
- Jiang, G.L.; Huang, S. The yin–yang of PR-domain family genes in tumorigenesis. Histol. Histopathol. 2000, 15, 109–117. [Google Scholar] [CrossRef]
- Kim, K.C.; Geng, L.; Huang, S. Inactivation of a histone methyltransferase by mutations in human cancers. Cancer Res. 2003, 63, 7619–7623. [Google Scholar] [PubMed]
- He, L.; Yu, J.X.; Liu, L.; Buyse, I.M.; Wang, M.S.; Yang, Q.C.; Nakagawara, A.; Brodeur, G.M.; Shi, Y.E.; Huang, S. RIZ1, but not the alternative RIZ2 product of the same gene, is underexpressed in breast cancer, and forced RIZ1 expression causes G2-M cell cycle arrest and/or apoptosis. Cancer Res. 1998, 58, 4238–4244. [Google Scholar] [PubMed]
- Rossi, M.; Abbondanza, C.; D’Arcangelo, A.; Gazzerro, P.; Medici, N.; Moncharmont, B.; Puca, G.A. The Zn-finger domain of RIZ protein promotes MCF-7 cell proliferation. Cancer Lett. 2004, 215, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Chambery, A.; Farina, A.; Di Maro, A.; Rossi, M.; Abbondanza, C.; Moncharmont, B.; Malorni, L.; Cacace, G.; Pocsfalvi, G.; Malorni, A.; et al. Proteomic analysis of MCF-7 cell lines expressing the zinc-finger or the proline-rich domain of retinoblastoma-interacting-zincfinger protein. J. Proteome Res. 2004, 5, 1176–1185. [Google Scholar] [CrossRef] [PubMed]
- Steele-Perkins, G.; Fang, W.; Yang, X.H.; Van Gele, M.; Carling, T.; Gu, J.; Buyse, I.M.; Fletcher, J.A.; Liu, J.; Bronson, R.; et al. Tumor formation and inactivation of RIZ1, an Rb-binding member of a nuclear protein methyltransferase superfamily. Genes Dev. 2001, 15, 2250–2262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piao, Z.; Fang, W.; Malkhosyan, S.; Kim, H.; Horii, A.; Perucho, M.; Huang, S. Frequent frameshift mutations of RIZ in sporadic gastrointestinal and endometrial carcinomas with microsatellite instability. Cancer Res. 2000, 60, 4701–4704. [Google Scholar]
- Jiang, G.L.; Huang, S. Adenovirus expressing RIZ1 in tumor suppressor gene therapy of microsatellite-unstable colorectal cancers. Cancer Res. 2001, 61, 1796–1798. [Google Scholar]
- Sakurada, K.; Furukawa, T.; Kato, Y.; Kayama, T.; Huang, S.; Horii, A. RIZ, the retinoblastoma protein interacting zinc finger gene, is mutated in genetically unstable cancers of the pancreas, stomach, and colorectum. Genes Chromosomes Cancer 2001, 30, 207–211. [Google Scholar] [CrossRef]
- Chadwick, R.B.; Jiang, G.L.; Bennington, G.A.; Yuan, B.; Johnson, C.K.; Stevens, M.W.; Niemann, T.H.; Peltomaki, P.; Huang, S.; de la Chapelle, A. Candidate tumor suppressor RIZ is frequently involved in colorectal carcinogenesis. Proc. Natl. Acad. Sci. USA 2000, 97, 2662–2667. [Google Scholar] [CrossRef] [Green Version]
- Maruvka, Y.E.; Mouw, K.W.; Karlic, R.; Parasuraman, P.; Kamburov, A.P.; Haradhvala, N.J.; Hess, J.M.; Rheinbay, E.; Brody, Y.; Koren, A.; et al. Analysis of somatic microsatellite indels identifies driver events in human tumors. Nat. Biotechnol. 2017, 35, 951–959. [Google Scholar] [CrossRef]
- Pandzic, T.; Rendo, V.; Lim, J.; Larsson, C.; Larsson, J.; Stoimenov, I.; Kundu, S.; Ali, M.A.; Hellström, M.; He, L.; et al. Somatic PRDM2 c.4467delA mutations in colorectal cancers control histone methylation and tumor growth. Oncotarget 2017, 8, 98646–98659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poetsch, M.; Dittberner, T.; Woenckhaus, C. Frameshift mutations of RIZ, but no point mutations in RIZ1 exons in malignant melanomas with deletions in 1p36. Oncogene 2002, 21, 3038–3042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, O.; Meguro, K.; Tohmiya, Y.; Funato, T.; Shibahara, S.; Sasaki, T. Nucleotide alteration of retinoblastoma protein-interacting zinc finger gene, RIZ, in human leukemia. Tohoku J. Exp. Med. 2002, 196, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Fang, W.; Piao, Z.; Simon, D.; Sheu, J.C.; Huang, S. Mapping of a minimal deleted region in human hepatocellular carcinoma to 1p36.13-p36.23 and mutational analysis of the RIZ (PRDM2) gene localized to the region. Genes Chromosomes Cancer 2000, 28, 269–275. [Google Scholar] [CrossRef]
- Fang, W.; Piao, Z.; Simon, D.; Sheu, J.C.; Perucho, M.; Huang, S. Preferential loss of a polymorphic RIZ allele in human hepatocellular carcinoma. Br. J. Cancer 2001, 84, 743–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mir, R.; Najar, I.A.; Guru, S.; Javaid, J.; Yadav, P.; Masroor, M.; Zuberi, M.; Farooq, S.; Bhat, M.; Gupta, N.; et al. A deletion polymorphism in the RIZ gene is associated with increased progression of imatinib treated chronic myeloid leukemia patients. Leuk. Lymphoma 2017, 58, 1694–1701. [Google Scholar] [CrossRef]
- Rossi, V.; Staibano, S.; Abbondanza, C.; Pasquali, D.; De Rosa, C.; Mascolo, M.; Bellastella, G.; Visconti, D.; De Bellis, A.; Moncharmont, B.; et al. Expression of RIZ1 protein (Retinoblastoma-interacting zinc-finger protein 1) in prostate cancer epithelial cells changes with cancer grade progression and is modulated in vitro by DHT and E2. J. Cell Physiol. 2009, 221, 771–777. [Google Scholar] [CrossRef]
- Yang, T.; Ren, C.; Jiang, A.; Yu, Z.; Li, G.; Wang, G.; Zhang, Q. RIZ1 is regulated by estrogen and suppresses tumor progression in endometrial cancer. Biochem. Biophys. Res. Commun. 2017, 489, 96–102. [Google Scholar] [CrossRef]
- Du, Y.; Carling, T.; Fang, W.; Piao, Z.; Sheu, J.C.; Huang, S. Hypermethylation in human cancers of the RIZ1 tumor suppressor gene, a member of a histone/protein methyltransferase superfamily. Cancer Res. 2001, 61, 8094–8099. [Google Scholar]
- Zhao, Z.; Hu, Y.; Shen, X.; Lao, Y.; Zhang, L.; Qiu, X.; Hu, J.; Gong, P.; Cui, H.; Lu, S.; et al. HBx represses RIZ1 expression by DNA methyltransferase 1 involvement in decreased miR-152 in hepatocellular carcinoma. Oncol. Rep. 2017, 37, 2811–2818. [Google Scholar] [CrossRef]
- Xue, Y.; Chen, R.; Du, W.; Yang, F.; Wei, X. RIZ1 and histone methylation status in pituitary adenomas. Tumour Biol. 2017, 39, 1010428317711794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.W.; Chan, A.; Kwong, D.L.; Wei, W.I.; Sham, J.S.; Yuen, A.P. Detection of hypermethylated RIZ1 gene in primary tumor, mouth, and throat rinsing fluid, nasopharyngeal swab, and peripheral blood of nasopharyngeal carcinoma patient. Clin. Cancer Res. 2003, 9, 1033–1038. [Google Scholar] [PubMed]
- Pastural, E.; Takahashi, N.; Dong, W.F.; Bainbridge, M.; Hull, A.; Pearson, D.; Huang, S.; Lowsky, R.; DeCoteau, J.F.; Geyer, C.R. RIZ1 repression is associated with insulin-like growth factor-1 signaling activation in chronic myeloid leukemia cell lines. Oncogene 2007, 26, 1586–1594. [Google Scholar] [CrossRef]
- Medici, N.; Abbondanza, C.; Nigro, V.; Rossi, V.; Piluso, G.; Belsito, A.; Gallo, L.; Roscigno, A.; Bontempo, P.; Puca, A.A.; et al. Identification of a DNA binding protein cooperating with estrogen receptor as RIZ (retinoblastoma interacting zinc finger protein). Biochem. Biophys. Res. Commun. 1999, 264, 983–989. [Google Scholar] [CrossRef]
- Abbondanza, C.; Medici, N.; Nigro, V.; Rossi, V.; Gallo, L.; Piluso, G.; Belsito, A.; Roscigno, A.; Bontempo, P.; Puca, A.A.; et al. The retinoblastoma-interacting zinc-finger protein RIZ is a downstream effector of estrogen action. Proc. Natl. Acad. Sci. USA 2000, 97, 3130–3135. [Google Scholar] [CrossRef]
- Gazzerro, P.; Abbondanza, C.; D’Arcangelo, A.; Rossi, M.; Medici, N.; Moncharmont, B.; Puca, G.A. Modulation of RIZ gene expression is associated to estradiol control of MCF-7 breast cancer cell proliferation. Exp. Cell Res. 2006, 312, 340–349. [Google Scholar] [CrossRef]
- Di Zazzo, E.; Porcile, C.; Bartollino, S.; Moncharmont, B. Critical Function of PRDM2 in the Neoplastic Growth of Testicular Germ Cell Tumors. Biology 2016, 5, 54. [Google Scholar] [CrossRef]
- Abbondanza, C.; De Rosa, C.; Ombra, M.N.; Aceto, F.; Medici, N.; Altucci, L.; Moncharmont, B.; Puca, G.A.; Porcellini, A.; Avvedimento, E.V.; et al. Highlighting chromosome loops in DNA-picked chromatin (DPC). Epigenetics 2011, 6, 979–986. [Google Scholar] [CrossRef] [Green Version]
- Bhat-Nakshatri, P.; Wang, G.; Collins, N.R.; Thomson, M.J.; Geistlinger, T.R.; Carroll, J.S.; Brown, M.; Hammond, S.; Srour, E.F.; Liu, Y.; et al. Estradiol-regulated microRNAs control estradiol response in breast cancer cells. Nucleic Acids Res. 2009, 37, 4850–4861. [Google Scholar] [CrossRef] [Green Version]
- Gazzerro, P.; Bontempo, P.; Schiavone, E.M.; Abbondanza, C.; Moncharmont, B.; Ignazio Armetta, I.; Medici, N.; De Simone, M.; Nola, E.; Puca, G.A.; et al. Differentiation of myeloid cell lines correlates with a selective expression of RIZ protein. Mol. Med. 2001, 7, 552–560. [Google Scholar] [CrossRef]
- Sun, W.; Qiao, L.; Liu, Q.; Chen, L.; Ling, B.; Sammynaiken, R.; Yang, J. Anticancer activity of the PR domain of tumor suppressor RIZ1. Int. J. Med. Sci. 2011, 8, 161–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, M.H.; Wang, Z.; Jiang, L.; Fu, H.L.; Gao, J.; Lin, X.B.; Zhang, C.L.; Liu, Z.Y.; Shi, Y.F.; Qiu, G.Z.; et al. The transducible TAT-RIZ1-PR protein exerts histone methyltransferase activity and tumor-suppressive functions in human malignant meningiomas. Biomaterials 2015, 56, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Zou, Y.; Hu, H.; Lu, C.; Sun, W.; Jiang, L.; Hu, G. RIZ1 negatively regulates ubiquitin-conjugating enzyme E2C/UbcH10 via targeting c-Myc in meningioma. Am. J. Transl. Res. 2017, 9, 2645–2655. [Google Scholar] [PubMed]
- Congdon, L.M.; Sims, J.K.; Tuzon, C.T.; Rice, J.C. The PR-Set7 binding domain of Riz1 is required for the H4K20me1-H3K9me1 trans-tail ‘histone code’ and Riz1 tumor suppressor function. Nucleic Acids Res. 2014, 42, 3580–3589. [Google Scholar] [CrossRef] [PubMed]
- Khurana, S.; Kruhlak, M.J.; Kim, J.; Tran, A.D.; Liu, J.; Nyswaner, K.; Shi, L.; Jailwala, P.; Sung, M.H.; Hakim, O.; et al. A macrohistone variant links dynamic chromatin compaction to BRCA1-dependent genome maintenance. Cell Rep. 2014, 8, 1049–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morishita, K.; Parker, D.S.; Mucenski, M.L.; Jenkins, N.A.; Copeland, N.G.; Ihle, J.N. Retroviral activation of a novel gene encoding a zinc finger protein in IL-3-dependent myeloid leukemia cell lines. Cell 1988, 54, 831–840. [Google Scholar] [CrossRef]
- Nucifora, G.; Laricchia-Robbio, L.; Senyuk, V. EVI1 and hematopoietic disorders: History and perspectives. Gene 2006, 368, 1–11. [Google Scholar] [CrossRef]
- Fears, S.; Mathieu, C.; Zeleznik-Le, N.; Huang, S.; Rowley, J.; Nucifora, G. Intergenic splicing of MDS1 and EVI1 occurs in normal tissues as well as in myeloid leukemia and produces a new member of the PR domain family. Proc. Natl. Acad. Sci. USA 1996, 93, 1642–1647. [Google Scholar] [CrossRef] [Green Version]
- Wieser, R. The oncogene and developmental regulator EVI1: Expression, biochemical properties, and biological functions. Gene 2007, 396, 346–357. [Google Scholar] [CrossRef]
- Suzukawa, K.; Parganas, E.; Gajjar, A.; Abe, T.; Takahashi, S.; Tani, K.; Asano, S.; Asou, H.; Kamada, N.; Yokota, J.; et al. Identification of a breakpoint cluster region 3’ of the ribophorin I gene at 3q21 associated with the transcriptional activation of the EVI1 gene in acute myelogenous leukemias with inv(3)(q21q26). Blood 1994, 84, 2681–2688. [Google Scholar] [CrossRef] [Green Version]
- Gröschel, S.; Lugthart, S.; Schlenk, R.F.; Valk, P.J.; Eiwen, K.; Goudswaard, C.; van Putten, W.J.; Kayser, S.; Verdonck, L.F.; Lübbert, M.; et al. High EVI1 expression predicts outcome in younger adult patients with acute myeloid leukemia and is associated with distinct cytogenetic abnormalities. J. Clin. Oncol. 2010, 28, 2101–2107. [Google Scholar] [CrossRef] [PubMed]
- Lugthart, S.; van Drunen, E.; van Norden, Y.; van Hoven, A.; Erpelinck, C.A.; Valk, P.J.; Beverloo, H.B.; Löwenberg, B.; Delwel, R. High EVI1 levels predict adverse outcome in acute myeloid leukemia: Prevalence of EVI1 overexpression and chromosome 3q26 abnormalities underestimated. Blood 2008, 111, 4329–4337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, X.; Wang, X.; Bi, K.; Jiang, G. The role of EVI-1 in normal hematopoiesis and myeloid malignancies (Review). Int. J. Oncol. 2015, 47, 2028–2036. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, E.; Wilson, M.P.; McGrath, K.E.; Li, A.J.; Frisch, B.J.; Palis, J.; Calvi, L.M.; Zhang, Y.; Perkins, A.S. EVI1 overexpression reprograms hematopoiesis via upregulation of Spi1 transcription. Nat. Commun. 2018, 9, 4239. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Tang, G.; Hu, S.; Patel, K.P.; Yin, C.C.; Wang, W.; Lin, P.; Toruner, G.A.; Ok, C.Y.; Gu, J.; et al. Deciphering the complexities of MECOM rearrangement-driven chromosomal aberrations. Cancer Genet. 2019, 233–234, 21–31. [Google Scholar] [CrossRef]
- Brooks, D.J.; Woodward, S.; Thompson, F.H.; Dos Santos, B.; Russell, M.; Yang, J.M.; Guan, X.Y.; Trent, J.; Alberts, D.S.; Taetle, R. Expression of the zinc finger gene EVI-1 in ovarian and other cancers. Br. J. Cancer 1996, 74, 1518–1525. [Google Scholar] [CrossRef]
- Yasui, K.; Konishi, C.; Gen, Y.; Endo, M.; Dohi, O.; Tomie, A.; Kitaichi, T.; Yamada, N.; Iwai, N.; Nishikawa, T.; et al. EVI1, a target gene for amplification at 3q26, antagonizes transforming growth factor-β-mediated growth inhibition in hepatocellular carcinoma. Cancer Sci. 2015, 106, 929–937. [Google Scholar] [CrossRef] [Green Version]
- Sattler, H.P.; Lensch, R.; Rohde, V.; Zimmer, E.; Meese, E.; Bonkhoff, H.; Retz, M.; Zwergel, T.; Bex, A.; Stoeckle, M.; et al. Novel amplification unit at chromosome 3q25-q27 in human prostate cancer. Prostate 2000, 45, 207–215. [Google Scholar] [CrossRef]
- Nanjundan, M.; Nakayama, Y.; Cheng, K.W.; Lahad, J.; Liu, J.; Lu, K.; Kuo, W.L.; Smith-McCune, K.; Fishman, D.; Gray, J.W.; et al. Amplification of MDS1/EVI1 and EVI1, located in the 3q26.2 amplicon, is associated with favorable patient prognosis in ovarian cancer. Cancer Res. 2007, 67, 3074–3084. [Google Scholar] [CrossRef] [Green Version]
- Dutta, P.; Bui, T.; Bauckman, K.A.; Keyomarsi, K.; Mills, G.B.; Nanjundan, M. EVI1 splice variants modulate functional responses in ovarian cancer cells. Mol. Oncol. 2013, 7, 647–668. [Google Scholar] [CrossRef]
- Morishita, K.; Parganas, E.; William, C.L.; Whittaker, M.H.; Drabkin, H.; Oval, J.; Taetle, R.; Valentine, M.B.; Ihle, J.N. Activation of EVI1 gene expression in human acute myelogenous leukemias by translocations spanning 300-400 kilobases on chromosome band 3q26. Proc. Natl. Acad. Sci. USA 1992, 89, 3937–3941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, S.; Yoshimi, A.; Shimabe, M.; Ichikawa, M.; Nakagawa, M.; Imai, Y.; Goyama, S.; Kurokawa, M. Evi-1 is a transcriptional target of mixed-lineage leukemia oncoproteins in hematopoietic stem cells. Blood 2011, 117, 6304–6314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louz, D.; van den Broek, M.; Verbakel, S.; Vankan, Y.; van Lom, K.; Joosten, M.; Meijer, D.; Löwenberg, B.; Delwel, R. Erythroid defects and increased retrovirally-induced tumor formation in Evi1 transgenic mice. Leukemia 2000, 14, 1876–1884. [Google Scholar] [CrossRef] [Green Version]
- Buonamici, S.; Li, D.; Chi, Y.; Zhao, R.; Wang, X.; Brace, L.; Ni, H.; Saunthararajah, Y.; Nucifora, G. EVI1 induces myelodysplastic syndrome in mice. J. Clin. Investig. 2004, 114, 713–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kustikova, O.S.; Schwarzer, A.; Stahlhut, M.; Brugman, M.H.; Neumann, T.; Yang, M.; Li, Z.; Schambach, A.; Heinz, N.; Gerdes, S.; et al. Activation of Evi1 inhibits cell cycle progression and differentiation of hematopoietic progenitor cells. Leukemia 2013, 27, 1127–1138. [Google Scholar] [CrossRef] [PubMed]
- Yoshimi, A.; Goyama, S.; Watanabe-Okochi, N.; Yoshiki, Y.; Nannya, Y.; Nitta, E.; Arai, S.; Sato, T.; Shimabe, M.; Nakagawa, M.; et al. Evi1 represses PTEN expression and activates PI3K/AKT/mTOR via interactions with polycomb proteins. Blood 2011, 117, 3617–3628. [Google Scholar] [CrossRef] [Green Version]
- Glass, C.; Wuertzer, C.; Cui, X.; Bi, Y.; Davuluri, R.; Xiao, Y.Y.; Wilson, M.; Owens, K.; Zhang, Y.; Perkins, A. Global Identification of EVI1 Target Genes in Acute Myeloid Leukemia. PLoS ONE 2013, 8, e67134. [Google Scholar] [CrossRef] [Green Version]
- Goyama, S.; Yamamoto, G.; Shimabe, M.; Sato, T.; Ichikawa, M.; Ogawa, S.; Chiba, S.; Kurokawa, M. Evi-1 is a critical regulator for hematopoietic stem cells and transformed leukemic cells. Cell Stem Cell 2008, 3, 207–220. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Stehling-Sun, S.; Lezon-Geyda, K.; Juneja, S.C.; Coillard, L.; Chatterjee, G.; Wuertzer, C.A.; Camargo, F.; Perkins, A.S. PR-domain-containing Mds1-Evi1 is critical for long-term hematopoietic stem cell function. Blood 2011, 118, 3853–3861. [Google Scholar] [CrossRef]
- Senyuk, V.; Premanand, K.; Xu, P.; Qian, Z.; Nucifora, G. The oncoprotein EVI1 and the DNA methyltransferase Dnmt3 co-operate in binding and de novo methylation of target DNA. PLoS One. 2011, 6, e20793. [Google Scholar] [CrossRef] [Green Version]
- Pradhan, A.K.; Halder, A.; Chakraborty, S. Physical and functional interaction ofthe proto-oncogene EVI1 and tumor suppressor gene HIC1 deregulates Bcl-xLmediated block in apoptosis. Int. J. Biochem. Cell Biol. 2014, 53, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Balgobind, B.V.; Lugthart, S.; Hollink, I.H.; Arentsen-Peters, S.T.; van Wering, E.R.; de Graaf, S.S.; Reinhardt, D.; Creutzig, U.; Kaspers, G.J.; de Bont, E.S.; et al. EVI1 overexpression in distinct subtypes of pediatric acute myeloid leukemia. Leukemia 2010, 24, 942–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jazaeri, A.A.; Ferriss, J.S.; Bryant, J.L.; Dalton, M.S.; Dutta, A. Evaluation of EVI1 and EVI1s (Delta324) as potential therapeutic targets in ovarian cancer. Gynecol. Oncol. 2010, 118, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Koos, B.; Bender, S.; Witt, H.; Mertsch, S.; Felsberg, J.; Beschorner, R.; Korshunov, A.; Riesmeier, B.; Pfister, S.; Paulus, W.; et al. The transcription factor evi-1 is overexpressed, promotes proliferation, and is prognostically unfavorable in infratentorial ependymomas. Clin. Cancer Res. 2011, 17, 3631–3637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Queisser, A.; Hagedorn, S.; Wang, H.; Schaefer, T.; Konantz, M.; Alavi, S.; Deng, M.; Vogel, W.; von Mässenhausen, A.; Kristiansen, G.; et al. Ecotropic viral integration site 1, a novel oncogene in prostate cancer. Oncogene 2017, 36, 1573–1584. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Schaefer, T.; Konantz, M.; Braun, M.; Varga, Z.; Paczulla, A.M.; Reich, S.; Jacob, F.; Perner, S.; Moch, H.; et al. Prominent Oncogenic Roles of EVI1 in Breast Carcinoma. Cancer Res. 2017, 77, 2148–2160. [Google Scholar] [CrossRef] [Green Version]
- Kurokawa, M.; Mitani, K.; Irie, K.; Matsuyama, T.; Takahashi, T.; Chiba, S.; Yazaki, Y.; Matsumoto, K.; Hirai, H. The oncoprotein Evi-1 represses TGF-beta signalling by inhibiting Smad3. Nature 1998, 394, 92–96. [Google Scholar] [CrossRef]
- Yoshimi, A.; Kurokawa, M. Evi1 forms a bridge between the epigenetic machinery and signaling pathways. Oncotarget 2011, 2, 575–586. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.Y.; Chen, F.Y.; Shen, L.J.; Wan, H.X.; Zhong, J.H. Arsenic trioxide induces apoptosis in the THP1 cell line by downregulating EVI-1. Exp. Ther. Med. 2014, 8, 85–90. [Google Scholar] [CrossRef] [Green Version]
- Kurokawa, M.; Mitani, K.; Yamagata, T.; Takahashi, T.; Izutsu, K.; Ogawa, S.; Moriguchi, T.; Nishida, E.; Yazaki, Y.; Hirai, H. The evi-1 oncoprotein inhibits c-Jun N-terminal kinase and prevents stress-induced cell death. EMBO J. 2000, 19, 2958–2968. [Google Scholar] [CrossRef] [Green Version]
- Yatsula, B.; Lin, S.; Read, A.J.; Poholek, A.; Yates, K.; Yue, D.; Hui, P.; Perkins, A.S. Identification of binding sites of EVI1 in mammalian cells. J. Biol. Chem. 2005, 280, 30712–30722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuasa, H.; Oike, Y.; Iwama, A.; Nishikata, I.; Sugiyama, D.; Perkins, A.; Mucenski, M.L.; Suda, T.; Morishita, K. Oncogenic transcription factor Evi1 regulates hematopoietic stem cell proliferation through GATA-2 expression. EMBO J. 2005, 24, 1976–1987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nayak, K.B.; Sajitha, I.S.; Kumar, T.R.S.; Chakraborty, S. Ecotropic viral integration site 1 promotes metastasis independent of epithelial mesenchymal transition in colon cancer cells. Cell Death Dis. 2018, 9, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Liang, Y.; Zheng, X.; Deng, X.; Huang, W.; Zhang, G. EVI1 promotes epithelial-to-mesenchymal transition, cancer stem cell features and chemo-/radioresistance in nasopharyngeal carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bard-Chapeau, E.A.; Jeyakani, J.; Kok, C.H.; Muller, J.; Chua, B.Q.; Gunaratne, J.; Batagov, A.; Jenjaroenpun, P.; Kuznetsov, V.A.; Wei, C.L.; et al. Ecotopic viral integration site 1 (EVI1) regulates multiple cellular processes important for cancer and is a synergistic partner for FOS protein in invasive tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 2168–2173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ripperger, T.; Hofmann, W.; Koch, J.C.; Shirneshan, K.; Haase, D.; Wulf, G.; Issing, P.R.; Karnebogen, M.; Schmidt, G.; Auber, B.; et al. MDS1 and EVI1 complex locus (MECOM): A novel candidate gene for hereditary hematological malignancies. Haematologica 2018, 103, e55–e58. [Google Scholar] [CrossRef] [Green Version]
- Choi, E.J.; Kim, M.S.; Song, S.Y.; Yoo, N.J.; Lee, S.H. Intratumoral Heterogeneity of Frameshift Mutations in MECOM Gene is Frequent in Colorectal Cancers with High Microsatellite Instability. Pathol. Oncol. Res. 2017, 23, 145–149. [Google Scholar] [CrossRef]
- Yang, X.H.; Huang, S. PFM1 (PRDM4), a new member of the PR-domain family, maps to a tumor suppressor locus on human chromosome 12q23-q24.1. Genomics 1999, 61, 319–325. [Google Scholar] [CrossRef]
- Yan, Z.; Xiong, Y.; Xu, W.; Li, M.; Cheng, Y.; Chen, F.; Ding, S.; Xu, H.; Zheng, G. Identification of recurrence-related genes by integrating microRNA and gene expression profiling of gastric cancer. Int. J. Oncol. 2012, 41, 2166–2174. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Dai, X.; Cao, X.; Yan, H.; Ji, X.; Zhang, H.; Shen, S.; Si, Y.; Zhang, H.; Chen, J.; et al. PRDM4 mediates YAP-induced cell invasion by activating leukocyte-specific integrin β2 expression. EMBO Rep. 2018, 19, e45180. [Google Scholar] [CrossRef]
- Deng, Q.; Huang, S. PRDM5 is silenced in human cancers and has growth suppressive activities. Oncogene 2004, 23, 4903–4910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, Y.; Toyota, M.; Kondo, Y.; Suzuki, H.; Imai, T.; Ohe-Toyota, M.; Maruyama, R.; Nojima, M.; Sasaki, Y.; Sekido, Y.; et al. PRDM5 identified as a target of epigenetic silencing in colorectal and gastric cancer. Clin. Cancer Res. 2007, 13, 4786–4794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahara, S.; Tahara, T.; Horiguchi, N.; Kato, T.; Shinkai, Y.; Yamashita, H.; Yamada, H.; Kawamura, T.; Terada, T.; Okubo, M.; et al. DNA methylation accumulation in gastric mucosa adjacent to cancer after Helicobacter pylori eradication. Int. J. Cancer 2019, 144, 80–88. [Google Scholar] [CrossRef] [Green Version]
- Galli, G.G.; Multhaupt, H.A.; Carrara, M.; de Lichtenberg, K.H.; Christensen, I.B.; Santoni-Rugiu, E.; Calogero, R.A.; Lund, A.H. Prdm5 suppresses Apc(Min)-driven intestinal adenomas and regulates monoacylglycerol lipase expression. Oncogene 2014, 33, 3342–3350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bond, C.E.; Bettington, M.L.; Pearson, S.A.; McKeone, D.M.; Leggett, B.A.; Whitehall, V.L. Methylation and expression of the tumour suppressor, PRDM5, in colorectal cancer and polyp subgroups. BMC Cancer 2015, 15, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.Y.; Chen, X.W.; Cheng, L.; Liu, Y.D.; Lou, G. DNA methylation and carcinogenesis of PRDM5 in cervical cancer. J. Cancer Res. Clin. Oncol. 2010, 136, 1821–1825. [Google Scholar] [CrossRef]
- Shu, X.S.; Geng, H.; Li, L.; Ying, J.; Ma, C.; Wang, Y.; Poon, F.F.; Wang, X.; Ying, Y.; Yeo, W.; et al. The epigenetic modifier PRDM5 functions as a tumor suppressor through modulating WNT/β-catenin signaling and is frequently silenced in multiple tumors. PLoS ONE 2011, 6, e27346. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.X.; Hu, R.C.; Tan, Y.L.; Liu, J.J.; Liu, W.E. Promoter methylation-mediated downregulation of PRDM5 contributes to the development of lung squamous cell carcinoma. Tumour Biol. 2014, 35, 4509–4516. [Google Scholar] [CrossRef]
- Tan, S.X.; Hu, R.C.; Liu, J.J.; Tan, Y.L.; Liu, W.E. Methylation of PRDM2, PRDM5 and PRDM16 genes in lung cancer cells. Int. J. Clin. Exp. Pathol. 2014, 7, 2305–2311. [Google Scholar]
- Tan, S.X.; Hu, R.C.; Xia, Q.; Tan, Y.L.; Liu, J.J.; Gan, G.X.; Wang, L.L. The methylation profiles of PRDM promoters in non-small cell lung cancer. Onco. Targets Ther. 2018, 11, 2991–3002. [Google Scholar] [CrossRef] [Green Version]
- Marzec-Kotarska, B.; Cybulski, M.; Kotarski, J.C.; Ronowicz, A.; Tarkowski, R.; Polak, G.; Antosz, H.; Piotrowski, A.; Kotarski, J. Molecular bases of aberrant miR-182 expression in ovarian cancer. Genes Chromosomes Cancer 2016, 55, 877–889. [Google Scholar] [CrossRef]
- Seehawer, M.; Heinzmann, F.; D’Artista, L.; Harbig, J.; Roux, P.F.; Hoenicke, L.; Dang, H.; Klotz, S.; Robinson, L.; Doré, G.; et al. Necroptosis microenvironment directs lineage commitment in liver cancer. Nature 2018, 562, 69–75. [Google Scholar] [CrossRef]
- Wang, L.; Ding, Q.Q.; Gao, S.S.; Yang, H.J.; Wang, M.; Shi, Y.; Cheng, B.F.; Bi, J.J.; Feng, Z.W. PRDM5 promotes the proliferation and invasion of murine melanoma cells through up-regulating JNK expression. Cancer Med. 2016, 5, 2558–2566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armengol, G.; Eissa, S.; Lozano, J.J.; Shoman, S.; Sumoy, L.; Caballín, M.R.; Knuutila, S. Genomic imbalances in Schistosoma-associated and non-Schistosoma-associated bladder carcinoma. An array comparative genomic hybridization analysis. Cancer Genet. Cytogenet. 2007, 177, 16–19. [Google Scholar] [CrossRef] [PubMed]
- Lindström, S.; Thompson, D.J.; Paterson, A.D.; Li, J.; Gierach, G.L.; Scott, C.; Stone, J.; Douglas, J.A.; dos-Santos-Silva, I.; Fernandez-Navarro, P.; et al. Genome-wide association study identifies multiple loci associated with both mammographic density and breast cancer risk. Nat. Commun. 2014, 5, 5303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Northcott, P.A.; Buchhalter, I.; Morrissy, A.S.; Hovestadt, V.; Weischenfeldt, J.; Ehrenberger, T.; Gröbner, S.; Segura-Wang, M.; Zichner, T.; Rudneva, V.A.; et al. The whole-genome landscape of medulloblastoma subtypes. Nature 2017, 547, 311–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basso, T.R.; Villacis, R.A.; Canto, L.M.; Alves, V.M.; Lapa, R.M.; Nóbrega, A.F.; Achatz, M.I.; Rogatto, S.R. Genomic profile of a Li-Fraumeni-like syndrome patient with a 45,X/46,XX karyotype, presenting neither mutations in TP53 nor clinical stigmata of Turner syndrome. Cancer Genet. 2015, 208, 341–344. [Google Scholar] [CrossRef]
- Lan, X.; Gao, H.; Wang, F.; Feng, J.; Bai, J.; Zhao, P.; Cao, L.; Gui, S.; Gong, L.; Zhang, Y. Whole-exome sequencing identifies variants in invasive pituitary adenomas. Oncol. Lett. 2016, 12, 2319–2328. [Google Scholar] [CrossRef]
- Wu, X.; Miao, J.; Jiang, J.; Liu, F. Analysis of methylation profiling data of hyperplasia and primary and metastatic endometrial cancers. Eur. J. Obstet. Gynecol. Reprod. Biol. 2017, 217, 161–166. [Google Scholar] [CrossRef]
- Chen, Z.; Gao, W.; Pu, L.; Zhang, L.; Han, G.; Zuo, X.; Zhang, Y.; Li, X.; Shen, H.; Wu, J.; et al. PRDM8 exhibits antitumor activities toward hepatocellular carcinoma by targeting NAP1L1. Hepatology 2018, 68, 994–1009. [Google Scholar] [CrossRef] [Green Version]
- Paigen, K.; Petkov, P.M. PRDM9 and Its Role in Genetic Recombination. Trends Genet. 2018, 34, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Alves, I.; Houle, A.A.; Hussin, J.G.; Awadalla, P. The impact of recombination on human mutation load and disease. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2017, 372, 20160465. [Google Scholar] [CrossRef] [PubMed]
- Altemose, N.; Noor, N.; Bitoun, E.; Tumian, A.; Imbeault, M.; Chapman, J.R.; Aricescu, A.R.; Myers, S.R. A map of human PRDM9 binding provides evidence for novel behaviors of PRDM9 and other zinc-finger proteins in meiosis. eLife 2017, 6, e28383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feichtinger, J.; Aldeailej, I.; Anderson, R.; Almutairi, M.; Almatrafi, A.; Alsiwiehri, N.; Griffiths, K.; Stuart, N.; Wakeman, J.A.; Larcombe, L.; et al. Meta-analysis of clinical data using human meiotic genes identifies a novel cohort of highly restricted cancer-specific marker genes. Oncotarget 2012, 3, 843–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussin, J.; Sinnett, D.; Casals, F.; Idaghdour, Y.; Bruat, V.; Saillour, V.; Healy, J.; Grenier, J.C.; de Malliard, T.; Busche, S.; et al. Rare allelic forms of PRDM9 associated with childhood leukemogenesis. Genome Res. 2013, 23, 419–430. [Google Scholar] [CrossRef] [Green Version]
- Woodward, E.L.; Olsson, M.L.; Johansson, B.; Paulsson, K. Allelic variants of PRDM9 associated with high hyperdiploid childhood acute lymphoblastic leukaemia. Br. J. Haematol. 2014, 166, 947–949. [Google Scholar] [CrossRef]
- Spinella, J.F.; Healy, J.; Saillour, V.; Richer, C.; Cassart, P.; Ouimet, M.; Sinnett, D. Whole-exome sequencing of a rare case of familial childhood acute lymphoblastic leukemia reveals putative predisposing mutations in Fanconi anemia genes. BMC Cancer 2015, 15, 539. [Google Scholar] [CrossRef] [Green Version]
- Zou, A.E.; Zheng, H.; Saad, M.A.; Rahimy, M.; Ku, J.; Kuo, S.Z.; Honda, T.K.; Wang-Rodriguez, J.; Xuan, Y.; Korrapati, A.; et al. The non-coding landscape of head and neck squamous cell carcinoma. Oncotarget 2016, 7, 51211–51222. [Google Scholar] [CrossRef]
- Ding, B.; Yan, L.; Zhang, Y.; Wang, Z.; Zhang, Y.; Xia, D.; Ye, Z.; Xu, H. Analysis of the role of mutations in the KMT2D histone lysine methyltransferase in bladder cancer. FEBS Open Bio 2019, 9, 693–706. [Google Scholar] [CrossRef] [Green Version]
- Houle, A.A.; Gibling, H.; Lamaze, F.C.; Edgington, H.A.; Soave, D.; Fave, M.J.; Agbessi, M.; Bruat, V.; Stein, L.D.; Awadalla, P. Aberrant PRDM9 expression impacts the pan-cancer genomic landscape. Genome Res. 2018, 28, 1611–1620. [Google Scholar] [CrossRef] [Green Version]
- Hofvander, J.; Tayebwa, J.; Nilsson, J.; Magnusson, L.; Brosjö, O.; Larsson, O.; Vult von Steyern, F.; Mandahl, N.; Fletcher, C.D.; Mertens, F. Recurrent PRDM10 gene fusions in undifferentiated pleomorphic sarcoma. Clin. Cancer Res. 2015, 21, 864–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puls, F.; Pillay, N.; Fagman, H.; Palin-Masreliez, A.; Amary, F.; Hansson, M.; Kindblom, L.G.; McCulloch, T.A.; Meligonis, G.; Muc, R.; et al. PRDM10-rearrangedSoft Tissue Tumor: A Clinicopathologic Study of 9 Cases. Am. J. Surg. Pathol. 2019, 43, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Lou, W.; Liu, J.; Ding, B.; Xu, L.; Fan, W. Identification of chemoresistance-associated miRNAs in breast cancer. Cancer Manag. Res. 2018, 10, 4747–4757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansouri, V.; Rezaei Tavirani, S.; Zadeh-Esmaeel, M.M.; Rostami-Nejad, M.; Rezaei-Tavirani, M. Comparative study of gastric cancer and chronic gastritis via network analysis. Gastroenterol. Hepatol. Bed Bench 2018, 11, 343–351. [Google Scholar]
- Rostami-Nejad, M.; Mansouri, V.; Mahmoud Robati, R.; Mohaghegh Shalmani, H.; Mahmoudi Lamouki, R.; Rezaei Tavirani, M. Network analysis of grade II into grade III transition in rectum cancer patients. Gastroenterol. Hepatol. Bed Bench 2018, 11, S118–S123. [Google Scholar]
- Wu, K.; Yin, X.; Jin, Y.; Liu, F.; Gao, J. Identification of aberrantly methylated differentially expressed genes in prostate carcinoma using integrated bioinformatics. Cancer Cell Int. 2019, 19, 51. [Google Scholar] [CrossRef] [Green Version]
- Zamanian Azodi, M.; Rezaei Tavirani, M.; Rezaei Tavirani, M.; Vafaee, R.; Rostami-Nejad, M. Nasopharyngeal Carcinoma Protein Interaction Mapping Analysis via Proteomic Approaches. Asian Pac. J. Cancer Prev. 2018, 19, 845–851. [Google Scholar]
- Zhang, L.; Huang, Y.; Ling, J.; Zhuo, W.; Yu, Z.; Shao, M.; Luo, Y.; Zhu, Y. Screening and function analysis of hub genes and pathways in hepatocellular carcinoma via bioinformatics approaches. Cancer Biomark. 2018, 22, 511–521. [Google Scholar] [CrossRef]
- Chen, N.; Hu, T.; Gui, Y.; Gao, J.; Li, Z.; Huang, S. Transcriptional regulation of Bcl-2 gene by the PR/SET domain family member PRDM10. PeerJ 2019, 7, e6941. [Google Scholar] [CrossRef]
- Chen, C.; Bartenhagen, C.; Gombert, M.; Okpanyi, V.; Binder, V.; Röttgers, S.; Bradtke, J.; Teigler-Schlegel, A.; Harbott, J.; Ginzel, S.; et al. Next-generation-sequencing-based risk stratification and identification of new genes involved in structural and sequence variations in near haploid lymphoblastic leukemia. Genes Chromosomes Cancer 2013, 52, 564–579. [Google Scholar] [CrossRef]
- Fog, C.K.; Asmar, F.; Côme, C.; Jensen, K.T.; Johansen, J.V.; Kheir, T.B.; Jacobsen, L.; Friis, C.; Louw, A.; Rosgaard, L.; et al. Loss of PRDM11 promotes MYC-driven lymphomagenesis. Blood 2015, 125, 1272–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.; Chang, Z.; Wu, C.; Zhu, Y.; Li, K.; Xu, Y. Identification of potential cancer-related pseudogenes in lung adenocarcinoma based on ceRNA hypothesis. Oncotarget 2017, 8, 59036–59047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolomietz, E.; Marrano, P.; Yee, K.; Thai, B.; Braude, I.; Kolomietz, A.; Chun, K.; Minkin, S.; Kamel-Reid, S.; Minden, M.; et al. Quantitative PCR identifies a minimal deleted region of 120 kb extending from the Philadelphia chromosome ABL translocation breakpoint in chronic myeloid leukemia with poor outcome. Leukemia 2003, 17, 1313–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reid, A.G.; Nacheva, E.P. A potential role for PRDM12 in the pathogenesis of chronic myeloid leukaemia with derivative chromosome 9 deletion. Leukemia 2004, 18, 178–180. [Google Scholar] [CrossRef] [Green Version]
- Huet, S.; Dulucq, S.; Chauveau, A.; Ménard, A.; Chomel, J.C.; Maisonneuve, H.; Legros, L.; Perrin, M.C.; Ferrant, E.; Moreilhon, C.; et al. Molecular characterization and follow-up of five CML patients with new BCR-ABL1 fusion transcripts. Genes Chromosomes Cancer 2015, 54, 595–605. [Google Scholar] [CrossRef]
- Zhang, Y.; Yan, L.; Yao, W.; Chen, K.; Xu, H.; Ye, Z. Integrated Analysis of Genetic Abnormalities of the Histone Lysine Methyltransferases in Prostate Cancer. Med. Sci. Monit. 2019, 25, 193–239. [Google Scholar] [CrossRef]
- Chen, Y.C.; Auer-Grumbach, M.; Matsukawa, S.; Zitzelsberger, M.; Themistocleous, A.C.; Strom, T.M.; Samara, C.; Moore, A.W.; Cho, L.T.; Young, G.T.; et al. Transcriptional regulator PRDM12 is essential for human pain perception. Nat. Genet. 2015, 47, 803–808. [Google Scholar] [CrossRef]
- Behrends, U.; Schneider, I.; Rössler, S.; Frauenknecht, H.; Golbeck, A.; Lechner, B.; Eigenstetter, G.; Zobywalski, C.; Müller-Weihrich, S.; Graubner, U.; et al. Novel tumor antigens identified by autologous antibody screening of childhood medulloblastoma cDNA libraries. Int. J. Cancer 2003, 106, 244–251. [Google Scholar] [CrossRef]
- Rubicz, R.; Zhao, S.; Geybels, M.; Wright, J.L.; Kolb, S.; Klotzle, B.; Bibikova, M.; Troyer, D.; Lance, R.; Ostrander, E.A.; et al. DNA methylation profiles in African American prostate cancer patients in relation to disease progression. Genomics 2019, 111, 10–16. [Google Scholar] [CrossRef]
- Zhang, L.; Cao, H.; He, T.; Yang, J.; Tao, H.; Wang, Y.; Hu, Q. Overexpression of PRDM13 inhibits glioma cells via Rho and GTP enzyme activation protein. Int. J. Mol. Med. 2018, 42, 966–974. [Google Scholar] [CrossRef] [Green Version]
- Nishikawa, N.; Toyota, M.; Suzuki, H.; Honma, T.; Fujikane, T.; Ohmura, T.; Nishidate, T.; Ohe-Toyota, M.; Maruyama, R.; Sonoda, T.; et al. Gene amplification and overexpression of PRDM14 in breast cancers. Cancer Res. 2007, 67, 9649–9657. [Google Scholar] [CrossRef] [Green Version]
- Moelans, C.B.; de Weger, R.A.; Monsuur, H.N.; Vijzelaar, R.; van Diest, P.J. Molecular profiling of invasive breast cancer by multiplex ligation-dependent probe amplification-based copy number analysis of tumor suppressor and oncogenes. Mod. Pathol. 2010, 23, 1029–1039. [Google Scholar] [CrossRef]
- Moelans, C.B.; de Wegers, R.A.; Monsuurs, H.N.; Maess, A.H.; van Diest, P.J. Molecular differences between ductal carcinoma in situ and adjacent invasive breast carcinoma: A multiplex ligation-dependent probe amplification study. Cell Oncol. 2011, 34, 475–482. [Google Scholar] [CrossRef] [Green Version]
- Moelans, C.B.; van der Groep, P.; Hoefnagel, L.D.C.; van de Vijver, M.J.; Wesseling, P.; Wesseling, J.; van der Wall, E.; van Diest, P.J. Genomic evolution from primary breast carcinoma to distant metastasis: Few copy number changes of breast cancer related genes. Cancer Lett. 2014, 344, 138–146. [Google Scholar] [CrossRef]
- Seki, Y. PRDM14 Is a Unique Epigenetic Regulator Stabilizing Transcriptional Networks for Pluripotency. Front. Cell Dev. Biol. 2018, 6, 12. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, H.; Hoshino, D.; Moriya, C.; Zembutsu, H.; Nishiyama, N.; Yamamoto, H.; Kataoka, K.; Imai, K. Silencing PRDM14 expression by an innovative RNAi therapy inhibits stemness, tumorigenicity, and metastasis of breast cancer. Oncotarget 2017, 8, 46856–46874. [Google Scholar] [CrossRef] [Green Version]
- Ou, M.; Li, S.; Tang, L. PRDM14: A Potential Target for Cancer Therapy. Curr. Cancer Drug Targets 2018, 18, 945–956. [Google Scholar] [CrossRef]
- Nandy, S.B.; Orozco, A.; Lopez-Valdez, R.; Roberts, R.; Subramani, R.; Arumugam, A.; Dwivedi, A.K.; Stewart, V.; Prabhakar, G.; Jones, S.; et al. Glucose insult elicits hyperactivation of cancer stem cells through miR-424-cdc42-prdm14 signalling axis. Br. J. Cancer 2017, 117, 1665–1675. [Google Scholar] [CrossRef] [Green Version]
- Moriya, C.; Taniguchi, H.; Nagatoishi, S.; Igarashi, H.; Tsumoto, K.; Imai, K. PRDM14 directly interacts with heat shock proteins HSP90α and glucose-regulated protein 78. Cancer Sci. 2018, 109, 373–383. [Google Scholar] [CrossRef]
- Dettman, E.J.; Justice, M.J. The zinc finger SET domain gene Prdm14 is overexpressed in lymphoblastic lymphomas with retroviral insertions at Evi32. PLoS ONE 2008, 3, e3823. [Google Scholar] [CrossRef] [Green Version]
- Dettman, E.J.; Simko, S.J.; Ayanga, B.; Carofino, B.L.; Margolin, J.F.; Morse, H.C.; Justice, M.J. Prdm14 initiates lymphoblastic leukemia after expanding a population of cells resembling common lymphoid progenitors. Oncogene 2011, 30, 2859–2873. [Google Scholar] [CrossRef] [Green Version]
- Simko, S.J.; Voicu, H.; Carofino, B.L.; Justice, M.J. Mouse Lymphoblastic Leukemias Induced by Aberrant Prdm14 Expression Demonstrate Widespread Copy Number Alterations Also Found in Human ALL. Cancers 2012, 4, 1050–1066. [Google Scholar] [CrossRef] [Green Version]
- Carofino, B.L.; Ayanga, B.; Justice, M.J. A mouse model for inducible overexpression of Prdm14 results in rapid-onset and highly penetrant T-cell acute lymphoblastic leukemia (T-ALL). Dis. Model. Mech. 2013, 6, 1494–1506. [Google Scholar] [CrossRef] [Green Version]
- Carofino, B.L.; Ayanga, B.; Tracey, L.J.; Brooke-Bisschop, T.; Justice, M.J. PRDM14 promotes RAG-dependent Notch1 driver mutations in mouse T-ALL. Biol. Open 2016, 5, 645–653. [Google Scholar] [CrossRef] [Green Version]
- Tracey, L.J.; Brooke-Bisschop, T.; Jansen, P.W.T.C.; Campos, E.I.; Vermeulen, M.; Justice, M.J. The pluripotency regulator PRDM14 requires hematopoietic regulator CBFA2T3 to initiate leukemia in mice. Mol. Cancer Res. 2019, 17, 1468–1479. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Meng, L.; Dong, W.; Shen, H.; Zhang, S.; Liu, Q.; Du, J. High expression of PRDM14 correlates with cell differentiation and is a novel prognostic marker in resected non-small cell lung cancer. Med. Oncol. 2013, 30, 605. [Google Scholar] [CrossRef]
- Bi, H.X.; Shi, H.B.; Zhang, T.; Cui, G. PRDM14 promotes the migration of human non-small cell lung cancer through extracellular matrix degradation in vitro. Chin. Med. J. 2015, 128, 373–377. [Google Scholar] [CrossRef]
- Lu, Y.; Wan, Z.; Zhang, X.; Zhong, X.; Rui, L.; Li, Z. PRDM14 inhibits 293T cell proliferation by influencing the G1/S phase transition. Gene 2016, 595, 180–186. [Google Scholar] [CrossRef]
- Baykara, O.; Bakir, B.; Buyru, N.; Kaynak, K.; Dalay, N. Amplification of chromosome 8 genes in lung cancer. J. Cancer 2015, 6, 270–275. [Google Scholar] [CrossRef] [Green Version]
- Moriya, C.; Taniguchi, H.; Miyata, K.; Nishiyama, N.; Kataoka, K.; Imai, K. Inhibition of PRDM14 expression in pancreatic cancer suppresses cancer stem-like properties and liver metastasis in mice. Carcinogenesis 2017, 38, 638–648. [Google Scholar] [CrossRef] [Green Version]
- Moriya, C.; Imai, K.; Taniguchi, H. PRDM14 is overexpressed in chronic pancreatitis prior to pancreatic cancer. FEBS Open Bio 2018, 8, 1733–1741. [Google Scholar] [CrossRef]
- Terashima, K.; Yu, A.; Chow, W.Y.; Hsu, W.C.; Chen, P.; Wong, S.; Hung, Y.S.; Suzuki, T.; Nishikawa, R.; Matsutani, M.; et al. Genome-wide analysis of DNA copy number alterations and loss of heterozygosity in intracranial germ cell tumors. Pediatr. Blood Cancer 2014, 61, 593–600. [Google Scholar] [CrossRef]
- Baltaci, E.; Karaman, E.; Dalay, N.; Buyru, N. Analysis of gene copy number changes in head and neck cancer. Clin. Otolaryngol. 2018, 43, 1004–1009. [Google Scholar] [CrossRef]
- Ruark, E.; Seal, S.; McDonald, H.; Zhang, F.; Elliot, A.; Lau, K.; Perdeaux, E.; Rapley, E.; Eeles, R.; Peto, J.; et al. Identification of nine new susceptibility loci for testicular cancer, including variants near DAZL and PRDM14. Nat. Genet. 2013, 45, 686–689. [Google Scholar] [CrossRef] [Green Version]
- Gell, J.J.; Zhao, J.; Chen, D.; Hunt, T.J.; Clark, A.T. PRDM14 is expressed in germ cell tumors with constitutive overexpression altering human germline differentiation and proliferation. Stem Cell Res. 2018, 27, 46–56. [Google Scholar] [CrossRef]
- Steenbergen, R.D.; Ongenaert, M.; Snellenberg, S.; Trooskens, G.; van der Meide, W.F.; Pandey, D.; Bloushtain-Qimron, N.; Polyak, K.; Meijer, C.J.; Snijders, P.J.; et al. Methylation-specific digital karyotyping of HPV16E6E7-expressing human keratinocytes identifies novel methylation events in cervical carcinogenesis. J. Pathol. 2013, 231, 53–62. [Google Scholar] [CrossRef]
- Snellenberg, S.; Cillessen, S.A.; Van Criekinge, W.; Bosch, L.; Meijer, C.J.; Snijders, P.J.; Steenbergen, R.-D. Methylation-mediated repression of PRDM14 contributes to apoptosis evasion in HPV-positive cancers. Carcinogenesis 2014, 35, 2611–2618. [Google Scholar] [CrossRef] [Green Version]
- Kitchen, M.O.; Bryan, R.T.; Emes, R.D.; Glossop, J.R.; Luscombe, C.; Cheng, K.K.; Zeegers, M.P.; James, N.D.; Devall, A.J.; Mein, C.A.; et al. Quantitative genome-wide methylation analysis of high-grade non-muscle invasive bladder cancer. Epigenetics 2016, 11, 237–246. [Google Scholar] [CrossRef] [Green Version]
- Ashktorab, H.; Shakoori, A.; Zarnogi, S.; Sun, X.; Varma, S.; Lee, E.; Shokrani, B.; Laiyemo, A.O.; Washington, K.; Brim, H. Reduced Representation Bisulfite Sequencing Determination of Distinctive DNA Hypermethylated Genes in the Progression to Colon Cancer in African Americans. Gastroenterol. Res. Pract. 2016, 2016, 2102674. [Google Scholar] [CrossRef] [Green Version]
- Hubers, A.J.; Brinkman, P.; Boksem, R.J.; Rhodius, R.J.; Witte, B.I.; Zwinderman, A.H.; Heideman, D.A.; Duin, S.; Koning, R.; Steenbergen, R.D.; et al. Combined sputum hypermethylation and eNose analysis for lung cancer diagnosis. J. Clin. Pathol. 2014, 67, 707–711. [Google Scholar] [CrossRef] [Green Version]
- Hubers, A.J.; Heideman, D.A.; Burgers, S.A.; Herder, G.J.; Sterk, P.J.; Rhodius, R.J.; Smit, H.J.; Krouwels, F.; Welling, A.; Witte, B.I.; et al. DNA hypermethylation analysis in sputum for the diagnosis of lung cancer: Training validation set approach. Br. J. Cancer 2015, 112, 1105–1113. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.; Fang, H.; Jiang, F. Integrating DNA methylation and microRNA biomarkers in sputum for lung cancer detection. Clin. Epigenetics 2016, 8, 109. [Google Scholar] [CrossRef] [Green Version]
- Bashyam, M.D.; Bair, R.; Kim, Y.H.; Wang, P.; Hernandez-Boussard, T.; Karikari, C.A.; Tibshirani, R.; Maitra, A.; Pollack, J.R. Array-based comparative genomic hybridization identifies localized DNA amplifications and homozygous deletions in pancreatic cancer. Neoplasia 2005, 7, 556–562. [Google Scholar] [CrossRef] [Green Version]
- Giallourakis, C.C.; Benita, Y.; Molinie, B.; Cao, Z.; Despo, O.; Pratt, H.E.; Zukerberg, L.R.; Daly, M.J.; Rioux, J.D.; Xavier, R.J. Genome-wide analysis of immune system genes by expressed sequence Tag profiling. J. Immunol. 2013, 190, 5578–5587. [Google Scholar] [CrossRef]
- Park, H.Y.; Lee, S.B.; Yoo, H.Y.; Kim, S.J.; Kim, W.S.; Kim, J.I.; Ko, Y.H. Whole-exome and transcriptome sequencing of refractory diffuse large B-cell lymphoma. Oncotarget 2016, 7, 86433–86445. [Google Scholar] [CrossRef] [Green Version]
- Mzoughi, S.; Zhang, J.; Hequet, D.; Teo, S.X.; Fang, H.; Xing, Q.R.; Bezzi, M.; Seah, M.K.Y.; Ong, S.L.M.; Shin, E.M.; et al. PRDM15 safeguards naive pluripotency by transcriptionally regulating WNT and MAPK-ERK signaling. Nat. Genet. 2017, 49, 1354–1363. [Google Scholar] [CrossRef]
- Mochizuki, N.; Shimizu, S.; Nagasawa, T.; Tanaka, H.; Taniwaki, M.; Yokota, J.; Morishita, K. A novel gene, MEL1, mapped to 1p36.3 is highly homologous to the MDS1/EVI1 gene and is transcriptionally activated in t(1;3)(p36;q21)-positive leukemia cells. Blood 2000, 96, 3209–3214. [Google Scholar] [CrossRef]
- Nishikata, I.; Sasaki, H.; Iga, M.; Tateno, Y.; Imayoshi, S.; Asou, N.; Nakamura, T.; Morishita, K. A novel EVI1 gene family, MEL1, lacking a PR domain (MEL1S) is expressed mainly in t(1;3)(p36;q21)-positive AML and blocks G-CSF-induced myeloid differentiation. Blood 2003, 102, 3323–3332. [Google Scholar] [CrossRef]
- Yoshida, M.; Nosaka, K.; Yasunaga, J.; Nishikata, I.; Morishita, K.; Matsuoka, M. Aberrant expression of the MEL1S gene identified in association with hypomethylation in adult T-cell leukemia cells. Blood 2004, 103, 2753–2760. [Google Scholar] [CrossRef] [Green Version]
- Shing, D.C.; Trubia, M.; Marchesi, F.; Radaelli, E.; Belloni, E.; Tapinassi, C.; Scanziani, E.; Mecucci, C.; Crescenzi, B.; Lahortiga, I.; et al. Overexpression of sPRDM16 coupled with loss of p53 induces myeloid leukemias in mice. J. Clin. Investig. 2007, 117, 3696–3707. [Google Scholar] [CrossRef] [Green Version]
- Sakai, I.; Tamura, T.; Narumi, H.; Uchida, N.; Yakushijin, Y.; Hato, T.; Fujita, S.; Yasukawa, M. Novel RUNX1-PRDM16 fusion transcripts in a patient with acute myeloid leukemia showing t(1;21)(p36;q22). Genes Chromosomes Cancer 2005, 44, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Stevens-Kroef, M.J.; Schoenmakers, E.F.; van Kraaij, M.; Huys, E.; Vermeulen, S.; van der Reijden, B.; van Kessel, A.G. Identification of truncated RUNX1 and RUNX1-PRDM16 fusion transcripts in a case of t(1;21)(p36;q22)-positive therapy-related AML. Leukemia 2006, 20, 1187–1189. [Google Scholar] [CrossRef] [PubMed]
- Hazourli, S.; Chagnon, P.; Sauvageau, M.; Fetni, R.; Busque, L.; Hébert, J. Overexpression of PRDM16 in the presence and absence of the RUNX1/PRDM16 fusion gene in myeloid leukemias. Genes Chromosomes Cancer 2006, 45, 1072–1076. [Google Scholar] [CrossRef]
- Roche-Lestienne, C.; Deluche, L.; Corm, S.; Tigaud, I.; Joha, S.; Philippe, N.; Geffroy, S.; Laï, J.L.; Nicolini, F.E.; Preudhomme, C.; et al. RUNX1 DNA-binding mutations and RUNX1-PRDM16 cryptic fusions in BCR-ABL+ leukemias are frequently associated with secondary trisomy 21 and may contribute to clonal evolution and imatinib resistance. Blood 2008, 111, 3735–3741. [Google Scholar] [CrossRef] [Green Version]
- Deluche, L.; Joha, S.; Corm, S.; Daudignon, A.; Geffroy, S.; Quief, S.; Villenet, C.; Kerckaert, J.P.; Laï, J.L.; Preudhomme, C.; et al. Cryptic and partial deletions of PRDM16 and RUNX1 without t(1;21)(p36;q22) and/or RUNX1-PRDM16 fusion in a case of progressive chronic myeloid leukemia: A complex chromosomal rearrangement of underestimated frequency in disease progression? Genes Chromosomes Cancer 2008, 47, 1110–1117. [Google Scholar] [CrossRef] [PubMed]
- Xinh, P.T.; Tri, N.K.; Nagao, H.; Nakazato, H.; Taketazu, F.; Fujisawa, S.; Yagasaki, F.; Chen, Y.Z.; Hayashi, Y.; Toyoda, A.; et al. Breakpoints at 1p36.3 in three MDS/AML(M4) patients with t(1;3)(p36;q21) occur in the first intron and in the 5’ region of MEL1. Genes Chromosomes Cancer 2003, 36, 313–316. [Google Scholar] [CrossRef]
- Wong, K.F.; Wong, M.L.; Tu, S.P. Dup(1)(p31.2p36.2) in acute myelomonocytic leukemia. Cancer Genet. Cytogenet. 2006, 165, 83–84. [Google Scholar] [CrossRef]
- Storlazzi, C.T.; Albano, F.; Guastadisegni, M.C.; Impera, L.; Mühlematter, D.; Meyer-Monard, S.; Wuillemin, W.; Rocchi, M.; Jotterand, M. Upregulation of MEL1 and FLJ42875 genes by position effect resulting from a t(1;2)(p36;p21) occurring during evolution of chronic myelomonocytic leukemia. Blood Cells Mol. Dis. 2008, 40, 452–455. [Google Scholar] [CrossRef]
- Quentin, S.; Cuccuini, W.; Ceccaldi, R.; Nibourel, O.; Pondarre, C.; Pagès, M.P.; Vasquez, N.; Dubois d’Enghien, C.; Larghero, J.; Peffault de Latour, R.; et al. Myelodysplasia and leukemia of Fanconi anemia are associated with a specific pattern of genomic abnormalities that includes cryptic RUNX1/AML1 lesions. Blood 2011, 117, e161–e170. [Google Scholar] [CrossRef] [Green Version]
- Duhoux, F.P.; Ameye, G.; Montano-Almendras, C.P.; Bahloula, K.; Mozziconacci, M.J.; Laibe, S.; Wlodarska, I.; Michaux, L.; Talmant, P.; Richebourg, S.; et al. PRDM16 (1p36) translocations define a distinct entity of myeloid malignancies with poor prognosis but may also occur in lymphoid malignancies. Br. J. Haematol. 2012, 156, 76–88. [Google Scholar] [CrossRef]
- Masetti, R.; Togni, M.; Astolfi, A.; Pigazzi, M.; Indio, V.; Rivalta, B.; Manara, E.; Rutella, S.; Basso, G.; Pession, A.; et al. Whole transcriptome sequencing of a paediatric case of de novo acute myeloid leukaemia with del(5q) reveals RUNX1-USP42 and PRDM16-SKI fusion transcripts. Br. J. Haematol. 2014, 166, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Jo, A.; Mitani, S.; Shiba, N.; Hayashi, Y.; Hara, Y.; Takahashi, H.; Tsukimoto, I.; Tawa, A.; Horibe, K.; Tomizawa, D.; et al. High expression of EVI1 and MEL1 is a compelling poor prognostic marker of pediatric AML. Leukemia 2015, 29, 1076–1083. [Google Scholar] [CrossRef] [PubMed]
- Shiba, N.; Ohki, K.; Kobayashi, T.; Hara, Y.; Yamato, G.; Tanoshima, R.; Ichikawa, H.; Tomizawa, D.; Park, M.J.; Shimada, A.; et al. High PRDM16 expression identifies a prognostic subgroup of pediatric acute myeloid leukaemia correlated to FLT3-ITD, KMT2A-PTD, and NUP98-NSD1: The results of the Japanese Paediatric Leukaemia/Lymphoma Study Group AML-05 trial. Br. J. Haematol. 2016, 172, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Yamato, G.; Yamaguchi, H.; Handa, H.; Shiba, N.; Kawamura, M.; Wakita, S.; Inokuchi, K.; Hara, Y.; Ohki, K.; Okubo, J.; et al. Clinical features and prognostic impact of PRDM16 expression in adult acute myeloid leukemia. Genes Chromosomes Cancer 2017, 56, 800–809. [Google Scholar] [CrossRef] [PubMed]
- Miyamura, T.; Moritake, H.; Nakayama, H.; Tanaka, S.; Tomizawa, D.; Shiba, N.; Saito, A.M.; Tawa, A.; Shimada, A.; Iwamoto, S.; et al. Clinical and biological features of paediatric acute myeloid leukaemia (AML) with primary induction failure in the Japanese Paediatric Leukaemia/Lymphoma Study Group AML-05 study. Br. J. Haematol. 2019, 185, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Barjesteh van Waalwijk van Doorn-Khosrovani, S.; Erpelinck, C.; Löwenberg, B.; Delwel, R. Low expression of MDS1-EVI1-like-1 (MEL1) and EVI1-like-1 (EL1) genes in favorable-risk acute myeloid leukemia. Exp. Hematol. 2003, 31, 1066–1072. [Google Scholar] [CrossRef]
- Yu, H.; Neale, G.; Zhang, H.; Lee, H.M.; Ma, Z.; Zhou, S.; Forget, B.G.; Sorrentino, B.P. Downregulation of Prdm16 mRNA is a specific antileukemic mechanism during HOXB4-mediated HSC expansion in vivo. Blood 2014, 124, 1737–1747. [Google Scholar] [CrossRef] [Green Version]
- Dong, S.; Chen, J. SUMOylation of sPRDM16 promotes the progression of acute myeloid leukemia. BMC Cancer 2015, 15, 893. [Google Scholar] [CrossRef]
- Du, Y.; Jenkins, N.A.; Copeland, N.G. Insertional mutagenesis identifies genes that promote the immortalization of primary bone marrow progenitor cells. Blood 2005, 106, 3932–3939. [Google Scholar] [CrossRef] [Green Version]
- Ott, M.G.; Schmidt, M.; Schwarzwaelder, K.; Stein, S.; Siler, U.; Koehl, U.; Glimm, H.; Kühlcke, K.; Schilz, A.; Kunkel, H.; et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat. Med. 2006, 12, 401–409. [Google Scholar] [CrossRef]
- Adair, J.E.; Beard, B.C.; Trobridge, G.D.; Neff, T.; Rockhill, J.K.; Silbergeld, D.L.; Mrugala, M.M.; Kiem, H.P. Extended survival of glioblastoma patients after chemoprotective HSC gene therapy. Sci. Transl. Med. 2012, 4, 133ra57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuo, H.; Goyama, S.; Kamikubo, Y.; Adachi, S. The subtype-specific features of EVI1 and PRDM16 in acute myeloid leukemia. Haematologica 2015, 100, e116–e117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eveillard, M.; Delaunay, J.; Richebourg, S.; Lodé, L.; Garand, R.; Wuillème, S.; Duhoux, F.; Antoine-Poirel, H.; Godon, C.; Béné, M.C. The closely related rare and severe acute myeloid leukemias carrying EVI1 or PRDM16 rearrangements share singular biological features. Haematologica 2015, 100, e114–e115. [Google Scholar] [CrossRef] [Green Version]
- Corrigan, D.J.; Luchsinger, L.L.; Justino de Almeida, M.; Williams, L.J.; Strikoudis, A.; Snoeck, H.W. PRDM16 isoforms differentially regulate normal and leukemic hematopoiesis and inflammatory gene signature. J. Clin. Investig. 2018, 128, 3250–3264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanochko, D.; Halabelian, L.; Henderson, E.; Savitsky, P.; Jain, H.; Marcon, E.; Duan, S.; Hutchinson, A.; Seitova, A.; Barsyte-Lovejoy, D.; et al. Direct interaction between the PRDM3 and PRDM16 tumor suppressors and the NuRD chromatin remodeling complex. Nucleic Acids Res. 2019, 47, 1225–1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Man, T.K.; Lu, X.Y.; Jaeweon, K.; Perlaky, L.; Harris, C.P.; Shah, S.; Ladanyi, M.; Gorlick, R.; Lau, C.C.; Rao, P.H. Genome-wide array comparative genomic hybridization analysis reveals distinct amplifications in osteosarcoma. BMC Cancer 2004, 4, 45. [Google Scholar] [CrossRef]
- Beck, A.H.; Lee, C.H.; Witten, D.M.; Gleason, B.C.; Edris, B.; Espinosa, I.; Zhu, S.; Li, R.; Montgomery, K.D.; Marinelli, R.J.; et al. Discovery of molecular subtypes in leiomyosarcoma through integrative molecular profiling. Oncogene 2010, 29, 845–854. [Google Scholar] [CrossRef] [Green Version]
- Cuppens, T.; Moisse, M.; Depreeuw, J.; Annibali, D.; Colas, E.; Gil-Moreno, A.; Huvila, J.; Carpén, O.; Zikán, M.; Matias-Guiu, X.; et al. Integrated genome analysis of uterine leiomyosarcoma to identify novel driver genes and targetable pathways. Int. J. Cancer 2018, 142, 1230–1243. [Google Scholar] [CrossRef] [Green Version]
- Burghel, G.J.; Lin, W.Y.; Whitehouse, H.; Brock, I.; Hammond, D.; Bury, J.; Stephenson, Y.; George, R.; Cox, A. Identification of candidate driver genes in common focal chromosomal aberrations of microsatellite stable colorectal cancer. PLoS ONE 2013, 8, e83859. [Google Scholar] [CrossRef]
- Mehrian-Shai, R.; Yalon, M.; Moshe, I.; Barshack, I.; Nass, D.; Jacob, J.; Dor, C.; Reichardt, J.K.; Constantini, S.; Toren, A. Identification of genomic aberrations in hemangioblastoma by droplet digital PCR and SNP microarray highlights novel candidate genes and pathways for pathogenesis. BMC Genom. 2016, 17, 56. [Google Scholar] [CrossRef] [Green Version]
- Takahata, M.; Inoue, Y.; Tsuda, H.; Imoto, I.; Koinuma, D.; Hayashi, M.; Ichikura, T.; Yamori, T.; Nagasaki, K.; Yoshida, M.; et al. SKI and MEL1 cooperate to inhibit transforming growth factor-beta signal in gastric cancer cells. J. Biol. Chem. 2009, 284, 3334–3344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bibi, F.; Ali, I.; Naseer, M.I.; Ali Mohamoud, H.S.; Yasir, M.; Alvi, S.A.; Jiman-Fatani, A.A.; Sawan, A.; Azhar, E.I. Detection of genetic alterations in gastric cancer patients from Saudi Arabia using comparative genomic hybridization (CGH). PLoS ONE 2018, 13, e0202576. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, W.; Wang, Y.; Zou, T.; Zhang, B.; Xu, Y.; Pang, T.; Hu, Q.; Chen, M.; Wang, L.; et al. Hypoxia-induced miR-214 expression promotes tumour cell proliferation and migration by enhancing the Warburg effect in gastric carcinoma cells. Cancer Lett. 2018, 414, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.L.; Qu, L.W.; Ma, L.; Zhou, Y.C.; Wang, G.Z.; Zhao, X.C.; Zhang, C.; Zhang, Y.F.; Wang, M.; Zhang, M.Y.; et al. Genome-wide identification of transcription factors that are critical to non-small cell lung cancer. Cancer Lett. 2018, 434, 132–143. [Google Scholar] [CrossRef]
- Lv, W.; Yu, X.; Li, W.; Feng, N.; Feng, T.; Wang, Y.; Lin, H.; Qian, B. Low expression of LINC00982 and PRDM16 is associated with altered gene expression, damaged pathways and poor survival in lung adenocarcinoma. Oncol. Rep. 2018, 40, 2698–2709. [Google Scholar] [CrossRef]
- Fei, L.R.; Huang, W.J.; Wang, Y.; Lei, L.; Li, Z.H.; Zheng, Y.W.; Wang, Z.; Yang, M.Q.; Liu, C.C.; Xu, H.T. PRDM16 functions as a suppressor of lung adenocarcinoma metastasis. J. Exp. Clin. Cancer Res. 2019, 38, 35. [Google Scholar] [CrossRef] [Green Version]
- Lei, Q.; Liu, X.; Fu, H.; Sun, Y.; Wang, L.; Xu, G.; Wang, W.; Yu, Z.; Liu, C.; Li, P.; et al. miR-101 reverses hypomethylation of the PRDM16 promoter to disrupt mitochondrial function in astrocytoma cells. Oncotarget 2016, 7, 5007–5022. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Wu, M. Epigenetic Mechanisms of Glioblastoma. In Glioblastoma; De Vleeschouwer, S., Ed.; Codon Publications: Brisbane, Australia, 2017. Available online: http://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/books/NBK469995/ (accessed on 8 April 2020).
- Peng, X.; Xue, H.; Lü, L.; Shi, P.; Wang, J.; Wang, J. Accumulated promoter methylation as a potential biomarker for esophageal cancer. Oncotarget 2017, 8, 679–691. [Google Scholar] [CrossRef]
- Deng, J.; Kong, W.; Mou, X.; Wang, S.; Zeng, W. Identifying novel candidate biomarkers of RCC based on WGCNA analysis. Pers. Med. 2018, 15, 381–394. [Google Scholar] [CrossRef]
- Tegeder, I.; Thiel, K.; Erkek, S.; Johann, P.D.; Berlandi, J.; Thatikonda, V.; Frühwald, M.C.; Kool, M.; Jeibmann, A.; Hasselblatt, M. Functional relevance of genes predicted to be affected by epigenetic alterations in atypical teratoid/rhabdoid tumors. J. Neurooncol. 2019, 141, 43–55. [Google Scholar] [CrossRef]
- Zhu, S.; Xu, Y.; Song, M.; Chen, G.; Wang, H.; Zhao, Y.; Wang, Z.; Li, F. PRDM16 is associated with evasion of apoptosis by prostatic cancer cells according to RNA interference screening. Mol. Med. Rep. 2016, 14, 3357–3361. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Parveen, M.; Basgen, J.M.; Fazel, S.; Meshesha, M.F.; Thames, E.C.; Moore, B.; Martinez, L.; Howard, C.B.; Vergnes, L.; et al. Increased Expression of Beige/Brown Adipose Markers from Host and Breast Cancer Cells Influence Xenograft Formation in Mice. Mol. Cancer Res. 2016, 14, 78–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elattar, S.; Dimri, M.; Satyanarayana, A. The tumor secretory factor ZAG promotes white adipose tissue browning and energy wasting. FASEB J. 2018, 32, 4727–4743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Zhu, L.; Bai, M.; Liu, Y.; Zhan, Y.; Deng, T.; Yang, H.; Sun, W.; Wang, X.; Zhu, K.; et al. Exosomal circRNA derived from gastric tumor promotes white adipose browning by targeting the miR-133/PRDM16 pathway. Int. J. Cancer 2019, 144, 2501–2515. [Google Scholar] [CrossRef] [PubMed]
- Avila-Fernandez, A.; Perez-Carro, R.; Corton, M.; Lopez-Molina, M.I.; Campello, L.; Garanto, A.; Fernandez-Sanchez, L.; Duijkers, L.; Lopez-Martinez, M.A.; Riveiro-Alvarez, R.; et al. Whole-exome sequencing reveals ZNF408 as a new gene associated with autosomal recessive retinitis pigmentosa with vitreal alterations. Hum. Mol. Genet. 2015, 24, 4037–4048. [Google Scholar] [CrossRef] [Green Version]
- Tsang, A.P.; Visvader, J.E.; Turner, C.A.; Fujiwara, Y.; Yu, C.; Weiss, M.J.; Crossley, M.; Orkin, S.H. FOG, a multitype zinc finger protein, acts as a cofactor for transcription factor GATA-1 in erythroid and megakaryocytic differentiation. Cell 1997, 90, 109–119. [Google Scholar] [CrossRef] [Green Version]
- Tsang, A.P.; Fujiwara, Y.; Hom, D.B.; Orkin, S.H. Failure of megakaryopoiesis and arrested erythropoiesis in mice lacking the GATA-1 transcriptional cofactor FOG. Genes Dev. 1998, 12, 1176–1188. [Google Scholar] [CrossRef] [Green Version]
- Marcucci, G.; Maharry, K.; Radmacher, M.D.; Mrózek, K.; Vukosavljevic, T.; Paschka, P.; Whitman, S.P.; Langer, C.; Baldus, C.D.; Liu, C.G.; et al. Prognostic significance of, and gene and microRNA expression signatures associated with, CEBPA mutations in cytogenetically normal acute myeloid leukemia with high-risk molecular features: A Cancer and Leukemia Group B Study. J. Clin. Oncol. 2008, 26, 5078–5087. [Google Scholar] [CrossRef] [Green Version]
- Buck, I.; Morceau, F.; Cristofanon, S.; Reuter, S.; Dicato, M.; Diederich, M. The inhibitory effect of the proinflammatory cytokine TNFalpha on erythroid differentiation involves erythroid transcription factor modulation. Int. J. Oncol. 2009, 34, 853–860. [Google Scholar]
- Yang, H.; Hui, H.; Wang, Q.; Li, H.; Zhao, K.; Zhou, Y.; Zhu, Y.; Wang, X.; You, Q.; Guo, Q.; et al. Wogonin induces cell cycle arrest and erythroid differentiation in imatinib-resistant K562 cells and primary CML cells. Oncotarget 2014, 5, 8188–8201. [Google Scholar] [CrossRef] [Green Version]
- Cai, Q.; Jeannet, R.; Hua, W.K.; Cook, G.J.; Zhang, B.; Qi, J.; Liu, H.; Li, L.; Chen, C.C.; Marcucci, G.; et al. CBFβ-SMMHC creates aberrant megakaryocyte-erythroid progenitors prone to leukemia initiation in mice. Blood 2016, 128, 1503–1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwara, T.; Sasaki, K.; Saito, K.; Hatta, S.; Ichikawa, S.; Kobayashi, M.; Okitsu, Y.; Fukuhara, N.; Onishi, Y.; Harigae, H. Forced FOG1 expression in erythroleukemia cells: Induction of erythroid genes and repression of myelo-lymphoid transcription factor PU.1. Biochem. Biophys. Res. Commun. 2017, 485, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Li, S.; Donnenberg, V.; Fu, J.; Gollin, S.M.; Ma, H.; Lu, C.; Stolz, D.B.; Mapara, M.Y.; Monaghan, S.A.; et al. Immunomodulatory drugs downregulate IKZF1 leading to expansion of hematopoietic progenitors with concomitant block of megakaryocytic maturation. Haematologica 2018, 103, 1688–1697. [Google Scholar] [CrossRef] [PubMed]
- Litchfield, K.; Holroyd, A.; Lloyd, A.; Broderick, P.; Nsengimana, J.; Eeles, R.; Easton, D.F.; Dudakia, D.; Bishop, D.T.; Reid, A.; et al. Identification of four new susceptibility loci for testicular germ cell tumour. Nat. Commun. 2015, 6, 8690. [Google Scholar] [CrossRef]
- Liu, H.; Zhao, H. Prognosis related miRNAs, DNA methylation, and epigenetic interactions in lung adenocarcinoma. Neoplasma 2019, 66, 487–493. [Google Scholar] [CrossRef]
- Rahane, C.S.; Kutzner, A.; Heese, K. Establishing a human adrenocortical carcinoma (ACC)-specific gene mutation signature. Cancer Genet. 2019, 230, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantor, A.B.; Orkin, S.H. Coregulation of GATA factors by the Friend of GATA (FOG) family of multitype zinc finger proteins. Semin. Cell. Dev. Biol. 2005, 16, 117–128. [Google Scholar] [CrossRef]
- Laitinen, M.P.; Anttonen, M.; Ketola, I.; Wilson, D.B.; Ritvos, O.; Butzow, R.; Heikinheimo, M. Transcription factors GATA-4 and GATA-6 and a GATA family cofactor, FOG-2, are expressed in human ovary and sex cord-derived ovarian tumors. J. Clin. Endocrinol. Metab. 2000, 85, 3476–3483. [Google Scholar]
- Efimenko, E.; Padua, M.B.; Manuylov, N.L.; Fox, S.C.; Morse, D.A.; Tevosian, S.G. The transcription factor GATA4 is required for follicular development and normal ovarian function. Dev. Biol. 2013, 381, 144–158. [Google Scholar] [CrossRef] [Green Version]
- Anttonen, M.; Unkila-Kallio, L.; Leminen, A.; Butzow, R.; Heikinheimo, M. High GATA-4 expression associates with aggressive behavior, whereas low anti-Müllerian hormone expression associates with growth potential of ovarian granulosa cell tumors. J. Clin. Endocrinol. Metab. 2005, 90, 6529–6535. [Google Scholar] [CrossRef] [Green Version]
- Virgone, C.; Cecchetto, G.; Ferrari, A.; Bisogno, G.; Donofrio, V.; Boldrini, R.; Collini, P.; Dall’Igna, P.; Alaggio, R. GATA-4 and FOG-2 expression in pediatric ovarian sex cord-stromal tumors replicates embryonal gonadal phenotype: Results from the TREP project. PLoS ONE 2012, 7, e45914. [Google Scholar] [CrossRef]
- Salonen, J.; Rajpert-De Meyts, E.; Mannisto, S.; Nielsen, J.E.; Graem, N.; Toppari, J.; Heikinheimo, M. Differential developmental expression of transcription factors GATA-4 and GATA-6, their cofactor FOG-2 and downstream target genes in testicular carcinoma in situ and germ cell tumors. Eur. J. Endocrinol. 2010, 162, 625–631. [Google Scholar] [PubMed] [Green Version]
- Manuylov, N.L.; Smagulova, F.O.; Tevosian, S.G. Fog2 excision in mice leads to premature mammary gland involution and reduced Esr1 gene expression. Oncogene 2007, 26, 5204–5213. [Google Scholar] [PubMed] [Green Version]
- Aumsuwan, P.; Khan, S.I.; Khan, I.A.; Ali, Z.; Avula, B.; Walker, L.A.; Shariat-Madar, Z.; Helferich, W.G.; Katzenellenbogen, B.S.; Dasmahapatra, A.K. The anticancer potential of steroidal saponin, dioscin, isolated from wild yam (Dioscorea villosa) root extract in invasive human breast cancer cell line MDA-MB-231 in vitro. Arch. Biochem. Biophys. 2016, 591, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.C.; Cohn, B.A.; Cirillo, P.M.; Santella, R.M.; Terry, M.B. DDT exposure during pregnancy and DNA methylation alterations in female offspring in the Child Health and Development Study. Reprod. Toxicol. 2019. [Google Scholar] [CrossRef]
- Hoene, V.; Fischer, M.; Ivanova, A.; Wallach, T.; Berthold, F.; Dame, C. GATA factors in human neuroblastoma: Distinctive expression patterns in clinical subtypes. Br. J. Cancer 2009, 101, 1481–1489. [Google Scholar] [CrossRef] [Green Version]
- Tsang, S.Y.; Mei, L.; Wan, W.; Li, J.; Li, Y.; Zhao, C.; Ding, X.; Pun, F.W.; Hu, X.; Wang, J.; et al. Glioma Association and Balancing Selection of ZFPM2. PLoS ONE 2015, 10, e0133003. [Google Scholar] [CrossRef] [Green Version]
- Vastrad, B.; Vastrad, C.; Godavarthi, A.; Chandrashekar, R. Molecular mechanisms underlying gliomas and glioblastoma pathogenesis revealed by bioinformatics analysis of microarray data. Med. Oncol. 2017, 34, 182. [Google Scholar] [CrossRef]
- Guan, D.; Tian, H. Integrated network analysis to explore the key genes regulated by parathyroid hormone receptor 1 in osteosarcoma. World J. Surg. Oncol. 2017, 15, 177. [Google Scholar] [CrossRef] [Green Version]
- Panagopoulos, I.; Gorunova, L.; Davidson, B.; Heim, S. Novel TNS3-MAP3K3 and ZFPM2-ELF5 fusion genes identified by RNA sequencing in multicystic mesothelioma with t(7;17)(p12;q23) and t(8;11)(q23;p13). Cancer Lett. 2015, 357, 502–509. [Google Scholar] [CrossRef]
- Karlsson, J.; Holmquist Mengelbier, L.; Elfving, P.; Gisselsson Nord, D. High-resolution genomic profiling of an adult Wilms’ tumor: Evidence for a pathogenesis distinct from corresponding pediatric tumors. Virchows Arch. 2011, 459, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.; Deng, X.; Kong, X.; Du, Y.; Li, L.; Zhu, H.; Wang, Y.; Xie, D.; Guha, S.; Li, Z.; et al. ZFPM2-AS1, a novel lncRNA, attenuates the p53 pathway and promotes gastric carcinogenesis by stabilizing MIF. Oncogene 2018, 37, 5982–5996. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Zhou, C.; Guo, K.; Li, Q.; Wang, Z. A novel seven-lncRNA signature for prognosis prediction in hepatocellular carcinoma. J. Cell Biochem. 2019, 120, 213–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.H.; Ruggiero, D.; Sorice, R.; Song, C.; Nutile, T.; Vernon Smith, A.; Concas, M.P.; Traglia, M.; Barbieri, C.; Ndiaye, N.C.; et al. Six Novel Loci Associated with Circulating VEGF Levels Identified by a Meta-analysis of Genome-Wide Association Studies. PLoS Genet. 2016, 12, e1005874. [Google Scholar] [CrossRef] [PubMed]
- Clara, J.A.; Monge, C.; Yang, Y.; Takebe, N. Targeting signalling pathways and the immune microenvironment of cancer stem cells—A clinical update. Nat. Rev. Clin. Oncol. 2019. [Google Scholar] [CrossRef]
- Qin, H.; Ruan, Z.H. The role of monoacylglycerol lipase (MAGL) in the cancer progress. Cell Biochem. Biophys. 2014, 70, 33–36. [Google Scholar] [CrossRef]
- Allali-Hassani, A.; Szewczyk, M.M.; Ivanochko, D.; Organ, S.L.; Bok, J.; Ho, J.S.Y.; Gay, F.P.H.; Li, F.; Blazer, L.; Eram, M.S.; et al. Discovery of a chemical probe for PRDM9. Nat. Commun. 2019, 10, 5759. [Google Scholar] [CrossRef]
- Wang, B.D.; Lee, N.H. Aberrant RNA Splicing in Cancer and Drug Resistance. Cancers 2018, 10, 458. [Google Scholar] [CrossRef] [Green Version]
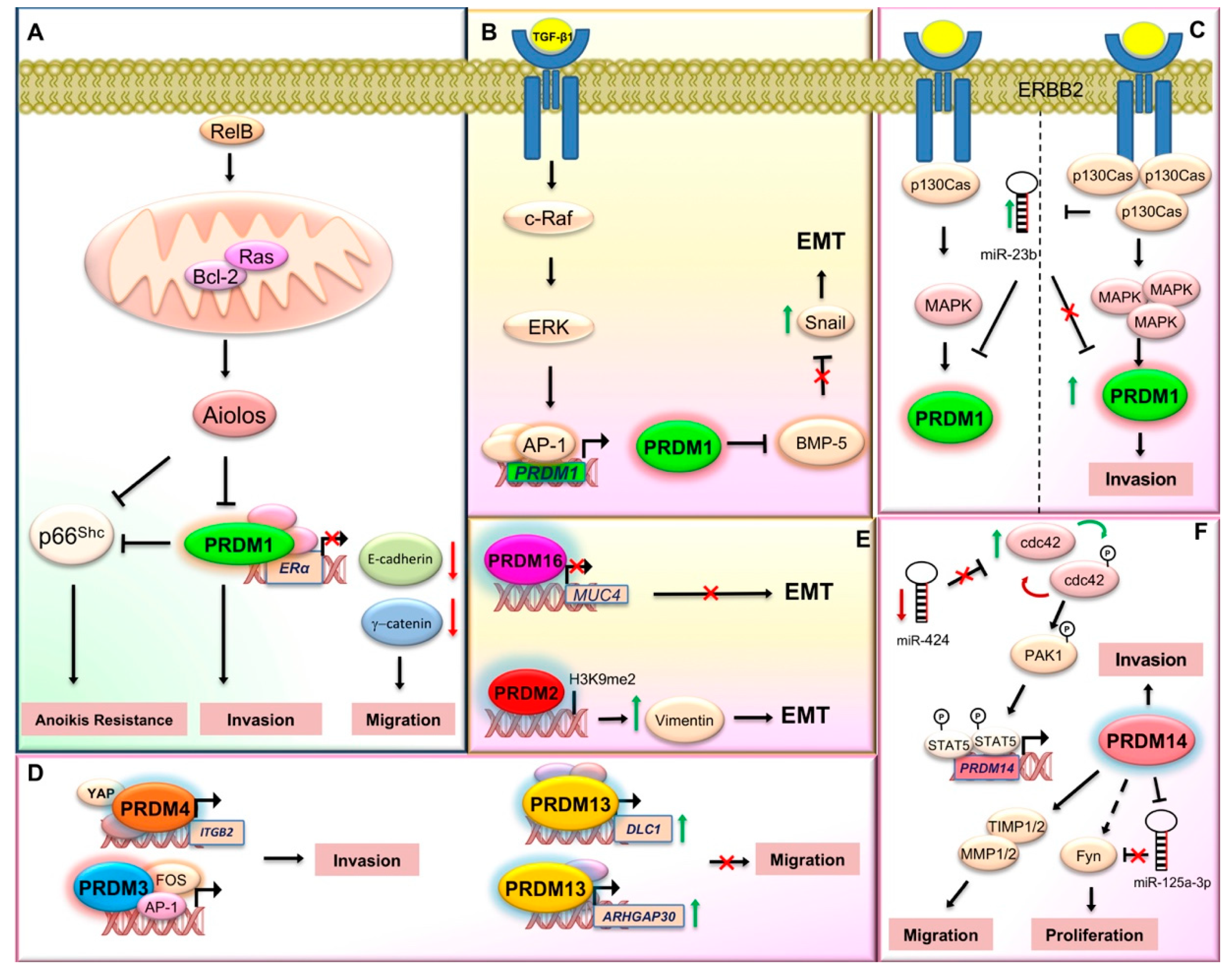
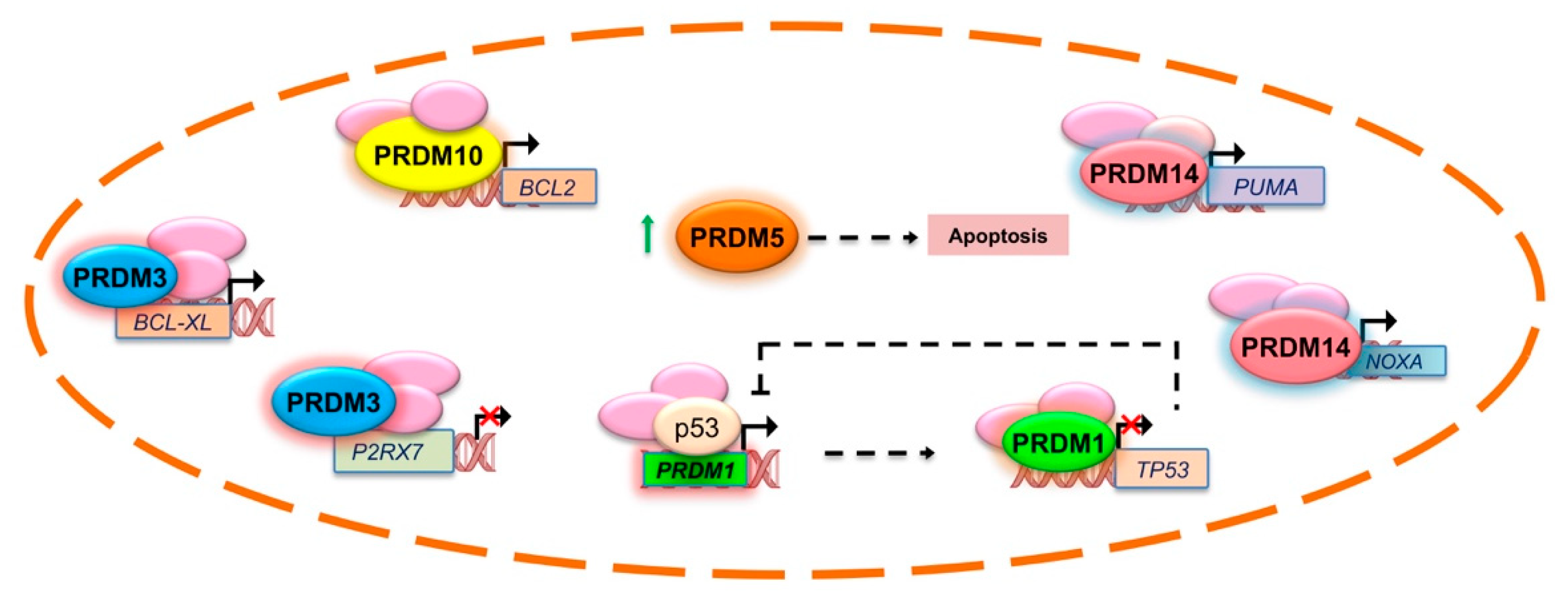
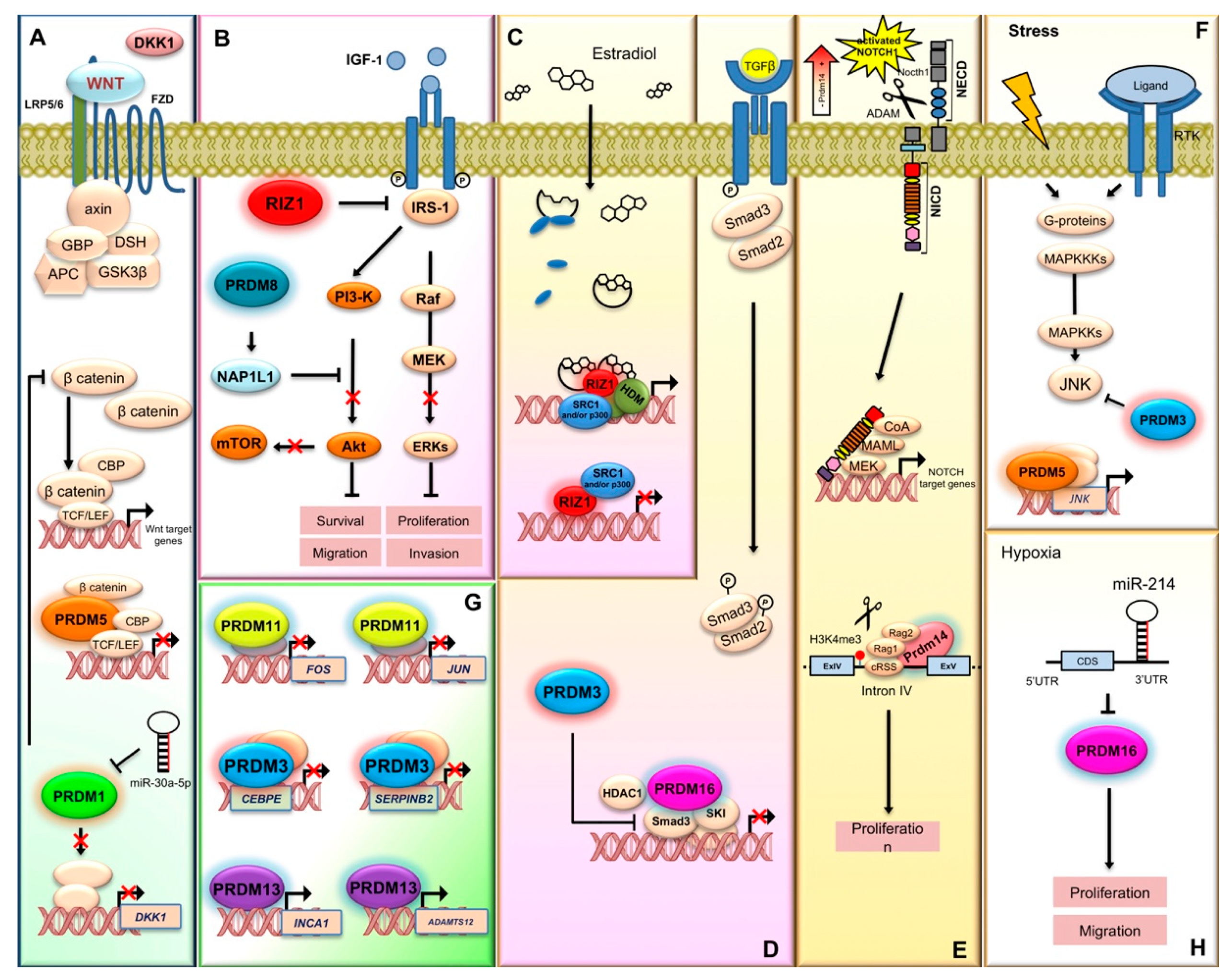



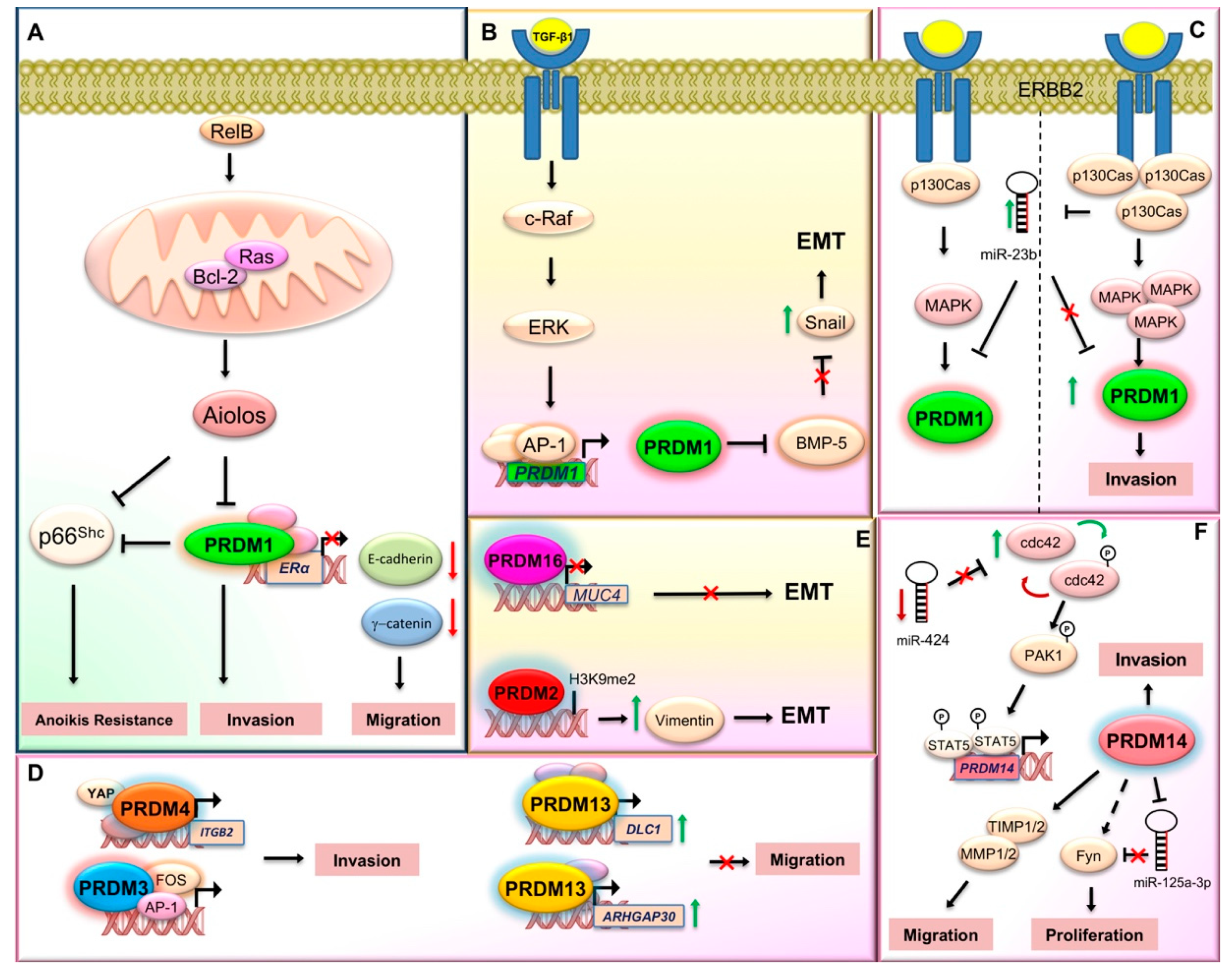{kind=link}
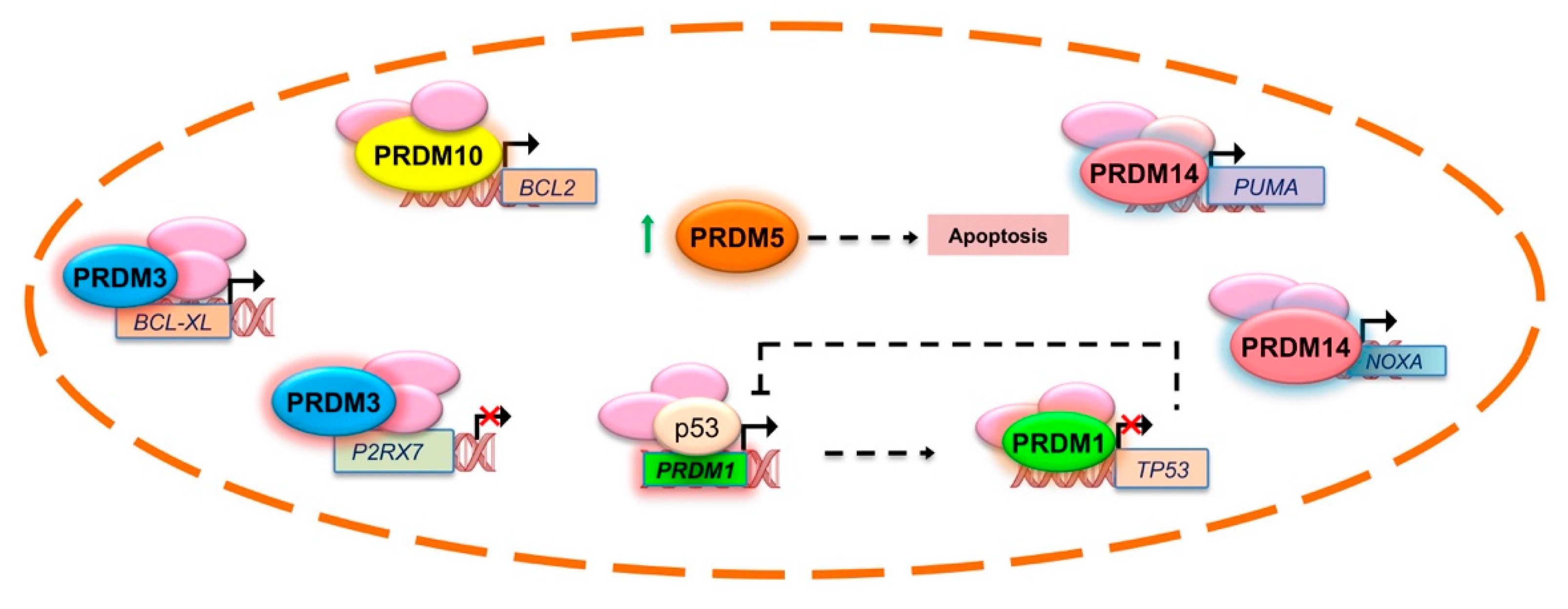{kind=link}
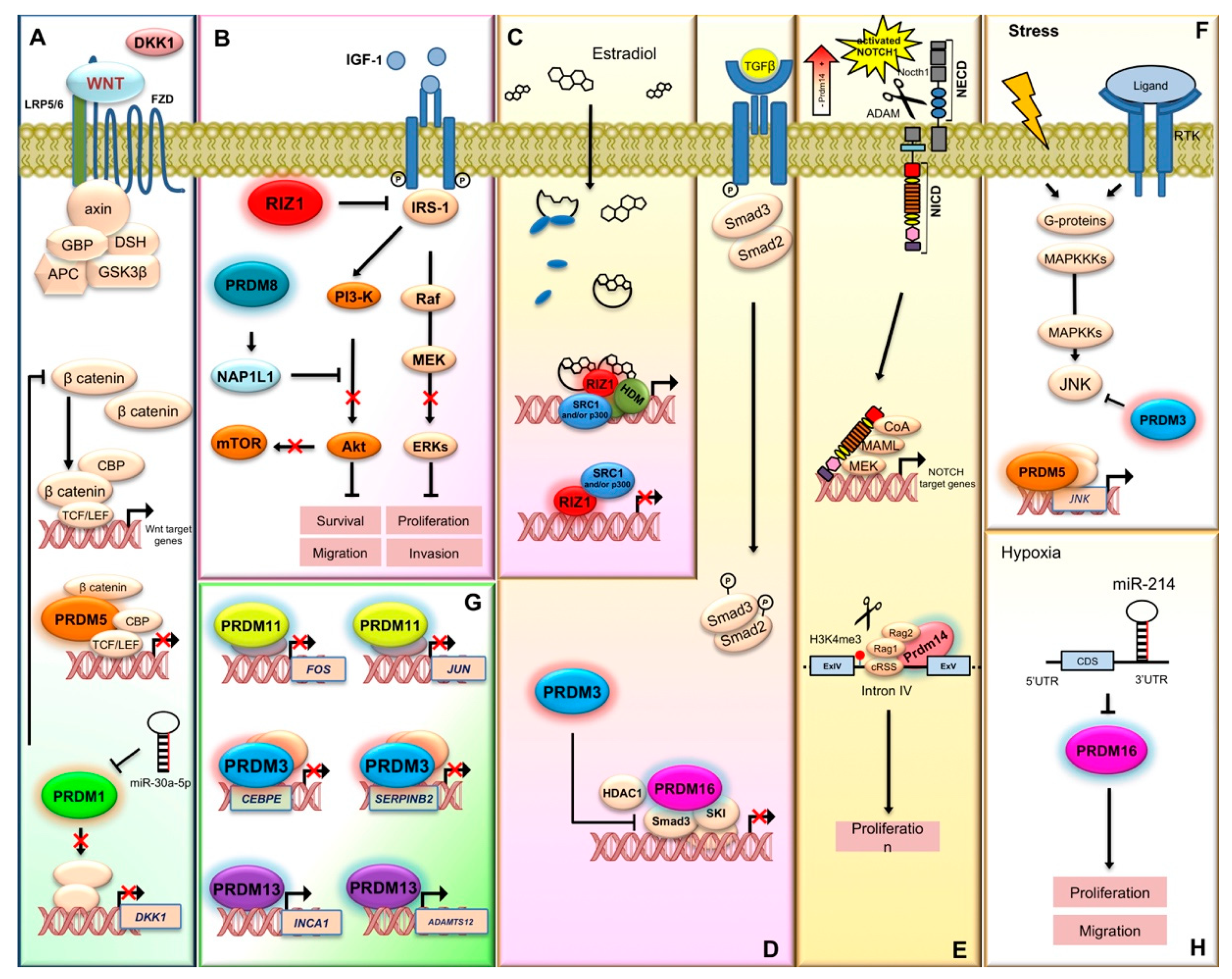{kind=link}
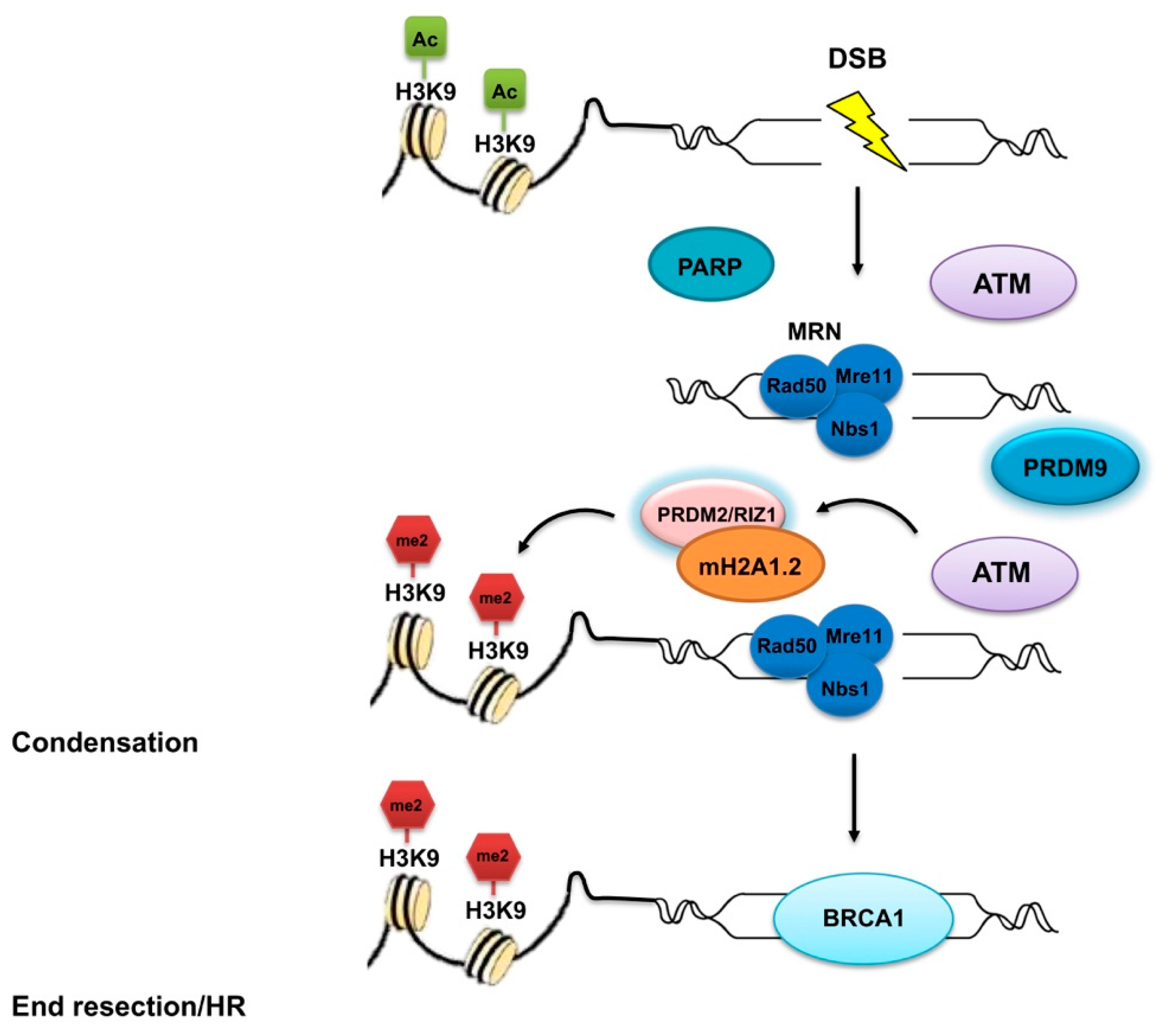{kind=link}
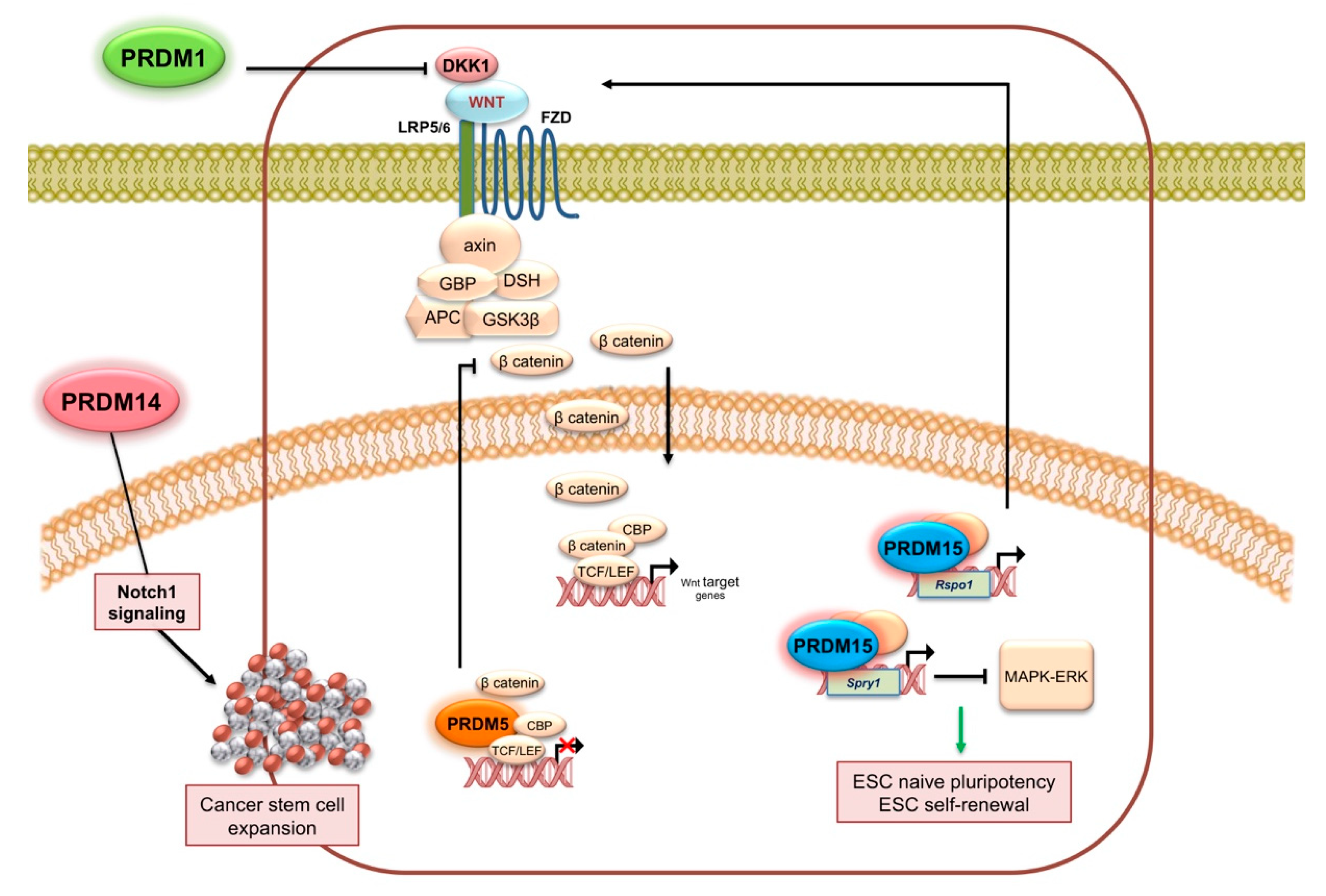{kind=link}
| Gene Symbol (Previous Symbols/Synonyms) | Cancer Type | Molecular Alteration | Putative Effect/Mechanism | References |
|---|---|---|---|---|
| PRDM1 (PRDI-BF1; BLIMP1) | Lymphoma (Diffuse large B cell lymphoma, extranodal NK (natural killer)/T-cell lymphoma) | Inactivating mutations, chromosomal deletion, and epigenetic silencing | Putative tumor suppressor gene. It is downregulated or silenced in human DLBCL (diffuse large B cell lymphoma) and other haematological malignancies. The activation of B cell lymphoma (Bcl)-2/Ras pathway stimulates RelB and p130Cas/ErbB2 invasion leading to its overexpression | [13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46] |
| Breast cancer | Upregulated | |||
| Lung cancer | Downregulated | |||
| Glioma | Downregulated | |||
| PRDM2 (RIZ; RIZ1; RIZ2; KMT8; MTB-ZF; HUMHOXY1; KMT8A) | Neuroblastoma, hepatoma, colorectal, ovarian, and breast cancers, chronic myelocytic leukemia, non-Hodgkin’s lymphoma, melanoma, parathyroid adenoma, Merkel cell carcinoma, and pheochromocytoma | Aberrant isoform expression Up/downregulated | The imbalance of its main protein isoforms, Riz1 and Riz2, (through promoter DNA methylation, frameshift, and missense mutations) may constitute an important cause of malignancy with the PR+ plus product commonly lost or downregulated and the PR− isoform always present at higher levels in cancer cells. It modulates estrogen receptor signaling in breast cancer | [3,45,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85] |
| Colorectal, gastric, endometrial, pancreatic, Microsatellite instability positive cancers | Frameshift mutations | |||
| Prostate, endometrial cancer | Polymorphisms | |||
| Breast carcinomas, liver tumors, colon and lung cancer | Methylation | |||
| MECOM (MDS1-EVI1; PRDM3; KMT8E) | Acute myeloid leukemia | Chromosomal rearrangements or proviral insertion | Tumor suppressor gene: short PR- isoform (EVI1) is overexpressed or rearranged in cancer | [7,45,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127] |
| Ovarian cancer | Downregulated | |||
| Colon cancer | Integrated bioinformatics and network analyses | |||
| Colorectal cancer | Frameshift mutation | |||
| PRDM4 (PFM1) | Ovarian, gastric, and pancreatic cancer | Deletion | Maps to frequently deleted locus (12q23-q24.1). It could contribute to YAP (yes-associated protein)-induced tumorigenesis possibly via mediating the expression of other YAP target genes, which finally contribute to cell invasion and metastasis promotion | [128,129,130] |
| Gastric cancer | Upregulated | |||
| PRDM5 (PFM2) | Breast and ovarian cancer, cervical carcinoma, liver carcinoma, gastric and colorectal cancer, lung cancer, nasopharyngeal and esophageal carcinoma | Silenced | Silenced in several human cancers through aberrant DNA methylation. Ectopic overexpression induced G2/M arrest and apoptosis in cancer cell lines. Its tumor suppressor function could be explained at least in part through negative regulation of aberrant Wnt/β-catenin signaling and oncogene expression | [131,132,133,134,135,136,137,138,139,140,141,142,143] |
| PRDM6 (PRISM; KMT8C) | Bladder cancer | Downregulated | Transcriptional repressor involved in the regulation of endothelial cell proliferation, survival and differentiation | [45,144,145,146] |
| Breast cancer | Susceptibility gene variants | |||
| PRDM7 (ZNF910) | Hepatocellular carcinoma | Upregulated | Potentially associated with the risk of developing cancer in a Li-Fraumeni-like syndrome patients without TP53 mutations | [45,147] |
| PRDM8 (KMT8D) | Pituitary adenomas | Downregulated | Its alterations are mostly associated with metastasis. Mechanistically, it suppresses the PI3K/AKT/mTOR signaling cascade through the regulation of nucleosome assembly protein 1-like 1. It could be a driver gene in pancreas adenocarcinoma | [45,148,149,150] |
| Endometrial cancer | Hypomethylated | |||
| Hepatocellular carcinoma | Downregulated | |||
| Pancreas adenocarcinoma | Frequently mutated | |||
| PRDM9 (MSBP3; PFM6; ZNF899; KMT8B) | Acute lymphoblastic leukemia and diffuse large B cell lymphoma | Frequent mutations and rare allelic variants | Key role in the mechanisms of homologous recombination. Indeed, it facilitates the association of hotspots with the chromosomal axis and affects the subsequent programmed DNA double-strand breaks initiation and repair. Rare allelic variants were associated with acute lymphoblastic leukemia | [6,45,151,152,153,154,155,156,157,158,159,160] |
| Head and neck squamous cell carcinoma, endometrial, esophageal, stomach and colon carcinomas, kidney and lung tumors and melanoma | ||||
| PRDM10 (KIAA1231; PFM7; MGC131802) | Soft tissue sarcoma | Gene fusions | Gene fusions were found in many cases of low-grade undifferentiated pleomorphic sarcoma. It could influence apoptosis by affecting Bcl-2 expression | [45,161,162,163,164,165,166,167,168,169] |
| Hepatocellular carcinoma, nasopharyngeal carcinoma, gastric cancer, rectum cancer | Integrated bioinformatics and network analyses | |||
| PRDM11 (PFM8) | Diffuse large B cell lymphoma | Non-synonymous coding mutations | Its deletion accelerated Myc-driven lymphomagenesis whereas overexpression induced apoptosis and delayed lymphoma onset in a mouse model. Part of a ceRNA (competitive endogenous RNA) triple (miR-21-5p-NKAPP1-PRDM11) associated with the poor prognosis of lung adenocarcinoma | [170,171,172] |
| Lung adenocarcinoma | Integrative systems biology approach | |||
| PRDM12 (PFM9) | Chronic myeloid leukemia | Chromosome rearrangements | Chromosome rearrangements in chronic myeloid leukemia. Its overexpression showed anti-proliferative properties in vitro | [45,173,174,175,176,177] |
| Prostate cancer, colon cancer | Upregulated | |||
| PRDM13 (PFM10) | Medulloblastoma | Immunotherapy target | Its overexpression was able to inhibit proliferation, migration, and invasion of malignant glioma cells. | [45,178,179,180] |
| Prostate cancer | Hypermethylated | |||
| Head and neck squamous cell carcinoma, bladder, kidney, lung, cervical, and colorectal cancers | Upregulated | |||
| PRDM14 | Lymphoblastic leukemia | Upregulated | Its aberrant high expression observed in human lymphoid malignancies, breast cancer, and other neoplasms may be ascribed to either gene amplification on chromosome 8q13 or copy number gain. Functionally, its requirement in the stemness phenotypes could also explain the involvement in the proliferation and migration of cancer cells. However, a dual role, as both oncogene and tumor suppressor gene, has been recently described in several human cancers and needs to be investigated | [181,182,183,184,185,186,187,188,189,190,191,192,193,194,195,196,197,198,199,200,201,202,203,204,205,206,207,208,209,210,211,212] |
| Breast cancer | Gene amplification/copy number gain | |||
| Lung cancer, head and neck cancer, germ cell tumors | ||||
| Cervical, bladder, colon, and lung cancers | Promoter methylation | |||
| PRDM15 (ZNF298; C21orf83) | Pancreatic cancer | Homozygous deletions | It modulates the transcription of upstream regulators of Wnt and MAPK-ERK signaling to safeguard naive pluripotency | [213,214,215,216] |
| Diffuse large B cell lymphoma | Recurrent mutations | |||
| PRDM16 (MEL1; PFM13; KIAA1675; MGC166915; KMT8F) | Myeloid leukemia | Aberrant isoform expression/gene fusion/mutations | As for other PRDM genes, two main products were identified, with the short PR-l isoform (sPRDM16) displaying oncogenic properties; indeed, this variant could induce myeloid leukemia in p53 knock-out KO mice and was responsible for transforming growth factor (TGF)-β resistance in leukemogenesis. PRDM16 gene fusions with RUNX1 and other partners could also contribute to these hematological malignancies. Further genetic and epigenetic alterations have been observed in brain and other solid tumors, where also the short isoform may function. Recently, a role in cancer cachexia has been suggested owing to its function in adipose browning | [45,217,218,219,220,221,222,223,224,225,226,227,228,229,230,231,232,233,234,235,236,237,238,239,240,241,242,243,244,245,246,247,248,249,250,251,252,253,254,255,256,257,258,259,260,261,262,263,264,265] |
| Prostate cancer | Aberrant isoform expression | |||
| Brain tumors | Upregulated by hypomethylation | |||
| Osteosarcoma, colon cancer, renal cell carcinoma | Gene amplification | |||
| Leiomyosarcoma, gastric, lung, and esophageal cancer | Gene deletion/reduced expression | |||
| Skin melanoma, endometrial carcinoma | Frequently mutated | |||
| ZNF408 (PRDM17; FLJ12827) | - | - | No associations have been found with cancer | [58,266] |
| ZFPM1 (FOG1; FOG; ZNF89A; ZC2HC11A) | Acute myeloid leukemia, chronic myeloid leukemia | Upregulated | Forced FOG1 (friend of GATA-1) expression in human erythroleukemia cells suggested an important role in inducing differentiation toward the erythroid lineage rather than the myelo-lymphoid one by repressing the expression of PU.1. Putative cancer driver gene in adrenocortical carcinoma since recurrent mutations (50%) with a hotspot region were found in this neoplasm. Frequent mutations were also observed in colon and rectum adenocarcinomas | [45,267,268,269,270,271,272,273,274,275,276,277] |
| Adrenocortical carcinoma, colon and rectum adenocarcinomas | Frequently mutated | |||
| Testicular germ cell tumors | Genome wide association studies | |||
| Lung adenocarcinoma | Upregulated by hypomethylation | |||
| ZFPM2 (FOG2; hFOG-2; ZNF89B; ZC2HC11B) | Ovarian tumors | Upregulated | Putative function as tumor suppressor gene. Mostly, it is downregulated and frequently mutated in many cancer types | [45,278,279,280,281,282,283,284,285,286,287,288,289,290,291,292,293,294,295] |
| Neuroblastoma | Downregulated | |||
| Mesothelioma | Fusion gene | |||
| Skin cutaneous melanoma, lung cancers uterine carcinosarcoma, esophageal carcinoma, stomach and rectum adenocarcinoma | Frequently mutated |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casamassimi, A.; Rienzo, M.; Di Zazzo, E.; Sorrentino, A.; Fiore, D.; Proto, M.C.; Moncharmont, B.; Gazzerro, P.; Bifulco, M.; Abbondanza, C. Multifaceted Role of PRDM Proteins in Human Cancer. Int. J. Mol. Sci. 2020, 21, 2648. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072648
Casamassimi A, Rienzo M, Di Zazzo E, Sorrentino A, Fiore D, Proto MC, Moncharmont B, Gazzerro P, Bifulco M, Abbondanza C. Multifaceted Role of PRDM Proteins in Human Cancer. International Journal of Molecular Sciences. 2020; 21(7):2648. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072648
Chicago/Turabian StyleCasamassimi, Amelia, Monica Rienzo, Erika Di Zazzo, Anna Sorrentino, Donatella Fiore, Maria Chiara Proto, Bruno Moncharmont, Patrizia Gazzerro, Maurizio Bifulco, and Ciro Abbondanza. 2020. "Multifaceted Role of PRDM Proteins in Human Cancer" International Journal of Molecular Sciences 21, no. 7: 2648. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072648