Extended Interactions between HIV-1 Viral RNA and tRNALys3 Are Important to Maintain Viral RNA Integrity


Abstract
: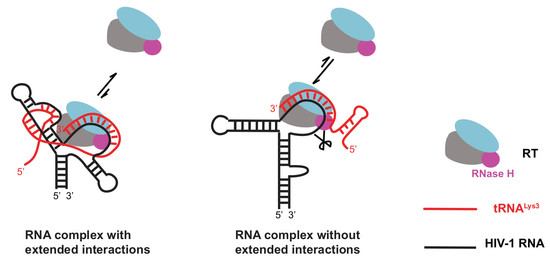
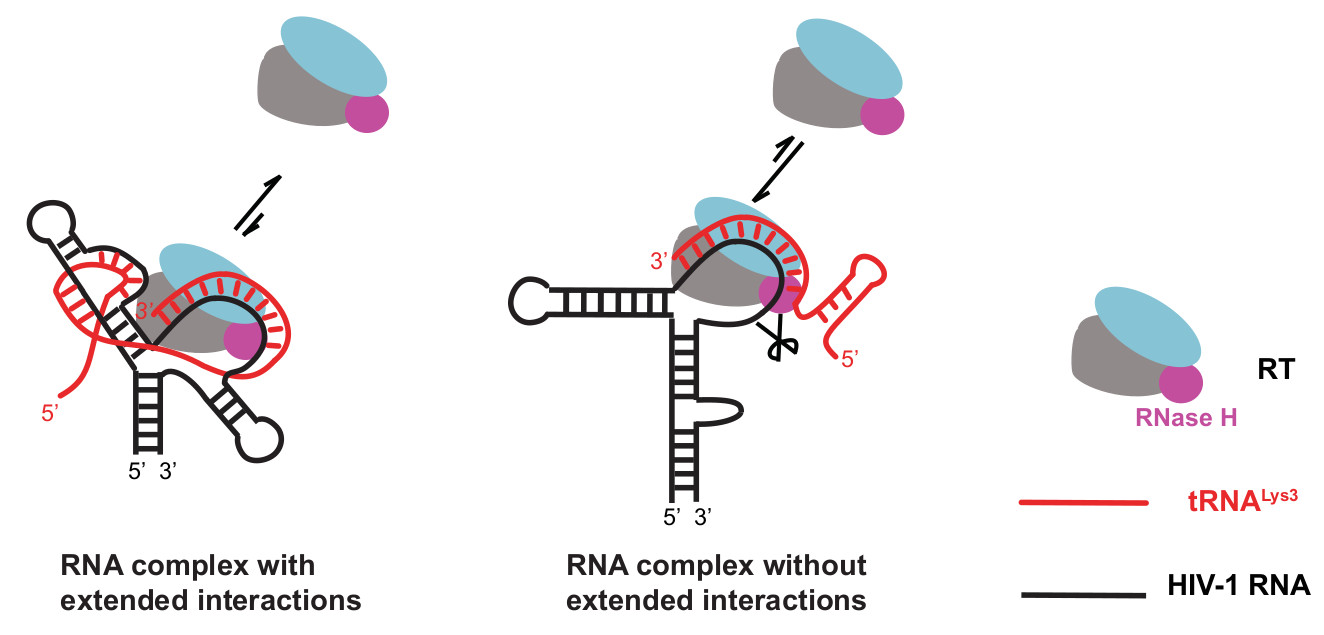{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
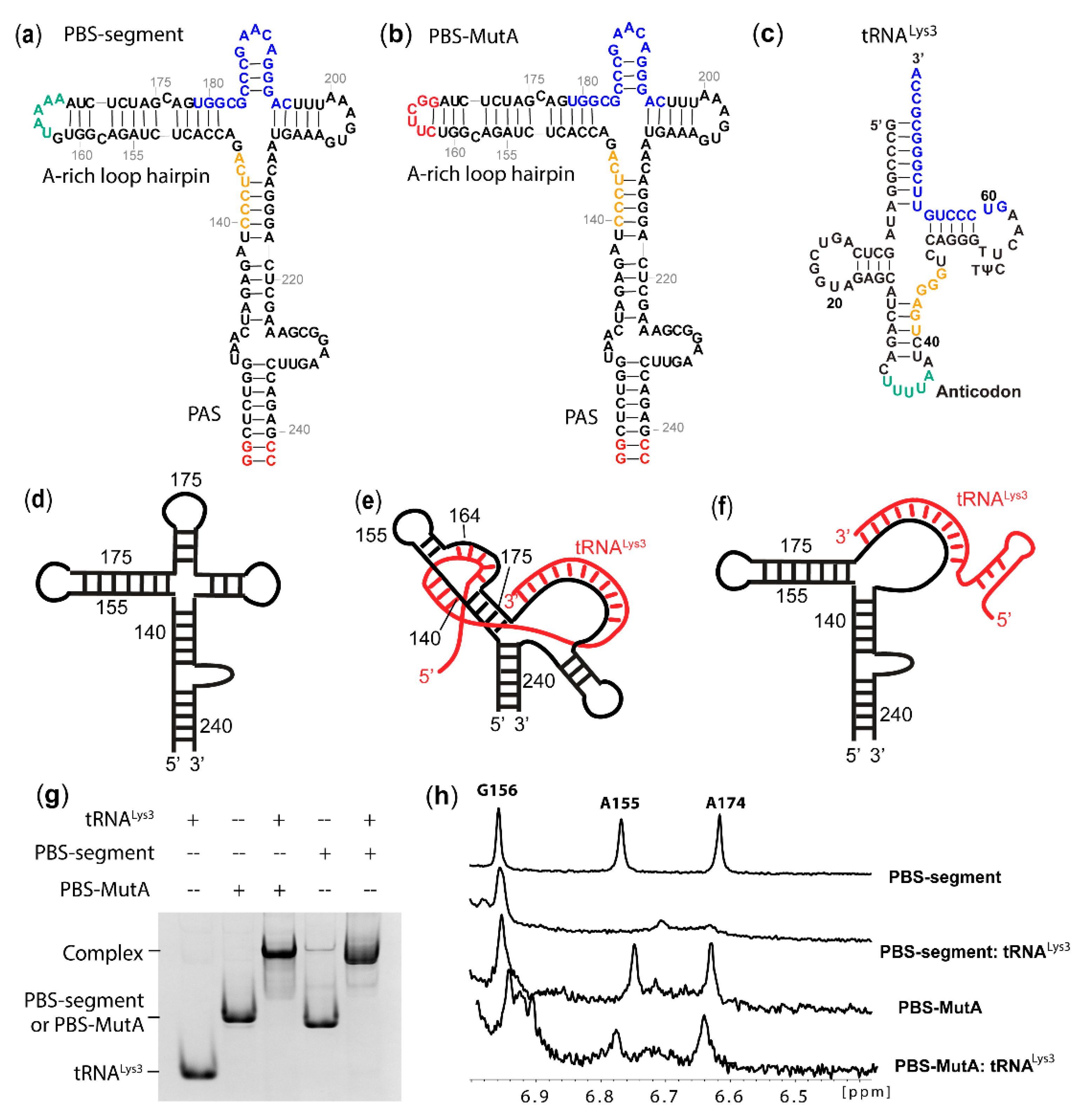
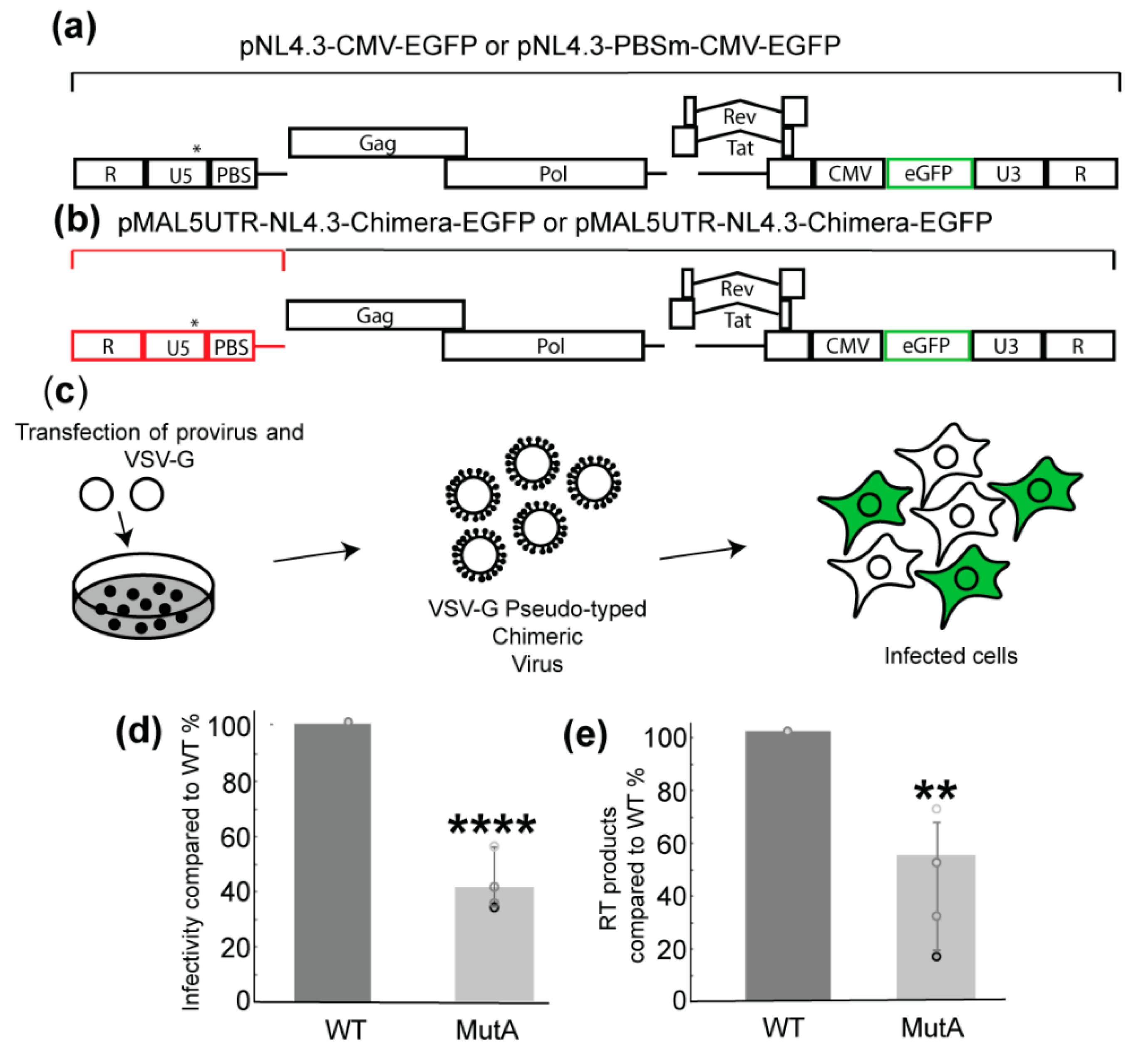
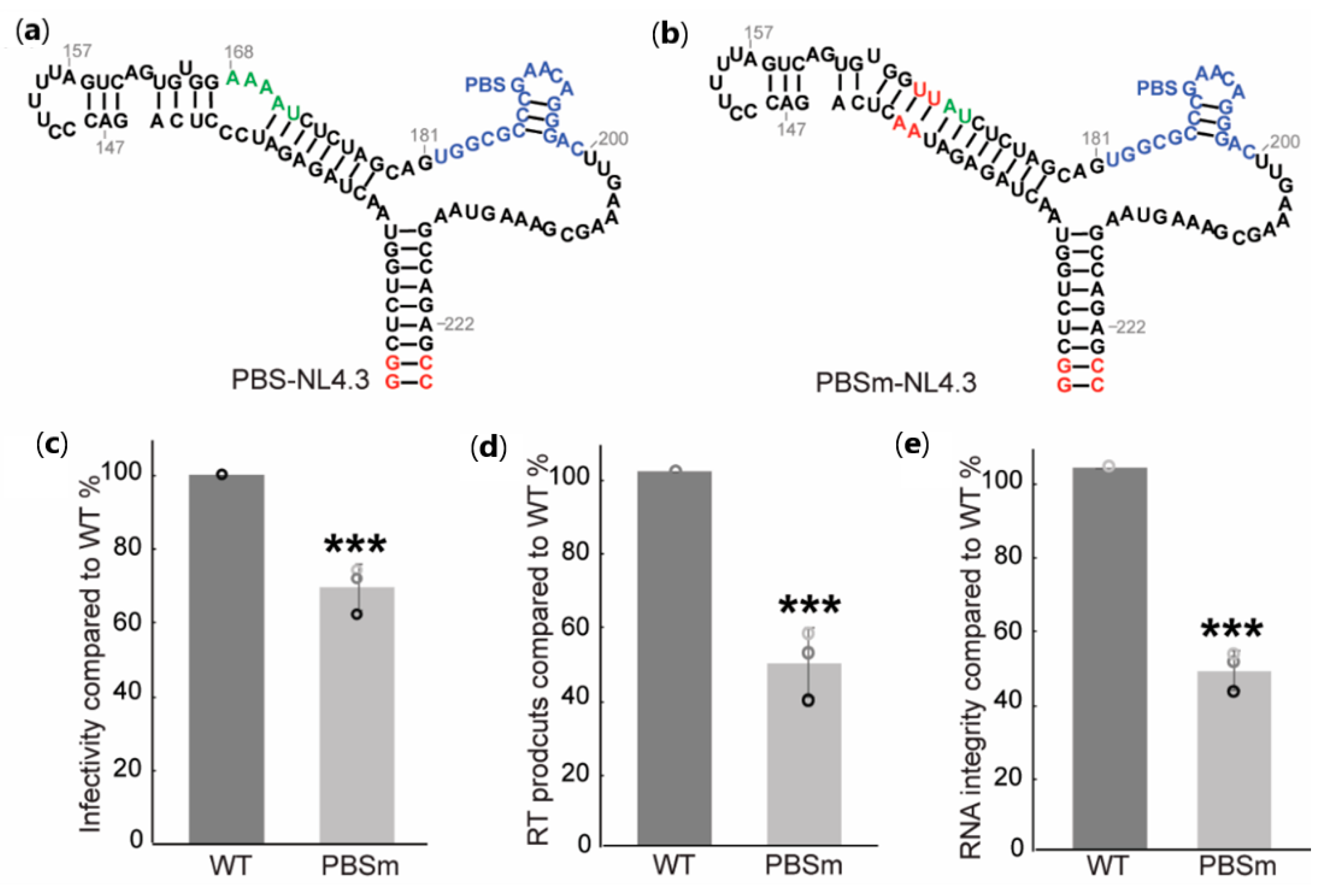
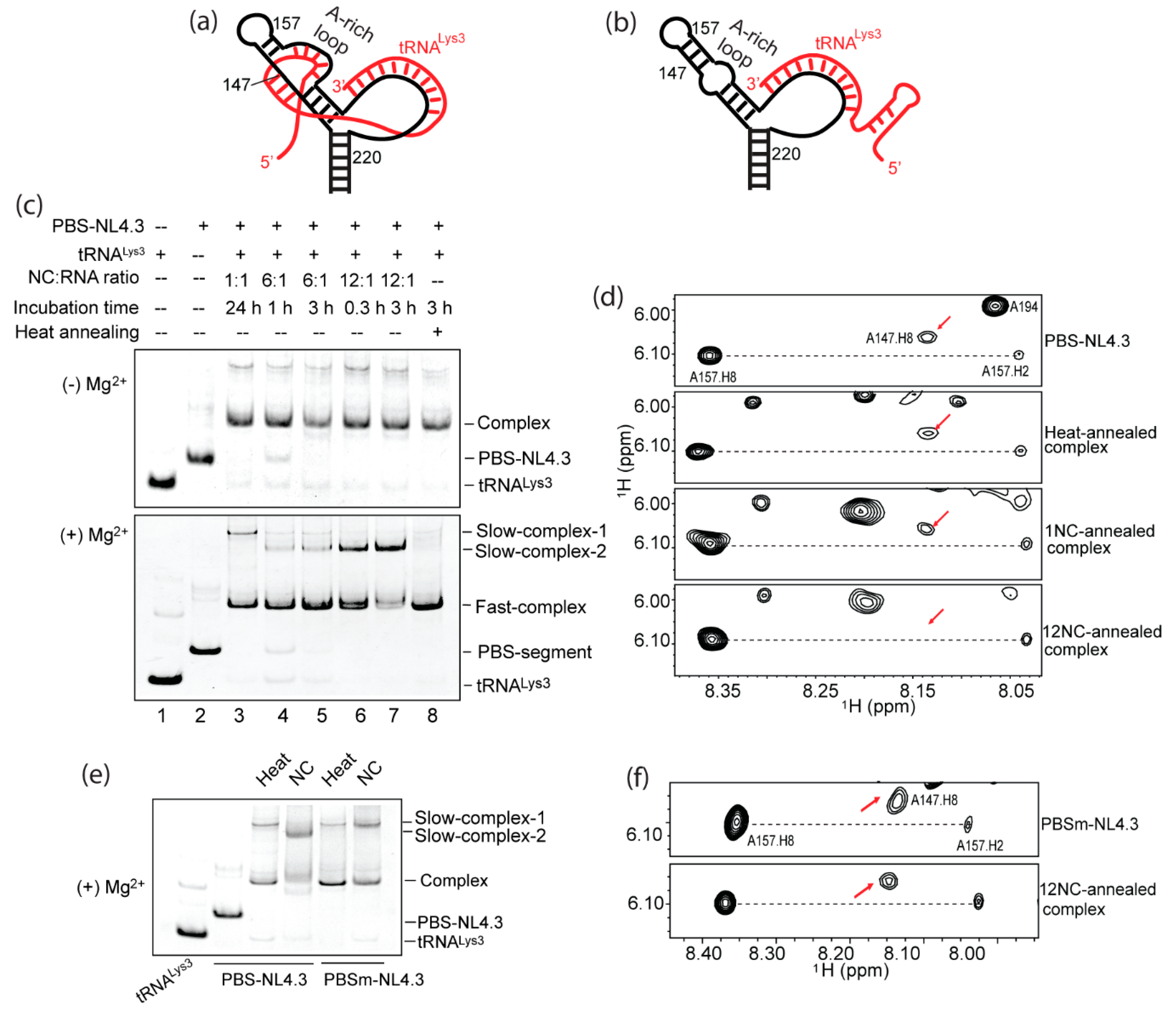
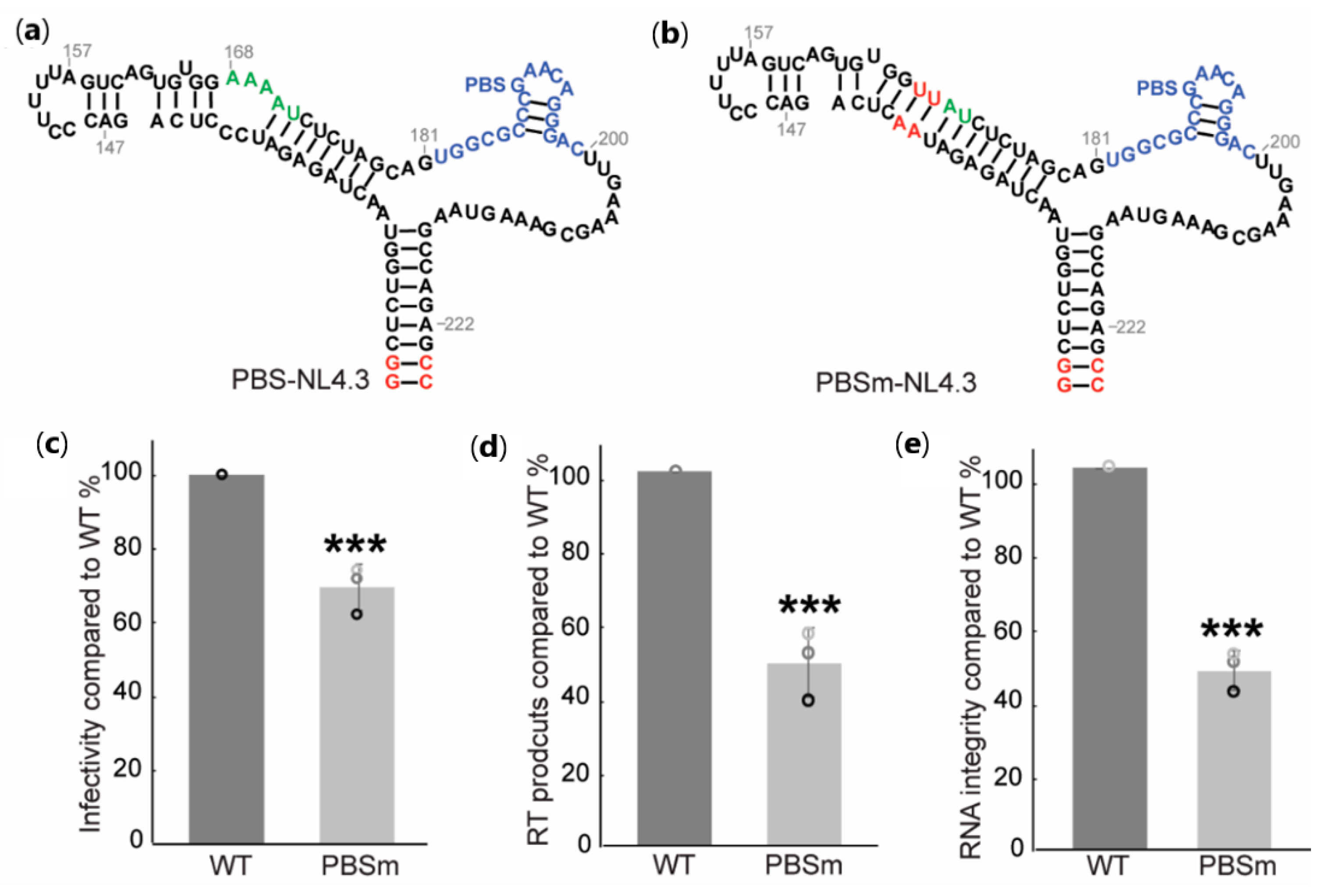
2.1. A-Rich Loop Mutations in the PBS Segment Disrupted Extended Interaction Between vRNA and tRNALys3 and Led to Reduced Infectivity
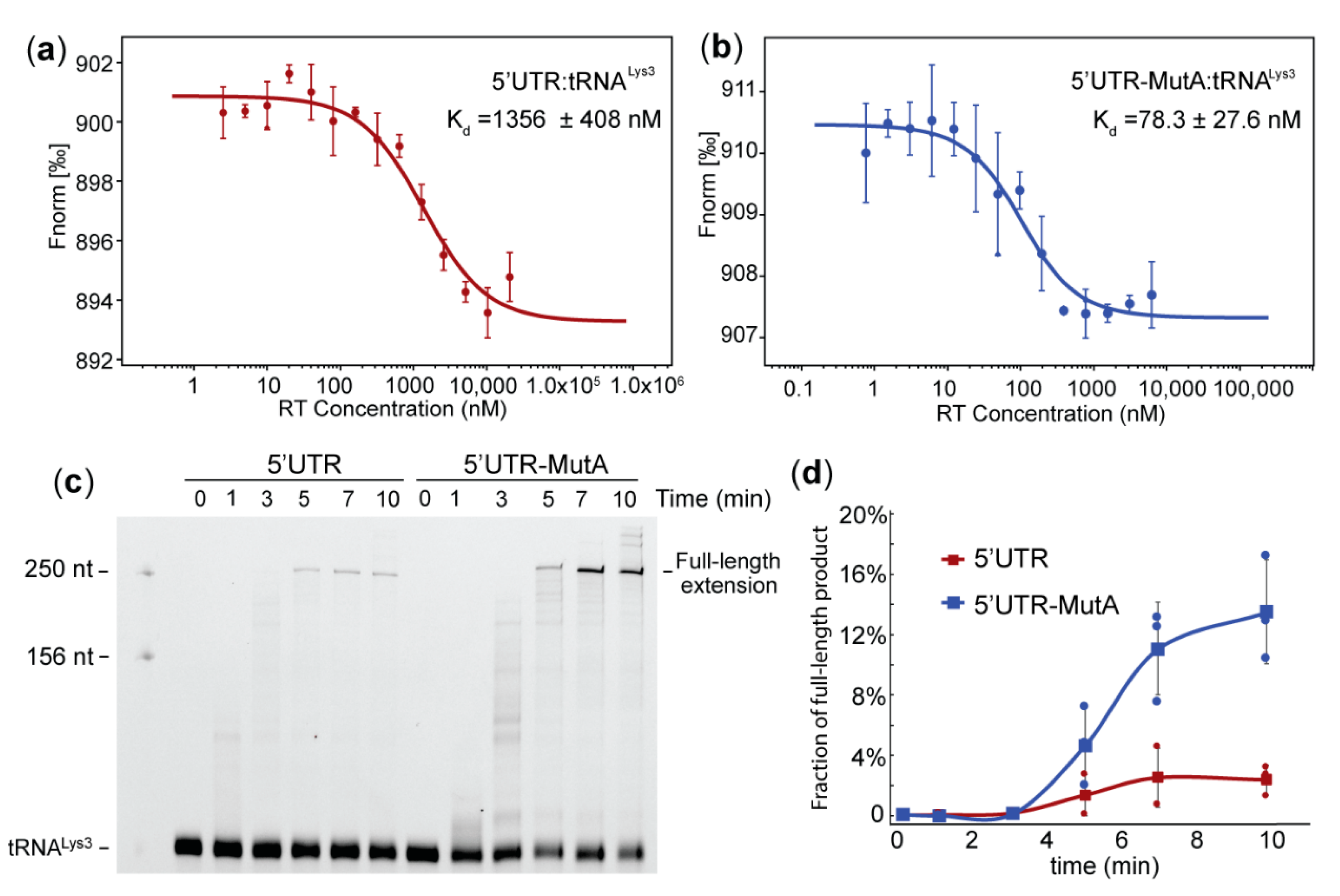
2.2. Mutation of the A-Rich Loop Increased the Affinity of RT Binding to the vRNA: tRNALys3 Complex and Enhanced the Primer Extension Efficiency
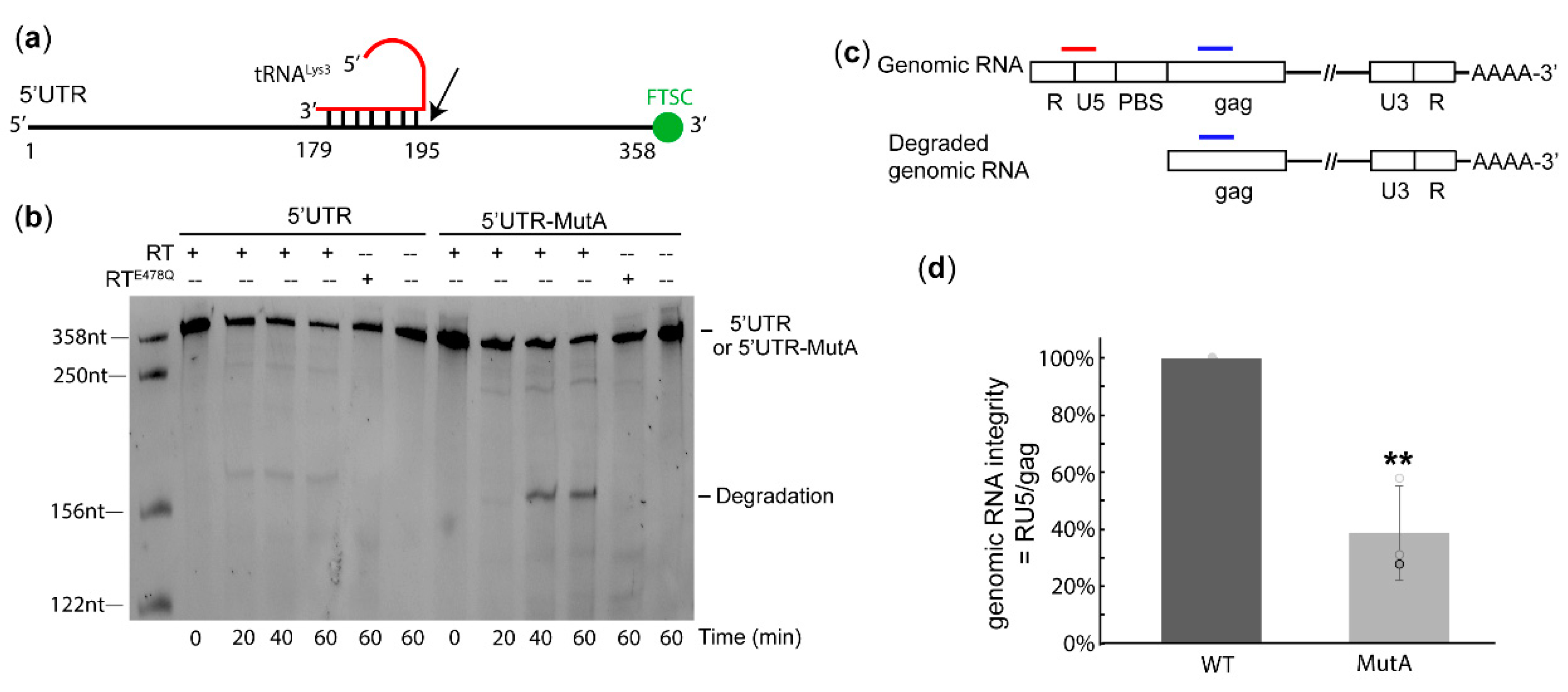
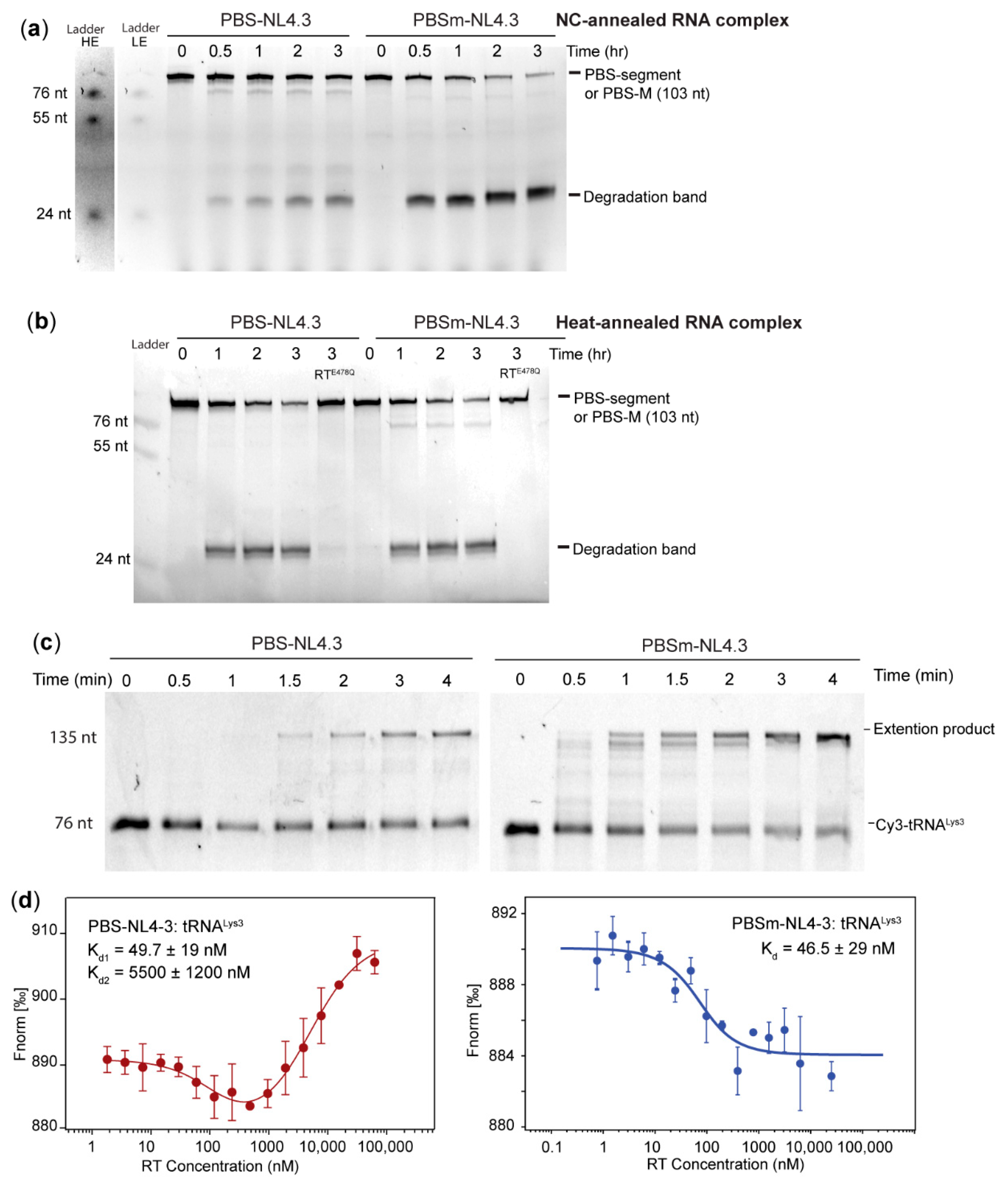
2.3. The A-Rich Loop:Anticodon Interaction Protected vRNA from Degradation by RT in the Absence of dNTP
2.4. The A-Rich Loop: Anticodon Interaction is Conserved in Other HIV-1 Subtypes to Protect vRNA Integrity
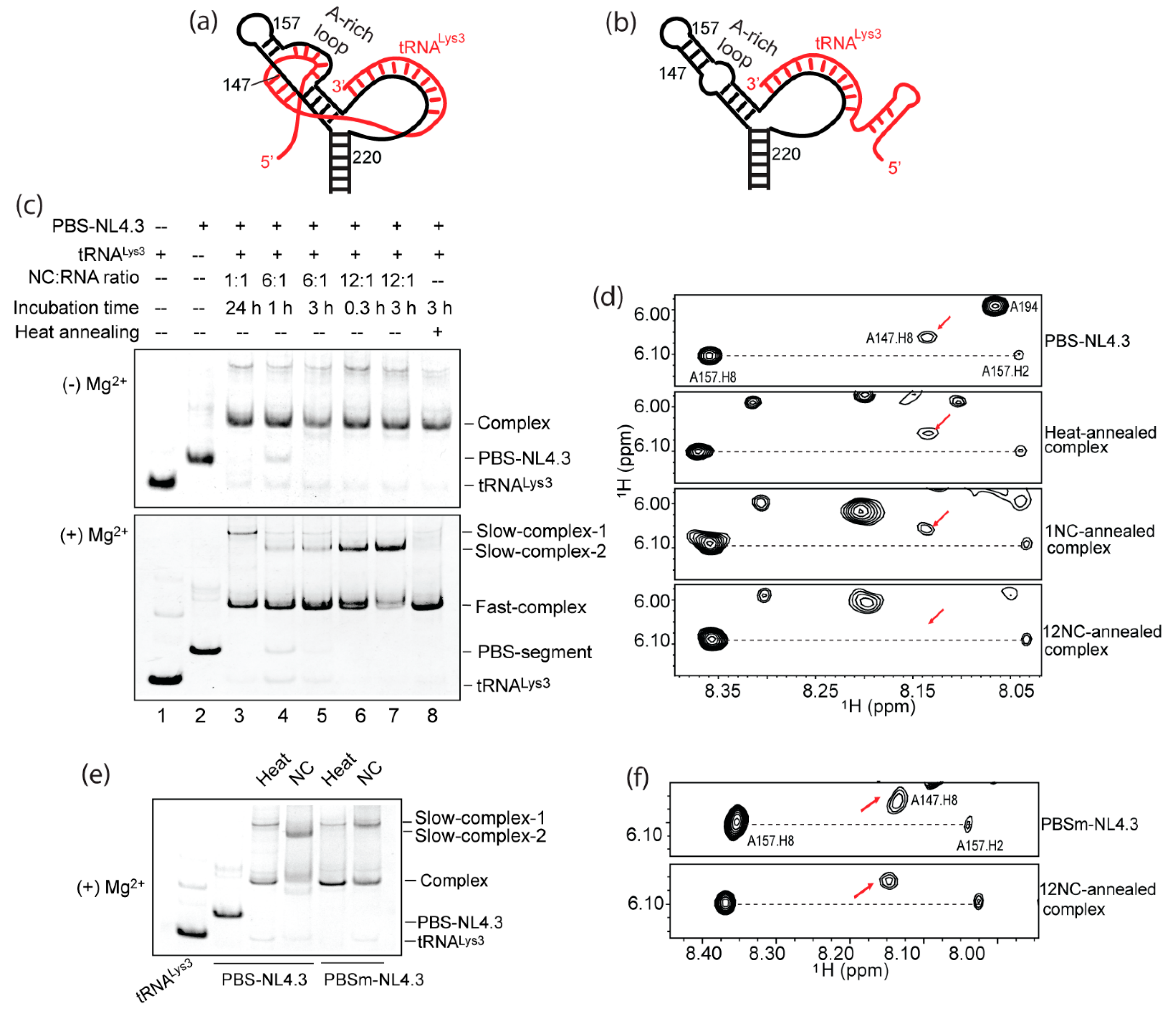
2.5. Formation of the A-Rich Loop:Anticodon Interaction Requires nucleocapsid (NC) Under In Vitro Conditions
2.6. The A-Rich Loop:Anticodon Interaction Protects the Viral RNA from the RNaseH Degradation of RT
3. Discussion
3.1. Biophysical Evidence Supports the Extended vRNA: tRNALys3 Interactions
3.2. The A-Rich Loop:Anticodon Interactions Protect Viral Genomic RNA as a Template for Reverse Transcription
3.3. NC Annealing and Heat Annealing Lead to Differently Folded RNA Complexes In Vitro
3.4. Rediscovery of the RNaseH Activity of RT
3.5. Affinity of RT to DNA/RNA, DNA/DNA, and RNA/RNA:A-Rich Loop Interaction Creates Hindrance for RT to Bind
4. Materials and Methods
4.1. Plasmids
4.2. In Vitro RNA Transcription
4.3. HIV-1 NC, RT, and RTE478Q Purification
4.4. RNA Fluorescence Labeling
4.5. NMR Experiments
4.6. RNA Annealing
4.7. Microscale Thermophoresis
4.8. Primer Extension Assay
4.9. In Vitro Degradation Assay
4.10. Cells and Viruses
4.11. Infectivity Assay
4.12. Quantification of cDNA Products in Infected Cells
4.13. RNA Integrity Assay
4.14. Statistics Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HIV-1 | Human immunodeficiency virus 1 |
| PBS | Primer binding site |
| vRNA | Viral RNA |
| RT | Reverse transcriptase |
| NC | Nucleocapsid |
| WT | Wild type |
| PAS | Primer activation signal |
| cDNA | Complementary DNA |
| dNTP | Deoxyribose nucleoside triphosphate |
| NMR | Nuclear magnetic resonance |
| PCR | Polymerase chain reaction |
| MST | Microscale thermophoresis |
| 5′UTR | 5′ untranslated region |
| FTSC | Fluorescein thiosemicarbazide |
| qRT-PCR | Real-Time Quantitative Reverse-Transcription PCR |
| qPCR | Quantitative PCR |
| EMSA | Electrophoretic mobility shift assay |
| smFRET | Single-molecule Förster resonance energy transfer |
References
- Baltimore, D. Viral RNA-dependent DNA Polymerase. Nature 1970, 226, 1209–1211. [Google Scholar] [CrossRef] [PubMed]
- Marquet, R.; Isel, C.; Ehresmann, C.; Ehresmann, B. tRNAs as primer of reverse transcriptases. Biochimie 1995, 77, 113–124. [Google Scholar] [CrossRef]
- Rhim, H.; Park, J.; Morrow, C.D. Deletions in the tRNA(Lys) primer-binding site of human immunodeficiency virus type 1 identify essential regions for reverse transcription. J. Virol. 1991, 65, 4555–4564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakefield, J.K.; Kang, S.M.; Morrow, C.D. Construction of a type 1 human immunodeficiency virus that maintains a primer binding site complementary to tRNA(His). J. Virol. 1996, 70, 966–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Kang, S.M.; LeBlanc, A.; Hajduk, S.L.; Morrow, C.D. Nucleotide sequences within the U5 region of the viral RNA genome are the major determinants for an human immunodeficiency virus type 1 to maintain a primer binding site complementary to tRNA(His). Virology 1996, 226, 306–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.-M.; Wakefield, J.K.; Morrow, C.D. Mutations in Both the U5 Region and the Primer-Binding Site Influence the Selection of the tRNA Used for the Initiation of HIV-1 Reverse Transcription. Virology 1996, 222, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Isel, C.; Lanchy, J.M.; Le Grice, S.F.J.; Ehresmann, C.; Ehresmann, B.; Marquet, R. Specific initiation and switch to elongation of human immunodeficiency virus type 1 reverse transcription require the post-transcriptional modifications of primer tRNA3Lys. EMBO J. 1996, 15, 917–924. [Google Scholar] [CrossRef]
- Liang, C.; Li, X.; Rong, L.; Inouye, P.; Quan, Y.; Kleiman, L.; Wainberg, M.A. The importance of the A-rich loop in human immunodeficiency virus type 1 reverse transcription and infectivity. J. Virol. 1997, 71, 5750–5757. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.M.; Zhang, Z.; Morrow, C.D. Identification of a human immunodeficiency virus type 1 that stably uses tRNA(Lys1,2) rather than tRNA(Lys,3) for initiation of reverse transcription. Virology 1999, 257, 95–105. [Google Scholar] [CrossRef] [Green Version]
- Das, A.T.; Klaver, B.; Berkhout, B. Reduced replication of human immunodeficiency virus type 1 mutants that use reverse transcription primers other than the natural tRNA(3Lys). J. Virol. 1995, 69, 3090–3097. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Mak, J.; Arts, E.J.; Gu, Z.; Kleiman, L.; Wainberg, M.A.; Parniak, M.A. Effects of alterations of primer-binding site sequences on human immunodeficiency virus type 1 replication. J. Virol. 1994, 68, 6198–6206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isel, C.; Marquet, R.; Keith, G.; Ehresmann, C.; Ehresmann, B. Modified nucleotides of tRNA3/(Lys) modulate primer/template loop-loop interaction in the initiation complex of HIV-1 reverse transcription. J. Biol. Chem. 1993, 268, 25269–25272. [Google Scholar] [PubMed]
- Goldschmidt, V.; Paillart, J.C.; Rigourd, M.; Ehresmann, B.; Aubertin, A.M.; Ehresmann, C.; Marquet, R. Structural variability of the initiation complex of HIV-1 reverse transcription. J. Biol. Chem. 2004, 279, 35923–35931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berkhout, B.; Schoneveld, I. Secondary structure of the HIV-2 leader RNA comprising the tRNA-primer binding site. Nucleic Acids Res. 1993, 21, 1171–1178. [Google Scholar] [CrossRef] [Green Version]
- Isel, C.; Westhof, E.; Massire, C.; Le Grice, S.F.J.; Ehresmann, B.; Ehresmann, C.; Marquet, R. Structural basis for the specificity of the initiation of HIV-1 reverse transcription. EMBO J. 1999, 18, 1038–1048. [Google Scholar] [CrossRef] [Green Version]
- Puglisi, E.V.; Puglisi, J.D. Secondary structure of the HIV reverse transcription initiation complex by NMR. J. Mol. Biol. 2011, 410, 863–874. [Google Scholar] [CrossRef] [Green Version]
- Tisne, C.; Roques, B.P.; Dardel, F. The Annealing Mechanism of HIV-1 Reverse Transcription Primer onto the Viral Genome. J. Biol. Chem. 2004, 279, 3588–3595. [Google Scholar] [CrossRef] [Green Version]
- Coey, A.; Larsen, K.; Puglisi, J.D.; Puglisi, E.V. Heterogeneous structures formed by conserved RNA sequences within the HIV reverse transcription initiation site. RNA 2016, 22, 1689–1698. [Google Scholar] [CrossRef] [Green Version]
- Dupuy, L.C.; Kelly, N.J.; Elgavish, T.E.; Harvey, S.C.; Morrow, C.D. Probing the Importance of tRNA Anticodon: Human Immunodeficiency Virus Type 1 (HIV-1) RNA Genome Complementarity with an HIV-1 That Selects tRNAGlu for Replication. J. Virol. 2003, 77, 8756–8764. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.M.; Zhang, Z.; Morrow, C.D. Identification of a sequence within U5 required for human immunodeficiency virus type 1 to stably maintain a primer binding site complementary to tRNA(Met). J. Virol. 1997, 71, 207–217. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Kang, S.; Li, Y.U.N.; Morrow, C.D. Genetic analysis of the U5-PBS of a novel HIV-1 reveals multiple interactions between the tRNA and RNA genome required for initiation of reverse transcription. RNA 1998, 4, 394–406. [Google Scholar] [PubMed]
- Ooms, M.; Cupac, D.; Abbink, T.E.M.; Huthoff, H.; Berkhout, B. The availability of the primer activation signal (PAS) affects the efficiency of HIV-1 reverse transcription initiation. Nucleic Acids Res. 2007, 35, 1649–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beerens, N.; Berkhout, B. The tRNA Primer Activation Signal in the Human Immunodeficiency Virus Type 1 Genome Is Important for Initiation and Processive Elongation of Reverse Transcription. J. Virol. 2002, 76, 2329–2339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beerens, N.; Groot, F.; Berkhout, B. Initiation of HIV-1 Reverse Transcription is Regulated by a Primer Activation Signal. J. Biol. Chem. 2001, 276, 31247–31256. [Google Scholar] [CrossRef] [Green Version]
- Huthoff, H.; Bugala, K.; Barciszewski, J.; Berkhout, B. On the importance of the primer activation signal for initiation of tRNA lys3-primed reverse transcription of the HIV-1 RNA genome. Nucleic Acids Res. 2003, 31, 5186–5194. [Google Scholar] [CrossRef] [Green Version]
- Goldschmidt, V.; Ehresmann, C.; Ehresmann, B.; Marquet, R. Does the HIV-1 primer activation signal interact with tRNA3Lys during the initiation of reverse transcription. Nucleic Acids Res. 2003, 31, 850–859. [Google Scholar] [CrossRef] [Green Version]
- Larsen, K.P.; Mathiharan, Y.K.; Kappel, K.; Coey, A.T.; Chen, D.H.; Barrero, D.; Madigan, L.; Puglisi, J.D.; Skiniotis, G.; Puglisi, E.V. Architecture of an HIV-1 reverse transcriptase initiation complex. Nature 2018, 557, 118–122. [Google Scholar] [CrossRef]
- Lange, M.J.; Burke, D.H. Screening inhibitory potential of anti-HIV RT RNA aptamers. Methods Mol. Biol. 2014, 1103, 11–29. [Google Scholar] [CrossRef]
- Mbisa, J.L.; Delviks-Frankenberry, K.A.; Thomas, J.A.; Gorelick, R.J.; Pathak, V.K. Real-Time PCR Analysis of HIV-1 Replication Post-entry Events. Methods Mol Biol. 2009, 485, 55–72. [Google Scholar] [CrossRef]
- Brown, J.D.; Kharytonchyk, S.; Chaudry, I.; Iyer, A.S.; Carter, H.; Becker, G.; Desai, Y.; Glang, L.; Choi, S.H.; Singh, K.; et al. Structural basis for transcriptional start site control of HIV-1 RNA fate. Science 2020, 368, 413–417. [Google Scholar] [CrossRef]
- Zhang, H.; Dornadula, G.; Pomerantz, R.J. Natural endogenous reverse transcription of HIV-1. J. Reprod. Immunol. 1998, 41, 255–260. [Google Scholar] [CrossRef]
- Lori, F.; di Marzo Veronese, F.; de Vico, A.L.; Lusso, P.; Reitz, M.S.; Gallo, R.C. Viral DNA carried by human immunodeficiency virus type 1 virions. J. Virol. 1992, 66, 5067–5074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trono, D. Partial reverse transcripts in virions from human immunodeficiency and murine leukemia viruses. J. Virol. 1992, 66, 4893–4900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, W.S.; Hughes, S.H. HIV-1 reverse transcription. Cold Spring Harb. Perspect. Med. 2012, 2, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Gotte, M.; Fackler, S.; Hermann, T.; Perola, E.; Cellai, L.; Gross, H.J.; Le Grice, S.F.; Heumann, H. HIV-1 reverse transcriptase-associated RNase H cleaves RNA/RNA in arrested complexes: Implications for the mechanism by which RNase H discriminates beween RNA/RNA and RNA/DNA. EMBO J. 1995, 14, 833–841. [Google Scholar] [CrossRef] [PubMed]
- Ben-artzi, H.; Zeelon, E.; Le-Grice, S.F.; Gorecki, M.; Panet, A. Characterization of the double stranded RNA dependent RNase activity associated with recombinant reverse transcriptases. Nucleic Acids Res. 1992, 20, 5115–5118. [Google Scholar] [CrossRef] [Green Version]
- Mizrahi, V.; Brooksbank, R.; Nkabinde, N. Mutagenesis of the Conserved Aspartic Acid 443, Glutamic Acid 478, Asparagine 494, and Aspartic Acid 498 Residues in the Ribonuclease H Domain of p66/p51 Human Immunodeficiency Virus Type I Reverse Transcriptase. J. Biol. Chem. 1994, 269, 19245–19249. [Google Scholar]
- DeStefano, J.J.; Wu, W.; Seehra, J.; McCoy, J.; Laston, D.; Albone, E.; Fay, P.J.; Bambara, R.A. Characterization of an RNase H deficient mutant of human immunodeficiency virus-1 reverse transcriptase having an aspartate to asparagine change at position 498. BBA Gene Struct. Expr. 1994, 1219, 380–388. [Google Scholar] [CrossRef]
- Lange, M.J.; Sharma, T.K.; Whatley, A.S.; Landon, L.A.; Tempesta, M.A.; Johnson, M.C.; Burke, D.H. Robust suppression of HIV replication by intracellularly expressed reverse transcriptase aptamers is independent of ribozyme processing. Mol. Ther. 2012, 20, 2304–2314. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Quan, Y.; Arts, E.J.; Li, Z.; Preston, B.D.; de Rocquigny, H.; Roques, B.P.; Darlix, J.L.; Kleiman, L.; Parniak, M.A.; et al. Human immunodeficiency virus Type 1 nucleocapsid protein (NCp7) directs specific initiation of minus-strand DNA synthesis primed by human tRNA(Lys3) in vitro: Studies of viral RNA molecules mutated in regions that flank the primer binding site. J. Virol. 1996, 70, 4996–5004. [Google Scholar] [CrossRef] [Green Version]
- Brulé, F.; Marquet, R.; Rong, L.; Wainberg, M.A.; Roques, B.P.; Le Grice, S.F.J.; Ehresmann, B.; Ehresmann, C. Structural and functional properties of the HIV-1 RNA-tRNA3Lys primer complex annealed by the nucleocapsid protein: Comparison with the heat-annealed complex. RNA 2002, 8, 8–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakefield, J.K.; Wolf, A.G.; Morrow, C.D. Human immunodeficiency virus type 1 can use different tRNAs as primers for reverse transcription but selectively maintains a primer binding site complementary to tRNA(3Lys). J. Virol. 1995, 69, 6021–6029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigourd, M.; Goldschmidt, V.; Brulé, F.; Morrow, C.D.; Ehresmann, B.; Ehresmann, C.; Marquet, R. Structure-function relationships of the initiation complex of HIV-1 reverse transcription: The case of mutant viruses using tRNAHis as primer. Nucleic Acids Res. 2003, 31, 5764–5775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beerens, N.; Berkhout, B. Switching the in vitro tRNA usage of HIV-1 by simultaneous adaptation of the PBS and PAS. RNA 2002, 8, 357–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beerens, N.; Jepsen, M.D.E.; Nechyporuk-Zloy, V.; Krüger, A.C.; Darlix, J.L.; Kjems, J.; Birkedal, V. Role of the primer activation signal in tRNA annealing onto the HIV-1 genome studied by single-molecule FRET microscopy. RNA 2013, 19, 517–526. [Google Scholar] [CrossRef] [Green Version]
- Goldschmidt, V.; Rigourd, M.; Ehresmann, C.; Le Grice, S.F.J.; Ehresmann, B.; Marquet, R. Direct and indirect contributions of RNA secondary structure elements to the initiation of HIV-1 reverse transcription. J. Biol. Chem. 2002, 277, 43233–43242. [Google Scholar] [CrossRef] [Green Version]
- Seif, E.; Niu, M.; Kleiman, L. In virio SHAPE analysis of tRNA(Lys3) annealing to HIV-1 genomic RNA in wild type and protease-deficient virus. Retrovirology 2015, 12, 40. [Google Scholar] [CrossRef] [Green Version]
- Miller, S.B.; Yildiz, F.Z.; Lo, J.A.; Wang, B.; D’Souza, V.M. A structure-based mechanism for tRNA and retroviral RNA remodelling during primer annealing. Nature 2014, 515, 591–595. [Google Scholar] [CrossRef] [Green Version]
- Coey, A.T.; Larsen, K.P.; Choi, J.; Barrero, D.J.; Puglisi, J.D.; Puglisi, E.V. Dynamic Interplay of RNA and Protein in the Human Immunodeficiency Virus-1 Reverse Transcription Initiation Complex. J. Mol. Biol. 2018, 430, 5137–5150. [Google Scholar] [CrossRef]
- Ben-Artzi, H.; Zeelon, E.; Gorecki, M.; Panet, A. Double-stranded RNA-dependent RNase activity associated with human immunodeficiency virus type 1 reverse transcriptase. Proc. Natl. Acad. Sci. USA 1992, 89, 927–931. [Google Scholar] [CrossRef] [Green Version]
- Hostomsky, Z.; Hudson, G.O.; Rahmati, S.; Hostomska, Z. RNase D, a reported new activity associated with HIV-1 reverse transcriptase, displays the same cleavage specificity as Escherichia coli RNase III. Nucleic Acids Res. 1992, 20, 5819–5824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Harada, B.T.; Miller, J.T.; Le Grice, S.F.J.; Zhuang, X. Initiation complex dynamics direct the transitions between distinct phases of early HIV reverse transcription. Nat. Struct. Mol. Biol. 2010, 17, 1453–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaccaro, J.A.; Singh, H.A.; Anderson, K.S. Initiation of minus-strand DNA synthesis by human immunodeficiency virus type 1 reverse transcriptase. Biochemistry 1999, 38, 15978–15985. [Google Scholar] [CrossRef] [PubMed]
- Kati, W.M.; Johnson, K.A.; Jerva, L.F.; Anderson, K.S. Mechanism and fidelity of HIV reverse transcriptase. J. Biol. Chem. 1992, 267, 25988–25997. [Google Scholar]
- Alizon, M.; Wain-Hobson, S.; Montagnier, L.; Sonigo, P. Genetic variability of the AIDS virus: Nucleotide sequence analysis of two isolates from African patients. Cell 1986, 46, 63–74. [Google Scholar] [CrossRef]
- Alam, K.K.; Chang, J.L.; Lange, M.J.; Nguyen, P.D.M.; Sawyer, A.W.; Burke, D.H. Poly-Target Selection Identifies Broad-Spectrum RNA Aptamers. Mol. Ther. Nucleic Acids 2018, 13, 605–619. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, P.D.M.; Zheng, J.; Gremminger, T.J.; Qiu, L.; Zhang, D.; Tuske, S.; Lange, M.J.; Griffin, P.R.; Arnold, E.; Chen, S.-J.; et al. Binding interface and impact on protease cleavage for an RNA aptamer to HIV-1 reverse transcriptase. Nucleic Acids Res. 2020, 48. [Google Scholar] [CrossRef]
- Lee, B.M.; De Guzman, R.N.; Turner, B.G.; Tjandra, N.; Summers, M.F. Dynamical behavior of the HIV-1 nucleocapsid protein. J. Mol. Biol. 1998, 279, 633–649. [Google Scholar] [CrossRef]
- Brady, S.; Singh, G.; Bolinger, C.; Song, Z.; Boeras, I.; Weng, K.; Trent, B.; Brown, W.C.; Singh, K.; Boris-Lawrie, K.; et al. Virion-associated, host-derived DHX9/RNA helicase A enhances the processivity of HIV-1 reverse transcriptase on genomic RNA. J. Biol. Chem. 2019, 294, 11473–11485. [Google Scholar] [CrossRef]
- Maciejewski, M.W.; Schuyler, A.D.; Gryk, M.R.; Moraru, I.I.; Romero, P.R.; Ulrich, E.L.; Eghbalnia, H.R.; Livny, M.; Delaglio, F.; Hoch, J.C. NMRbox: A Resource for Biomolecular NMR Computation. Biophys. J. 2017, 112, 1529–1534. [Google Scholar] [CrossRef] [Green Version]
- Hill, K.J.; Rogers, L.C.; Njenda, D.T.; Burke, D.H.; Sarafianos, S.G.; Sönnerborg, A.; Neogi, U.; Singh, K. Strain-specific Effect on Biphasic DNA Binding by HIV-1 Integrase. Aids 2019, 33, 588–592. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gremminger, T.; Song, Z.; Ji, J.; Foster, A.; Weng, K.; Heng, X. Extended Interactions between HIV-1 Viral RNA and tRNALys3 Are Important to Maintain Viral RNA Integrity. Int. J. Mol. Sci. 2021, 22, 58. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010058
Gremminger T, Song Z, Ji J, Foster A, Weng K, Heng X. Extended Interactions between HIV-1 Viral RNA and tRNALys3 Are Important to Maintain Viral RNA Integrity. International Journal of Molecular Sciences. 2021; 22(1):58. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010058
Chicago/Turabian StyleGremminger, Thomas, Zhenwei Song, Juan Ji, Avery Foster, Kexin Weng, and Xiao Heng. 2021. "Extended Interactions between HIV-1 Viral RNA and tRNALys3 Are Important to Maintain Viral RNA Integrity" International Journal of Molecular Sciences 22, no. 1: 58. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010058