
Time-of-Day-Dependent Effects of Bromocriptine to Ameliorate Vascular Pathology and Metabolic Syndrome in SHR Rats Held on High Fat Diet


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
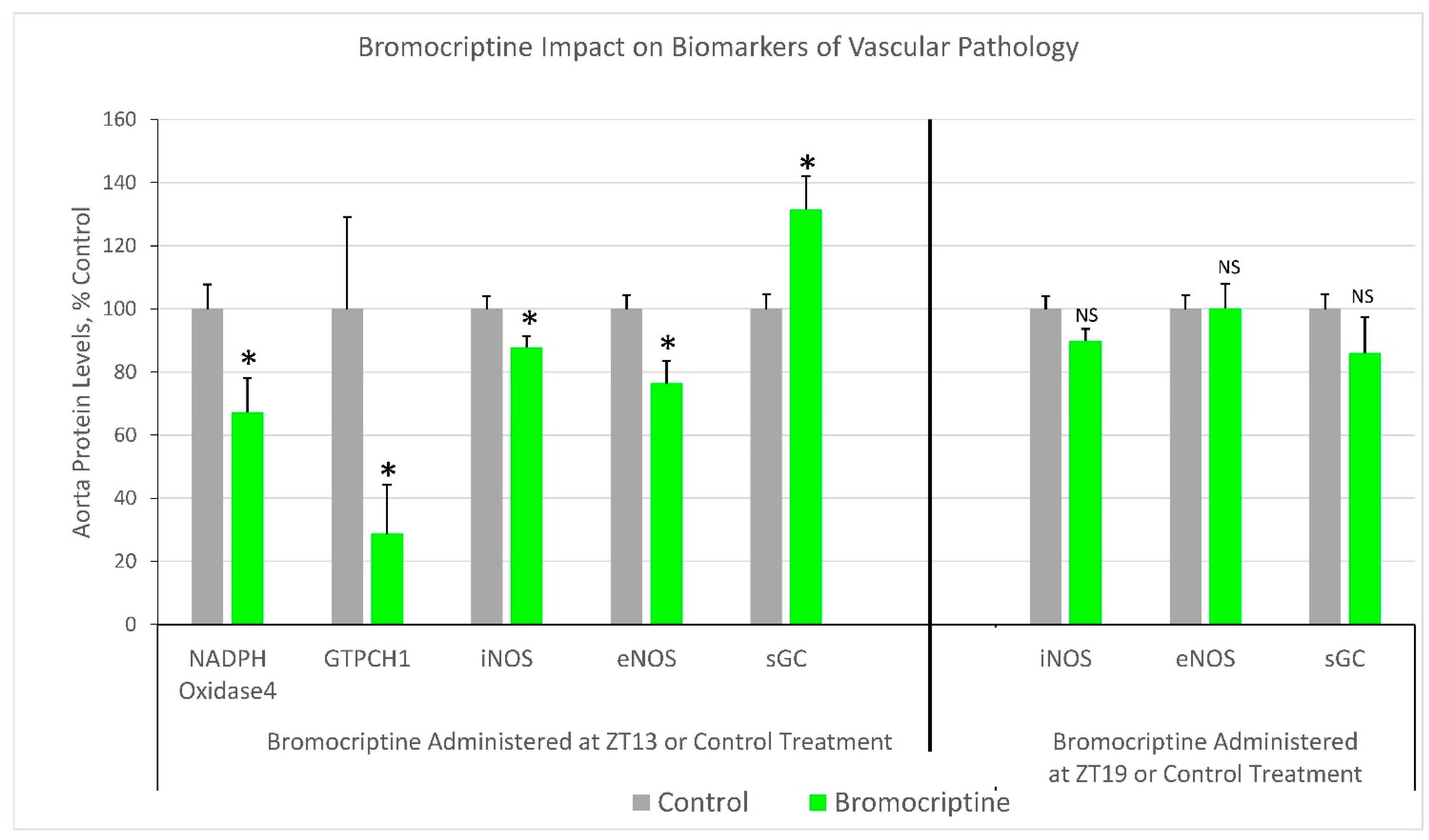
2.1. Effect of ZT 13 versus ZT 19 Bromocriptine Treatment on Aortic Levels of Enzymes Regulating Tissue Redox Status
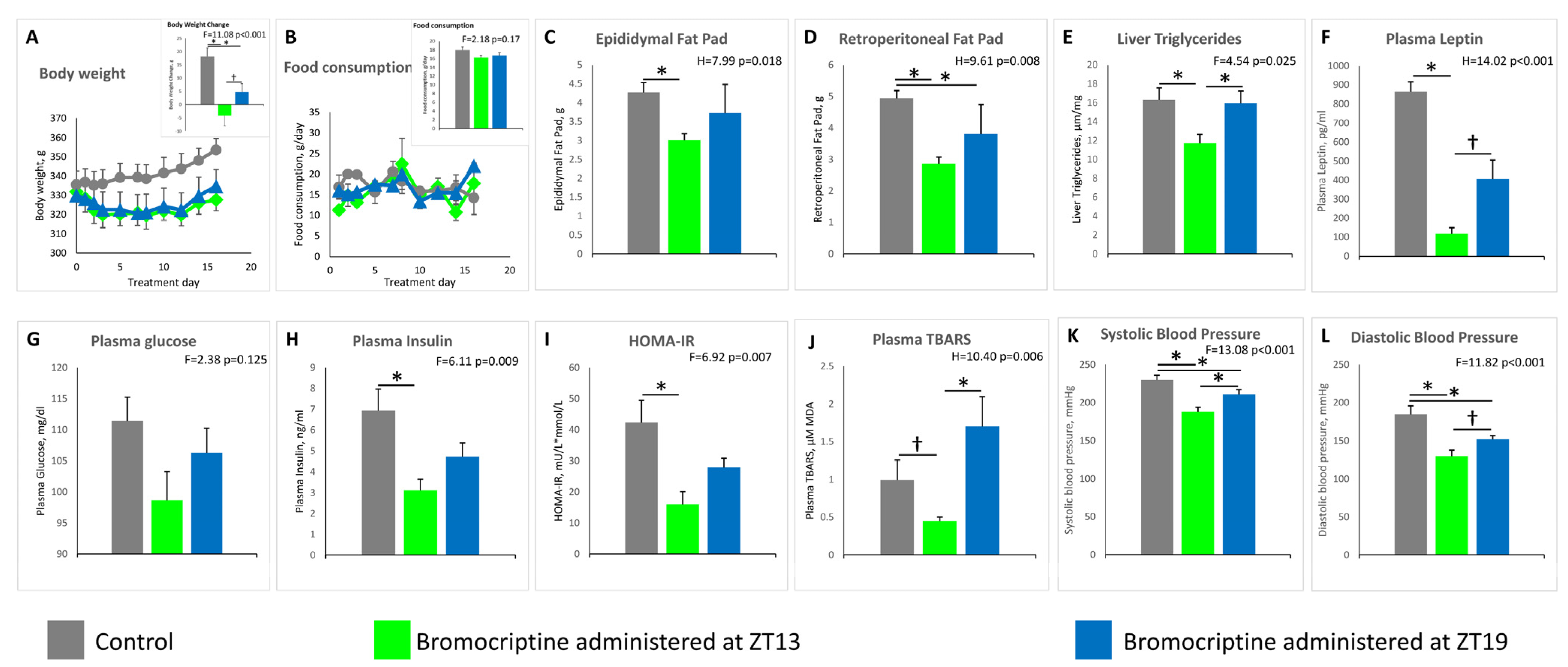
2.2. Effect of ZT 13 versus ZT 19 Bromocriptine Treatment on Cardiometabolic Parameters
2.3. Effect of ZT 13 versus ZT 19 Bromocriptine Treatment on MBH AgRP/NPY, Glial Cell Neuronal Support Factor, and Neuronal Plasticity Genes mRNA Levels
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Animals
5.2. Experimental Design
5.3. Rationale for Assessing Vascular ROS/RNS Pathology from Isolated Aorta Tissue
5.4. Western Blot Analyses of Aortic Proteins
5.5. Quantitative PCR Analysis of MBH mRNAs
- Gfap (glial fibrillary acidic protein) ThermoFisher, Waltham, MA, USA Cat# Rn00566603_m1
- Mef2c (myocyte enhancer factor 2C) ThermoFisher, Waltham, MA, USA Cat# Rn01494040_m1
- NPY (neuropeptide Y) ThermoFisher, Waltham, MA, USA Cat# Rn01410145_m1
- AgRP (agouti related neuropeptide) ThermoFisher, Waltham, MA, USA Cat# Rn01431703_g1
- S100a10 (S100 calcium binding protein A10) ThermoFisher, Waltham, MA, USA Cat# Rn01409218_m1
- Aqp4 (aquaporin 4) ThermoFisher, Waltham, MA, USA Cat# Rn00563196_m1
- Bmp4 (bone morphogenetic protein 4) ThermoFisher, Waltham, MA, USA Cat# Rn00432087_m1
- The relative expression of genes was calculated using the 2−ΔΔCq method.
5.6. Biochemical Assays of Blood Samples and Analysis of Liver Lipid Content
5.7. Blood Pressure (BP) Measurements
5.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Raskin, A.; Cincotta, A.H. Bromocriptine-QR therapy for the management of type 2 diabetes mellitus: Developmental basis and therapeutic profile summary. Expert Rev. Endocrinol. Metab. 2016, 11, 113–148. [Google Scholar] [CrossRef] [Green Version]
- Luo, S.; Zhang, Y.; Ezrokhi, M.; Li, Y.; Tsai, T.H.; Cincotta, A.H. Circadian peak dopaminergic activity response at the biological clock pacemaker (suprachiasmatic nucleus) area mediates the metabolic responsiveness to a high-fat diet. J. Neuroendocrinol. 2018, 30, e12563. [Google Scholar] [CrossRef] [Green Version]
- Stoelzel, C.R.; Zhang, Y.; Cincotta, A.H. Circadian-timed dopamine agonist treatment reverses high-fat diet-induced diabetogenic shift in ventromedial hypothalamic glucose sensing. Endocrinol. Diabetes Metab. 2020, 3, e00139. [Google Scholar] [CrossRef]
- La Fleur, S.E. Daily rhythms in glucose metabolism: Suprachiasmatic nucleus output to peripheral tissue. J. Neuroendocrinol. 2003, 15, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Kalsbeek, A.; Bruinstroop, E.; Yi, C.X.; Klieverik, L.P.; La Fleur, S.E.; Fliers, E. Hypothalamic control of energy metabolism via the autonomic nervous system. Ann. N. Y. Acad. Sci. 2010, 1212, 114–129. [Google Scholar] [CrossRef]
- Opland, D.M.; Leinninger, G.M.; Myers, M.G., Jr. Modulation of the mesolimbic dopamine system by leptin. Brain Res. 2010, 1350, 65–70. [Google Scholar] [CrossRef] [Green Version]
- Geiger, B.M.; Haburcak, M.; Avena, N.M.; Moyer, M.C.; Hoebel, B.G.; Pothos, E.N. Deficits of mesolimbic dopamine neurotransmission in rat dietary obesity. Neuroscience 2009, 159, 1193–1199. [Google Scholar] [CrossRef] [Green Version]
- Geiger, B.M.; Behr, G.G.; Frank, L.E.; Caldera-Siu, A.D.; Beinfeld, M.C.; Kokkotou, E.G.; Pothos, E.N. Evidence for defective mesolimbic dopamine exocytosis in obesity-prone rats. FASEB J. 2008, 22, 2740–2746. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Luo, J.; Cincotta, A.H. Suprachiasmatic nuclei monoamine metabolism of glucose tolerant versus intolerant hamsters. Neuroreport 1999, 10, 2073–2077. [Google Scholar] [CrossRef]
- Luo, S.; Luo, J.; Meier, A.H.; Cincotta, A.H. Dopaminergic neurotoxin administration to the area of the suprachiasmatic nuclei induces insulin resistance. Neuroreport 1997, 8, 3495–3499. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Ezrokhi, M.; Cominos, N.; Tsai, T.H.; Stoelzel, C.R.; Trubitsyna, Y.; Cincotta, A.H. Experimental dopaminergic neuron lesion at the area of the biological clock pacemaker, suprachiasmatic nuclei (SCN) induces metabolic syndrome in rats. Diabetol. Metab. Syndr. 2021, 13, 11. [Google Scholar] [CrossRef] [PubMed]
- Chamarthi, B.; Vinik, A.; Ezrokhi, M.; Cincotta, A.H. Circadian-timed quick-release bromocriptine lowers elevated resting heart rate in patients with type 2 diabetes mellitus. Endocrinol. Diabetes Metab. 2020, 3, e00101. [Google Scholar] [CrossRef]
- Ezrokhi, M.; Luo, S.; Trubitsyna, Y.; Cincotta, A.H. Neuroendocrine and metabolic components of dopamine agonist amelioration of metabolic syndrome in SHR rats. Diabetol. Metab. Syndr. 2014, 6, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bina, K.G.; Cincotta, A.H. Dopaminergic agonists normalize elevated hypothalamic neuropeptide Y and corticotropin-releasing hormone, body weight gain, and hyperglycemia in ob/ob mice. Neuroendocrinology 2000, 71, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Gaziano, J.M.; Cincotta, A.H.; O’onnor, C.M.; Ezrokhi, M.; Rutty, D.; Ma, Z.J.; Scranton, R.E. Randomized clinical trial of quick-release bromocriptine among patients with type 2 diabetes on overall safety and cardiovascular outcomes. Diabetes Care 2010, 33, 1503–1508. [Google Scholar] [CrossRef] [Green Version]
- Gaziano, J.M.; Cincotta, A.H.; Vinik, A.; Blonde, L.; Bohannon, N.; Scranton, R. Effect of bromocriptine-QR (a quick-release formulation of bromocriptine mesylate) on major adverse cardiovascular events in type 2 diabetes subjects. J. Am. Heart Assoc. 2012, 1, e002279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chamarthi, B.; Gaziano, J.M.; Blonde, L.; Vinik, A.; Scranton, R.E.; Ezrokhi, M.; Rutty, D.; Cincotta, A.H. Timed Bromocriptine-QR Therapy Reduces Progression of Cardiovascular Disease and Dysglycemia in Subjects with Well-Controlled Type 2 Diabetes Mellitus. J. Diabetes Res. 2015, 2015, 157698. [Google Scholar] [CrossRef] [Green Version]
- Chamarthi, B.; Ezrokhi, M.; Rutty, D.; Cincotta, A.H. Impact of bromocriptine-QR therapy on cardiovascular outcomes in type 2 diabetes mellitus subjects on metformin. Postgrad. Med. 2016, 128, 761–769. [Google Scholar] [CrossRef]
- Aziz, I.S.; McMahon, A.M.; Friedman, D.; Rabinovich-Nikitin, I.; Kirshenbaum, L.A.; Martino, T.A. Circadian influence on inflammatory response during cardiovascular disease. Curr. Opin. Pharmacol. 2020, 57, 60–70. [Google Scholar] [CrossRef]
- Man, A.W.C.; Li, H.; Xia, N. Circadian Rhythm: Potential Therapeutic Target for Atherosclerosis and Thrombosis. Int. J. Mol. Sci. 2021, 22, 676. [Google Scholar] [CrossRef] [PubMed]
- Prasai, M.J.; George, J.T.; Scott, E.M. Molecular clocks, type 2 diabetes and cardiovascular disease. Diab. Vasc. Dis. Res. 2008, 5, 89–95. [Google Scholar] [CrossRef] [Green Version]
- Young, M.E.; Bray, M.S. Potential role for peripheral circadian clock dyssynchrony in the pathogenesis of cardiovascular dysfunction. Sleep Med. 2007, 8, 656–667. [Google Scholar] [CrossRef] [Green Version]
- Reilly, D.F.; Westgate, E.J.; FitzGerald, G.A. Peripheral circadian clocks in the vasculature. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1694–1705. [Google Scholar] [CrossRef] [Green Version]
- Maemura, K.; Takeda, N.; Nagai, R. Circadian rhythms in the CNS and peripheral clock disorders: Role of the biological clock in cardiovascular diseases. J. Pharmacol. Sci. 2007, 103, 134–138. [Google Scholar] [CrossRef] [Green Version]
- Kudo, T.; Horikawa, K.; Shibata, S. Circadian rhythms in the CNS and peripheral clock disorders: The circadian clock and hyperlipidemia. J. Pharmacol. Sci. 2007, 103, 139–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, P.L. eNOS, metabolic syndrome and cardiovascular disease. Trends Endocrinol. Metab. 2009, 20, 295–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzik, T.J.; Mussa, S.; Gastaldi, D.; Sadowski, J.; Ratnatunga, C.; Pillai, R.; Channon, K.M. Mechanisms of increased vascular superoxide production in human diabetes mellitus: Role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation 2002, 105, 1656–1662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, A.K.; Ravussin, E.; Johannsen, D.L.; Stull, A.J.; Cefalu, W.T.; Johnson, W.D. Endothelial Dysfunction: An Early Cardiovascular Risk Marker in Asymptomatic Obese Individuals with Prediabetes. Br. J. Med. Med. Res. 2012, 2, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Forstermann, U.; Munzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef] [Green Version]
- Williams, I.L.; Wheatcroft, S.B.; Shah, A.M.; Kearney, M.T. Obesity, atherosclerosis and the vascular endothelium: Mechanisms of reduced nitric oxide bioavailability in obese humans. Int. J. Obes. Relat. Metab. Disord. 2002, 26, 754–764. [Google Scholar] [CrossRef] [Green Version]
- Shinozaki, K.; Ayajiki, K.; Kashiwagi, A.; Masada, M.; Okamura, T. Malfunction of vascular control in lifestyle-related diseases: Mechanisms underlying endothelial dysfunction in the insulin-resistant state. J. Pharmacol. Sci. 2004, 96, 401–405. [Google Scholar] [CrossRef] [Green Version]
- Maiese, K.; Morhan, S.D.; Chong, Z.Z. Oxidative stress biology and cell injury during type 1 and type 2 diabetes mellitus. Curr. Neurovasc Res. 2007, 4, 63–71. [Google Scholar] [CrossRef] [Green Version]
- Kagota, S.; Yamaguchi, Y.; Tanaka, N.; Kubota, Y.; Kobayashi, K.; Nejime, N.; Nakamura, K.; Kunitomo, M.; Shinozuka, K. Disturbances in nitric oxide/cyclic guanosine monophosphate system in SHR/NDmcr-cp rats, a model of metabolic syndrome. Life Sci. 2006, 78, 1187–1196. [Google Scholar] [CrossRef]
- Lopez-Farre, A.; Rodriguez-Feo, J.A.; Garcia-Colis, E.; Gomez, J.; Lopez-Blaya, A.; Fortes, J.; de Andres, R.; Rico, L.; Casado, S. Reduction of the soluble cyclic GMP vasorelaxing system in the vascular wall of stroke-prone spontaneously hypertensive rats: Effect of the alpha1 -receptor blocker doxazosin. J. Hypertens 2002, 20, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Chamiot-Clerc, P.; Renaud, J.F.; Safar, M.E. Pulse pressure, aortic reactivity, and endothelium dysfunction in old hypertensive rats. Hypertension 2001, 37, 313–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mason, R.P.; Kubant, R.; Jacob, R.F.; Walter, M.F.; Boychuk, B.; Malinski, T. Effect of nebivolol on endothelial nitric oxide and peroxynitrite release in hypertensive animals: Role of antioxidant activity. J. Cardiovasc. Pharmacol. 2006, 48, 862–869. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Yoshikawa, N.; Kagota, S.; Nakamura, K.; Haginaka, J.; Kunitomo, M. Elevated circulating levels of markers of oxidative-nitrative stress and inflammation in a genetic rat model of metabolic syndrome. Nitric Oxide 2006, 15, 380–386. [Google Scholar] [CrossRef]
- Kalinowski, L.; Malinski, T. Endothelial NADH/NADPH-dependent enzymatic sources of superoxide production: Relationship to endothelial dysfunction. Acta Biochim. Pol. 2004, 51, 459–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paravicini, T.M.; Touyz, R.M. NADPH oxidases, reactive oxygen species, and hypertension: Clinical implications and therapeutic possibilities. Diabetes Care 2008, 31 (Suppl. S2), S170–S180. [Google Scholar] [CrossRef] [Green Version]
- Cai, H. NAD(P)H oxidase-dependent self-propagation of hydrogen peroxide and vascular disease. Circ. Res. 2005, 96, 818–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, S.; Lei, H.; Qin, H.; Xia, Y. Molecular mechanisms of endothelial NO synthase uncoupling. Curr. Pharm. Des. 2014, 20, 3548–3553. [Google Scholar] [CrossRef] [PubMed]
- Ginnan, R.; Guikema, B.J.; Halligan, K.E.; Singer, H.A.; Jourd’heuil, D. Regulation of smooth muscle by inducible nitric oxide synthase and NADPH oxidase in vascular proliferative diseases. Free Radic Biol. Med. 2008, 44, 1232–1245. [Google Scholar] [CrossRef] [Green Version]
- Upmacis, R.K.; Crabtree, M.J.; Deeb, R.S.; Shen, H.; Lane, P.B.; Benguigui, L.E.; Maeda, N.; Hajjar, D.P.; Gross, S.S. Profound biopterin oxidation and protein tyrosine nitration in tissues of ApoE-null mice on an atherogenic diet: Contribution of inducible nitric oxide synthase. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H2878–H2887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharawy, N.; Lehmann, C. Molecular mechanisms by which iNOS uncoupling can induce cardiovascular dysfunction during sepsis: Role of posttranslational modifications (PTMs). Life Sci. 2020, 255, 117821. [Google Scholar] [CrossRef] [PubMed]
- Oliveira-Paula, G.H.; Lacchini, R.; Tanus-Santos, J.E. Inducible nitric oxide synthase as a possible target in hypertension. Curr. Drug Targets 2014, 15, 164–174. [Google Scholar] [CrossRef]
- Mungrue, I.N.; Gros, R.; You, X.; Pirani, A.; Azad, A.; Csont, T.; Schulz, R.; Butany, J.; Stewart, D.J.; Husain, M. Cardiomyocyte overexpression of iNOS in mice results in peroxynitrite generation, heart block, and sudden death. J. Clin. Investig. 2002, 109, 735–743. [Google Scholar] [CrossRef]
- Buttery, L.D.; Springall, D.R.; Chester, A.H.; Evans, T.J.; Standfield, E.N.; Parums, D.V.; Yacoub, M.H.; Polak, J.M. Inducible nitric oxide synthase is present within human atherosclerotic lesions and promotes the formation and activity of peroxynitrite. Lab. Investig. 1996, 75, 77–85. [Google Scholar]
- Thum, T.; Fraccarollo, D.; Schultheiss, M.; Froese, S.; Galuppo, P.; Widder, J.D.; Tsikas, D.; Ertl, G.; Bauersachs, J. Endothelial nitric oxide synthase uncoupling impairs endothelial progenitor cell mobilization and function in diabetes. Diabetes 2007, 56, 666–674. [Google Scholar] [CrossRef] [Green Version]
- Munzel, T.; Daiber, A.; Ullrich, V.; Mulsch, A. Vascular consequences of endothelial nitric oxide synthase uncoupling for the activity and expression of the soluble guanylyl cyclase and the cGMP-dependent protein kinase. Publication and Online Material. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1551–1557. [Google Scholar] [CrossRef]
- Landmesser, U.; Dikalov, S.; Price, S.R.; McCann, L.; Fukai, T.; Holland, S.M.; Mitch, W.E.; Harrison, D.G. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J. Clin. Investig. 2003, 111, 1201–1209. [Google Scholar] [CrossRef]
- Bendall, J.K.; Alp, N.J.; Warrick, N.; Cai, S.; Adlam, D.; Rockett, K.; Yokoyama, M.; Kawashima, S.; Channon, K.M. Stoichiometric relationships between endothelial tetrahydrobiopterin, endothelial NO synthase (eNOS) activity, and eNOS coupling in vivo: Insights from transgenic mice with endothelial-targeted GTP cyclohydrolase 1 and eNOS overexpression. Circ. Res. 2005, 97, 864–871. [Google Scholar] [CrossRef]
- Schulz, E.; Jansen, T.; Wenzel, P.; Daiber, A.; Munzel, T. Nitric oxide, tetrahydrobiopterin, oxidative stress, and endothelial dysfunction in hypertension. Antioxid. Redox. Signal. 2008, 10, 1115–1126. [Google Scholar] [CrossRef]
- Kalra, S.P.; Kalra, P.S. NPY and cohorts in regulating appetite, obesity and metabolic syndrome: Beneficial effects of gene therapy. Neuropeptides 2004, 38, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Denis, R.G.P.; Joly-Amado, A.; Cansell, C.; Castel, J.; Martinez, S.; Delbes, A.S.; Luquet, S. Central orchestration of peripheral nutrient partitioning and substrate utilization: Implications for the metabolic syndrome. Diabetes Metab. 2014, 40, 191–197. [Google Scholar] [CrossRef]
- Stofkova, A.; Skurlova, M.; Kiss, A.; Zelezna, B.; Zorad, S.; Jurcovicova, J. Activation of hypothalamic NPY, AgRP, MC4R, AND IL-6 mRNA levels in young Lewis rats with early-life diet-induced obesity. Endocr. Regul. 2009, 43, 99–106. [Google Scholar] [PubMed]
- Marcelin, G.; Jo, Y.H.; Li, X.; Schwartz, G.J.; Zhang, Y.; Dun, N.J.; Lyu, R.M.; Blouet, C.; Chang, J.K.; Chua, S., Jr. Central action of FGF19 reduces hypothalamic AGRP/NPY neuron activity and improves glucose metabolism. Mol. Metab. 2014, 3, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Aleixandre de Artinano, A.; Miguel Castro, M. Experimental rat models to study the metabolic syndrome. Br. J. Nutr. 2009, 102, 1246–1253. [Google Scholar] [CrossRef] [Green Version]
- Lassegue, B.; Clempus, R.E. Vascular NAD(P)H oxidases: Specific features, expression, and regulation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 285, R277–R297. [Google Scholar] [CrossRef] [Green Version]
- Bruckdorfer, R. The basics about nitric oxide. Mol. Asp. Med. 2005, 26, 3–31. [Google Scholar] [CrossRef]
- Cai, H. Hydrogen peroxide regulation of endothelial function: Origins, mechanisms, and consequences. Cardiovasc. Res. 2005, 68, 26–36. [Google Scholar] [CrossRef] [Green Version]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radi, R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848. [Google Scholar] [CrossRef] [Green Version]
- Shimokawa, H. Reactive oxygen species in cardiovascular health and disease: Special references to nitric oxide, hydrogen peroxide, and Rho-kinase. J. Clin. Biochem. Nutr. 2020, 66, 83–91. [Google Scholar] [CrossRef] [Green Version]
- Byon, C.H.; Heath, J.M.; Chen, Y. Redox signaling in cardiovascular pathophysiology: A focus on hydrogen peroxide and vascular smooth muscle cells. Redox Biol. 2016, 9, 244–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook-Mills, J.M. Hydrogen peroxide activation of endothelial cell-associated MMPs during VCAM-1-dependent leukocyte migration. Cell Mol. Biol. 2006, 52, 8–16. [Google Scholar] [PubMed]
- Laude, K.; Cai, H.; Fink, B.; Hoch, N.; Weber, D.S.; McCann, L.; Kojda, G.; Fukai, T.; Schmidt, H.H.; Dikalov, S.; et al. Hemodynamic and biochemical adaptations to vascular smooth muscle overexpression of p22phox in mice. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H7–H12. [Google Scholar] [CrossRef] [Green Version]
- Cai, H.; Davis, M.E.; Drummond, G.R.; Harrison, D.G. Induction of endothelial NO synthase by hydrogen peroxide via a Ca(2+)/calmodulin-dependent protein kinase II/janus kinase 2-dependent pathway. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1571–1576. [Google Scholar] [CrossRef] [Green Version]
- Drummond, G.R.; Cai, H.; Davis, M.E.; Ramasamy, S.; Harrison, D.G. Transcriptional and posttranscriptional regulation of endothelial nitric oxide synthase expression by hydrogen peroxide. Circ. Res. 2000, 86, 347–354. [Google Scholar] [CrossRef]
- Zou, M.H.; Cohen, R.; Ullrich, V. Peroxynitrite and vascular endothelial dysfunction in diabetes mellitus. Endothelium 2004, 11, 89–97. [Google Scholar] [CrossRef]
- Ishii, M.; Shimizu, S.; Wajima, T.; Hagiwara, T.; Negoro, T.; Miyazaki, A.; Tobe, T.; Kiuchi, Y. Reduction of GTP cyclohydrolase I feedback regulating protein expression by hydrogen peroxide in vascular endothelial cells. J. Pharmacol. Sci. 2005, 97, 299–302. [Google Scholar] [CrossRef] [Green Version]
- Hool, L.C.; Corry, B. Redox control of calcium channels: From mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2007, 9, 409–435. [Google Scholar] [CrossRef]
- Kimura, S.; Zhang, G.X.; Nishiyama, A.; Shokoji, T.; Yao, L.; Fan, Y.Y.; Rahman, M.; Abe, Y. Mitochondria-derived reactive oxygen species and vascular MAP kinases: Comparison of angiotensin II and diazoxide. Hypertension 2005, 45, 438–444. [Google Scholar] [CrossRef] [Green Version]
- Yoshioka, J.; Schreiter, E.R.; Lee, R.T. Role of thioredoxin in cell growth through interactions with signaling molecules. Antioxid. Redox Signal. 2006, 8, 2143–2151. [Google Scholar] [CrossRef]
- Griendling, K.K.; Sorescu, D.; Lassegue, B.; Ushio-Fukai, M. Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2175–2183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandes, R.P. Role of NADPH oxidases in the control of vascular gene expression. Antioxid. Redox. Signal. 2003, 5, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Rey, F.E.; Pagano, P.J. The reactive adventitia: Fibroblast oxidase in vascular function. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1962–1971. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.K. Novel NAD(P)H oxidases in the cardiovascular system. Heart 2004, 90, 491–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Torres, I.; Manzano-Pech, L.; Rubio-Ruiz, M.E.; Soto, M.E.; Guarner-Lans, V. Nitrosative Stress and Its Association with Cardiometabolic Disorders. Molecules 2020, 25, 2555. [Google Scholar] [CrossRef] [PubMed]
- Umar, S.; van der Laarse, A. Nitric oxide and nitric oxide synthase isoforms in the normal, hypertrophic, and failing heart. Mol. Cell Biochem. 2010, 333, 191–201. [Google Scholar] [CrossRef]
- Bahadoran, Z.; Mirmiran, P.; Ghasemi, A.; Kashfi, K. Type 2 Diabetes and Cancer: The Nitric Oxide Connection. Crit. Rev. Oncog. 2019, 24, 235–242. [Google Scholar] [CrossRef]
- Otani, H. The role of nitric oxide in myocardial repair and remodeling. Antioxid. Redox. Signal. 2009, 11, 1913–1928. [Google Scholar] [CrossRef]
- Goyal, P.; Weissmann, N.; Grimminger, F.; Hegel, C.; Bader, L.; Rose, F.; Fink, L.; Ghofrani, H.A.; Schermuly, R.T.; Schmidt, H.H.; et al. Upregulation of NAD(P)H oxidase 1 in hypoxia activates hypoxia-inducible factor 1 via increase in reactive oxygen species. Free Radic. Biol. Med. 2004, 36, 1279–1288. [Google Scholar] [CrossRef] [PubMed]
- San Martin, A.; Du, P.; Dikalova, A.; Lassegue, B.; Aleman, M.; Gongora, M.C.; Brown, K.; Joseph, G.; Harrison, D.G.; Taylor, W.R.; et al. Reactive oxygen species-selective regulation of aortic inflammatory gene expression in Type 2 diabetes. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H2073–H2082. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Ni, Z.; Oveisi, F.; Trnavsky-Hobbs, D.L. Effect of antioxidant therapy on blood pressure and NO synthase expression in hypertensive rats. Hypertension 2000, 36, 957–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cincotta, A.H. Hypothalamic role in the insulin resistance syndrome. In Insulin Resistance and Insulin Resistance Syndrome, Frontiers in Animal Diabetes Research Series; Hansen, B., Shafrir, E., Eds.; Taylor and Francis: London, UK, 2002; pp. 271–312. [Google Scholar]
- Wei, W.; Pham, K.; Gammons, J.W.; Sutherland, D.; Liu, Y.; Smith, A.; Kaczorowski, C.C.; O’Connell, K.M. Diet composition, not calorie intake, rapidly alters intrinsic excitability of hypothalamic AgRP/NPY neurons in mice. Sci. Rep. 2015, 5, 16810. [Google Scholar] [CrossRef] [Green Version]
- Dalvi, P.S.; Chalmers, J.A.; Luo, V.; Han, D.Y.; Wellhauser, L.; Liu, Y.; Tran, D.Q.; Castel, J.; Luquet, S.; Wheeler, M.B.; et al. High fat induces acute and chronic inflammation in the hypothalamus: Effect of high-fat diet, palmitate and TNF-alpha on appetite-regulating NPY neurons. Int. J. Obes. 2017, 41, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Levin, E.; Yom-Tov, Y.; Hefetz, A.; Kronfeld-Schor, N. Changes in diet, body mass and fatty acid composition during pre-hibernation in a subtropical bat in relation to NPY and AgRP expression. J. Comp. Physiol. B 2013, 183, 157–166. [Google Scholar] [CrossRef]
- Niswender, K.D.; Baskin, D.G.; Schwartz, M.W. Insulin and its evolving partnership with leptin in the hypothalamic control of energy homeostasis. Trends Endocrinol. Metab. 2004, 15, 362–369. [Google Scholar] [CrossRef]
- Schwartz, M.W.; Baskin, D.G.; Bukowski, T.R.; Kuijper, J.L.; Foster, D.; Lasser, G.; Prunkard, D.E.; Porte, D., Jr.; Woods, S.C.; Seeley, R.J.; et al. Specificity of leptin action on elevated blood glucose levels and hypothalamic neuropeptide Y gene expression in ob/ob mice. Diabetes 1996, 45, 531–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belgardt, B.F.; Okamura, T.; Bruning, J.C. Hormone and glucose signalling in POMC and AgRP neurons. J. Physiol. 2009, 587 Pt 22, 5305–5314. [Google Scholar] [CrossRef]
- Morton, G.J.; Gelling, R.W.; Niswender, K.D.; Morrison, C.D.; Rhodes, C.J.; Schwartz, M.W. Leptin regulates insulin sensitivity via phosphatidylinositol-3-OH kinase signaling in mediobasal hypothalamic neurons. Cell Metab. 2005, 2, 411–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wauman, J.; Tavernier, J. Leptin receptor signaling: Pathways to leptin resistance. Front. Biosci. 2011, 16, 2771–2793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myers, M.G., Jr.; Leibel, R.L.; Seeley, R.J.; Schwartz, M.W. Obesity and leptin resistance: Distinguishing cause from effect. Trends Endocrinol. Metab. 2010, 21, 643–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munzberg, H.; Flier, J.S.; Bjorbaek, C. Region-specific leptin resistance within the hypothalamus of diet-induced obese mice. Endocrinology 2004, 145, 4880–4889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohno, D.; Yada, T. Arcuate NPY neurons sense and integrate peripheral metabolic signals to control feeding. Neuropeptides 2012, 46, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Samodien, E.; Pheiffer, C.; Erasmus, M.; Mabasa, L.; Louw, J.; Johnson, R. Diet-induced DNA methylation within the hypothalamic arcuate nucleus and dysregulated leptin and insulin signaling in the pathophysiology of obesity. Food Sci. Nutr. 2019, 7, 3131–3145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volkow, N.D.; Wang, G.J.; Telang, F.; Fowler, J.S.; Thanos, P.K.; Logan, J.; Alexoff, D.; Ding, Y.S.; Wong, C.; Ma, Y.; et al. Low dopamine striatal D2 receptors are associated with prefrontal metabolism in obese subjects: Possible contributing factors. Neuroimage 2008, 42, 1537–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Giessen, E.; la Fleur, S.E.; Eggels, L.; de Bruin, K.; van den Brink, W.; Booij, J. High fat/carbohydrate ratio but not total energy intake induces lower striatal dopamine D2/3 receptor availability in diet-induced obesity. Int. J. Obes. 2013, 37, 754–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horvath, T.L.; Sarman, B.; Garcia-Caceres, C.; Enriori, P.J.; Sotonyi, P.; Shanabrough, M.; Borok, E.; Argente, J.; Chowen, J.A.; Perez-Tilve, D.; et al. Synaptic input organization of the melanocortin system predicts diet-induced hypothalamic reactive gliosis and obesity. Proc. Natl. Acad. Sci. USA 2010, 107, 14875–14880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bentsen, M.A.; Rausch, D.M.; Mirzadeh, Z.; Muta, K.; Scarlett, J.M.; Brown, J.M.; Herranz-Perez, V.; Baquero, A.F.; Thompson, J.; Alonge, K.M.; et al. Transcriptomic analysis links diverse hypothalamic cell types to fibroblast growth factor 1-induced sustained diabetes remission. Nat. Commun. 2020, 11, 4458. [Google Scholar] [CrossRef]
- Barbosa, A.C.; Kim, M.S.; Ertunc, M.; Adachi, M.; Nelson, E.D.; McAnally, J.; Richardson, J.A.; Kavalali, E.T.; Monteggia, L.M.; Bassel-Duby, R.; et al. MEF2C, a transcription factor that facilitates learning and memory by negative regulation of synapse numbers and function. Proc. Natl. Acad. Sci. USA 2008, 105, 9391–9396. [Google Scholar] [CrossRef] [Green Version]
- Kalsbeek, A.; Palm, I.F.; La Fleur, S.E.; Scheer, F.A.; Perreau-Lenz, S.; Ruiter, M.; Kreier, F.; Cailotto, C.; Buijs, R.M. SCN outputs and the hypothalamic balance of life. J. Biol. Rhythm. 2006, 21, 458–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albrecht, U. Orchestration of gene expression and physiology by the circadian clock. J. Physiol. Paris 2006, 100, 243–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maywood, E.S.; O’Neill, J.; Wong, G.K.; Reddy, A.B.; Hastings, M.H. Circadian timing in health and disease. Prog. Brain. Res. 2006, 153, 253–269. [Google Scholar] [PubMed]
- Manfredini, R.; Fabbian, F.; Manfredini, F.; Salmi, R.; Gallerani, M.; Bossone, E. Chronobiology in aortic diseases—“Is this really a random phenomenon?”. Prog. Cardiovasc. Dis. 2013, 56, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Hofman, M.A. The brain’s calendar: Neural mechanisms of seasonal timing. Biol. Rev. Camb. Philos Soc. 2004, 79, 61–77. [Google Scholar] [CrossRef]
- Meier, A.H.; Cincotta, A. Circadian rhythms regulate the expression of the thrifty genotype/phenotype. Diabetes Rev. 1996, 4, 464–487. Available online: www.veroscience.com/pubs.html (accessed on 21 May 2021).
- Meier, A.H.; Davis, K.B. Diurnal variations of the fattening response to prolactin in the white-throated sparrow, Zonotrichia albicollis. Gen. Comp. Endocrinol. 1967, 8, 110–114. [Google Scholar] [CrossRef]
- Meier, A.H. Temporal synergism of prolactin and adrenal steroids. Gen. Comp. Endocrinol. 1972, 3, 499–508. [Google Scholar] [CrossRef]
- Meier, A.H.; Trobec, T.N.; Joseph, M.M.; John, T.M. Temporal synergism of prolactin and adrenal steroids in the regulation of fat stores. Proc. Soc. Exp. Biol. Med. 1971, 137, 408–415. [Google Scholar] [CrossRef]
- Cincotta, A.H.; Wilson, J.M.; DeSouza, C.J.; Meier, A.H. Properly timed injections of cortisol and prolactin produce long-term reductions in obesity, hyperinsulinaemia and insulin resistance in the Syrian hamster (Mesocricetus auratus). J. Endocrinol. 1989, 120, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Roelfsema, F.; Pijl, H. Phase difference between serum prolactin and cortisol rhythms is related to body mass index in humans. J. Clin. Endocrinol. Metab. 2012, 97, E2293–E2296. [Google Scholar] [CrossRef] [Green Version]
- Meier, A.H.; Martin, D.D.; MacGregor, R., 3rd. Temporal synergism of corticosterone and prolactin controlling gonadal growth in sparrows. Science 1971, 173, 1240–1242. [Google Scholar] [CrossRef] [PubMed]
- Meier, A.H.; Russo, A.C. Circadian organization of the avian annual cycle. In Current Ornithology; Johnston, R.E., Ed.; Plenum: New York, NY, USA, 1984; Volume 2, pp. 303–343. [Google Scholar]
- Cincotta, A.H.; Schiller, B.C.; Landry, R.J.; Herbert, S.J.; Miers, W.R.; Meier, A.H. Circadian neuroendocrine role in age-related changes in body fat stores and insulin sensitivity of the male Sprague-Dawley rat. Chronobiol. Int. 1993, 10, 244–258. [Google Scholar] [CrossRef]
- Meier, A.H.; Fivizzani, A.J.; Spieler, R.E.; Horseman, N.D. Circadian hormone basis for seasonal conditions in the gulf killifish, Fundulus grandis. In Comparative Endocrinology; Gaillard, P.J., Boer, H.H., Eds.; Elsevier/North Holland Biomedical Press: Amsterdam, The Netherlands, 1978; pp. 141–144. [Google Scholar]
- Meier, A.H.; Ferrell, B.R.; Miller, L.J. Circadian components of the circannual mechanism in the white-throated sparrow. In Acta XVII Congressus Internationalis Ornithologia; Nohring, R., Ed.; Verlag Der Deutschen Ornithologen-Gesellschaft: Berlin, Germany, 1981; Volume 1, pp. 453–462. [Google Scholar]
- Moore, K.E.; Demarest, K.T.; Johnston, C.A. Influence of prolactin on dopaminergic neuronal systems in the hypothalamus. Fed. Proc. 1980, 39, 2912–2916. [Google Scholar] [PubMed]
- Telegdy, G.; Vermes, I. Effect of adrenocorical hormones on activity of the serotonergic system in limbic structures. Neuroendocrinology 1975, 18, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Meier, A.H. Circadian basis for neuroendocrine regulation. In Proceedings International Symposium on Rhythms in Fishes; Ali, M.A., Ed.; Plenum: New York, NY, USA, 1993; pp. 109–126. [Google Scholar]
- Wilson, J.M.; Meier, A.H. Resetting the annual cycle with timed daily injections of 5-hydroxytryptophan and L-dihydroxyphenylalanine in Syrian hamsters. Chronobiol. Int. 1989, 6, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.J.; Meier, A.H. Temporal synergism of neurotransmitter-affecting drugs influences seasonal conditions in sparrows. J. Interdiscipl. Cycle Res. 1982, 14, 75–84. [Google Scholar] [CrossRef]
- Emata, A.C.; Meier, A.H.; Spieler, R.E. Temporal variations in gonadal and body fat responses to daily injections of 5-hydroxytryptophan (5-HTP) and dihydroxyphenylalanine (DOPA) in the gulf killifish, Fundulus grandis. J. Exp. Zool. 1985, 233, 29–34. [Google Scholar] [CrossRef]
- Davis, J.F.; Tracy, A.L.; Schurdak, J.D.; Tschop, M.H.; Lipton, J.W.; Clegg, D.J.; Benoit, S.C. Exposure to elevated levels of dietary fat attenuates psychostimulant reward and mesolimbic dopamine turnover in the rat. Behav. Neurosci. 2008, 122, 1257–1263. [Google Scholar] [CrossRef] [Green Version]
- York, D.A.; Teng, L.; Park-York, M. Effects of dietary fat and enterostatin on dopamine and 5-hydroxytrytamine release from rat striatal slices. Brain Res. 2010, 1349, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Tellez, L.A.; Medina, S.; Han, W.; Ferreira, J.G.; Licona-Limon, P.; Ren, X.; Lam, T.T.; Schwartz, G.J.; de Araujo, I.E. A gut lipid messenger links excess dietary fat to dopamine deficiency. Science 2013, 341, 800–802. [Google Scholar] [CrossRef]
- Scislowski, P.W.; Tozzo, E.; Zhang, Y.; Phaneuf, S.; Prevelige, R.; Cincotta, A.H. Biochemical mechanisms responsible for the attenuation of diabetic and obese conditions in ob/ob mice treated with dopaminergic agonists. Int. J. Obes. Relat. Metab. Disord. 1999, 23, 425–431. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Jetton, T.L.; Lubkin, M.; Meier, A.H.; Cincotta, A.H. Bromocriptine/SKF38393 ameliorates islet dysfunction in the diabetic (db/db) mouse. Cell Mol. Life Sci. 1998, 54, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Cincotta, A.H.; Scislowski, P.; Phaneuf, S.; Prevelige, R.; Meier, A.H.; Joslin, J. Dopamine agonist treatment ameliorates the obese hyperglycemic condition in lethal yellow (A y/a) mice. Diabetes 1998, 47, A318. [Google Scholar]
- Framnes-DeBoer, S.N.; Bakke, E.; Yalamanchili, S.; Peterson, H.; Sandoval, D.A.; Seeley, R.J.; Arble, D.M. Bromocriptine improves glucose tolerance independent of circadian timing, prolactin, or the melanocortin-4 receptor. Am. J. Physiol. Endocrinol. Metab. 2020, 318, E62–E71. [Google Scholar] [CrossRef] [PubMed]
- Kok, P.; Roelfsema, F.; Frolich, M.; Meinders, A.E.; Pijl, H. Prolactin release is enhanced in proportion to excess visceral fat in obese women. J. Clin. Endocrinol. Metab. 2004, 89, 4445–4449. [Google Scholar] [CrossRef]
- Auriemma, R.S.; De Alcubierre, D.; Pirchio, R.; Pivonello, R.; Colao, A. The effects of hyperprolactinemia and its control on metabolic diseases. Expert Rev. Endocrinol. Metab. 2018, 13, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Bernabeu, I.; Casanueva, F.F. Metabolic syndrome associated with hyperprolactinemia: A new indication for dopamine agonist treatment? Endocrine 2013, 44, 273–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berinder, K.; Nystrom, T.; Hoybye, C.; Hall, K.; Hulting, A.L. Insulin sensitivity and lipid profile in prolactinoma patients before and after normalization of prolactin by dopamine agonist therapy. Pituitary 2011, 14, 199–207. [Google Scholar] [CrossRef]
- Foss, M.C.; Paula, F.J.; Paccola, G.M.; Piccinato, C.E. Peripheral glucose metabolism in human hyperprolactinaemia. Clin. Endocrinol. 1995, 43, 721–726. [Google Scholar] [CrossRef]
- Serri, O.; Beauregard, H.; Rasio, E.; Hardy, J. Decreased sensitivity to insulin in women with microprolactinomas. Fertil. Steril. 1986, 45, 572–574. [Google Scholar] [CrossRef]
- Dos Santos Silva, C.M.; Barbosa, F.R.; Lima, G.A.; Warszawski, L.; Fontes, R.; Domingues, R.C.; Gadelha, M.R. BMI and metabolic profile in patients with prolactinoma before and after treatment with dopamine agonists. Obesity 2011, 19, 800–805. [Google Scholar] [CrossRef]
- Park, S.; Kang, S.; Lee, H.W.; Ko, B.S. Central prolactin modulates insulin sensitivity and insulin secretion in diabetic rats. Neuroendocrinology 2012, 95, 332–343. [Google Scholar] [CrossRef]
- Cincotta, A.H.; Meier, A.H.; Cincotta, M., Jr. Bromocriptine improves glycaemic control and serum lipid profile in obese Type 2 diabetic subjects: A new approach in the treatment of diabetes. Expert Opin. Investig. Drugs 1999, 8, 1683–1707. [Google Scholar] [CrossRef] [PubMed]
- Cincotta, A.H.; Knisely, T.L.; Landry, R.J.; Miers, W.R.; Gutierrez, P.J.; Esperanza, P.; Meier, A.H. The immunoregulatory effects of prolactin in mice are time of day dependent. Endocrinology 1995, 136, 2163–2171. [Google Scholar] [CrossRef]
- Debruyne, J.P.; Noton, E.; Lambert, C.M.; Maywood, E.S.; Weaver, D.R.; Reppert, S.M. A clock shock: Mouse CLOCK is not required for circadian oscillator function. Neuron 2006, 50, 465–477. [Google Scholar] [CrossRef] [Green Version]
- Dardente, H. Synchronization and genetic redundancy in circadian clocks. Med. Sci. 2008, 24, 270–276. [Google Scholar]
- DeBruyne, J.P.; Weaver, D.R.; Reppert, S.M. CLOCK and NPAS2 have overlapping roles in the suprachiasmatic circadian clock. Nat. Neurosci. 2007, 10, 543–545. [Google Scholar] [CrossRef] [Green Version]
- Bahler, L.; Verberne, H.J.; Brakema, E.; Tepaske, R.; Booij, J.; Hoekstra, J.B.; Holleman, F. Bromocriptine and insulin sensitivity in lean and obese subjects. Endocr. Connect. 2016, 5, 44–52. [Google Scholar] [CrossRef]
- Caravaggio, F.; Borlido, C.; Hahn, M.; Feng, Z.; Fervaha, G.; Gerretsen, P.; Nakajima, S.; Plitman, E.; Chung, J.K.; Iwata, Y.; et al. Reduced insulin sensitivity is related to less endogenous dopamine at D2/3 receptors in the ventral striatum of healthy nonobese humans. Int. J. Neuropsychopharmacol. 2015, 18, pyv014. [Google Scholar] [CrossRef]
- Ter Horst, K.W.; Lammers, N.M.; Trinko, R.; Opland, D.M.; Figee, M.; Ackermans, M.T.; Booij, J.; van den Munckhof, P.; Schuurman, P.R.; Fliers, E.; et al. Striatal dopamine regulates systemic glucose metabolism in humans and mice. Sci. Transl. Med. 2018, 10, eaar3752. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.J.; Thomas, G.N.; Xu, Z.L.; Fang, J.Q.; Critchley, J.A.; Tomlinson, B. An affected pedigree member analysis of linkage between the dopamine D2 receptor gene TaqI polymorphism and obesity and hypertension. Int. J. Cardiol. 2005, 102, 111–116. [Google Scholar] [CrossRef] [Green Version]
- Volkow, N.D.; Tomasi, D.; Wang, G.J.; Telang, F.; Fowler, J.S.; Logan, J.; Benveniste, H.; Kim, R.; Thanos, P.K.; Ferre, S. Evidence that sleep deprivation downregulates dopamine D2R in ventral striatum in the human brain. J. Neurosci. 2012, 32, 6711–6717. [Google Scholar] [CrossRef] [Green Version]
- Alghasham, A.; Rasheed, N. Stress-mediated modulations in dopaminergic system and their subsequent impact on behavioral and oxidative alterations: An update. Pharm. Biol. 2014, 52, 368–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leproult, R.; Holmback, U.; Van Cauter, E. Circadian misalignment augments markers of insulin resistance and inflammation, independently of sleep loss. Diabetes 2014, 63, 1860–1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheer, F.A.; Hilton, M.F.; Mantzoros, C.S.; Shea, S.A. Adverse metabolic and cardiovascular consequences of circadian misalignment. Proc. Natl. Acad. Sci. USA 2009, 106, 4453–4458. [Google Scholar] [CrossRef] [Green Version]
- Innes, K.E.; Vincent, H.K.; Taylor, A.G. Chronic stress and insulin resistance-related indices of cardiovascular disease risk, part I: Neurophysiological responses and pathological sequelae. Altern. Ther. Health Med. 2007, 13, 46–52. [Google Scholar]
- Innes, K.E.; Vincent, H.K.; Taylor, A.G. Chronic stress and insulin resistance-related indices of cardiovascular disease risk, part 2: A potential role for mind-body therapies. Altern. Ther. Health Med. 2007, 13, 44–51. [Google Scholar] [PubMed]



 —increases;
—increases;  —decreases;
—decreases;  —stimulates;
—stimulates;  —inhibits;
—inhibits;  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  leads to;
leads to;  ,
,  collectively leads to these collective events. Enzyme levels and cardiometabolic parameters measured in the study are highlighted in yellow.
—increases; —decreases; —stimulates; —inhibits; , , , , , , , , , leads to; , collectively leads to these collective events. Enzyme levels and cardiometabolic parameters measured in the study are highlighted in yellow.
collectively leads to these collective events. Enzyme levels and cardiometabolic parameters measured in the study are highlighted in yellow.
—increases; —decreases; —stimulates; —inhibits; , , , , , , , , , leads to; , collectively leads to these collective events. Enzyme levels and cardiometabolic parameters measured in the study are highlighted in yellow.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ezrokhi, M.; Zhang, Y.; Luo, S.; Cincotta, A.H. Time-of-Day-Dependent Effects of Bromocriptine to Ameliorate Vascular Pathology and Metabolic Syndrome in SHR Rats Held on High Fat Diet. Int. J. Mol. Sci. 2021, 22, 6142. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22116142
Ezrokhi M, Zhang Y, Luo S, Cincotta AH. Time-of-Day-Dependent Effects of Bromocriptine to Ameliorate Vascular Pathology and Metabolic Syndrome in SHR Rats Held on High Fat Diet. International Journal of Molecular Sciences. 2021; 22(11):6142. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22116142
Chicago/Turabian StyleEzrokhi, Michael, Yahong Zhang, Shuqin Luo, and Anthony H. Cincotta. 2021. "Time-of-Day-Dependent Effects of Bromocriptine to Ameliorate Vascular Pathology and Metabolic Syndrome in SHR Rats Held on High Fat Diet" International Journal of Molecular Sciences 22, no. 11: 6142. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22116142