
Mutational Analysis of Redβ Single Strand Annealing Protein: Roles of the 14 Lysine Residues in DNA Binding and Recombination In Vivo

Abstract
:1. Introduction
2. Results
2.1. Mapping of the 14 Lysine Residues onto a Structural Model of Redβ
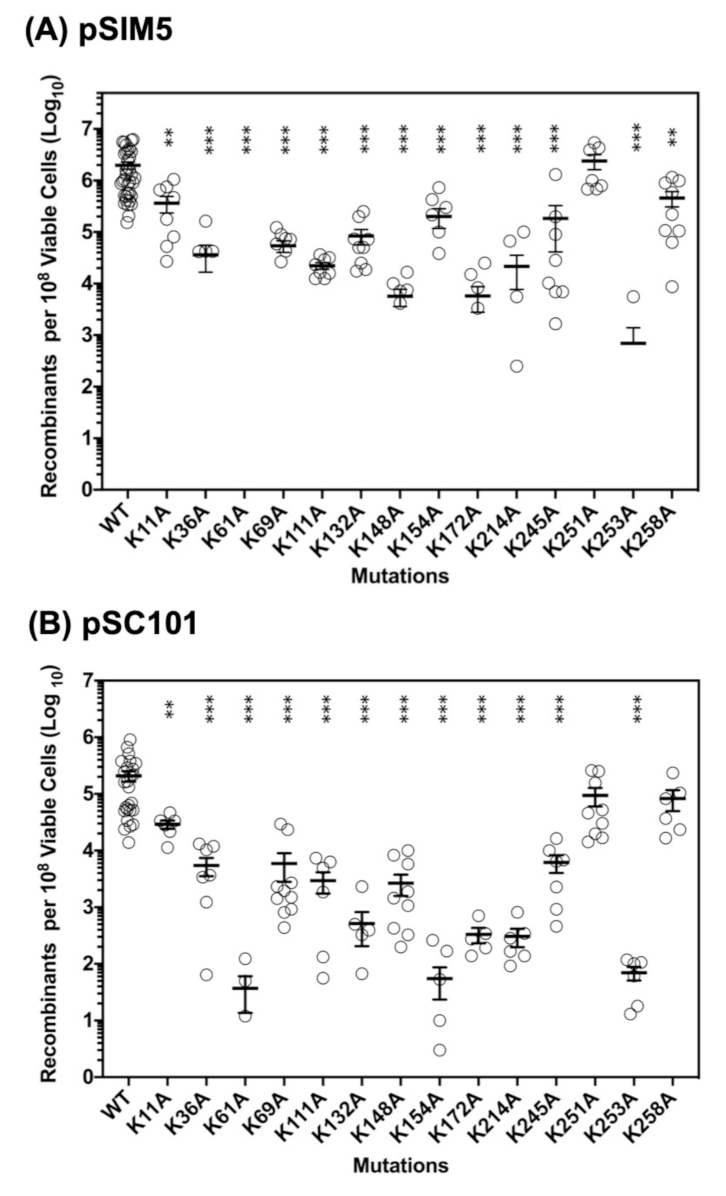
2.2. Roles of the 14 Lysine Residues in ssDNA Recombination In Vivo
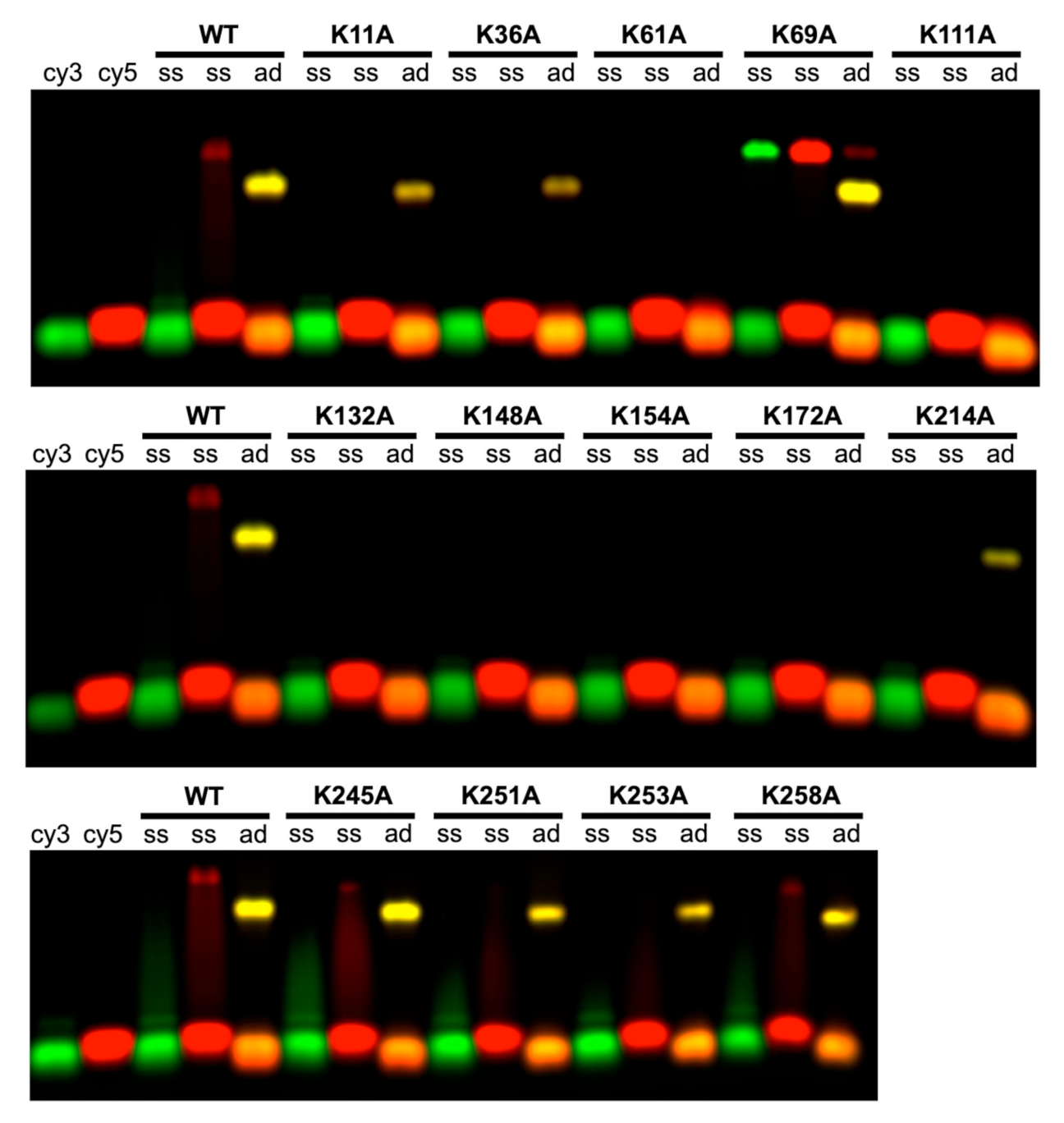
2.3. Roles of the 14 Lysine Residues in Forming the Complex with Annealed Duplex Intermediate
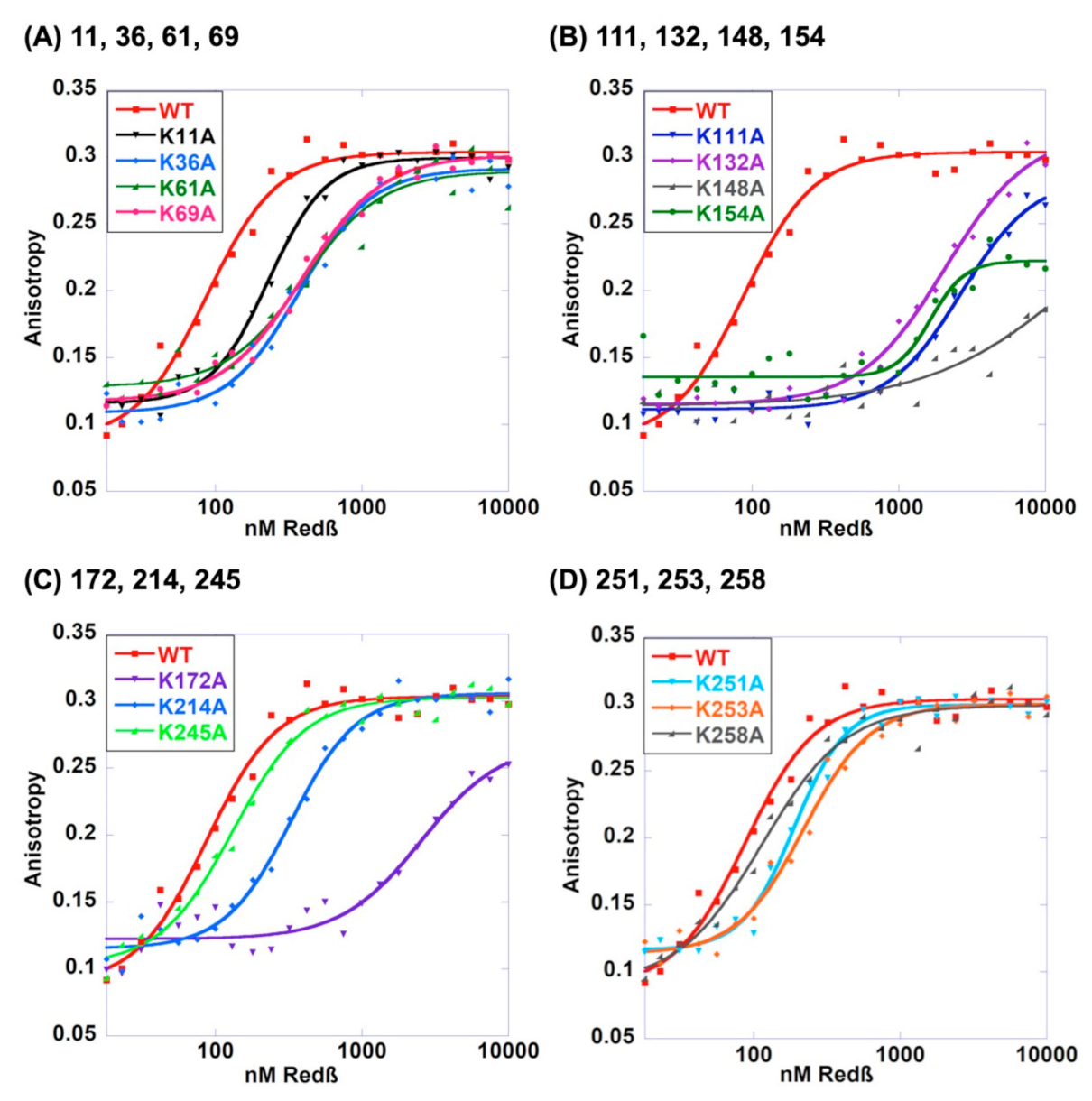
2.4. Roles of the 14 Lysines in Binding to ssDNA Substrate
3. Discussion
4. Materials and Methods
4.1. In Vivo DNA Recombination Assays
4.2. Western Blots
4.3. Expression and Purification of the 14 Lysine to Alanine Redβ Variants
4.4. Gel-Based Assay for Binding to Annealed Duplex Product
4.5. Fluorescence Anisotropy Assay for Binding to ssDNA Substrate
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CTD | C-terminal domain |
| DNA | deoxyribonucleic acid |
| dsDNA | double-stranded DNA |
| DTT | dithiothreitol |
| FAM | fluorescein |
| IPTG | isopropyl β-D-1-thiogalactopyranoside |
| NHS | N-hydroxysuccinimide |
| NTD | N-terminal domain |
| OD600 | optical density at 600 nm |
| PCR | polymerase chain reaction |
| SDS-PAGE | sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| SSA | single strand annealing |
| SSB | E. coli single-stranded DNA binding protein |
| ssDNA | single-stranded DNA |
| TRIS | Tris(hydroxymethyl)aminomethane |
References
- Iyer, L.M.; Koonin, E.V.; Aravind, L. Classification and evolutionary history of the single-strand annealing proteins, RecT, Redβ, ERF, and RAD52. BMC Genom. 2002, 3, 8. [Google Scholar] [CrossRef] [Green Version]
- Mott, C.; Symington, L.S. RAD51-independent inverted-repeat recombination by a strand-annealing mechanism. DNA Repair 2011, 10, 408–415. [Google Scholar] [CrossRef] [Green Version]
- McIlwraith, M.J.; West, S.C. DNA repair synthesis facilitates RAD52-mediated second-end capture during DSB repair. Mol. Cell 2007, 29, 510–516. [Google Scholar] [CrossRef]
- Singleton, M.R.; Wentzell, L.M.; Liu, Y.; West, S.C.; Wigley, D.B. Structure of the single-strand annealing domain of human RAD52 protein. Proc. Natl. Acad. Sci. USA 2002, 99, 13492–13497. [Google Scholar] [CrossRef] [Green Version]
- Kagawa, W.; Kurumizaka, H.; Ishitani, R.; Fukai, S.; Nureki, O.; Shibata, T.; Yokoyama, S. Crystal structure of the homologous-pairing domain from the human Rad52 recombinase in the undecameric form. Mol. Cell 2002, 10, 359–371. [Google Scholar] [CrossRef]
- Makharashvili, N.; Koroleva, O.; Bera, S.; Grandgenett, D.P.; Korolev, S. A novel structure of DNA repair protein RecO from Deinococcus Radiodurans. Structure 2004, 12, 1881–1889. [Google Scholar] [CrossRef] [PubMed]
- Sugimann-Marangos, S.; Junop, M.S. The structure of DdrB from Deinococcus: A new fold for single-stranded DNA binding proteins. Nucleic Acids Res. 2010, 38, 3432–3440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mapelli, M.; Panjikar, P.; Tucker, P.A. The crystal structure of the Herpes Simplex Virus 1 ssDNA-binding protein suggests the structural basis for flexible, cooperative single stranded DNA binding. J. Biol. Chem. 2005, 280, 2990–2997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stasiak, A.Z.; Larquet, E.; Stasiak, A.; Müller, S.; Engel, A.; Van Dyck, E.; West, S.C.; Egelman, E.H. The human Rad52 protein exists as a heptameric ring. Curr. Biol. 2000, 10, 337–340. [Google Scholar] [CrossRef] [Green Version]
- Sugiman-Marangos, S.N.; Peel, J.K.; Weiss, Y.M.; Ghirlando, R.; Junop, M.S. Crystal structure of the DdrB/ssDNA complex from Deinococcus radiodurans reveals a DNA binding surface involving higher-order oligomeric states. Nucleic Acids Res. 2013, 41, 9934–9944. [Google Scholar] [CrossRef] [Green Version]
- Tolun, G.; Makhov, A.M.; Ludtke, S.J.; Griffith, J.D. Details of ssDNA annealing revealed by an HSV-1 ICP8-ssDNA binary complex. Nucleic Acids Res. 2013, 41, 5927–5937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makhov, A.M.; Sen, A.; Yu, X.; Simon, M.N.; Griffith, J.D.; Egelman, E.H. The bipolar filaments formed by herpes simplex virus type SSB/recombination protein (ICP8) suggest a mechanism for DNA annealing. J. Mol. Biol. 2009, 386, 273–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saotome, M.; Saito, K.; Yasuda, T.; Ohtomo, H.; Sugiyama, S.; Nishimura, Y.; Kurumizaka, H.; Kagawa, W. Structural basis of homology-directed DNA repair mediated by RAD52. iScience 2018, 3, 50–62. [Google Scholar] [CrossRef] [Green Version]
- Erler, A.; Wegmann, S.; Ellie-Caille, C.; Bradshaw, C.R.; Maresca, M.; Seidel, R.; Habermann, B.; Muller, D.J.; Stewart, A.F. Conformational adaptability of Redβ during DNA annealing and implications for its structural relationship with Rad52. J. Mol. Biol. 2009, 391, 586–598. [Google Scholar] [CrossRef]
- Lopes, A.; Amarir-Bouhram, J.; Faure, G.; Petit, M.-A.; Guerois, R. Detection of novel recombinases in bacteriophage genomes unveils Rad52, Rad51, and Gp2.5 remote homologs. Nucleic Acids Res. 2010, 38, 3952–3962. [Google Scholar] [CrossRef]
- Matsubara, K.; Malay, A.D.; Curtis, F.A.; Sharples, G.J.; Heddle, J.G. Structural and functional characterization of the Redβ recombinase from bacteriophage λ. PLoS ONE 2013, 8, e78869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; Salazar, G.A.; Tate, J.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar]
- Roy, D.; Huguet, K.T.; Grenier, F.; Burrus, V. IncC conjugative plasmids and SXT/R391 elements repair double-strand breaks caused by CRISPR-Cas during conjugation. Nucleic Acids Res. 2020, 48, 8815–8827. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.Y.; Ho, J.W.; Huang, J.D.; Watt, R.M. Functional characterization of an alkaline exonuclease and single strand annealing protein from the SXT genetic element of Vibrio cholera. BMC Mol. Biol. 2011, 12, 16. [Google Scholar] [CrossRef] [Green Version]
- Muyrers, J.P.P.; Zhang, Y.; Buchholz, F.; Stewart, A.F. RecE/RecT and Redα/Redβ initiate double-stranded break repair by specifically interacting with their respective partners. Genes Dev. 2000, 14, 1971–1982. [Google Scholar] [PubMed]
- Vellani, T.S.; Myers, R.S. Bacteriophage SPP1 Chu is an alkaline exonuclease in the SynExo family of viral two-component recombinases. J. Bacteriol. 2003, 185, 2465–2474. [Google Scholar] [CrossRef] [Green Version]
- Caldwell, B.J.; Bell, C.E. Structure and mechanism of the Red recombination system of bacteriophage λ. Prog. Biophys. Mol. Biol. 2019, 147, 33–46. [Google Scholar] [CrossRef]
- Kovall, R.; Matthews, B.W. Toroidal structure of λ-exonuclease. Science 2011, 277, 1824–1827. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; McCabe, K.A.; Bell, C.E. Crystal structures of λ exonuclease in complex with DNA suggest an electrostatic ratchet mechanism for processivity. Proc. Natl. Acad. Sci. USA 2011, 108, 11872–11877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, C.E.; Bell, C.E. Domain structure of the Redβ single-strand annealing protein: The C-terminal domain is required for fine-tuning DNA-binding properties, interaction with the exonuclease partner, and recombination in vivo. J. Mol. Biol. 2016, 428, 561–578. [Google Scholar] [CrossRef] [Green Version]
- Caldwell, B.J.; Zakharova, E.; Filsinger, G.T.; Wannier, T.M.; Hempfling, J.P.; Chun-Der, L.; Pei, D.; Church, G.M.; Bell, C.E. Crystal structure of the Redβ C-terminal domain in complex with λ exonuclease reveals and unexpected homology with λ Orf and an interaction with Escherichia coli single-stranded DNA binding protein. Nucleic Acids Res. 2019, 47, 1950–1963. [Google Scholar] [CrossRef] [Green Version]
- Maxwell, K.L.; Reed, P.; Zhang, R.G.; Beasley, S.; Walmsley, A.R.; Curtis, F.A.; Joachimiak, A.; Edwards, A.M.; Sharples, G.J. Functional similarities between phage λ Orf and Escherichia coli RecFOR in initiation of genetic exchange. Proc. Natl. Acad. Sci. USA 2005, 102, 11260–11265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellis, H.M.; Yu, D.; DiTizio, T.; Court, D.L. High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides. Proc. Natl. Acad. Sci. USA 2001, 98, 6742–6746. [Google Scholar] [CrossRef] [Green Version]
- Copeland, N.G.; Jenkins, N.A.; Court, D.L. Recombineering: A powerful new tool for mouse functional genomics. Nat. Rev. Genet. 2001, 2, 769–779. [Google Scholar] [CrossRef]
- Muyrers, J.P.; Zhang, Y.; Stewart, A.F. Techniques: Recombinogenic engineering- new options for cloning and manipulating DNA. Trends Biochem. Sci. 2001, 26, 325–331. [Google Scholar] [CrossRef]
- Wang, H.H.; Isaacs, F.J.; Carr, P.A.; Sun, Z.Z.; Xu, G.; Forest, C.R.; Church, G.M. Programming cells by multiplex genome engineering and accelerated evolution. Nature 2009, 460, 894–898. [Google Scholar] [CrossRef]
- Karakousis, G.; Ye, N.; Li, Z.; Chiu, S.K.; Reddy, G.; Radding, C.M. The beta protein of phage λ binds preferentially to an intermediate in DNA renaturation. J. Mol. Biol. 1998, 276, 721–731. [Google Scholar] [CrossRef]
- Passy, S.I.; Yu, X.; Li, Z.; Radding, C.M.; Egelman, E.H. Rings and filaments of beta protein from bacteriophage lambda suggest a superfamily of recombination proteins. Proc. Natl. Acad. Sci. USA 1999, 96, 4279–4284. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Xing, X.; Wisler, J.W.; Dalton, J.T.; Bell, C.E. Domain structure and DNA binding regions of β protein from bacteriophage λ. J. Biol. Chem. 2006, 281, 25205–25214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kagawa, W.; Kagawa, A.; Saito, K.; Ikawa, S.; Shibata, T.; Kurumizaka, H.; Yokoyama, S. Identification of a second DNA binding site in the human Rad52 protein. J. Biol. Chem. 2008, 283, 24264–24273. [Google Scholar] [CrossRef] [Green Version]
- Datta, S.; Costantino, N.; Court, D.L. A set of recombineering plasmids for gram-negative bacteria. Gene 2006, 379, 109–115. [Google Scholar] [CrossRef]
- Wang, J.; Sarov, M.; Rientjes, J.; Fu, J.; Hollak, H.; Kranz, H.; Xie, W.; Stewart, A.F.; Zhang, Y. An improved recombineering approach by adding RecA to λ Red recombination. Mol. Biotechnol. 2006, 32, 43–53. [Google Scholar] [CrossRef]
- Subramaniam, S.; Erler, A.; Fu, J.; Kranz, A.; Tang, J.; Gopalswamy, M.; Ramakrishnan, S.; Keller, A.; Grundmeier, G.; Müller, D.; et al. DNA annealing by Redβ is insufficient for homologous recombination and the additional requirements involve intra- and inter-molecular interactions. Sci. Rep. 2016, 6, 34525. [Google Scholar] [CrossRef]
- Ander, M.; Subramaniam, S.; Fahmy, K.; Stewart, A.F.; Schäffer, E.A. single-strand annealing protein clamps DNA to detect and secure homology. PLoS Biol. 2015, 13, e1002213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caldwell, B.J.; Norris, A.; Zakharova, E.; Smith, C.E.; Wheat, C.T.; Choudhary, D.; Sotomayor, M.; Wysocki, V.H.; Bell, C.E. Oligomeric complexes formed by Redβ single strand annealing protein in its different DNA bound states. Nucleic Acids Res. 2021, 49, 3441–3460. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| In Vitro DNA Binding | In Vivo Recombination (% of WT) | ||||
|---|---|---|---|---|---|
| Redβ Variant | ssDNA 1 | Annealed Duplex 2 | pSIM5 | pSC101 | |
| (Kd, nM) | (n) | ||||
| WT | 110 ± 70 | 2.5 ± 0.9 | + | 100 | 100 |
| K11A | 230 ± 40 | 2.2 ± 0.1 | + | 17 | 14 |
| K36A | 530 ± 310 | 1.6 ± 0.2 | + | 1.7 | 2.6 |
| K61A | 390 ± 10 | 1.6 ± 0.01 | - | 0 | 0.02 |
| K69A | 410 ± 150 | 1.4 ± 0.4 | + | 2.5 | 2.8 |
| K111A | 2400 ± 560 | 1.6 ± 0.4 | - | 1.0 | 1.4 |
| K132A | 1770 ± 600 | 1.3 ± 0.2 | - | 3.9 | 0.43 |
| K148A | 14,100 ± 14,500 | 0.87 ± 0.1 | - | 0.26 | 1.3 |
| K154A | 2100 ± 1100 | 1.8 ± 1.5 | - | 9.2 | 0.03 |
| K172A | 2600 ± 240 | 1.4 ± 0.2 | - | 0.26 | 0.16 |
| K214A | 310 ± 90 | 1.7 ± 0.2 | + | 0.99 | 0.15 |
| K245A | 160 ± 40 | 1.7± 0.2 | + | 9.5 | 3.0 |
| K251A | 190 ± 30 | 2.3 ± 0.4 | + | 110 | 60 |
| K253A | 210 ± 30 | 1.8 ± 0.2 | + | 0.03 | 0.03 |
| K258A | 110 ± 40 | 1.9 ± 0.4 | + | 21 | 40 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zakharova, K.; Caldwell, B.J.; Ta, S.; Wheat, C.T.; Bell, C.E. Mutational Analysis of Redβ Single Strand Annealing Protein: Roles of the 14 Lysine Residues in DNA Binding and Recombination In Vivo. Int. J. Mol. Sci. 2021, 22, 7758. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147758
Zakharova K, Caldwell BJ, Ta S, Wheat CT, Bell CE. Mutational Analysis of Redβ Single Strand Annealing Protein: Roles of the 14 Lysine Residues in DNA Binding and Recombination In Vivo. International Journal of Molecular Sciences. 2021; 22(14):7758. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147758
Chicago/Turabian StyleZakharova, Katerina, Brian J. Caldwell, Shalya Ta, Carter T. Wheat, and Charles E. Bell. 2021. "Mutational Analysis of Redβ Single Strand Annealing Protein: Roles of the 14 Lysine Residues in DNA Binding and Recombination In Vivo" International Journal of Molecular Sciences 22, no. 14: 7758. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147758