
A Panel of rSNPs Demonstrating Allelic Asymmetry in Both ChIP-seq and RNA-seq Data and the Search for Their Phenotypic Outcomes through Analysis of DEGs

Abstract
:1. Introduction
2. Results
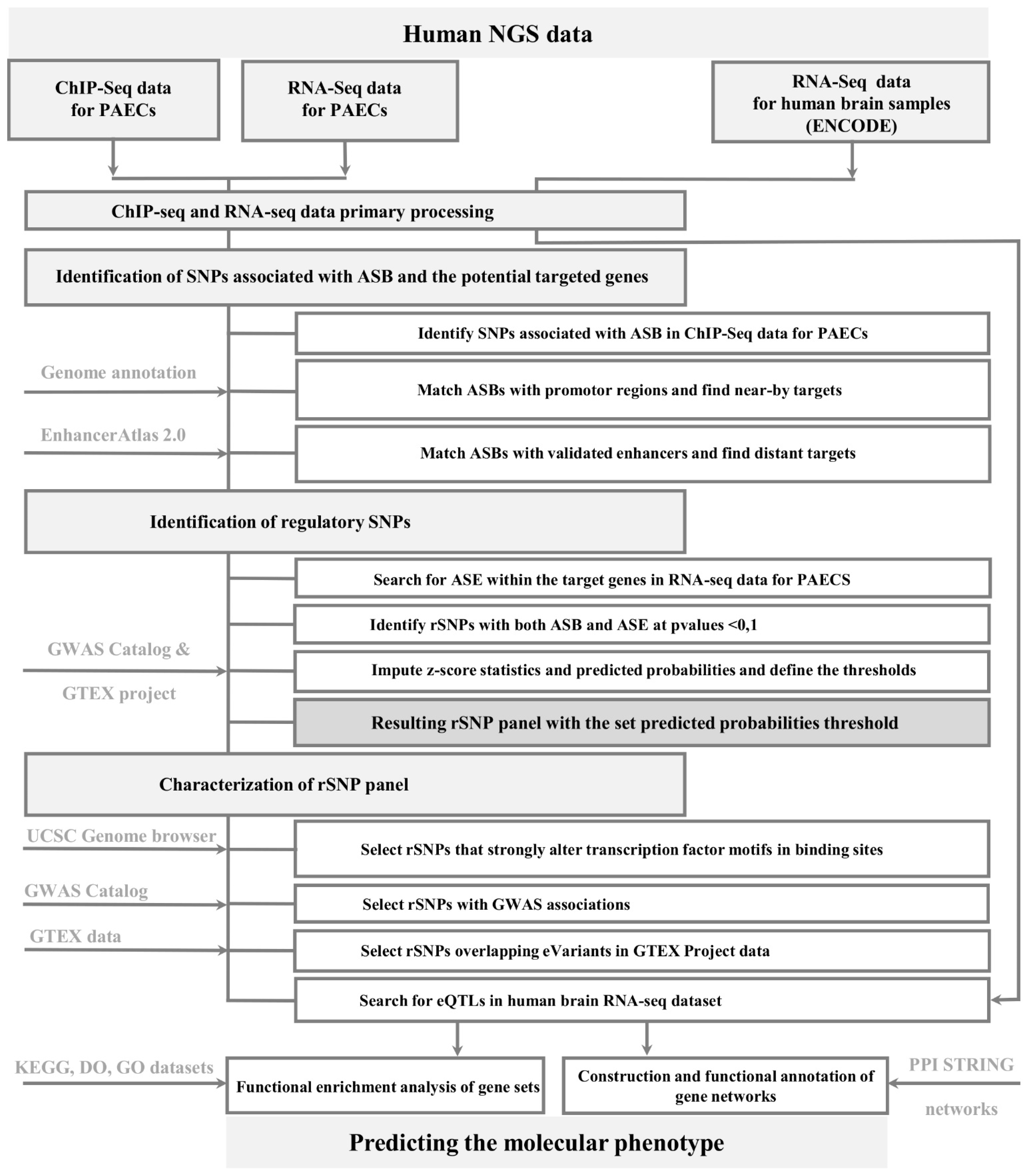
2.1. Workflow for rSNP Identification
2.1.1. General Description
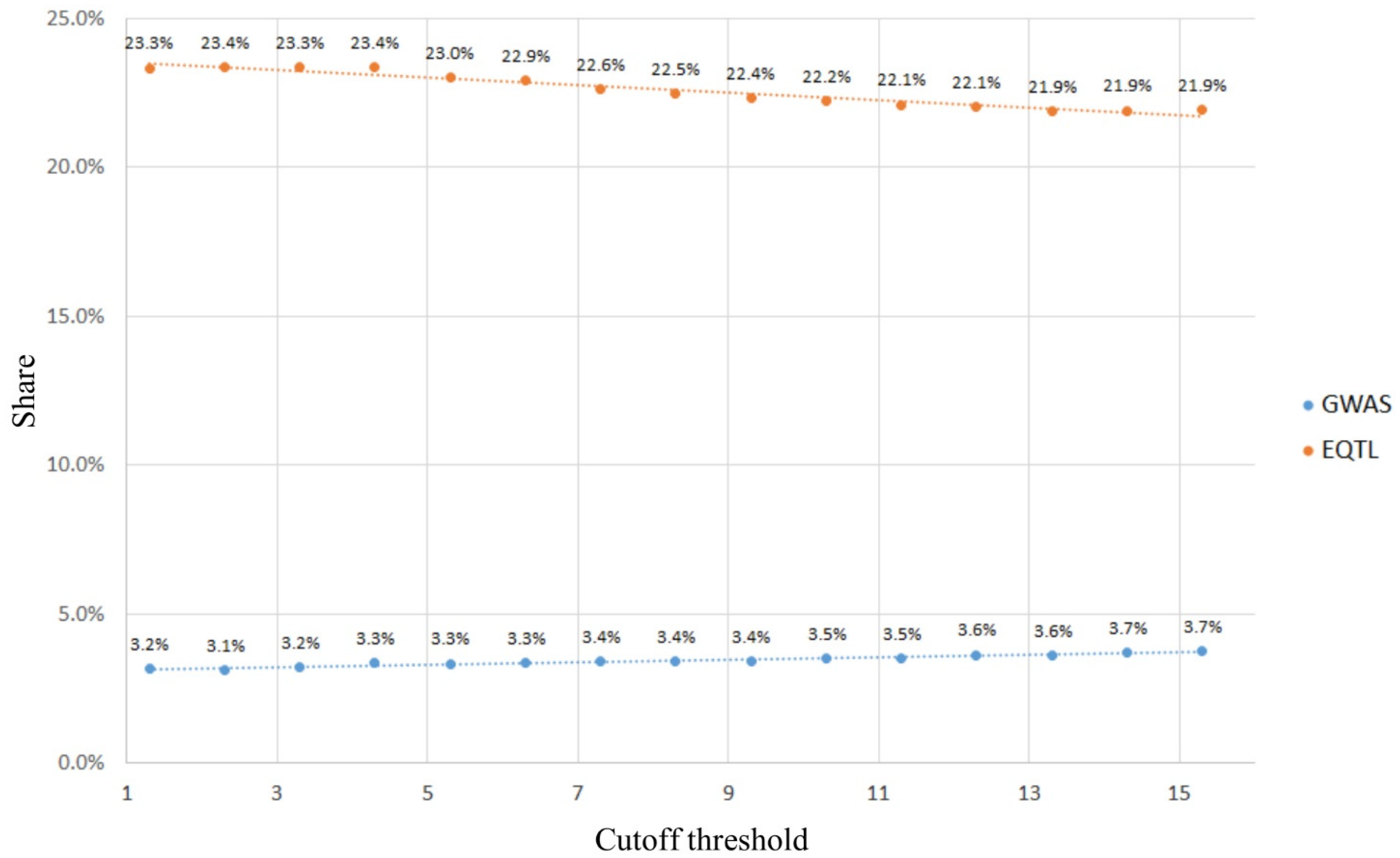
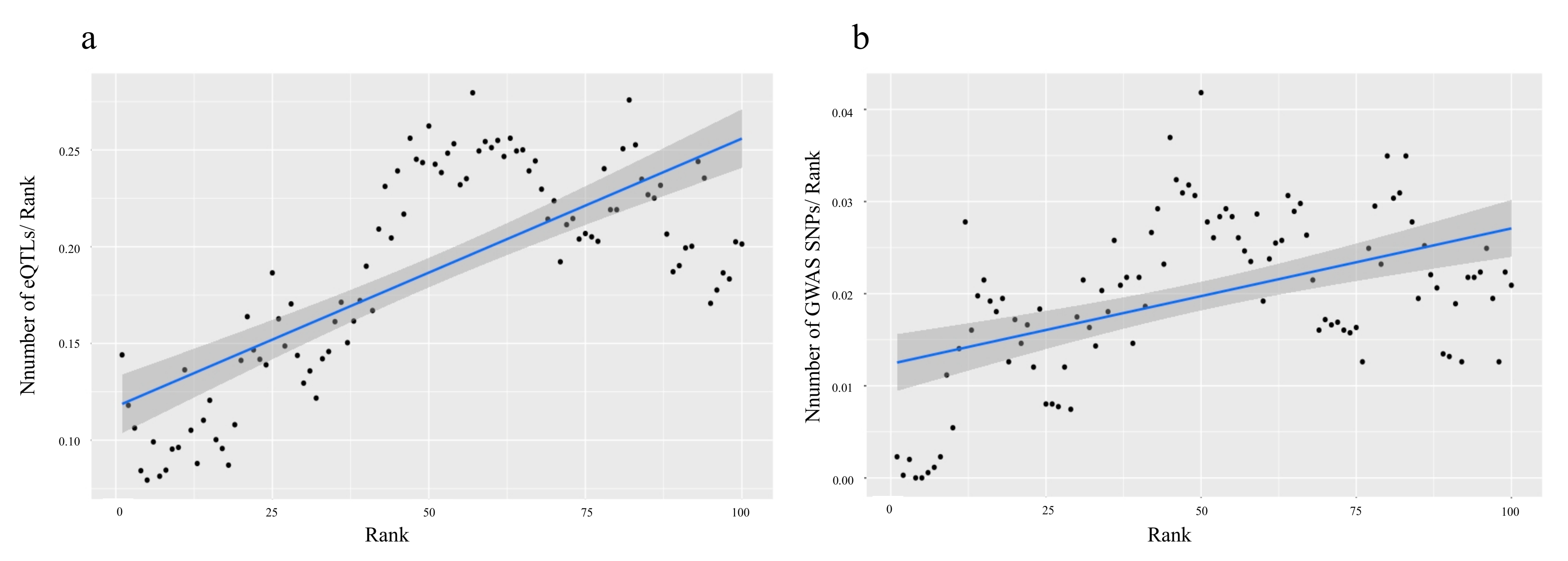
2.1.2. Using GWAS and eQTLs to Define Cutoff Thresholds
2.1.3. Setting Predicted Probabilities from Logistic Regression
2.2. Characterization of the Resulting rSNP Panel
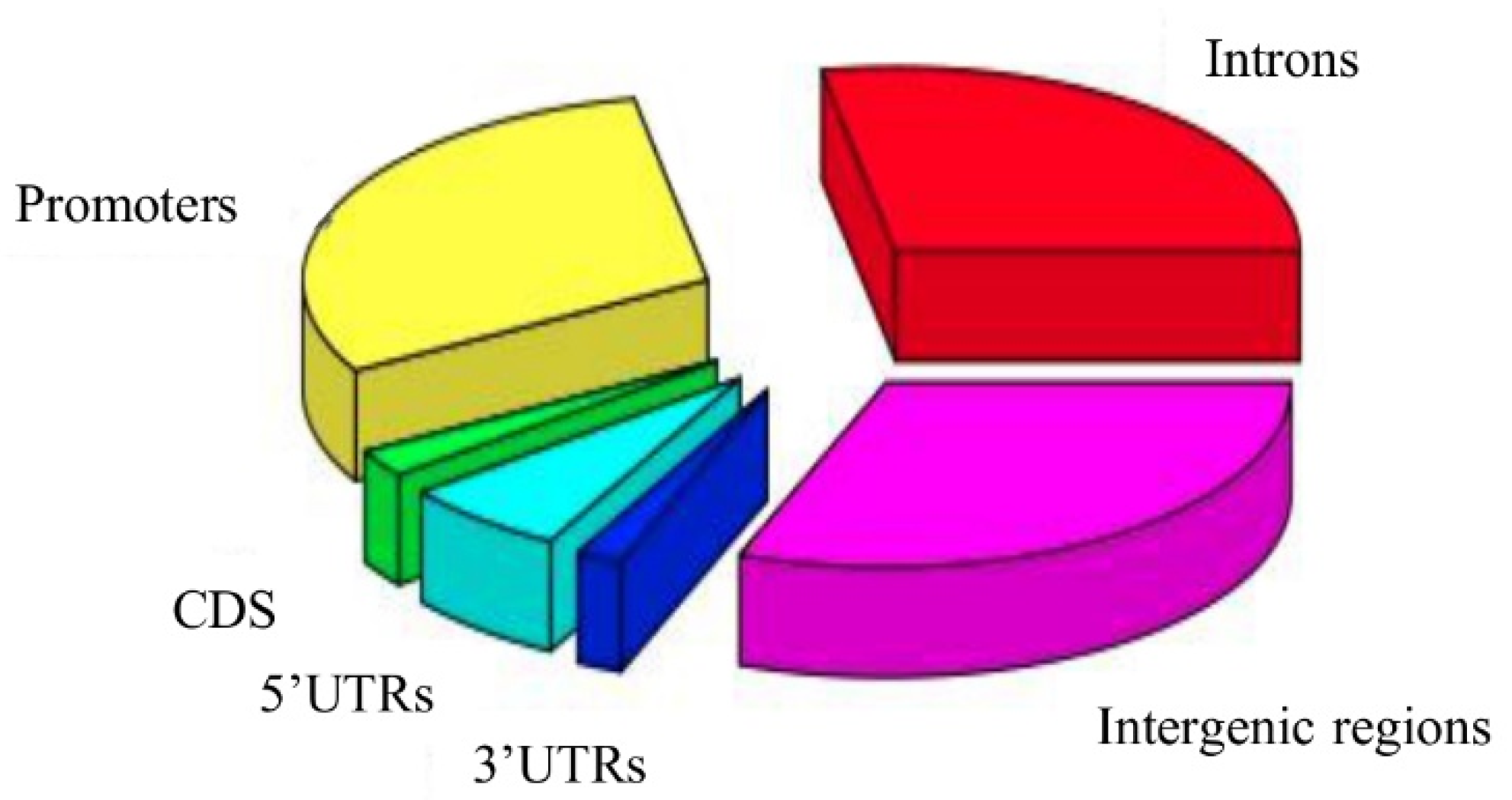
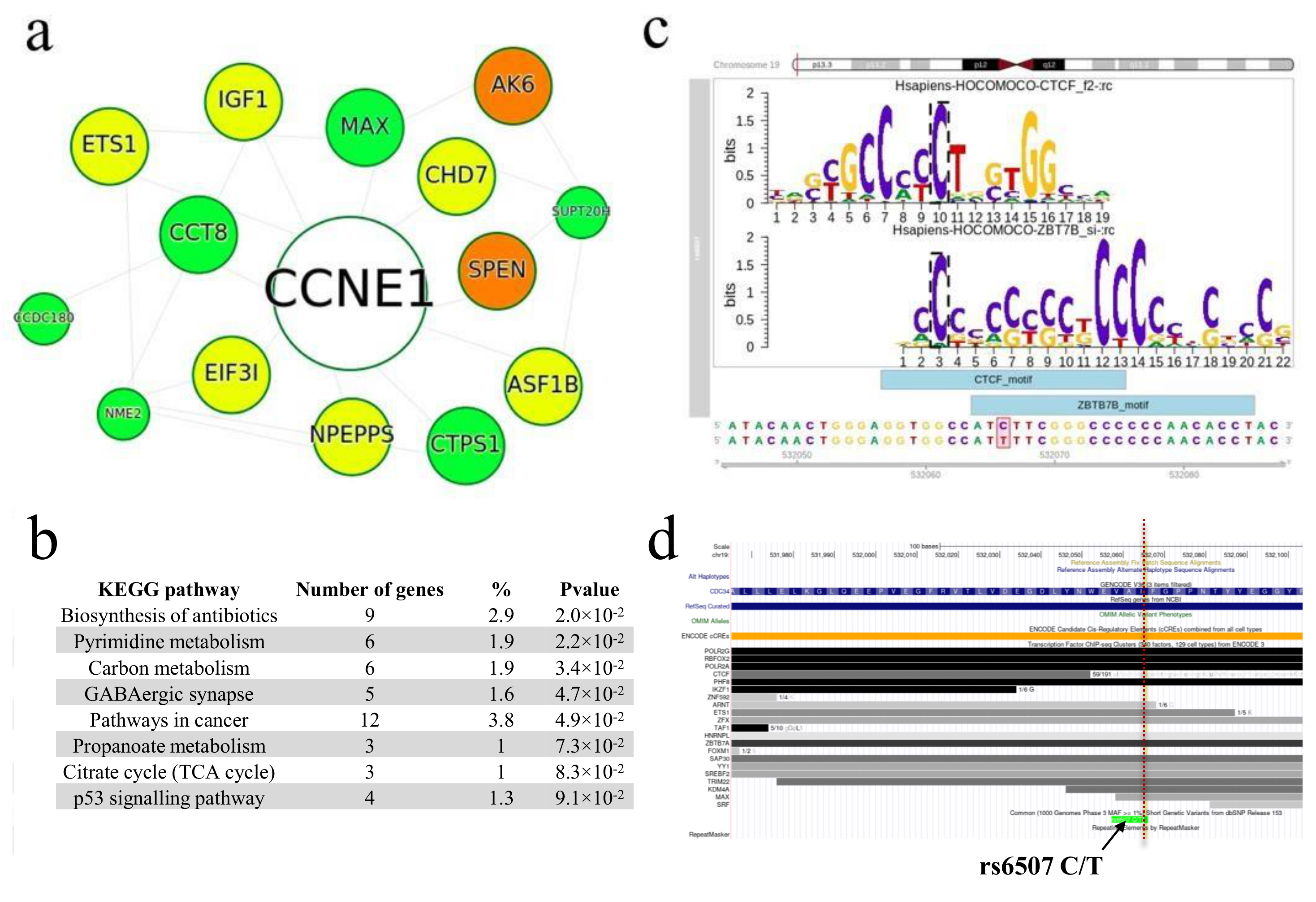
2.2.1. Search for rSNPs within Known TF Binding Motifs
2.2.2. Overlapping with GWAS Variants
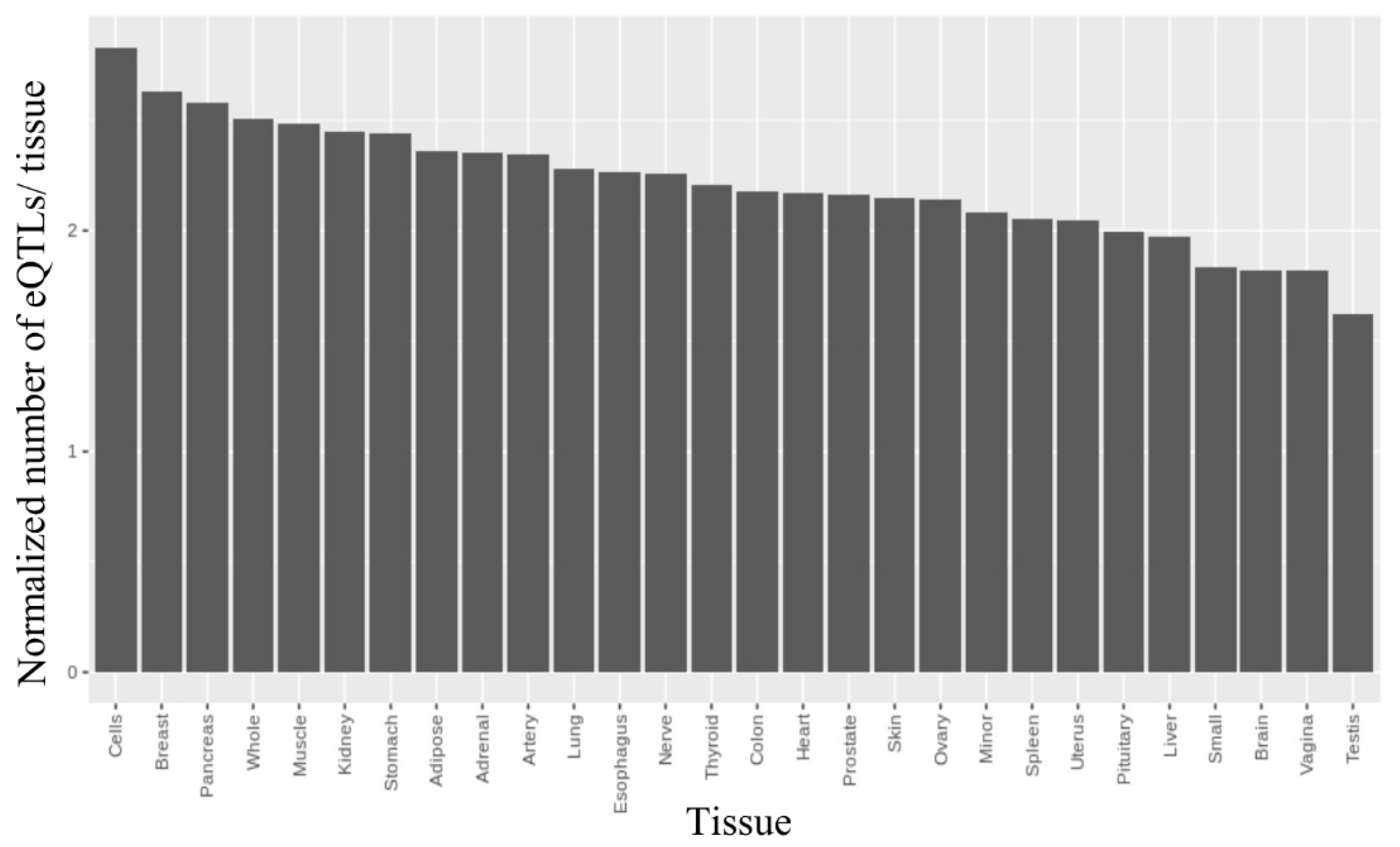
2.2.3. Finding rSNPs in GTExeQTLCollection
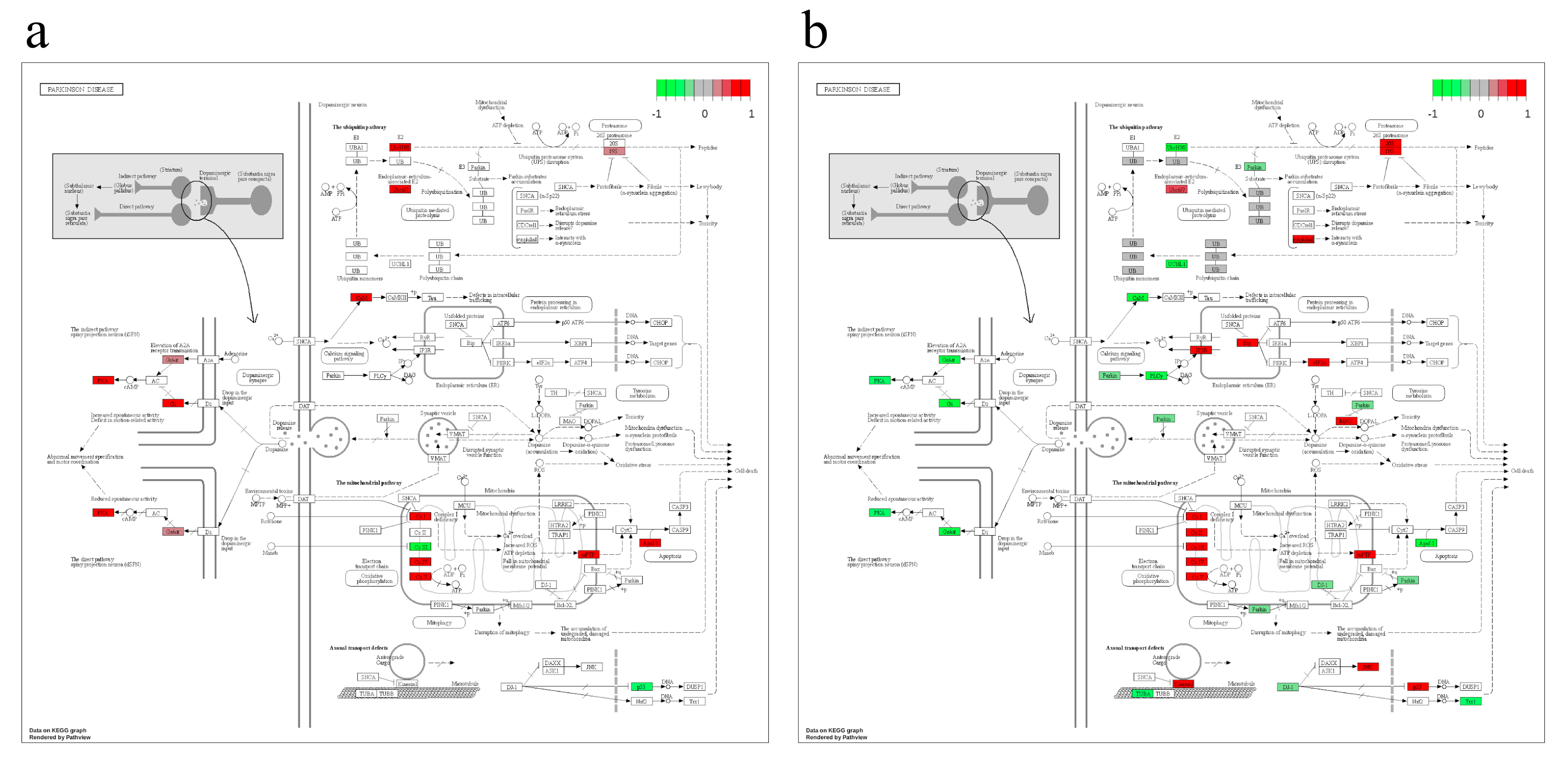
2.3. Assessing eQTLs in Human Brain RNA-seq Dataset
3. Discussion
4. Materials and Methods
4.1. Human NGS Data
4.2. Open Access Resources
4.3. NGS Data Preprocessing
4.3.1. Quality Filtering
4.3.2. Genomic Alignment and SNP Calling
4.4. Assessing Allele-Specific Binding and Expression Events
4.5. Z-Test
4.6. Evaluation of Linked SNP Pairs by HAMMING Distance
4.7. Transcription Factor Motif Disruption Analysis
4.8. Differential Expression Analysis
4.9. Construction of Protein–Protein Interaction Networks and Functional Annotation
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- The International SNP Map Working Group. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nat. Cell Biol. 2001, 409, 928–933. [Google Scholar] [CrossRef] [Green Version]
- Claussnitzer, M.; Cho, J.H.; Collins, R.; Cox, N.J.; Dermitzakis, E.T.; Hurles, M.E.; Kathiresan, S.; Kenny, E.E.; Lindgren, C.M.; MacArthur, D.G.; et al. A brief history of human disease genetics. Nat. Cell Biol. 2020, 577, 179–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar] [CrossRef] [Green Version]
- Edwards, S.L.; Beesley, J.; French, J.D.; Dunning, A.M. Beyond GWASs: Illuminating the dark road from association to function. Am. J. Hum. Genet. 2013, 93, 779–797. [Google Scholar] [CrossRef] [Green Version]
- Lappalainen, T. Functional genomics bridges the gap between quantitative genetics and molecular biology. Genome Res. 2015, 25, 1427–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Wu, D.; Jiang, D.; Zhang, X.; Wu, T.; Cui, J.; Qian, M.; Zhao, J.; Oesterreich, S.; Sun, W.; et al. A sequential methodology for the rapid identification and characterization of breast cancer-associated functional SNPs. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef]
- Farh, K.K.-H.; Marson, A.; Zhu, J.; Kleinewietfeld, M.; Housley, W.J.; Beik, S.; Shoresh, N.; Whitton, H.; Ryan, R.J.H.; Shishkin, A.A.; et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nat. Cell Biol. 2015, 518, 337–343. [Google Scholar] [CrossRef]
- Welter, D.; MacArthur, J.; Morales, J.; Burdett, A.; Hall, P.; Junkins, H.; Klemm, A.; Flicek, P.; Manolio, T.; Hindorff, L.; et al. The NHGRI GWAS catalog, A curated resource of SNP-trait associations. Nucleic Acids Res. 2014, 42, D1001–D1006. [Google Scholar] [CrossRef]
- Fang, J.; Jia, J.; Makowski, M.; Xu, M.; Wang, Z.; Zhang, T.; Hoskins, J.W.; Choi, J.; Han, Y.; Zhang, M.; et al. Functional characterization of a multi-cancer risk locus on chr5p15.33 reveals regulation of TERT by ZNF148. Nat. Commun. 2017, 8, 15034. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Xia, J.; Sipeky, C.; Dong, X.-M.; Zhang, Q.; Yang, Y.; Zhang, P.; Cruz, S.P.; Zhang, K.; Zhu, J.; et al. Biology and clinical implications of the 19q13 aggressive prostate cancer susceptibility locus. Cell 2018, 174, 576–589.e18. [Google Scholar] [CrossRef] [Green Version]
- Prestel, M.; Prell-Schicker, C.; Webb, T.; Malik, R.; Lindner, B.; Ziesch, N.; Rex-Haffner, M.; Röh, S.; Viturawong, T.; Lehm, M.; et al. The atherosclerosis risk variant rs2107595 mediates allele-specific transcriptional regulation of HDAC9 via E2F3 and Rb1. Stroke 2019, 50, 2651–2660. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ma, R.; Liu, B.; Kong, J.; Lin, H.; Yu, X.; Wang, R.; Li, L.; Gao, M.; Zhou, B.; et al. SNP rs17079281 decreases lung cancer risk through creating an YY1-binding site to suppress DCBLD1 expression. Oncogene 2020, 39, 4092–4102. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Hayes, J.E.; Xu, X.; Gao, X.; Mehta, D.; Lilja, H.G.; Klein, R.J. Validation of prostate cancer risk variants rs10993994 and rs7098889 by CRISPR/Cas9 mediated genome editing. Gene 2021, 768, 145265. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Lou, J.; Cai, Y.; Rao, M.; Lu, Z.; Zhu, Y.; Zou, D.; Peng, X.; Wang, H.; Zhang, M.; et al. Risk SNP-mediated enhancer–promoter interaction drives colorectal cancer through both FADS2 and AP002754.2. Cancer Res. 2020, 80, 1804–1818. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.W.; Patro, C.P.K.; Zhu, J.J.; Dampier, C.H.; Plummer, S.J.; Kuscu, C.; Adli, M.; Lau, C.; Lai, R.K.; Casey, G. A functional variant on 20q13.33 related to glioma risk alters enhancer activity and modulates expression of multiple genes. Hum. Mutat. 2021, 42, 77–88. [Google Scholar] [CrossRef]
- Corces, M.R.; Shcherbina, A.; Kundu, S.; Gloudemans, M.J.; Frésard, L.; Granja, J.M.; Louie, B.H.; Eulalio, T.; Shams, S.; Bagdatli, S.T.; et al. Single-cell epigenomic analyses implicate candidate causal variants at inherited risk loci for Alzheimer’s and Parkinson’s diseases. Nat. Genet. 2020, 52, 1158–1168. [Google Scholar] [CrossRef]
- Guo, L.; Du, Y.; Qu, S.; Wang, J. rVarBase: An updated database for regulatory features of human variants. Nucleic Acids Res. 2016, 44, D888–D893. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Cheng, F.; Jia, P.; Cox, N.; Denny, J.C.; Zhao, Z. An integrative functional genomics framework for effective identification of novel regulatory variants in genome–phenome studies. Genome Med. 2018, 10, 1–15. [Google Scholar] [CrossRef]
- Jones, M.R.; Peng, P.-C.; Coetzee, S.G.; Tyrer, J.; Reyes, A.L.P.; Corona, R.I.; Davis, B.; Chen, S.; Dezem, F.; Seo, J.-H.; et al. Ovarian cancer risk variants are enriched in histotype-specific enhancers and disrupt transcription factor binding sites. Am. J. Hum. Genet. 2020, 107, 622–635. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, Y.; Li, X.; Liu, J.; Huo, Y.; Wang, J.; Liu, Z.; Li, M.; Luo, X.-J. Regulatory mechanisms of major depressive disorder risk variants. Mol. Psychiatry 2020, 25, 1926–1945. [Google Scholar] [CrossRef] [PubMed]
- Zeng, B.; Lloyd-Jones, L.R.; Montgomery, G.W.; Metspalu, A.; Esko, T.; Franke, L.; Võsa, U.; Claringbould, A.; Brigham, K.L.; Quyyumi, A.A.; et al. Comprehensive multiple eQTL detection and its application to GWAS interpretation. Genetics 2019, 212, 905–918. [Google Scholar] [CrossRef] [PubMed]
- Barbeira, A.; Bonazzola, R.; Gamazon, E.R.; Liang, Y.; Park, Y.; Kim-Hellmuth, S.; Wang, G.; Jiang, Z.; Zhou, D.; Hormozdiari, F.; et al. Exploiting the GTEx resources to decipher the mechanisms at GWAS loci. Genome Biol. 2021, 22, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Zhang, T.; Vu, A.; Ablain, J.; Makowski, M.M.; Colli, L.M.; Xu, M.; Hennessey, R.C.; Yin, J.; Rothschild, H.; et al. Massively parallel reporter assays of melanoma risk variants identify MX2 as a gene promoting melanoma. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef]
- Zhang, P.; Xia, J.; Zhu, J.; Gao, P.; Tian, Y.-J.; Du, M.; Guo, Y.-C.; Suleman, S.; Zhang, Q.; Kohli, M.; et al. High-throughput screening of prostate cancer risk loci by single nucleotide polymorphisms sequencing. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Qiu, Y.; dos Santos, A.M.R.; Yin, Y.; Li, Y.E.; Vinckier, N.; Nariai, N.; Benaglio, P.; Raman, A.; Li, X.; et al. Systematic analysis of binding of transcription factors to noncoding variants. Nature 2021, 591, 147–151. [Google Scholar] [CrossRef]
- Maurano, M.T.; Haugen, E.; Sandstrom, R.; Vierstra, J.; Shafer, A.; Kaul, R.; Stamatoyannopoulos, J.A. Large-scale identification of sequence variants influencing human transcription factor occupancy in vivo. Nat. Genet. 2015, 47, 1393–1401. [Google Scholar] [CrossRef] [Green Version]
- Cavalli, M.; Pan, G.; Nord, H.; Wallerman, O.; Arzt, E.W.; Berggren, O.; Elvers, I.; Eloranta, M.-L.; Rönnblom, L.; Toh, K.L.; et al. Allele-specific transcription factor binding to common and rare variants associated with disease and gene expression. Qual. Life Res. 2016, 135, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, M.; Pan, G.; Nord, H.; Arzt, E.W.; Wallerman, O.; Wadelius, C. Allele-specific transcription factor binding in liver and cervix cells unveils many likely drivers of GWAS signals. Genomics 2016, 107, 248–254. [Google Scholar] [CrossRef]
- Cavalli, M.; Baltzer, N.; Umer, H.M.; Grau, J.; Lemnian, I.; Pan, G.; Wallerman, O.; Spalinskas, R.; Sahlén, P.; Grosse, I.; et al. Allele specific chromatin signals, 3D interactions, and motif predictions for immune and B cell related diseases. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef]
- Kumar, S.; Ambrosini, G.; Bucher, P. SNP2TFBS—A database of regulatory SNPs affecting predicted transcription factor binding site affinity. Nucleic Acids Res. 2017, 45, D139–D144. [Google Scholar] [CrossRef] [Green Version]
- Law, W.D.; Fogarty, E.A.; Vester, A.; Antonellis, A. A genome-wide assessment of conserved SNP alleles reveals a panel of regulatory SNPs relevant to the peripheral nerve. BMC Genom. 2018, 19, 311. [Google Scholar] [CrossRef] [Green Version]
- GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 2020, 369, 1318–1330. [Google Scholar] [CrossRef]
- Korbolina, E.E.; Brusentsov, I.I.; Bryzgalov, L.O.; Leberfarb, E.Y.; Degtyareva, A.O.; Merkulova, T.I. Novel approach to functional SNPs discovery from genome-wide data reveals promising variants for colon cancer risk. Hum. Mutat. 2018, 39, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Benaglio, P.; D’Antonio-Chronowska, A.; Ma, W.; Yang, F.; Greenwald, W.W.Y.; Donovan, M.K.R.; DeBoever, C.; Li, H.; Drees, F.; Singhal, S.; et al. Allele-specific NKX2-5 binding underlies multiple genetic associations with human electrocardiographic traits. Nat. Genet. 2019, 51, 1506–1517. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Feng, W.; Lu, Z.; Yu, C.Y.; Shao, W.; Nakshatri, H.; Reiter, J.L.; Gao, H.; Chu, X.; Wang, Y.; et al. RegSNPs-ASB: A computational framework for identifying allele-specific transcription factor binding from ATAC-seq data. Front. Bioeng. Biotechnol. 2020, 8, 886. [Google Scholar] [CrossRef]
- Ernst, J.; Kheradpour, P.; Mikkelsen, T.S.; Shoresh, N.; Ward, L.; Epstein, C.B.; Zhang, X.; Wang, L.; Issner, R.; Coyne, M.; et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nat. Cell Biol. 2011, 473, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Bryzgalov, L.O.; Antontseva, E.V.; Matveeva, M.Y.; Shilov, A.G.; Kashina, E.V.; Mordvinov, V.A.; Merkulova, T.I. Detection of regulatory SNPs in human genome using ChIP-seq ENCODE data. PLoS ONE 2013, 8, e78833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryzgalov, L.O.; Korbolina, E.E.; Brusentsov, I.I.; Leberfarb, E.Y.; Bondar, N.P.; Merkulova, T.I. Novel functional variants at the GWAS-implicated loci might confer risk to major depressive disorder, bipolar affective disorder and schizophrenia. BMC Neurosci. 2018, 19, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upender, M.B.; Habermann, J.K.; McShane, L.M.; Korn, E.L.; Barrett, J.C.; Difilippantonio, M.J.; Ried, T. Chromosome transfer induced aneuploidy results in complex dysregulation of the cellular transcriptome in immortalized and cancer cells. Cancer Res. 2004, 64, 6941–6949. [Google Scholar] [CrossRef] [Green Version]
- Prochownik, E.V. C-Myc: Linking transformation and genomic instability. Curr. Mol. Med. 2008, 8, 446–458. [Google Scholar] [CrossRef]
- Gazdar, A.F.; Gao, B.; Minna, J.D. Lung cancer cell lines: Useless artifacts or invaluable tools for medical science? Lung Cancer 2010, 68, 309–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Garcia, A. Genomic instability of surgical sample and cancer-initiating cell lines from human glioblastoma. Front. Biosci. 2012, 17, 1469–1479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes-Palomares, A.; Gu, M.; Grubert, F.; Berest, I.; Sa, S.; Kasowski, M.; Arnold, C.; Shuai, M.; Srivas, R.; Miao, Y.; et al. Remodeling of active endothelial enhancers is associated with aberrant gene-regulatory networks in pulmonary arterial hypertension. Nat. Commun. 2020, 11, 1673. [Google Scholar] [CrossRef] [PubMed]
- Ramaker, R.C.; Bowling, K.M.; Lasseigne, B.N.; Hagenauer, M.; Hardigan, A.A.; Davis, N.S.; Gertz, J.; Cartagena, P.M.; Walsh, D.M.; Vawter, M.P.; et al. Post-mortem molecular profiling of three psychiatric disorders. Genome Med. 2017, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaykin, D.V. Optimally weighted Z-test is a powerful method for combining probabilities in meta-analysis. J. Evol. Biol. 2011, 24, 1836–1841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasza, J.; Wolfe, R. Interpretation of commonly used statistical regression models. Respirology 2013, 19, 14–21. [Google Scholar] [CrossRef] [Green Version]
- Gao, T.; Qian, J. EnhancerAtlas 2.0: An updated resource with enhancer annotation in 586 tissue/cell types across nine species. Nucleic Acids Res. 2019, 48, D58–D64. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.-T.; Liu, S.; Liu, L.; Ma, R.-R.; Gao, P. EGR1-mediated transcription of lncRNA-HNF1A-AS1 promotes cell cycle progression in gastric cancer. Cancer Res. 2018, 78, 5877–5890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brodsky, M.C. Congenital nystagmus and its congeners. J. Binocul. Vis. Ocul. Motil. 2020, 70, 63–70. [Google Scholar] [CrossRef]
- Fang, C.; Wang, Z.; Han, C.; Safgren, S.L.; Helmin, K.A.; Adelman, E.R.; Serafin, V.; Basso, G.; Eagen, K.P.; Gaspar-Maia, A.; et al. Cancer-specific CTCF binding facilitates oncogenic transcriptional dysregulation. Genome Biol. 2020, 21, 1–30. [Google Scholar] [CrossRef]
- Talebi, M.; Talebi, M.; Kakouri, E.; Farkhondeh, T.; Pourbagher-Shahri, A.M.; Tarantilis, P.A.; Samarghandian, S. Tantalizing role of p53 molecular pathways and its coherent medications in neurodegenerative diseases. Int. J. Biol. Macromol. 2021, 172, 93–103. [Google Scholar] [CrossRef]
- Castel, S.E.; Levy-Moonshine, A.; Mohammadi, P.; Banks, E.; Lappalainen, T. Tools and best practices for data processing in allelic expression analysis. Genome Biol. 2015, 16, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Van Arensbergen, J.; Pagie, L.; Fitzpatrick, V.D.; De Haas, M.; Baltissen, M.P.; Comoglio, F.; Van Der Weide, R.H.; Teunissen, H.; Võsa, U.; Franke, L.; et al. High-throughput identification of human SNPs affecting regulatory element activity. Nat. Genet. 2019, 51, 1160–1169. [Google Scholar] [CrossRef] [PubMed]
- Harvey, C.T.; Moyerbrailean, G.A.; Davis, G.O.; Wen, X.; Luca, F.; Pique-Regi, R. QuASAR: Quantitative allele-specific analysis of reads. Bioinformatics 2014, 31, 1235–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abramov, S.; Boytsov, A.; Bykova, D.; Penzar, D.D.; Yevshin, I.; Kolmykov, S.K.; Fridman, M.V.; Favorov, A.V.; Vorontsov, I.E.; Baulin, E.; et al. Landscape of allele-specific transcription factor binding in the human genome. Nat. Commun. 2021, 12, 1–15. [Google Scholar] [CrossRef]
- Brodie, A.; Azaria, J.R.; Ofran, Y. How far from the SNP may the causative genes be? Nucleic Acids Res. 2016, 44, 6046–6054. [Google Scholar] [CrossRef] [PubMed]
- Gamazon, E.R.; Zwinderman, A.H.; Cox, N.J.; Denys, D.; Derks, E.M. Multi-tissue transcriptome analyses identify genetic mechanisms underlying neuropsychiatric traits. Nat. Genet. 2019, 51, 933–940. [Google Scholar] [CrossRef]
- Van Der Wijst, M.G.; Brugge, H.; de Vries, D.H.; Deelen, P.; Swertz, M.A.; Franke, L. Single-cell RNA sequencing identifies celltype-specific cis-eQTLs and co-expression QTLs. Nat. Genet. 2018, 50, 493–497. [Google Scholar] [CrossRef] [Green Version]
- Gerring, Z.F.; Lupton, M.K.; Edey, D.; Gamazon, E.R.; Derks, E.M. An analysis of genetically regulated gene expression across multiple tissues implicates novel gene candidates in Alzheimer’s disease. Alzheimer Res. Ther. 2020, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- GTEx Consortium. Genetic effects on gene expression across human tissues. Nat. Cell Biol. 2017, 550, 204–213. [Google Scholar] [CrossRef]
- Stolze, L.K.; Conklin, A.C.; Whalen, M.B.; Rodríguez, M.L.; Õunap, K.; Selvarajan, I.; Toropainen, A.; Örd, T.; Li, J.; Eshghi, A.; et al. Systems genetics in human endothelial cells identifies non-coding variants modifying enhancers, expression, and complex disease traits. Am. J. Hum. Genet. 2020, 106, 748–763. [Google Scholar] [CrossRef]
- The 1000 Genomes Project Consortium; Auton, A.; Abecasis, G.R.; Altshuler, D.M.; Durbin, R.M.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Donnelly, P.; Eichler, E.E.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Frankish, A.; Diekhans, M.; Ferreira, A.-M.; Johnson, R.; Jungreis, I.; Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.; Armstrong, J.; et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019, 47, D766–D773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuno, A.; Okada, Y. Biological characterization of expression quantitative trait loci (eQTLs) showing tissue-specific opposite directional effects. Eur. J. Hum. Genet. 2019, 27, 1745–1756. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10. [Google Scholar] [CrossRef]
- Wang, C.; Kao, W.-H.; Hsiao, C.K. Using hamming distance as information for SNP-sets clustering and testing in disease association studies. PLoS ONE 2015, 10, e0135918. [Google Scholar] [CrossRef] [PubMed]
- Postovalov, S.; Berikov, V.; Bryzgalov, L.; Korbolina, E. On the relationship between regulatory and exomic DNA markers. In Proceedings of the 2020 Ural Symposium on Biomedical Engineering, Radioelectronics and Information Technology (USBEREIT), Yekaterinburg, Russia, 14–15 May 2020; pp. 97–100. [Google Scholar]
- Coetzee, S.G.; Coetzee, G.A.; Hazelett, D.J. MotifbreakR: An R/bioconductor package for predicting variant effects at transcription factor binding sites: Figure 1. Bioinformatics 2015, 31, 3847–3849. [Google Scholar] [CrossRef] [Green Version]
- Kulakovskiy, I.V.; Vorontsov, I.E.; Yevshin, I.S.; Sharipov, R.N.; Fedorova, A.D.; Rumynskiy, E.I.; Medvedeva, Y.A.; Magana-Mora, A.; Bajic, V.B.; Papatsenko, D.A.; et al. HOCOMOCO: Towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-seq analysis. Nucleic Acids Res. 2017, 46, D252–D259. [Google Scholar] [CrossRef]
- Davis, C.A.; Hitz, B.C.; Sloan, C.A.; Chan, E.T.; Davidson, J.M.; Gabdank, I.; Hilton, J.A.; Jain, K.; Baymuradov, U.K.; Narayanan, A.K.; et al. The encyclopedia of DNA elements (ENCODE): Data portal update. Nucleic Acids Res. 2018, 46, D794–D801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. ClusterProfiler: An R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- The Gene Ontology Consortium. The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 2021, 49, D325–D334. [Google Scholar] [CrossRef] [PubMed]
- Schriml, L.M.; Mitraka, E.; Munro, J.; Tauber, B.; Schor, M.; Nickle, L.; Felix, V.; Jeng, L.; Bearer, C.; Lichenstein, R.; et al. Human disease ontology 2018 update: Classification, content and workflow expansion. Nucleic Acids Res. 2019, 47, D955–D962. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef]
- Hochberg, Y.; Benjamini, Y. More powerful procedures for multiple significance testing. Stat. Med. 1990, 9, 811–818. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Regression Coefficient | Std. Error | p-Value | Sign |
|---|---|---|---|---|
| |log2FC1| | −0.547335 | 0.009193 | <2 × 10−16 | *** |
| |log2FC2| | −0.022103 | 0.008592 | 0.0101 | * |
| |log2FC1/FC2| | −0.125600 | 0.010718 | <2 × 10−16 | *** |
| Identified SNPs | n | Overlapping with All GTEX eQTLs, % | Overlapping withthe GTExeQTLs with p-Value < 0.1, % | Positions Contained in GWAS Catalog, % |
|---|---|---|---|---|
| Heterozygous SNPs | ~4.3 × 106 | 13 | 8 | 2.1 |
| SNPs with ASB (p-value < 0.1) | 58,191 | 15 | 10 | 2.5 |
| SNPs with ASE (p-value < 0.1) | 230,553 | 15 | 10 | 2.7 |
| SNPs with both ASB and ASE (both p-values < 0.1) | 20,321 | 23 | 18 | 3.0 |
| SNPs with both ASB and ASE (z-test p-values < 0.0005) | 14,898 | 23 | 18 | 3.1 |
| SNPs selected by predicted probabilities, pp > 0.1929408 | 14,543 | 26 | 20 | 3.5 |
| SNPs selected by log regression and z-score | 10,318 | 26 | 21 | 3.7 |
| rs7289432 22chr:19171209 | cSNP (rs738904) 22chr:19179872 | Total Number of Genotypes in 1000 Genomes | ||
|---|---|---|---|---|
| CC | AC | AA | ||
| AA | 1084 | 5 | 0 | 1089 |
| AG | 9 | 1042 | 5 | 1056 |
| GG | 0 | 7 | 352 | 359 |
| Total number of genotypes in 1000 Genomes | 1093 | 1054 | 357 | 2504 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korbolina, E.E.; Bryzgalov, L.O.; Ustrokhanova, D.Z.; Postovalov, S.N.; Poverin, D.V.; Damarov, I.S.; Merkulova, T.I. A Panel of rSNPs Demonstrating Allelic Asymmetry in Both ChIP-seq and RNA-seq Data and the Search for Their Phenotypic Outcomes through Analysis of DEGs. Int. J. Mol. Sci. 2021, 22, 7240. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147240
Korbolina EE, Bryzgalov LO, Ustrokhanova DZ, Postovalov SN, Poverin DV, Damarov IS, Merkulova TI. A Panel of rSNPs Demonstrating Allelic Asymmetry in Both ChIP-seq and RNA-seq Data and the Search for Their Phenotypic Outcomes through Analysis of DEGs. International Journal of Molecular Sciences. 2021; 22(14):7240. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147240
Chicago/Turabian StyleKorbolina, Elena E., Leonid O. Bryzgalov, Diana Z. Ustrokhanova, Sergey N. Postovalov, Dmitry V. Poverin, Igor S. Damarov, and Tatiana I. Merkulova. 2021. "A Panel of rSNPs Demonstrating Allelic Asymmetry in Both ChIP-seq and RNA-seq Data and the Search for Their Phenotypic Outcomes through Analysis of DEGs" International Journal of Molecular Sciences 22, no. 14: 7240. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147240