
Brain Protein Expression Profile Confirms the Protective Effect of the ACTH(4–7)PGP Peptide (Semax) in a Rat Model of Cerebral Ischemia–Reperfusion

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Morphology of Brain Tissues in the Experimental Groups
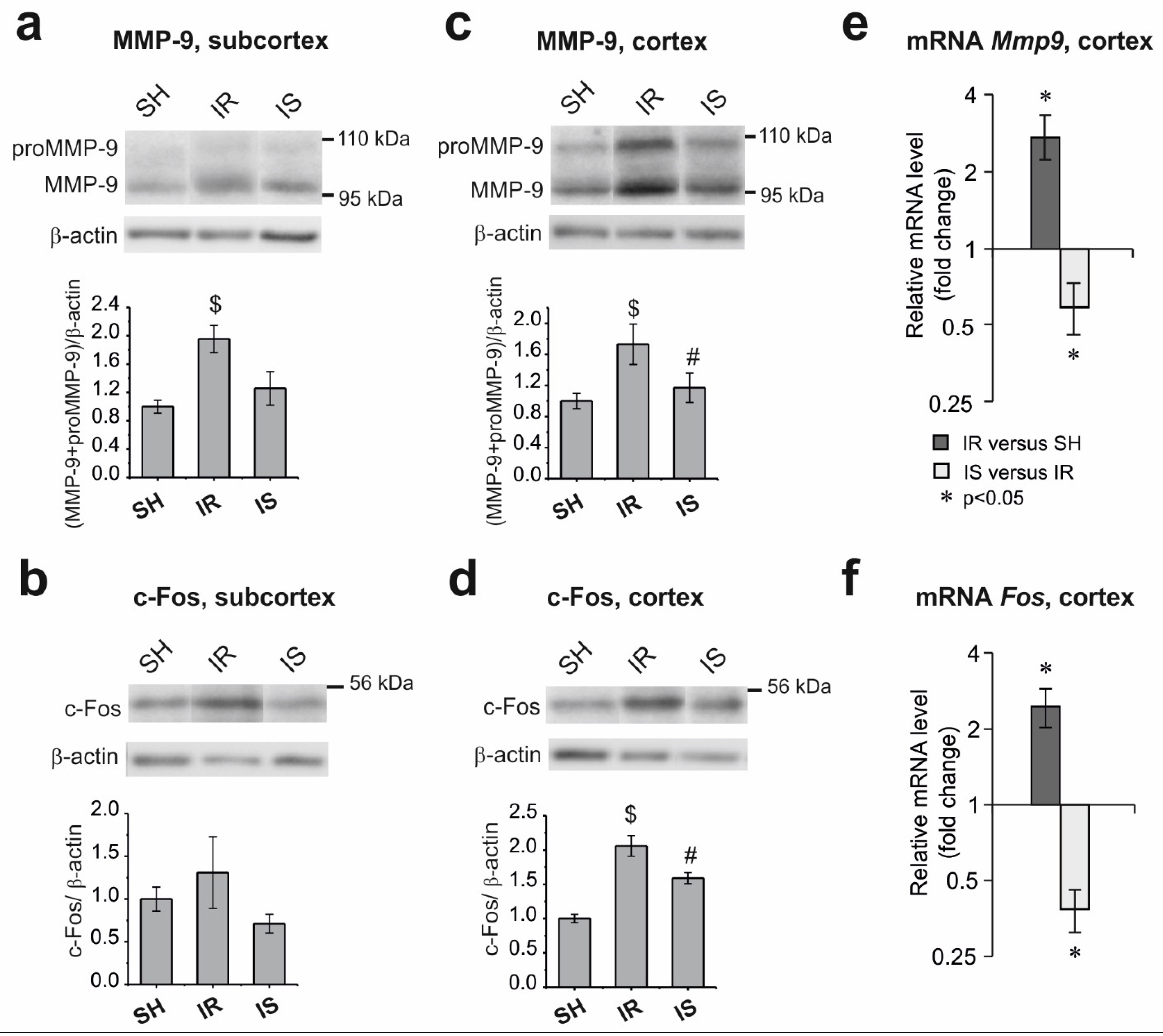
2.2. Changes in the Levels of pJNK, pCREB, MMP-9, and c-Fos in the Subcortical Structures and Cortex of the Rat Brain under the Conditions of Ischemia–Reperfusion
2.3. Changes in the mRNA Level of the Mmp9 and Fos Genes in the Frontoparietal Cortical Area Adjacent to the Ischemic Damage Zone
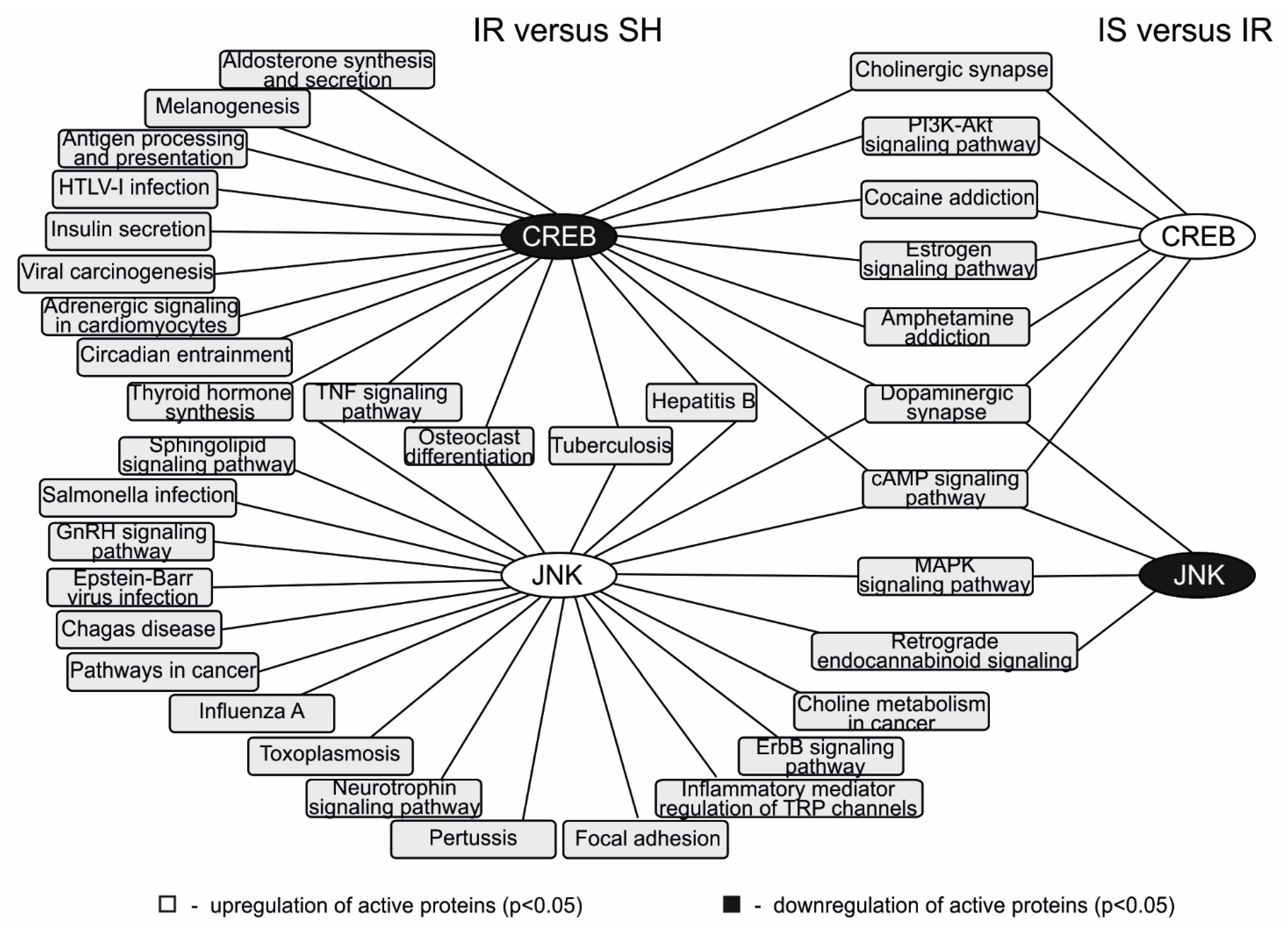
2.4. Analysis of the Involvement of the JNK, CREB, MMP-9, and c-Fos Proteins in the Signaling Pathways That Were Activated during Ischemia and the Semax Treatment
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. The tMCAO Model and Semax Administration
4.3. RNA Isolation
4.4. cDNA Synthesis
4.5. Real-Time RT–PCR
4.6. Data Analysis of Real-Time RT–PCR and Statistics
4.7. Protein Level Analysis by Immunodetection (Western Blotting)
4.8. Histological Examination of the Rat Brains
4.9. Analysis of pJNK and pCREB Involvement in Signaling Pathways
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Wied, D. Behavioral pharmacology of neuropeptides related to melanocortins and the neurohypophyseal hormones. Eur. J. Pharmacol. 1999, 375, 1–11. [Google Scholar] [CrossRef]
- Spaccapelo, L.; Bitto, A.; Galantucci, M.; Ottani, A.; Irrera, N.; Minutoli, L.; Altavilla, D.; Novellino, E.; Grieco, P.; Zaffe, D.; et al. Melanocortin MC4 receptor agonists counteract late inflammatory and apoptotic responses and improve neuronal functionality after cerebral ischemia. Eur. J. Pharmacol. 2011, 670, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Spaccapelo, L.; Galantucci, M.; Neri, L.; Contri, M.; Pizzala, R.; D’Amico, R.; Ottani, A.; Sandrini, M.; Zaffe, D.; Giuliani, D.; et al. Up-regulation of the canonical Wnt-3A and Sonic hedgehog signaling underlies melanocortin-induced neurogenesis after cerebral ischemia. Eur. J. Pharmacol. 2013, 707, 78–86. [Google Scholar] [CrossRef]
- Mykicki, N.; Herrmann, A.M.; Schwab, N.; Deenen, R.; Sparwasser, T.; Limmer, A.; Wachsmuth, L.; Klotz, L.; Köhrer, K.; Faber, C.; et al. Melanocortin-1 receptor activation is neuroprotective in mouse models of neuroinflammatory disease. Sci. Transl. Med. 2016, 8, 362. [Google Scholar] [CrossRef] [Green Version]
- Giuliani, D.; Ottani, A.; Neri, L.; Zaffe, D.; Grieco, P.; Jochem, J.; Cavallini, G.M.; Catania, A.; Guarini, S. Multiple beneficial effects of melanocortin MC4 receptor agonists in experimental neurodegenerative disorders: Therapeutic perspectives. Prog. Neurobiol. 2017, 148, 40–56. [Google Scholar] [CrossRef]
- Leone, S.; Noera, G.; Bertolini, A. Developments and new vistas in the field of melanocortins. Biomol. Concepts 2015, 6, 361–382. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, D.; Minutoli, L.; Ottani, A.; Spaccapelo, L.; Bitto, A.; Galantucci, M.; Altavilla, D.; Squadrito, F.; Guarini, S. Melanocortins as potential therapeutic agents in severe hypoxic conditions. Front. Neuroendocrinol. 2012, 33, 179–193. [Google Scholar] [CrossRef]
- Gusev, E.I.; Martynov, M.Y.; Kostenko, E.V.; Petrova, L.V.; Bobyreva, S.N. The efficacy of semax in the tretament of patients at different stages of ischemic stroke. Zh. Nevrol. Psikhiatr. Im. S S Korsakova 2018, 118, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Asmarin, I.P.; Nezavibat’ko, V.N.; Miasoedov, N.F.; Kamenskiĭ, A.A.; Grivennikov, I.A.; Ponomareva-Stepnaia, M.A.; Andreeva, L.A.; Kaplan, A.I.; Koshelev, V.B.; Riasina, T.V. A nootropic adrenocorticotropin analog 4-10-semax (l5 years experience in its design and study). Zh. Vyssh. Nerv. Deiat. Im. I P Pavlova 1997, 47, 420–430. [Google Scholar]
- Eremin, K.O.; Kudrin, V.S.; Saransaari, P.; Oja, S.S.; Grivennikov, I.A.; Myasoedov, N.F.; Rayevsky, K.S. Semax, an ACTH(4-10) analogue with nootropic properties, activates dopaminergic and serotoninergic brain systems in rodents. Neurochem. Res. 2005, 30, 1493–1500. [Google Scholar] [CrossRef]
- Grivennikov, J.A.; Dolotov, O.V.; Gol’Dina, Y.I. Peptide factors in proliferation, differentiation, and viability of the cells of mammalian nervous system. Mol. Biol. 1999, 33, 120–126. [Google Scholar]
- Storozhevykh, T.P.; Tukhbatova, G.R.; Senilova, Y.E.; Pinelis, V.G.; Andreeva, L.A.; Myasoyedov, N.F. Effects of semax and its Pro-Gly-Pro fragment on calcium homeostasis of neurons and their survival under conditions of glutamate toxicity. Bull. Exp. Biol. Med. 2007, 143, 601–604. [Google Scholar] [CrossRef]
- Dmitrieva, V.G.; Povarova, O.V.; Skvortsova, V.I.; Limborska, S.A.; Myasoedov, N.F.; Dergunova, L.V. Semax and Pro-Gly-Pro activate the transcription of neurotrophins and their receptor genes after cerebral ischemia. Cell. Mol. Neurobiol. 2010, 30, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Stavchansky, V.V.; Yuzhakov, V.V.; Botsina, A.Y.; Skvortsova, V.I.; Bondurko, L.N.; Tsyganova, M.G.; Limborska, S.A.; Myasoedov, N.F.; Dergunova, L.V. The effect of Semax and its C-end peptide PGP on the morphology and proliferative activity of rat brain cells during experimental ischemia: A pilot study. J. Mol. Neurosci. 2011, 45, 177–185. [Google Scholar] [CrossRef]
- Medvedeva, E.V.; Dmitrieva, V.G.; Povarova, O.V.; Limborska, S.A.; Skvortsova, V.I.; Myasoedov, N.F.; Dergunova, L.V. The peptide semax affects the expression of genes related to the immune and vascular systems in rat brain focal ischemia: Genome-wide transcriptional analysis. BMC Genom. 2014, 15, 228. [Google Scholar] [CrossRef] [Green Version]
- Medvedeva, E.V.; Dmitrieva, V.G.; Limborska, S.A.; Myasoedov, N.F.; Dergunova, L.V. Semax, an analog of ACTH(4-7), regulates expression of immune response genes during ischemic brain injury in rats. Mol. Genet. Genom. 2017, 292, 635–653. [Google Scholar] [CrossRef]
- Filippenkov, I.B.; Stavchansky, V.V.; Denisova, A.E.; Yuzhakov, V.V.; Sevan’kaeva, L.E.; Sudarkina, O.Y.; Dmitrieva, V.G.; Gubsky, L.V.; Myasoedov, N.F.; Limborska, S.A.; et al. Novel insights into the protective properties of acth(4-7)pgp (semax) peptide at the transcriptome level following cerebral ischaemia– reperfusion in rats. Genes 2020, 11, 681. [Google Scholar] [CrossRef]
- Zhong, C.; Yang, J.; Xu, T.; Xu, T.; Peng, Y.; Wang, A.; Wang, J.; Peng, H.; Li, Q.; Ju, Z.; et al. Serum matrix metalloproteinase-9 levels and prognosis of acute ischemic stroke. Neurology 2017, 89, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Borsello, T.; Bonny, C. Use of cell-permeable peptides to prevent neuronal degeneration. Trends Mol. Med. 2004, 10, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Chung, L. A Brief Introduction to the Transduction of Neural Activity into Fos Signal. Dev. Reprod. 2015, 19, 61–67. [Google Scholar] [CrossRef]
- Wu, X.; Fu, S.; Liu, Y.; Luo, H.; Li, F.; Wang, Y.; Gao, M.; Cheng, Y.; Xie, Z. NDP-MSH binding melanocortin-1 receptor ameliorates neuroinflammation and BBB disruption through CREB/Nr4a1/NF-κB pathway after intracerebral hemorrhage in mice. J. Neuroinflammation 2019, 16. [Google Scholar] [CrossRef]
- Dergunova, L.V.; Filippenkov, I.B.; Stavchansky, V.V.; Denisova, A.E.; Yuzhakov, V.V.; Mozerov, S.A.; Gubsky, L.V.; Limborska, S.A. Genome-wide transcriptome analysis using RNA-Seq reveals a large number of differentially expressed genes in a transient MCAO rat model. BMC Genom. 2018, 19, 655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software Environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Myasoedov, N.F.; Skvortsova, V.I.; Nasonov, E.L.; Zhuravleva, E.Y.; Grivennikov, I.A.; Arsenyeva, E.L.; Sukhanov, I.I. Investigation of mechanisms of neuroprotective effect of semax in acute period of ischemic stroke. Zh. Nevrol. Psikhiatr. Im. S S Korsakova 1999, 99, 15–19. [Google Scholar]
- Gusev, E.; Skvortsova, V.; Chukanova, E. Semax in prevention of disease progress and development of exacerbations in patients with cerebrovascular insufficiency. Zh. Nevrol. Psikhiatr. Im. S S Korsakova 2005, 105, 35–40. [Google Scholar] [PubMed]
- Umnov, R.S.; Lin’kova, N.S.; Khavinson, V.K. [Neuroprotective effects of peptides bioregulators in people of various age]. Adv. Gerontol. 2013, 26, 671–678. [Google Scholar]
- Repici, M.; Centeno, C.; Tomasi, S.; Forloni, G.; Bonny, C.; Vercelli, A.; Borsello, T. Time-course of c-Jun N-terminal kinase activation after cerebral ischemia and effect of D-JNKI1 on c-Jun and caspase-3 activation. Neuroscience 2007, 150, 40–49. [Google Scholar] [CrossRef]
- Zheng, J.; Dai, Q.; Han, K.; Hong, W.; Jia, D.; Mo, Y.; Lv, Y.; Tang, H.; Fu, H.; Geng, W. JNK-IN-8, a c-Jun N-terminal kinase inhibitor, improves functional recovery through suppressing neuroinflammation in ischemic stroke. J. Cell. Physiol. 2020, 235, 2792–2799. [Google Scholar] [CrossRef]
- Manning, A.M.; Davis, R.J. Targeting JNK for therapeutic benefit: From junk to gold? Nat. Rev. Drug Discov. 2003, 2, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Fernandez, M.; Bellolio, M.F.; Stead, L.G. Matrix metalloproteinase-9 as a marker for acute ischemic stroke: A systematic review. J. Stroke Cerebrovasc. Dis. 2011, 20, 47–54. [Google Scholar] [CrossRef]
- Liang, K.C.; Lee, C.W.; Lin, W.N.; Lin, C.C.; Wu, C.B.; Luo, S.F.; Yang, C.M. Interleukin-1β induces MMP-9 expression via p42/p44 MAPK, p38 MAPK, JNK, and nuclear factor-κB signaling pathways in human tracheal smooth muscle cells. J. Cell. Physiol. 2007, 211, 759–770. [Google Scholar] [CrossRef]
- Lin, C.C.; Tseng, H.W.; Hsieh, H.L.; Lee, C.W.; Wu, C.Y.; Cheng, C.Y.; Yang, C.M. Tumor necrosis factor-α induces MMP-9 expression via p42/p44 MAPK, JNK, and nuclear factor-κB in A549 cells. Toxicol. Appl. Pharmacol. 2008, 229, 386–398. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.H.; Park, S.W.; Kapadia, R.; Vemuganti, R. Role of transcription factors in mediating post-ischemic cerebral inflammation and brain damage. Neurochem. Int. 2007, 50, 1014–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rylski, M.; Kaczmarek, L. AP-1 targets in the brain. Front. Biosci. 2004, 9, 8–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, A.J.; Ling, G.; Berti, R.; Moffett, J.R.; Yao, C.; Lu, X.M.; Dave, J.R.; Tortella, F.C. Treatment with the snail peptide CGX-1007 reduces DNA damage and alters gene expression of c-fos and bcl-2 following focal ischemic brain injury in rats. Exp. Brain Res. 2003, 153, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Borsello, T.; Clarkel, P.G.H.; Hirt, L.; Vercelli, A.; Repici, M.; Schorderet, D.F.; Bogousslavsky, J.; Bonny, C. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat. Med. 2003, 9, 1180–1186. [Google Scholar] [CrossRef]
- Sakamoto, K.; Karelina, K.; Obrietan, K. CREB: A multifaceted regulator of neuronal plasticity and protection. J. Neurochem. 2011, 116, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Volakakis, N.; Kadkhodaei, B.; Joodmardi, E.; Wallis, K.; Panman, L.; Silvaggi, J.; Spiegelman, B.M.; Perlmann, T. NR4A orphan nuclear receptors as mediators of CREB-dependent neuroprotection. Proc. Natl. Acad. Sci. USA 2010, 107, 12317–12322. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.; Nagata, E.; Suzuki, S.; Dembo, T.; Nogawa, S.; Fukuuchi, Y. Immunohistochemical analysis of cyclic AMP response element binding protein phosphorylation in focal cerebral ischemia in rats. Brain Res. 1999, 818, 520–526. [Google Scholar] [CrossRef]
- Guan, X.; Wang, Y.; Kai, G.; Zhao, S.; Huang, T.; Li, Y.; Xu, Y.; Zhang, L.; Pang, T. Cerebrolysin ameliorates focal cerebral ischemia injury through neuroinflammatory inhibition via CREB/PGC-1α pathway. Front. Pharmacol. 2019, 10. [Google Scholar] [CrossRef]
- Lin, Y.; Chen, F.; Zhang, J.; Wang, T.; Wei, X.; Wu, J.; Feng, Y.; Dai, Z.; Wu, Q. Neuroprotective effect of resveratrol on ischemia/reperfusion injury in rats through TRPC6/CREB pathways. J. Mol. Neurosci. 2013, 50, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, A.R.; Almeida, H.; Gouveia, A.M. Intracellular signaling mechanisms of the melanocortin receptors: Current state of the art. Cell. Mol. Life Sci. 2015, 72, 1331–1345. [Google Scholar] [CrossRef]
- Ayata, C. Spreading depression and neurovascular coupling. Stroke 2013, 44. [Google Scholar] [CrossRef] [Green Version]
- Ayata, C.; Lauritzen, M. Spreading depression, spreading depolarizations, and the cerebral vasculature. Physiol. Rev. 2015, 95, 953–993. [Google Scholar] [CrossRef] [Green Version]
- Dreier, J.P.; Fabricius, M.; Ayata, C.; Sakowitz, O.W.; William Shuttleworth, C.; Dohmen, C.; Graf, R.; Vajkoczy, P.; Helbok, R.; Suzuki, M.; et al. Recording, analysis, and interpretation of spreading depolarizations in neurointensive care: Review and recommendations of the COSBID research group. J. Cereb. Blood Flow Metab. 2017, 37, 1595–1625. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Strong, A.J.; Dohmen, C.; Sakowitz, O.W.; Vollmar, S.; Sué, M.; Kracht, L.; Hashemi, P.; Bhatia, R.; Yoshimine, T.; et al. Spreading depolarizations cycle around and enlarge focal ischaemic brain lesions. Brain 2010, 133, 1994–2006. [Google Scholar] [CrossRef] [Green Version]
- Mendonça, B.P.; Cardoso, J.D.S.; Michels, M.; Vieira, A.C.; Wendhausen, D.; Manfredini, A.; Singer, M.; Dal-Pizzol, F.; Dyson, A. Neuroprotective effects of ammonium tetrathiomolybdate, a slow-release sulfide donor, in a rodent model of regional stroke. Intensive Care Med. Exp. 2020, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, H.M.; Zang, M.; Zhang, Q.; Shi, R.B.; Shi, X.J.; Mamtilahun, M.; Liu, C.; Luo, L.L.; Tian, X.; Zhang, Z.; et al. Farnesoid X receptor knockout protects brain against ischemic injury through reducing neuronal apoptosis in mice. J. Neuroinflammation 2020, 17. [Google Scholar] [CrossRef]
- Tanaka, Y.; Nakagomi, N.; Doe, N.; Nakano-Doi, A.; Sawano, T.; Takagi, T.; Matsuyama, T.; Yoshimura, S.; Nakagomi, T. Early Reperfusion Following Ischemic Stroke Provides Beneficial Effects, Even After Lethal Ischemia with Mature Neural Cell Death. Cells 2020, 9, 1374. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, P.; Ali, M.; Tabassum, H.; Parvez, S. Post-ischemic administration of dopamine D2 receptor agonist reduces cell death by activating mitochondrial pathway following ischemic stroke. Life Sci. 2020, 261. [Google Scholar] [CrossRef]
- Chomczynski, P.; Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfaffl, M.W.; Horgan, G.W.; Dempfle, L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002, 30, e36. [Google Scholar] [CrossRef]
- Pfaffl, M.W.; Tichopad, A.; Prgomet, C.; Neuvians, T.P. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper--Excel-based tool using pair-wise correlations. Biotechnol. Lett. 2004, 26, 509–515. [Google Scholar] [CrossRef]
- Gallagher, S.R. One-dimensional SDS gel electrophoresis of proteins. Curr. Protoc. Mol. Biol. 2012, 10. [Google Scholar] [CrossRef]
- Garcia, J.H.; Liu, K.F.; Ho, K.L. Neuronal necrosis after middle cerebral artery occlusion in wistar rats progresses at different time intervals in the caudoputamen and the cortex. Stroke 1995, 26, 636–642. [Google Scholar] [CrossRef]
- Li, Y.; Powers, C.; Jiang, N.; Chopp, M. Intact, injured, necrotic and apoptotic cells after focal cerebral ischemia in the rat. J. Neurol. Sci. 1998, 156, 119–132. [Google Scholar] [CrossRef]
- Lipton, P. Ischemic cell death in brain neurons. Physiol. Rev. 1999, 79, 1431–1568. [Google Scholar] [CrossRef] [PubMed]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates, 6th ed.; Academic Press: San-Diego, CA, USA, 2007. [Google Scholar]
- Chen, S.; Zhao, L.; Sherchan, P.; Ding, Y.; Yu, J.; Nowrangi, D.; Tang, J.; Xia, Y.; Zhang, J.H. Activation of melanocortin receptor 4 with RO27-3225 attenuates neuroinflammation through AMPK/JNK/p38 MAPK pathway after intracerebral hemorrhage in mice. J. Neuroinflammation 2018, 15. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/sra/PRJNA491404 (accessed on 25 May 2021). [CrossRef] [Green Version]
- National Center for Biotechnology Information. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/Traces/study/?acc=SRP148632 (accessed on 25 May 2021).




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sudarkina, O.Y.; Filippenkov, I.B.; Stavchansky, V.V.; Denisova, A.E.; Yuzhakov, V.V.; Sevan’kaeva, L.E.; Valieva, L.V.; Remizova, J.A.; Dmitrieva, V.G.; Gubsky, L.V.; et al. Brain Protein Expression Profile Confirms the Protective Effect of the ACTH(4–7)PGP Peptide (Semax) in a Rat Model of Cerebral Ischemia–Reperfusion. Int. J. Mol. Sci. 2021, 22, 6179. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126179
Sudarkina OY, Filippenkov IB, Stavchansky VV, Denisova AE, Yuzhakov VV, Sevan’kaeva LE, Valieva LV, Remizova JA, Dmitrieva VG, Gubsky LV, et al. Brain Protein Expression Profile Confirms the Protective Effect of the ACTH(4–7)PGP Peptide (Semax) in a Rat Model of Cerebral Ischemia–Reperfusion. International Journal of Molecular Sciences. 2021; 22(12):6179. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126179
Chicago/Turabian StyleSudarkina, Olga Yu., Ivan B. Filippenkov, Vasily V. Stavchansky, Alina E. Denisova, Vadim V. Yuzhakov, Larisa E. Sevan’kaeva, Liya V. Valieva, Julia A. Remizova, Veronika G. Dmitrieva, Leonid V. Gubsky, and et al. 2021. "Brain Protein Expression Profile Confirms the Protective Effect of the ACTH(4–7)PGP Peptide (Semax) in a Rat Model of Cerebral Ischemia–Reperfusion" International Journal of Molecular Sciences 22, no. 12: 6179. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126179