
Single- and Two-Electron Reduction of Nitroaromatic Compounds by Flavoenzymes: Mechanisms and Implications for Cytotoxicity

Abstract
:1. Introduction
2. Redox Properties of Nitroaromatic Compounds and Their Reduction Products
→ O2N-ArCH2· + CH3-N(CH2CH2Cl)2.
3. Mechanisms of Reduction in Nitroaromatic Compounds by Flavoenzymes
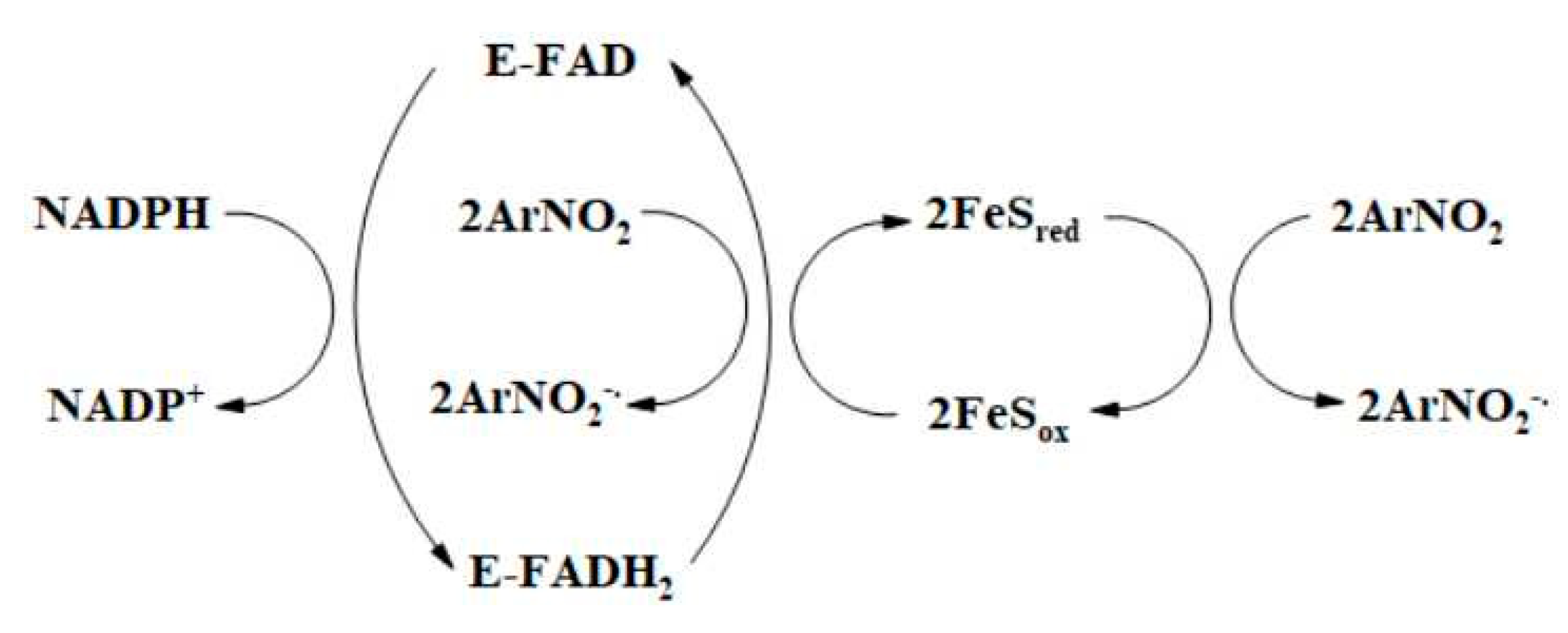
3.1. Single- and Mixed Single- and Two-Electron Reduction in Nitroaromatic Compounds by Flavoenzymes Dehydrogenases-Electrontransferases
3.2. Two-Electron Reduction in Nitroaromatic Compounds by NAD(P)H:Quinone Oxidoreductase (NQO1) and Bacterial Nitroreductases

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Redox Potential vs. NHE, pH 7.0 | Rate Constants of Electron (Hydride) Transfer, pH 7.0 |
|---|---|---|
| Rat liver NAD(P)H:quinone oxidoreductase | −0.159 V (FAD/FADH−), −0.201 V (FAD/FAD−·) [138] | >1000 s−1 (NADPH to FAD) [138]; kcat = 0.1–2300 s−1, kcat/Km = 3 × 102–5.4 × 108 M−1s−1 (quinones, steady-state) [139]; kcat = 0.04–75 s−1, kcat/Km = 5.0 × 101–5.7 × 105 M−1s−1 (ArNO2, steady-state) [141] |
| E. coli NfsB | >1000 s−1 (NADH to FMN) [142]; kcat = 225 s−1, kcat/Km = 1.5 × 105 M−1s−1 (nitrofurazone), kcat/Km = 650 M−1s−1 (nitrobenzene, steady-state) [143]; kcat = 140 s−1, kcat/Km = 7.0 × 103 M−1s−1 (CB-1954, steady-state) [144]; kcat = 60 s−1, kcat/Km = 1.3 × 104 M−1s−1 (PR-104A, steady-state) [145] | |
| E. cloacae NR-B | −0.190 V (FMN/FMNH−) [146] | >1000 s−1 (NADH to ArNO2, steady-state) [39]; 700 s−1 (NADH to FMN), 1.9 s−1 (FMNH− to p-nitrobenzoic acid, 4 °C) [147] |
| E. coli NfsA | −0.215 V (FMN/FMNH−) [148] | >400 s−1 (NADPH to FMN), kcat = 14–180 s−1, kcat/Km = 104–7.9 × 106 M−1s−1 (ArNO2, steady-state) [149] |
| M. smegmatis MsPnBA | −0.190 V (FMN/FMNH−) [150] | kcat = 3.4–19.2 s−1, kcat/Km = 1.2 × 103–1.6 × 105 M−1s−1 (ArNO2, steady-state) [150] |
| Peroxiredoxin- nitroreductase hybrid enzyme, Thermotoga maritima | −0.185 V (FMN/FMNH−, pH 8.0) [151] | 146 s−1 (NADH to FMN), kcat ≤ 15 s−1, kcat/Km = 101–5.8 × 105 M−1s−1 (ArNO2, steady-state) [152] |
3.3. Single- and Two-Electron Reduction in Nitroaromatic Compounds by Flavoenzymes Disulfide Reductases
4. Role of Enzymatic Reduction in Nitroaromatic Compounds in Their Cytotoxicity/Therapeutic Action
4.1. Role of Bioreductive Processes in Mammalian Cell Cytotoxicity of Nitroaromatics
| Compounds and Cells | Structure-Activity Relationships |
|---|---|
| Aerobic Conditions | |
| Nitroimidazoles, nitrofurans, nitrobenzenes, Chinese hamster V79–379A cells [238,239]; nitrobenzenes, nitrofurans, nitrobenzimidazoles, bovine leukemia virus-transformed lamb kidney fibroblast FLK cells [144,240,243]; nitrobenzenes, nitrofurans, nitrothiophenes, murine hepatoma MH22a cells, human colon carcinoma HCT116 cells [13,71] | ∆log cL50/∆E17 = −8 ÷ −11 V−1, ∆log cL50/∆log P(D) = 0 ÷ −0.3; CB-1954 (14), SN-23682 (19) and their derivatives follow the same correlation [240] |
| Nitrobenzenes, FLK cells [245], primary mice splenocytes [241] | ∆log cL50/∆E17 = −7.5 ÷ 8.5 V−1, increased cytotoxicity of amino and hydroxylamino metabolites of TNT (4) |
| Derivatives of nitracrine (49), CHO-AA8 cells [242] | The absence of a relationship between E17 and cytotoxicity |
| 4-(Alkylamino)-5-nitroquinolines, Chinese hamster ovary CHO-AA8 cells [249] | Cytotoxicity roughly increases with the rate of drug-stimulated O2 consumption |
| N,N-bis(2-chloroethyl)-N-methyl-N-(2-nitrobenzyl) ammonium chloride and its derivatives, CHO-AA8 cells [250] | Alkylating bioreductively activated leaving group does not affect the cytotoxicity, the relationship between E17 and cytotoxicity is absent |
| Derivatives of NitroCBI (54), Skov3 and HT29 cells [23] | The relationship between E17 and cytotoxicity is absent |
| Nitrofurans, nitrothiophenes, nitropyrroles and nitropyrazoles with alkyl-N-aziridine or alkyloxirane side chains, V79–379A cells [251,252,253] | Alkylating side chains increase the cytotoxicity, the relationship between E17 and cytotoxicity is absent |
| Hypoxic Conditions | |
| Nitroimidazoles, nitrofurans, V79–379A cells [254] | ∆log cL50/∆E17 = −10 V−1 |
| 4-(Alkylamino)-5-nitroquinolines, CHO-AA8 cells [249] | Cytotoxicity and hypoxic selectivity roughly increase with the rate of drug-stimulated O2 consumption |
| Nitrofurans and nitrothiophenes with alkyl-N-aziridine or alkyloxirane side chains, V79–379A cells [251,252,253] | Alkylating side chains do not increase hypoxic selectivity |
| N,N-bis(2-chloroethyl)-N-methyl-N-(2-nitrobenzyl) ammonium chloride and its derivatives, CHO-AA8 cells [250] | Alkylating leaving groups increase cytotoxicity, no relationship between E17 and cytotoxicity or hypoxic selectivity |
| Nitrochloromethylbenzindolines (nitroCBIs, 56), Skov3 and HT29 cells [23] | Hypoxic toxicity does not correlate with E17, hypoxic selectivity roughly increases with E17 |
4.2. Role of Bioreductive Processes in Antibacterial and Antiparasitic Action of Nitroaromatics
| Cell. Signaling Protein | Effect | Compound, Cells |
|---|---|---|
| p53 | Upregulation | Furazolidone, human hepatocytes LO2 [265]; phenylnitrobenzene B16, human cervical carcinoma HeLa [269] |
| Upregulation, downregulation of mutant p53 | Nitrofuran NSC59984, colorectal cancer [270] | |
| Activation of phosphorylation | 1-Nitropyrene, human hepatoma HepG2 [271] | |
| p21 | Upregulation | Indolin-2-one-subsituted nitrobenzene HO1-02, esophageal cancer [271]; 1,3-dinitrobenzene, rat Sertoli cells [272] |
| Downregulation | Furazolidone, HepG2 [267] | |
| Transcription factor STAT-3 | Inhibition of phosphorylation | Nifuroxazide, human myeloma [273] |
| Bax/Bcl-2 ratio | Upregulation | Phenylnitrobenzene B16, HeLa [269]; 1,3-dinitrobenzene, rat Sertoli cells [272]; furazolidone, HepG2 [267]; N-pentyl-furantoin, human promyelocytic leukemia HL-60 [274]; 2-(2-(5-nitrofuran-2-yl)-vinyl)-quinoline derivative, non-small cell lung cancer H1299 [275]; nifuroxazide, human breast cancer MCF-6 [276] |
| N-Myc | Downregulation | Nifurtimox, neuroblastoma cells [247] |
| Caspases | Upregulation | Tetryl, HeLa [216]; HO1-02, esophageal cancer [266]; furazolidone, HepG2 [267]; nifuroxazide, MCF-6 [276]; CB-1954, NR-transfected ovarian carcinoma SKOV3 [277]; TNT, HepG2 [278] |
| JNK | Activation of phosphorylation | 1,3-dinitrobenzene, rat Sertoli cells [272] |
| p38 | 2-(2-(5-Nitrofuran-2-yl)-vinyl)-quinoline derivative H1299 [275] | |
| ERK | Inhibition of phosphorylation | Nifurtimox, neuroblastoma cells [248]; 5-nitroimidazole DYT-40, human glyoma U251 and U87 |
| Activation of phosphorylation | 1,3-dinitrobenzene, rat Sertoli cells [272] | |
| AKT kinase | Inhibition of phosphorylation | 2-(2-(5-Nitrofuran-2-yl)-vinyl)-quinoline derivative, H1299 [275]; 5-nitroimidazole DYT-40, U251 and U87 [279]; nifurtimox, human neuroblastoma [280] |
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
| No. | Compound | E17 (V) a | E17(calc.) (V) b |
|---|---|---|---|
| Nitrobenzenes | |||
| 1 | 2,4,6-Trinitrophenyl-N-nitramino-ethylnitrate (pentryl) | - | −0.175 |
| 2 | 2,4,6-Trinitrophenyl-N-methylnitramine (tetryl) | - | −0.191 |
| 3 | N-Methylpicramide | - | −0.247 |
| 4 | 2,4,6-Trinitrotoluene (TNT) | −0.253 c | −0.279 |
| 5 | 1,4-Dinitrobenzene | −0.257 | −0.240 |
| 6 | 1,2-Dinitrobenzene | −0.287 | −0.311 |
| 7 | 3,5-Dinitrobenzamide | - | −0.311 |
| 8 | 4-Nitrobenzaldehyde | −0.325 | −0.342 |
| 9 | 3,5-Dinitrobenzoic acid | −0.344 | −0.336 |
| 10 | 1,3-Dinitrobenzene | −0.348 | −0.332 |
| 11 | 2-Hydroxylamino-4,6-dinitrotoluene | - | −0.351 |
| 12 | 4-Nitroacetophenone | −0.355 | −0.356 |
| 13 | 2-((2-Bromoethyl)(2,4-dinitro-6-((2-(phosphonooxy)ethyl)carbamoyl)phenyl)amino)ethyl methanesulfonate (PR-104A) | −0.366 d | |
| 14 | 5-(Aziridin-1-yl)-2,4-dinitrobenzamide (CB-1954) | −0.385 | −0.352 |
| 15 | 5,5-Dimethyl-3-(4-nitro-3-(trifluoromethyl)phenyl) imidazoline-2,4-dione (nilutamide) | - | −0.399 |
| 16 | 2-Methyl-N-(4-nitro-3-(trifluoromethyl)phenyl) propanamide (flutamide) | - | −0.408 |
| 17 | 2-Amino-4,6-dinitrotoluene | −0.417 c | −0.423 |
| 18 | 4-Nitrobenzoic acid | −0.425 | −0.423 |
| 19 | 5-(Bis(2,2′-chloroethyl)amino)-2,4-dinitrobenzamide (SN-23682) | −0.425 | −0.398 |
| 20 | 4-Hydroxylamino-2,6-dinitrotoluene | - | −0.429 |
| 21 | 4-Amino-2,6-dinitrotoluene | −0.449 c | −0.453 |
| 22 | Nitrobenzene | −0.485 | −0.489 |
| 23 | D-(-)-threo-2-dichloroacetamido-1-(4-nitrophenyl)-1,3- propandiol (chloramphenicol) | −0.546 e | |
| Nitrofurans and nitrothiophenes | |||
| 24 | 2-(5′-Nitrofurylvinyl)-quinoline-4-carboxydiethylamino- 1-methyl-but-1-ylamide (chinifur, quinifuryl) | −0.225 f | −0.221 |
| 25 | 5-Nitro-2-furaldoxime (nifuroxime) | −0.255 | −0.279 |
| 26 | N-(5-nitro-2-furfurylindene)-1-aminohydantoin (nitrofurantoin) | −0.265 | −0.263 |
| 27 | 4-(5-Nitrofurfurylidenamino)-3-methyltiomorpholine- 1,1-dioxide (nifurtimox) | −0.260 g | −0.285 |
| 28 | 2-Nitrothiophene-5-aldehyde | −0.260 h | |
| 29 | 2-Nitrothiophene-5-aldoxime | −0.280 h | |
| 30 | 2-Nitrothiophene-5-carboxymorpholide | −0.305 h | |
| 31 | 2-Nitrofuran | −0.330 | |
| 32 | 2-Nitrothiophene | −0.390 | |
| Nitroimidazoles | |||
| 33 | 1-Methyl-2-nitroimidazole-5-carboxamide | −0.321 | |
| 34 | N-Benzyl-2-(2-nitro-1H-imidazol-1-yl)acetamide (benznidazole) | −0.380 | |
| 35 | 1-Methoxy-3-(2-nitroimidazol-1-yl)propan-2-ol (misonidazole) | −0.389 | |
| 36 | 1-(Methyl-2-nitro-1H-imidazole-5-yl)-methyl-N,N’-bis(2- bromoethyl)phosphorodiamidate (TH-302, evofosfamide) | −0.407 i | |
| 37 | 2-Nitroimidazole | −0.418 | |
| 38 | 1-Methyl-2-(5-amino-1,3,4-thiadazole)- 5-nitroimidazole (megazol) | −0.438 g | |
| 39 | 1-Methyl-2-((4-(methylthio)phenoxy)methyl)-5-nitro-1H- imidazole (fexinidazole) | −0.458 | |
| 40 | 2-Methyl-5-nitroimidazole-1-ethanol (metronidazole) | −0.486 | |
| 41 | 4-Nitroimidazole | ≤−0.527 | |
| Nitrobenzimidazoles | |||
| 42 | 4,5,6,7-Tetranitrobenzimidazolone | - | −0.197 |
| 43 | 4,5,6-Trinitrobenzimidazolone | - | −0.224 |
| 44 | 5,6-Dinitrobenzimidazolone | - | −0.273 |
| 45 | 2-Nitrobenzimidazole | −0.300 | - |
| 46 | 5-Nitrobenzimidazolone | - | −0.355 |
| Miscellaneous | |||
| 47 | 4-Nitropyridine | −0.190 | |
| 48 | 4,6-Dinitrobenzofuroxane | - | −0.258 |
| 49 | 5-Nitro-4-(3′-dimethylaminopropylamino)-quinoline (nitraquine) | −0.286 j | |
| 50 | 1-Nitro-9-(3′-dimethylaminopropylamino)-acridine (nitracrine) | −0.303 j | −0.285 |
| 51 | 1-N-alkyl-3-nitro-1,2,4-triazines | −0.310–−0.330 k | |
| 52 | 2-((5-Nitrothiazol-2-yl)carbamoyl)phenyl acetate (nitazoxanide) | −0.380 | |
| 53 | 5-Nitrothiazole | −0.400 | |
| 54 | 5-Amino-3-nitro-1,2,4-triazole (ANTA) | - | −0.466 |
| 55 | 3-Nitro-1,2,4-triazolone (NTO) | - | −0.472 |
| 56 | 1-(Chloromethyl)-3-(5-(2-(dimethylamino)-ethoxy)indol- 2-carbonyl)-5-nitro-1,2-dihydro-3H-benzo[e]-indole (representative of nitroCBIs) | −0.512 l | |
| 57 | 2-Nitro-6((4-(trifluoromethoxy)benzyl)oxy)-6,7- dihydro-5H-imidazo [2,1-b][1,3]oxazine (PA-824) | −0.534 m | |
Appendix B
References
- Spain, J.C. Biodegradation of nitroaromatic compounds. Annu. Rev. Microbiol. 1995, 49, 523–555. [Google Scholar] [CrossRef]
- Purohit, V.; Basu, A.K. Mutagenicity of nitroaromatic compounds. Chem. Res. Toxicol. 2000, 13, 674–692. [Google Scholar] [CrossRef] [PubMed]
- Roldan, M.D.; Perez-Reinado, E.; Castillo, F.; Moreno-Vivian, E. Reduction of polynitroaromatic compounds: The bacterial nitroreductases. FEMS Microbiol. Rev. 2008, 32, 474–500. [Google Scholar] [CrossRef] [Green Version]
- Kovacic, P.; Somanathan, R. Nitroaromatic compounds: Environmental toxicity, carcinogenicity, mutagenicity, therapy and mechanism. J. Appl. Toxicol. 2014, 34, 810–824. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Mishra, K.; Ramanathan, G. Bioremediation of nitroaromatic compounds. In Wastewater Treatment Engineering; Samer, M., Ed.; IntechOpen: London, UK, 2015; pp. 51–83. [Google Scholar]
- Nepali, K.; Lee, H.-J.; Liou, J.-P. Nitro-group-containing drugs. J. Med. Chem. 2019, 62, 2851–2893. [Google Scholar] [CrossRef] [PubMed]
- Denny, W.A. Nitroreductase-based GDEPT. Curr. Pharm. Des. 2002, 8, 1349–1361. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.M.; Little, R.F.; Mowday, A.M.; Rich, M.H.; Chan-Hyans, J.V.E.; Copp, J.N.; Smaill, J.B.; Patterson, A.V.; Ackerley, D.F. Nitroreductase gene-directed enzyme prodrug therapy: Insights and advances toward clinical utility. Biochem. J. 2015, 471, 131–153. [Google Scholar] [CrossRef]
- Williams, R.E.; Rathbone, D.A.; Scrutton, N.S.; Bruce, N.C. Biotransformation of explosives by the old yellow enzyme family of flavoenzymes. Appl. Environ. Microbiol. 2004, 70, 3566–3574. [Google Scholar] [CrossRef] [Green Version]
- Wardman, P. Reduction potentials of one-electron couples involving free radicals in aqueous solutions. J. Phys. Chem. Ref. Data 1989, 18, 1637–1755. [Google Scholar] [CrossRef] [Green Version]
- Šarlauskas, J.; Nivinskas, H.; Anusevičius, Ž.; Misevičienė, L.; Marozienė, A.; Čėnas, N. Estimation of single-electron reduction potentials (E17) of nitroaromatic compounds according to the kinetics of their single-electron reduction by flavoenzymes. Chemija 2006, 17, 31–37. [Google Scholar]
- Šarlauskas, J.; Lesanavičius, M.; Aliverti, A.; Čėnas, N. Reduction of nitroaromatic explosives by Plasmodium falciparum ferredoxin: NADP+ reductase: Estimation of their single-electron reduction potentials. In Proceedings of the Abstracts of the 21st Seminar on New Trends in Research of Energetic Materials, Pardubice, Czech Republic, 18–20 April 2018; Pachman, J., Šelešovský, J., Eds.; University of Pardubice: Pardubice, Czech Republic, 2018; p. 135. [Google Scholar]
- Nemeikaitė-Čėnienė, A.; Marozienė, A.; Misevičienė, L.; Tamulienė, J.; Yantsevich, A.V.; Čėnas, N.K. Flavoenzyme-catalyzed single-electron reduction of nitroaromatic antiandrogens: Implications for their cytotoxicity. Free Radic. Res. 2021, 55, 246–254. [Google Scholar] [CrossRef]
- Riefler, R.G.; Smets, B.F. Enzymatic reduction of 2,4,6-trinitrotoluene and related nitroarenes: Kinetics linked to one-electron redox potentials. Environ. Sci. Technol. 2000, 34, 3900–3906. [Google Scholar] [CrossRef]
- Prosser, G.A.; Copp, J.N.; Mowday, A.M.; Guise, C.P.; Syddall, S.P.; Williams, E.M.; Horvat, C.N.; Swe, P.M.; Ashoorzadeh, A.; Denny, W.A.; et al. Creation and screening of a multi-family bacterial oxidoreductase library to discover novel nitroreductases that efficiently activate the bioreductive prodrugs CB1954 and PR-104A. Biochem. Pharmacol. 2013, 85, 1091–1103. [Google Scholar] [CrossRef]
- Kapoor, S.; Varshney, L. Redox reactions of chloramphenicol and some aryl peroxyl radicals in aqueous solution: A pulse radiolytic study. J. Phys. Chem. A 1997, 101, 7778–7782. [Google Scholar] [CrossRef]
- Grellier, P.; Šarlauskas, J.; Anusevičius, Ž.; Marozienė, A.; Houee-Levin, C.; Schrevel, J.; Čėnas, N. Antiplasmodial activity of nitroaromatic and quinoidal compounds: Redox potential vs. inhibition of erythrocyte glutathione reductase. Arch. Biochem. Biophys. 2001, 393, 199–206. [Google Scholar] [CrossRef]
- Viode, C.; de Albuquerque, C.; Chauviere, G.; Houee-Levin, C.; Perie, J. Comparative study by pulse radiolysis of the radical anion derived from compounds used in Chaga’s disease therapy. New J. Chem. 1997, 21, 1331–1338. [Google Scholar]
- Breccia, A.; Busi, F.; Gattavechia, E.; Tamba, M. Reactivity of nitro-thiophene derivatives with electron and oxygen radicals studied by pulse radiolysis and polarographic techniques. Radiat. Environ. Biophys. 1990, 29, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Evans, J.W.; Bhupatti, D.; Banica, M.; Lan, L.; Lorente, G.; Duan, J.-X.; Cai, X.; Mowday, A.M.; Guise, C.P.; et al. Molecular and cellular pharmacology of the hypoxia-activated prodrug TH-302. Mol. Cancer Ther. 2012, 11, 740–751. [Google Scholar] [CrossRef] [Green Version]
- Wilson, W.R.; Siim, B.G.; Denny, W.A.; van Zijl, P.L.; Taylor, M.L.; Chambers, D.M.; Roberts, P.B. 5-Nitro-4- (N,N-dimethyl-aminopropylamino)quinoline (5-nitraquine), a new DNA-affinic hypoxic cell radiosensitizer and bioreductive agent: Comparison with nitracrine. Radiat. Res. 1992, 131, 257–265. [Google Scholar] [CrossRef]
- Fielden, E.M.; Adams, G.E.; Cole, S.; Naylor, M.A.; O’Neill, P.; Stephens, M.A.; Stratford, I.J. Assessment of a range of novel nitro-aromatic radiosensitizers and bioreductive drugs. Int. J. Radiat. Oncol. Biol. Phys. 1992, 22, 707–711. [Google Scholar] [CrossRef]
- Tercel, M.; Atwell, G.J.; Yang, S.; Stevenson, R.J.; Botting, K.J.; Boyd, M.; Smith, E.; Anderson, R.F.; Denny, W.A.; Wilson, W.R.; et al. Hypoxia-activated prodrugs: Substituent effects on the properties of nitro seco-1,2,9,9a- tetrahydrocyclopropa[c]benz[e]indol-4-one (nitroCBI) prodrugs of DNA minor groove alkylating agents. J. Med. Chem. 2009, 52, 7258–7272. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.M.; Blaser, A.; Anderson, R.F.; Shinde, S.; Franzblau, S.G.; Ma, Z.; Denny, W.A.; Palmer, B.D. Synthesis, reduction potentials, and antitubercular activity of ring A/B analogues of the bioreductive drug (6S)-2-nitro-6-{[4-trifluoro methoxy)benzyl]oxy}-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine (PA-824). J. Med. Chem. 2009, 52, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Neta, P.; Simic, M.G.; Hoffman, M.Z. Pulse-radiolysis and electron spin resonance studies of nitroaromatic radical anions. Optical absorption spectra, kinetics, and one-electron reduction potentials. J. Phys. Chem. 1976, 80, 2018–2023. [Google Scholar] [CrossRef]
- Meotner, M.; Neta, P. Kinetics of electron transfer from nitroaromatic radical anions in aqueos solutions. Effects of temperature and steric configuration. J. Phys. Chem. 1986, 90, 4648–4650. [Google Scholar] [CrossRef]
- Guissany, A.; Henry, A.; Lougmani, N.; Hickel, B. Kinetic studies of four types of nitroheterocyclic radicals by pulse radiolysis: Correlation of pharmaceutical properties to decay rates. Free Radic. Biol. Med. 1990, 8, 173–189. [Google Scholar] [CrossRef]
- Marcus, R.; Sutin, N. Electron transfers in chemistry and biology. Biochim. Biophys. Acta 1985, 811, 265–322. [Google Scholar] [CrossRef]
- Grampp, G.; Jaenicke, W. ESR-spectroscopic investigation of the parallel electron and proton exchange between quinones and their radicals: Part I. Measurements at 298 K. J. Electroanal. Chem. 1987, 229, 297–303. [Google Scholar] [CrossRef]
- Mauk, A.G.; Scott, R.A.; Gray, H.B. Distances of electron transfer to and from metalloprotein redox sites in reactions with inorganic complexes. J. Am. Chem. Soc. 1980, 102, 4360–4363. [Google Scholar] [CrossRef]
- La-Scalea, M.A.; Menezes, C.M.S.; Juliao, M.S.S.; Chung, M.C.; Serrano, S.H.P.; Ferreira, E.I. Voltammetric behavior of nitrofurazone and its hydroxymethyl prodrug with potential anti-chagas activity. J. Braz. Chem. Soc. 2005, 16, 774–782. [Google Scholar] [CrossRef] [Green Version]
- Gal, M.; Kolivoška, W.; Ambrova, M.; Hiveš, J.; Sokolova, R. Correlation of the first reduction potential of selected radiosensitizers determined by cyclic voltammetry with theoretical calculations. Collect. Czechoslov. Chem. Commun. 2011, 76, 937–946. [Google Scholar] [CrossRef]
- Phillips, K.L.; Sandler, S.I.; Chiu, P.C. A method to calculate the one-electron reduction potentials for nitroaromatic compounds based on gas-phase quantum mechanics. J. Comput. Chem. 2011, 32, 226–239. [Google Scholar] [CrossRef]
- Fareghi-Alamdari, R.; Zandi, F.; Keshavarz, M.H. A new model for prediction of one electron reduction potential of nitroaryl compounds. Z. Anorg. Allg. Chem. 2015, 641, 2641–2648. [Google Scholar] [CrossRef]
- Salter-Blanc, A.J.; Bylaska, E.J.; Johnston, H.J.; Tratnyek, P.G. Predicting reduction rates of energetic nitroaromatic compounds using calculated one-electron reduction potentials. Environ. Sci. Technol. 2015, 49, 3778–3786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gooch, A.; Sizochenko, N.; Sviatenko, L.; Gorb, L.; Leszczynski, J. A quantum chemical based toxicity study of estimated reduction potential and hydrophobicity in series of nitroaromatic compounds. SAR QSAR Environ. Res. 2017, 28, 133–150. [Google Scholar] [CrossRef]
- Holtzman, J.; Crankshaw, D.; Peterson, F.; Polnaszek, C. The kinetics of the aerobic reduction of nitrofurantoin by NADPH-cytochrome P-450 c reductase. Mol. Pharmacol. 1981, 20, 669–673. [Google Scholar] [PubMed]
- Darchen, A.; Moinet, C. Mecanisme e.c.e. de reduction du para-dinitrobenzene en para-nitrophenylhydroxylamine. J. Electroanal. Chem. 1977, 78, 81–88. [Google Scholar] [CrossRef]
- Nivinskas, H.; Koder, R.L.; Anusevičius, Ž.; Šarlauskas, J.; Miller, A.F.; Čėnas, N. Quantitative structure-activity relationships in two-electron reduction of nitroaromatic compounds by Enterobacter cloacae NAD(P)H: Nitroreductase. Arch. Biochem. Biophys. 2001, 385, 170–178. [Google Scholar] [CrossRef] [Green Version]
- Laviron, E.; Vallat, A.; Meuner-Prest, R. The reduction mechanisms of aromatic nitrocompounds in aqueous medium. Part V. The reduction of nitrosobenzene between pH 0.4 and 13. J. Electroanal. Chem. 1994, 379, 427–435. [Google Scholar] [CrossRef]
- Valiauga, B.; Misevičienė, L.; Rich, M.H.; Ackerley, D.F.; Šarlauskas, J.; Čėnas, N. Mechanism of two-/four-electron reduction of nitroaromatics by oxygen-insensitive nitroreductases: The role of a non-enzymatic reduction step. Molecules 2018, 23, 1672. [Google Scholar] [CrossRef] [Green Version]
- Uršič, S.; Vrčik, V.; Ljubas, D.; Vinkovič, I. Interaction of L-ascorbate with substituted nitrosobenzenes. Role of the ascorbate 2-OH group in antioxidant reactions. New J. Chem. 1998, 22, 221–223. [Google Scholar] [CrossRef]
- Leung, K.H.; Yao, M.; Stearns, R.; Chiu, S.K. Mechanisms of bioactivation and covalent binding of 2,4,6-trinitrotoluene. Chem. Biol. Interact. 1995, 97, 37–51. [Google Scholar] [CrossRef]
- Becker, A.R.; Sternson, L.A. Oxidation of phenylhydroxylamine in aqueous solution: A model for study of the carcinogenic effect of primary aromatic amines. Proc. Natl. Acad. Sci. USA 1981, 78, 2003–2007. [Google Scholar] [CrossRef] [Green Version]
- Corbett, M.D.; Corbett, B.R. Bioorganic chemistry of the arylhydroxylamine and nitrosoarene functional groups. In Biodegradation of Nitroaromatic Compounds; Spain, J.C., Ed.; Plenum Press: New York, NY, USA, 1995; pp. 151–181. [Google Scholar]
- Helsby, N.A.; Atwell, G.J.; Yang, S.; Palmer, B.D.; Anderson, R.F.; Pullen, S.M.; Ferry, D.M.; Hogg, A.; Wilson, W.R.; Denny, W.A. Aziridinyldinitrobenzamides: Synthesis, and structure-activity relationships for activation by E. coli nitroreductase. J. Med. Chem. 2004, 47, 3295–3307. [Google Scholar] [CrossRef] [PubMed]
- Hay, M.P.; Sykes, B.M.; Denny, W.A.; Wilson, W.R. A 2-nitroimidazole carbomate prodrug of 5-amido-1- (chloromethyl)-3-[(5,6,7-trimethoxyindol-2-yl)carbonyl]-1,2-dihydro-3H-benz[e]indole (amino-seco-cbi-tmi) for use with ADEPT and GDEPT. Bioorg. Med. Chem. Lett. 1999, 9, 2237–2242. [Google Scholar] [CrossRef]
- Sykes, B.M.; Hay, M.P.; Bohinc-Herceg, D.; Helsby, N.A.; O’Connor, C.J.; Denny, W.A. Leaving group effects in reductively triggered fragmentation of 4-nitrobenzyl carbamates. J. Chem. Soc. Perkin Trans. 2000, 1, 1601–1608. [Google Scholar] [CrossRef]
- Borch, R.F.; Liu, J.; Schmidt, J.P.; Markovits, J.T.; Joswig, C.; Gipp, J.J.; Mulcahy, R.T. Synthesis and evaluation of nitroheterocyclic phosphoramidates as hypoxia-selective alkylating agents. J. Med. Chem. 2000, 43, 2258–2265. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Van Valckenborg, E.; Xu, D.; Menu, E.; De Raeve, H.; De Bruyne, E.; Xu, S.; Van Camp, B.; Handisides, D.; Hart, C.P.; et al. Synergistic induction of apoptosis in multiple myeloma cells by bortezomib and hypoxia-activated prodrug TH-302, in vivo and in vitro. Mol. Cancer Ther. 2013, 12, 1763–1773. [Google Scholar] [CrossRef] [Green Version]
- Gruber, T.D.; Krishnamurthy, C.; Grimm, J.B.; Tadross, M.R.; Wysocki, L.M.; Gartner, Z.J.; Lavis, L.D. Cell-specific chemical delivery using a selective nitroreductase-nitroaryl pair. ACS Chem. Biol. 2018, 13, 2888–2896. [Google Scholar] [CrossRef] [Green Version]
- Cyr, A.; Laviron, E.; Lessard, J. Electrochemical behavior of nitrobenzene and phenylhydroxylamine on copper rotating disk electrodes. J. Electroanal. Chem. 1989, 263, 69–78. [Google Scholar] [CrossRef]
- Iwase, K.; Fujinani, N.; Hashimoto, K.; Kamiya, K.; Nakanishi, S. Cooperative electrocatalytic reduction of nitrobenzene to aniline in aqueous solution by copper modified covalent triazine network. Chem. Lett. 2018, 47, 304–307. [Google Scholar] [CrossRef]
- Clarke, E.D.; Wardman, P.; Goulding, K.H. Anaerobic reduction of nitroimidazoles by reduced flavin mononucleotide and by xanthine oxidase. Biochem. Pharmacol. 1980, 29, 2684–2687. [Google Scholar] [CrossRef]
- Porter, T.D.; Kasper, C.B. Coding nucleotide sequence of rat NADPH-cytochrome P-450 reductase cDNA and identification of flavin-binding domains. Proc. Natl. Acad. Sci. USA 1985, 83, 973–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Roberts, D.L.; Paschke, R.; Shea, T.M.; Masters, B.S.S.; Kim, J.-J.P. Three-dimensional structure of NADPH- cytochrome P450 reductase: Prototype for FMN- and FAD-containing enzymes. Proc. Natl. Acad. Sci. USA 1997, 94, 8411–8416. [Google Scholar] [CrossRef] [Green Version]
- Oprian, D.D.; Coon, M.J. Oxidation-reduction states of FMN and FAD in NADPH-cytochrome P-450 reductase during the reduction by NADPH. J. Biol. Chem. 1982, 257, 8935–8944. [Google Scholar] [CrossRef]
- Gutierrez, A.; Paine, M.; Wolf, C.R.; Scrutton, N.S.; Roberts, G.C.K. Relaxation kinetics of cytochrome P450 reductase: Internal electron transfer is limited by conformational change and regulated by coenzyme binding. Biochemistry 2002, 41, 4626–4637. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P. Oxidation-reduction properties of rat liver cytochromes P-450 and NADPH-cytochrome P-450 reductase related to catalysis in reconstituted systems. Biochemistry 1983, 22, 2811–2820. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Sligar, S.G. Modulation of the cytochrome P-450 reductase redox potential by the phospholipid bilayer. Biochemistry 2009, 48, 12104–12112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubbard, P.A.; Shen, A.L.; Paschke, R.; Kasper, C.B.; Kim, J.-J.P. NADPH-cytochrome P450 oxidoreductase. Structural basis for hydride and electron transfer. J. Biol. Chem. 2001, 276, 29163–29170. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.T.; Smith, S.M.E.; Weinberg, J.B.; Montgomery, H.J.; Newman, E.; Guillemette, J.G.; Ghosh, D.K.; Roman, L.J.; Martasek, P.; Salerno, J.C. Thermodynamics of oxidation-reduction reactions in mammalian nitric-oxide synthase isoforms. J. Biol. Chem. 2004, 279, 18759–18766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, T.; Martasek, P.; Omura, T.; Masters, B.S.S. Rapid kinetic studies of electron transfer in the three isoforms of nitric oxide synthase. Biochem. Biophys. Res. Commun. 1999, 265, 184–188. [Google Scholar] [CrossRef]
- Anusevičius, Ž.; Nivinskas, H.; Šarlauskas, J.; Sari, M.A.; Boucher, J.-L.; Čėnas, N. Single-electron reduction of quinone and nitroaromatic xenobiotics by recombinant rat neuronal nitric oxide synthase. Acta Biochim. Pol. 2013, 60, 207–222. [Google Scholar] [CrossRef]
- Cooper, C.E.; Ioannidis, N.; D’mello, R.; Poole, R.K. Haem, flavin and oxygen interactions in Hmp, a flavohemoglobin from Escherichia coli. Biochem. Soc. Trans. 1994, 22, 709–713. [Google Scholar] [CrossRef]
- Nobre, L.S.; Goncalves, V.L.; Saraiva, L.M. Flavohemoglobin of Staphylococcus aureus. Methods Enzymol. 2008, 436, 203–216. [Google Scholar]
- Moussaoui, M.; Misevičienė, L.; Anusevičius, Ž.; Marozienė, A.; Lederer, F.; Baciou, L.; Čėnas, N. Quinones and nitroaromatic compounds as substrates of Staphylococcus aureus flavohemoglobin. Free Radic. Biol. Med. 2018, 123, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Orna, M.; Mason, R. Correlation of kinetic parameters of nitroreductase enzymes with redox properties of nitroaromatic compounds. J. Biol. Chem. 1989, 264, 12379–12384. [Google Scholar] [CrossRef]
- Čėnas, N.; Anusevičius, Ž.; Bironaitė, D.; Bachmanova, G.I.; Archakov, A.I.; Öllinger, K. The electron transfer reactions of NADPH-cytochrome P450 reductase with nonphysiological oxidants. Arch. Biochem. Biophys. 1994, 315, 400–406. [Google Scholar] [CrossRef]
- Nemeikaitė-Čėnienė, A.; Šarlauskas, J.; Jonušienė, V.; Marozienė, A.; Misevičienė, L.; Yantsevich, A.V.; Čėnas, N. Kinetics of flavoenzyme-catalyzed reduction of tirapazamine derivatives: Implications for their proxidant cytotoxicity. Int. J. Mol. Sci. 2019, 20, 4602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemeikaitė-Čėnienė, A.; Šarlauskas, J.; Jonušienė, V.; Misevičienė, L.; Marozienė, A.; Yantsevich, A.V.; Čėnas, N. QSARs in proxidant mammalian cell cytotoxicity of nitroaromatic compounds: The roles of compound lipophilicity and cytochrome P-450 and DT-diaphorase-catalyzed reactions. Chemija 2020, 31, 170–177. [Google Scholar] [CrossRef]
- Li, H.; Lin, D.; Peng, Y.; Zheng, J. Oxidative bioactivation of nitrofurantoin in rat liver microsomes. Xenobiotica 2017, 47, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Le Campion, L.; Delaforge, M.; Noel, J.P.; Ouazzani, J. Metabolism of 14C-labelled 5-nitro-1,2,4-triazol-3-one by rat liver microsomes. Evidence for the participation of cytochromes P450. Eur. J. Biochem. 1996, 248, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P.; Chun, Y.-J.; Kim, D.; Gillam, E.M.J.; Shimada, T. Cytochrome P450 1B1: A target for inhibition in anticarcinogenesis. Mutat. Res. 2003, 523–524, 173–182. [Google Scholar] [CrossRef]
- Pochapski, T.C.; Wong, N.; Zhuang, Y.; Futcher, J.; Pandelia, M.-E.; Teitz, D.R.; Colthart, A.M. NADH reduction of nitroaromatics as a probe for residual ferric form high spin in a cytochrome P450. Biochim. Biophys. Acta 2018, 1866, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Daff, S. NO synthase: Structures and mechanism. Nitric Oxide 2010, 23, 1–11. [Google Scholar] [CrossRef]
- Guan, Z.W.; Kamatani, D.; Kimura, S.; Iyanagi, T. Mechanistic studies on the intramolecular one-electron transfer between the two flavins in the human neuronal nitric oxide synthase and inducible nitric oxide synthase flavin domains. J. Biol. Chem. 2003, 278, 30859–30868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haque, M.M.; Tejero, J.; Bayachou, M.; Kenney, T.; Stuehr, D.J. A cross-domain charge interaction governs the activity of NO synthase. J. Biol. Chem. 2018, 293, 4545–4554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.J.; Yamamoto, K.; Guan, Z.W.; Kimura, T.; Iyanagi, T. Human neuronal nitric oxide synthase can catalyze one-electron reduction of adriamycin: Role of flavin domain. Arch. Biochem. Biophys. 2004, 427, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Ask, K.; Dijols, S.; Giroud, C.; Casse, L.; Frapart, Y.M.; Sari, M.A.; Kim, K.S.; Stuehr, D.J.; Mansuy, D.; Camus, P.; et al. Reduction of nilutamide by NO synthases: Implications for the adverse effects of this nitroaromatic antiandrogen drug. Chem. Res. Toxicol. 2003, 16, 1547–1554. [Google Scholar] [CrossRef]
- Chandor, A.; Dijols, S.; Ramassamy, B.; Frapart, Y.; Mansuy, D.; Stuehr, D.; Helsby, N.; Boucher, J.L. Metabolic activation of the antitumor drug 5-(Aziridin-1-yl)-2,4-dinitrobenzamide (CB1954) by NO synthases. Chem. Res. Toxicol. 2008, 21, 836–843. [Google Scholar] [CrossRef]
- Miller, R.T. Dinitrobenzene-mediated production of peroxynitrite by neuronal nitric oxide synthase. Chem. Res. Toxicol. 2002, 15, 927–934. [Google Scholar] [CrossRef]
- Kumagai, Y.; Kikushima, M.; Nakai, Y.; Shimojo, N.; Kanimoto, M. Neuronal nitric oxide synthase (NNOS) catalyzes one-electron reduction of 2,4,6-trinitrotoluene, resulting in decreased nitric oxide production and increased nNOS gene expression: Implications for oxidative stress. Free Radic. Biol. Med. 2004, 37, 350–357. [Google Scholar] [CrossRef]
- Gardner, A.M.; Martin, L.A.; Gardner, P.R. Steady-state and transient kinetics of Escherichia coli nitric oxide dioxygenase (flavohemoglobin). The B10 tyrosine hydroxyl is essential for dioxygen binding and catalysis. J. Biol. Chem. 2000, 275, 12581–12589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ermler, U.; Siddiqui, R.A.; Cramm, R.; Salzman, A.L. Crystal structure of the flavohemoglobin from Alcaligenes euthrophus at 1.75 Å resolution. EMBO J. 1995, 14, 6067–6077. [Google Scholar] [CrossRef]
- Hanukoglu, I. Interfaces in FAD and NADP binding adrenodoxin reductase—A ubiquitous enzyme. J. Mol. Evol. 2017, 85, 205–218. [Google Scholar] [CrossRef]
- Lambeth, J.D.; Kamin, H. Adrenodoxin reductase and adrenodoxin. Mechanism of reduction of ferricyanide and cytochrome c. J. Biol. Chem. 1977, 252, 2908–2917. [Google Scholar] [CrossRef]
- Lambeth, J.D.; Seybert, D.W.; Kamin, H. Ionic effects on adrenal steroidgenetic electron transport. The role of adrenodoxin as an electron shuttle. J. Biol. Chem. 1979, 254, 7255–7264. [Google Scholar] [CrossRef]
- Hurley, J.K.; Morales, R.; Martinez-Julvez, M.; Brodie, T.B.; Medina, M.; Gomez-Moreno, C.; Tollin, G. Structure-function relationships in Anabaena ferredoxin/ferredoxin:NADP+ reductase electron transfer: Insight from site-directed mutagenesis, transient absorption spectroscopy and X-ray crystallography. Biochim. Biophys. Acta 2002, 1554, 5–21. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Julvez, M.; Nogues, I.; Faro, M.; Hurtley, J.K.; Brodie, T.B.; Sanz-Apariccio, J.A.; Stankovich, M.T.; Medina, M.; Tollin, G.; Gomez-Moreno, C. Role of a cluster of hydrophobic residues near the FAD cofactor in Anabaena PCC7119 ferredoxin-NADP+ reductase for optimal complex formation and electron transfer to ferredoxin. J. Biol. Chem. 2001, 276, 27498–27510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faro, M.; Gómez-Moreno, C.; Stankovich, M.; Medina, M. Role of critical charged residues in reduction potential modulation of ferredoxin-NADP+ reductase. Eur. J. Biochem. 2002, 269, 2656–2661. [Google Scholar] [CrossRef] [Green Version]
- Kimata-Ariga, Y.; Kurisu, G.; Kusunoki, M.; Aoki, S.; Sato, K.; Kobayashi, T.; Kita, K.; Horii, T.; Hase, T. Cloning and characterization of ferredoxin and ferredoxin-NADP+ reductase from human malaria parasite. J. Biochem. 2007, 141, 421–428. [Google Scholar] [CrossRef]
- Balconi, E.; Pennati, A.; Crobu, D.; Pandini, V.; Cerutti, R.; Zanetti, G.; Aliverti, A. The ferredoxin-NADP+ reductase/ ferredoxin electron transfer system of Plasmodium falciparum. FEBS J. 2009, 276, 3825–3836. [Google Scholar] [CrossRef]
- Sled, V.D.; Rudnitzky, N.I.; Hatefi, Y.; Ohnishi, T. Thermodynamic analysis of flavin in mitochondrial NADH:ubiquinone oxidoreductase (complex I). Biochemistry 1994, 33, 10069–10075. [Google Scholar] [CrossRef] [PubMed]
- Ingledew, W.J.; Ohnishi, T. An analysis of some thermodynamic properties of iron-sulfur centres in site I of mitochondria. Biochem. J. 1980, 186, 111–117. [Google Scholar] [CrossRef] [Green Version]
- Dooijewaard, G.; Slater, E.C. Steady-state kinetics of high molecular weight (type-I) NADH dehydrogenase. Biochim. Biophys. Acta 1976, 440, 1–15. [Google Scholar] [CrossRef]
- Čėnas, N.K. Reactions of complex I of mitochondrial electron-transport chain with artificial electron acceptors. Ukr. Biokhimicheskii Zhurnal 1989, 61, 23–29. (In Russian) [Google Scholar]
- Fedor, J.G.; Jones, A.J.Y.; Di Luca, A.; Kaila, V.R.I.; Hirst, J. Correlating kinetic and structural data on ubiquinone binding and reduction by respiratory complex I. Proc. Natl. Acad. Sci. USA 2017, 114, 12737–12742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, K.; Miura, S.; Ichikawa, Y.; Tagawa, S. Interactions of NADPH-adrenodoxin reductase with NADP+ as studied by pulse-radiolysis. Biochemistry 1995, 34, 12932–12936. [Google Scholar] [CrossRef]
- Čėnas, N.K.; Marcinkevičienė, J.A.; Kulys, J.J.; Usanov, S.A. A negative cooperativity between NADPH and adrenodoxin on binding to NADPH:adrenodoxin reductase. FEBS Lett. 1990, 259, 338–340. [Google Scholar] [CrossRef] [Green Version]
- Marcinkevičienė, J.; Čėnas, N.; Kulys, J.; Usanov, S.A.; Sukhova, N.M.; Seleznieva, I.S.; Gryazev, V.F. Nitroreductase reactions of the NADPH–adrenodoxin reductase. Biomed. Biochim. Acta 1990, 49, 167–172. [Google Scholar]
- Fischer, F.; Raimondi, D.; Aliverti, A.; Zanetti, G. Mycobacterium tuberculosis FprA, a novel bacterial NADPH-ferredoxin reductase. Eur. J. Biochem. 2002, 269, 3005–3013. [Google Scholar] [CrossRef]
- McLean, K.; Scrutton, N.S.; Munro, A.W. Kinetic, spectroscopic and thermodynamic characterization of the Mycobacterium tuberculosis adrenodoxin reductase homologue FprA. Biochem. J. 2003, 372, 317–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serre, L.; Vellieux, F.M.; Medina, M.; Gomez-Moreno, C.; Fontecilla-Camps, J.C.; Frey, M. X-ray structure of the ferredoxin:NADP+ reductase from the cyanobacterium Anabaena PCC7119 at 1.8 Å resolution, and crystallographic studies of NADP+ binding at 2.25 Å resolution. J. Mol. Biol. 1996, 263, 20–39. [Google Scholar] [CrossRef]
- Kimata-Ariga, Y.; Yuasa, S.; Saitoh, T.; Fukuyama, H.; Hase, T. Plasmodium-specific basic amino acid residues important for the interaction with ferredoxin on the surface of ferredoxin-NADP+ reductase. J. Biochem. 2018, 164, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Milani, M.; Balconi, E.; Aliverti, A.; Mastrangelo, E.; Seeber, F.; Bolognesi, M.; Zanetti, G. Ferredoxin-NADP+ reductase from Plasmodium falciparum undergoes NADP+-dependent dimerization and inactivation: Functional and crystallographic analysis. J. Mol. Biol. 2007, 367, 501–513. [Google Scholar] [CrossRef]
- Anusevičius, Ž.; Martinez Julvez, M.; Genzor, C.G.; Nivinskas, H.; Gomez-Moreno, C.; Čėnas, N. Electron transfer reactions of Anabaena PCC 7119 ferredoxin:NADP+ reductase with nonphysiological oxidants. Biochim. Biophys. Acta 1997, 1320, 247–255. [Google Scholar] [CrossRef] [Green Version]
- Grellier, P.; Marozienė, A.; Nivinskas, H.; Šarlauskas, J.; Aliverti, A.; Čėnas, N. Antiplasmodial activity of quinones: Roles of aziridinyl substituents and the inhibition of Plasmodium falciparum glutathione reductase. Arch. Biochem. Biophys. 2010, 494, 32–39. [Google Scholar] [CrossRef]
- Lesanavičius, M.; Aliverti, A.; Šarlauskas, J.; Čėnas, N. Reactions of Plasmodium falciparum ferredoxin:NADP+ oxidoreductase with redox cycling xenobiotics: A mechanistic study. Int. J. Mol. Sci. 2020, 21, 3234. [Google Scholar] [CrossRef]
- Vidakovic, M.; Crossnoe, C.R.; Neidre, C.; Kim, K.; Krause, K.L.; Germanas, J.P. Reactivity of reduced [2Fe-2S] ferredoxin parallels host susceptibility to nitroimidazoles. Antimicrob. Agents Chemother. 2003, 47, 302–308. [Google Scholar] [CrossRef] [Green Version]
- Shah, M.M.; Spain, J.C. Elimination of nitrite from the explosive 2,4,6-trinitrophenylmethylnitramine (tetryl) catalyzed by ferredoxin NADP oxidoreductase from spinach. Biochem. Biophys. Res. Commun. 1996, 220, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.E. The NADH:ubiquinone oxidoreductase (complex I) of respiratory chain. Quart. Rev. Biophys. 1992, 25, 253–324. [Google Scholar] [CrossRef] [PubMed]
- Hirst, J. Mitochondrial complex I. Annu. Rev. Biochem. 2013, 82, 551–557. [Google Scholar] [CrossRef]
- Agip, A.-N.A.; Blaza, J.N.; Fedor, J.G.; Hirst, J. Mammalian respiratory complex I through the lens of cryo-EM. Annu. Rev. Biophys. 2019, 48, 165–184. [Google Scholar] [CrossRef]
- Bironaitė, D.A.; Čėnas, N.K.; Kulys, J.J. The rotenone-insensitive reduction of quinones and nitrocompounds by mitochondrial NADH:ubiquinone reductase. Biochim. Biophys. Acta 1991, 1060, 203–209. [Google Scholar] [CrossRef]
- Čėnas, N.K.; Bironaitė, D.A.; Kulys, J.J. On the mechanism of rotenone-insensitive reduction of quinones by mitochondrial NADH-ubiquinone reductase: The high-affinity binding of NAD+ and NADH to the reduced enzyme form. FEBS Lett. 1991, 284, 192–194. [Google Scholar] [CrossRef] [Green Version]
- Bironaitė, D.A.; Čėnas, N.K.; Anusevičius, Ž.J.; Medentsev, A.G.; Akimenko, V.K.; Usanov, A.A. Fungal quinone pigments as oxidizers and inhibitors of mitochondrial NADH-ubiquinone reductase. Arch. Biochem. Biophys. 1992, 297, 253–257. [Google Scholar] [CrossRef]
- Hrdy, I.; Hirt, R.P.; Dolezal, P.; Bardonová, L.; Foster, P.G.; Tachezy, J.; Embley, T.M. Trichomonas hydrogenosomes containthe NADH dehydrogenase module of mitochondrial complex I. Nature 2004, 432, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Do, P.M.; Angerhofer, A.; Hrdy, I.; Bardonova, L.; Ingram, L.O.; Shanmugam, K.T. Engineering Escherichia coli for fermentative dihydrogen production: Potential role of NADH-ferredoxin oxidoreductase from the hydrogenosome of anaerobic protozoa. Appl. Biochem. Biotechnol. 2009, 153, 21–33. [Google Scholar] [CrossRef]
- Graves, K.J.; Novak, J.; Secor, W.E.; Kissinger, P.J.; Schwebke, J.R.; Muzny, C.A. A systematic review of the literature on mechanisms of 5-nitroimidazole resistance in Trichomonas vaginalis. Parasitology 2020, 147, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Yarlett, N.; Gorrell, T.E.; Marczak, R.; Müller, G. Reduction of nitroimidazole derivatives by hydrogenosomal extracts of Trichomonas vaginalis. Mol. Biochem. Pharmacol. 1985, 14, 29–40. [Google Scholar] [CrossRef]
- Smutná, T.; Pilarová, K.; Tarábek, J.; Tachezy, J.; Hrdý, I. Novel functions of an iron-sulfur flavoprotein from Trichomonas vaginalis hydrogenosomes. Antimicrob. Agents Chemother. 2014, 58, 3224–3232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- St. Maurice, M.; Cremades, N.; Croxen, M.A.; Sisson, G.; Sancho, J.; Hoffman, P.S. Flavodoxin:quinone reductase (FqrB): A redox partner of Pyruvate: Ferredoxin oxidoreductase that reversibly couples pyruvate oxidation to NADPH production in Helicobacter pylori and Campylobacter jejuni. J. Bacteriol. 2007, 189, 4764–4773. [Google Scholar] [CrossRef] [Green Version]
- Galano, J.J.; Alias, M.; Perez, R.; Velazquez-Campoy, A.; Hoffman, P.S.; Sancho, J. Improved flavodoxin inhibitors with potential therapeutic effects against Helicobacter pylori infection. J. Med. Chem. 2013, 56, 6248–6258. [Google Scholar] [CrossRef] [PubMed]
- Leitsch, D.; Burgess, A.G.; Dunn, L.A.; Krauer, K.G.; Tan, K.; Duchene, M.; Upcroft, P.; Eckmann, L.; Upcroft, J.A. Pyruvate: Ferredoxin oxidoreductase and thioredoxin reductase are involved in 5-nitroimidazole activation while flavin metabolism is linked to 5-nitroimidazole resistance in Giardia lamblia. J. Antimicrob. Chemother. 2011, 66, 1756–1765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wardman, P.; Anderson, R.F.; Clarke, E.D.; Jones, N.R.; Milchinton, A.I.; Patel, K.B.; Stratford, I.J.; Watts, M.E. Reduction of nitroimidazoles in model chemical and biological systems. III. Effects of basic substituents in nitroimidazole sidechains. Int. J. Radiat. Oncol. Biol. Phys. 1982, 8, 777–780. [Google Scholar] [CrossRef]
- O’Connor, C.J.; McLennan, D.J.; Sutton, B.M.; Denny, W.A.; Wilson, W.R. Effect of reduction potential on the rate of reduction of nitroacridines by xanthine oxidase and by dihydro-flavin mononucleotide. J. Chem. Soc. Perkin Trans. 1991, 951–954. [Google Scholar] [CrossRef]
- Batelli, M.G.; Polito, L.; Bortolotti, M.; Bolognesi, A. Xanthine oxidoreductase in drug metabolism: Beyond a role as a detoxifying enzyme. Curr. Med. Chem. 2016, 23, 4027–4036. [Google Scholar] [CrossRef] [Green Version]
- Harris, C.M.; Massey, V. The oxidative half-reaction of xanthine dehydrogenase with NAD: Reaction kinetics and steady-state mechanism. J. Biol. Chem. 1997, 272, 28335–28341. [Google Scholar] [CrossRef] [Green Version]
- Kutcher, W.W.; McCalla, D.R. Aerobic reduction of 5-nitro-2-furaldehyde by rat liver xanthine dehydrogenase. Biochem. Pharmacol. 1984, 33, 799–805. [Google Scholar] [CrossRef]
- Ueda, O.; Sugihara, K.; Ohta, S.; Kitamura, K. Involvement of molybdenum hydroxylases in reductive metabolism of nitro polycyclic aromatic hydrocarbons in mammalian skin. Drug Metab. Dispos. 2005, 33, 1312–1318. [Google Scholar] [CrossRef]
- Anoz-Carbonell, E.; Timson, D.J.; Pey, A.L.; Medina, M. The catalytic cycle of the antioxidant and cancer associated human NQO1 enzyme: Hydride transfer, conformational dynamics and functional cooperativity. Antioxidants 2020, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Fitzsimmons, A.; Workman, P.; Grever, M.; Paull, K.; Camalier, R.; Lewis, A.D. Reductase enzyme expression across the National Cancer Institute tumor cell line panel—Correlation with sensitivity to mitomycin C and EO9. J. Natl. Cancer Inst. 1996, 88, 259–269. [Google Scholar] [CrossRef]
- Li, R.; Bianchet, M.A.; Talalay, P.; Amzel, L.M. The three-dimensional structure of NAD(P)H:quinone reductase, a flavoprotein involved in cancer chemoprotection and chemotherapy: Mechanism of the two-electron reduction. Proc. Natl. Acad. Sci. USA 1995, 92, 8846–8850. [Google Scholar] [CrossRef] [Green Version]
- Faig, M.; Bianchet, M.A.; Talalay, P.; Chen, S.; Winski, S.; Ross, D.; Amzel, L.M. Structures of recombinant human and mouse NAD(P)H: Quinone oxidoreductases: Species comparison and structural changes with substrate binding and release. Proc. Natl. Acad. Sci. USA 2000, 97, 3177–3182. [Google Scholar] [CrossRef]
- Bianchet, M.A.; Faig, A.; Amzel, L.M. Structure and mechanism of NAD[P}H:quinone acceptor oxidoreductases (NQO). Meth. Enzymol. 2004, 382, 144–174. [Google Scholar]
- Asher, G.; Dym, O.; Tsvetkov, P.; Adler, J.; Shaul, Y. The crystal structure of NAD(P)H quinone oxidoreductase 1 in complex with its potent inhibitor dicoumarol. Biochemistry 2006, 45, 6372–6378. [Google Scholar] [CrossRef]
- Tedeschi, G.; Chen, S.; Massey, V. DT-diaphorase. Redox potential, steady-state, and rapid reaction studies. J. Biol. Chem. 1995, 270, 1198–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anusevičius, Ž.; Šarlauskas, J.; Čėnas, N. Two-electron reduction of quinones by rat liver NAD(P)H: Quinone oxidoreductase: Quantitative structure-activity relationships. Arch. Biochem. Biophys. 2002, 404, 254–262. [Google Scholar] [CrossRef]
- Hubig, S.; Rathore, R.; Kochi, J. Steric control of electron transfer. Changeover from outer-sphere to inner-sphere mechanisms in arene/quinone redox pairs. J. Am. Chem. Soc. 1999, 121, 617–626. [Google Scholar] [CrossRef]
- Misevičienė, L.; Anusevičius, Ž.; Šarlauskas, J.; Čėnas, N. Reduction of nitroaromatic compounds by NAD(P)H:quinone oxidoreductase (NQO1): The role of electron-accepting potency and structural parameters in the substrate specificity. Acta Biochim. Pol. 2006, 53, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Day, M.A.; Jarrom, D.; Christofferson, A.J.; Graziano, A.E.; Anderson, J.L.R.; Searle, P.F.; Hyde, E.I.; White, S.A. The structures of E. coli NfsA bound to the antibiotic nitrofurantoin: To 1,4-benzoquinone and FMN. Biochem. J. 2021, 478, 2601–2617. [Google Scholar] [CrossRef]
- Race, P.R.; Lovering, A.L.; Green, R.M.; Ossor, A.; White, S.A.; Searle, P.F.; Wrighton, C.J.; Hyde, E.I. Structural and mechanistic studies of Escherichia coli nitroreductase with the antibiotic nitrofurazone: Reversed binding orientations in different redox states of the enzyme. J. Biol. Chem. 2005, 280, 13256–13264. [Google Scholar] [CrossRef] [Green Version]
- Jarrom, D.; Jaberipour, M.; Guise, C.P.; Daff, S.N.; White, S.A.; Searle, P.F.; Hyde, E.I. Steady-state and stopped-flow kinetic studies of three Escherichia coli NfsB mutants with enhanced activity for the prodrug CB1954. Biochemistry 2009, 48, 7665–7672. [Google Scholar] [CrossRef]
- Prosser, G.A.; Copp, J.N.; Syddall, S.P.; Williams, E.M.; Smaill, J.B.; Wilson, W.R.; Patterson, A.V.; Ackerley, D.F. Discovery and evaluation of Escherichia coli nitroreductases that activate the anti-cancer prodrug CB1954. Biochem. Pharmacol. 2010, 79, 678–687. [Google Scholar] [CrossRef]
- Koder, R.L.; Haynes, C.A.; Rodgers, M.E.; Rodgers, D.W.; Miller, A.F. Flavin thermodynamics explain the oxygen insensitivity of enteric nitroreductases. Biochemistry 2002, 41, 14197–14205. [Google Scholar] [CrossRef]
- Pitsawong, W.; Hoben, J.P.; Miller, A.F. Understanding the broad substrate repertoire of nitroreductase based on its kinetic mechanism. J. Biol. Chem. 2014, 289, 15203–15214. [Google Scholar] [CrossRef] [Green Version]
- Valiauga, B. Studies of Reduction Mechanisms of Quinones and Nitroaromatic Compounds by Flavoenzymes Dehydrogenases-Transhydrogenases. Ph.D. Thesis, Vilnius University, Vilnius, Lithuania, 2020. [Google Scholar]
- Valiauga, B.; Williams, E.M.; Ackerley, D.F.; Čėnas, N. Reduction of quinones and nitroaromatic compounds by Escherichia coli nitroreductase A (NfsA): Characterization of kinetics and substrate specificity. Arch. Biochem. Biophys. 2017, 624, 14–22. [Google Scholar] [CrossRef]
- Miller, A.F.; Park, J.T.; Ferguson, K.L.; Pitsawong, W.; Bommarius, A.S. Informing efforts to develop nitroreductase for amine production. Molecules 2018, 23, 211. [Google Scholar] [CrossRef] [Green Version]
- Couturier, J.; Prosper, P.; Winger, A.M.; Hecker, A.; Hirasawa, M.; Knaff, D.B.; Gans, P.; Jacquot, J.-P.; Navazza, A.; Haouz, A.; et al. In the absence of thioredoxins, what are the reductants for peroxiredoxins in Thermotoga maritima? Antioxid. Redox Signal. 2013, 18, 1613–1622. [Google Scholar] [CrossRef] [PubMed]
- Anusevičius, Ž.; Misevičienė, L.; Šarlauskas, J.; Rouhier, N.; Jacquot, J.-P.; Čėnas, N. Quinone- and nitroreductase reactions of Thermotoga maritima peroxiredoxin-nitroreductase hybrid enzyme. Arch. Biochem. Biophys. 2012, 528, 50–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Wu, K.; Zhang, D.; Sherman, M.; Knox, R.; Yang, C.S. Molecular characterization of binding of substrates and inhibitors to DT-diaphorase: Combined approach involving site-directed mutagenesis, inhibitor-binding analysis, and computer modeling. Mol. Pharmacol. 1999, 56, 272–278. [Google Scholar] [CrossRef]
- Anusevičius, Ž.; Šarlauskas, J.; Nivinskas, H.; Segura-Aguilar, J.; Čėnas, N. DT-diaphorase catalyzes N-denitration and redox cycling of tetryl. FEBS Lett. 1998, 436, 144–148. [Google Scholar] [CrossRef] [Green Version]
- Šarlauskas, J.; Dičkancaitė, E.; Nemeikaitė, A.; Anusevičius, Ž.; Nivinskas, H.; Segura-Aguilar, J.; Čėnas, N. Nitrobenzimidazoles as substrates for DT-diaphorase and redox cycling compounds: Their enzymatic reactions and cytotoxicity. Arch. Biochem. Biophys. 1997, 346, 219–229. [Google Scholar] [CrossRef]
- Akiva, E.; Copp, J.N.; Tokuriki, N.; Babbitt, P.C. Evolutionary and molecular foundations of multiple contemporary functions of the nitroreductase superfamily. Proc. Natl. Acad. Sci. USA 2017, 114, E9549–E9558. [Google Scholar] [CrossRef] [Green Version]
- Paterson, E.; Boucher, S.; Lambert, I. Regulation of the nfsA Gene in Escherichia coli by SoxS. J. Bacteriol. 2002, 184, 51–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zenno, S.; Koike, H.; Kumar, A.N.; Jayaraman, R.; Tanokura, M.; Saigo, K. Biochemical characterization of NfsA, the Escherichia coli major nitroreductase exhibiting a high amino acid sequence homology to Frp, a Vibrio harveyi flavin oxidoreductase. J. Bacteriol. 1996, 178, 4508–4514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zenno, S.; Kobori, T.; Tanokura, M.; Saigo, K. Conversion of NfsA, the major Escherichia coli nitroreductase, to a flavin reductase with an activity similar to that of Frp, a flavin reductase in Vibrio harveyi, by a single amino acid substitution. J. Bacteriol. 1998, 180, 422–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parkinson, G.; Skelly, J.; Neidle, S. Crystal structure of FMN-dependent nitroreductase from Escherichia coli B: A prodrug-activating enzyme. J. Med. Chem. 2000, 43, 3624–3631. [Google Scholar] [CrossRef]
- Johansson, E.; Parkinson, G.N.; Denny, W.A.; Neidle, S. Studies on the nitroreductase prodrug-activating system. Crystal structures of complexes with the inhibitor dicoumarol and dinitrobenzamide prodrugs and of the enzyme active form. J. Med. Chem. 2003, 46, 4009–4020. [Google Scholar] [CrossRef]
- Anlezark, G.M.; Melton, R.G.; Sherwood, R.F.; Wilson, W.R.; Denny, W.A.; Palmer, B.D.; Knox, R.J.; Friedlos, F.; Williams, A. Bioactivation of dinitrobenzamide mustards by an E. coli B nitroreductase. Biochem. Pharmacol. 1995, 50, 609–618. [Google Scholar] [CrossRef]
- Koder, R.L.; Miller, A.F. Steady state kinetic mechanism, stereospecificity, substrate and inhibitor specificity of Enterobacter cloacae nitroreductase. Biochim. Biophys. Acta 1998, 1387, 394–405. [Google Scholar] [CrossRef]
- Nivinskas, H.; Staškevičienė, S.; Šarlauskas, J.; Koder, R.L.; Miller, A.F.; Čėnas, N. Two-electron reduction of quinones by Enterobacter cloacae NAD(P)H:nitroreductase: Quantitative structure-activity relationships. Arch. Biochem. Biophys. 2002, 403, 249–258. [Google Scholar] [CrossRef]
- Yanto, Y.; Hall, M.; Bommarius, A.S. Nitroreductase from Salmonella typhimurium: Characterization and catalytic activity. Org. Biomol. Chem. 2010, 8, 1826–1832. [Google Scholar] [CrossRef]
- Kobori, T.; Sasaki, H.; Lee, W.C.; Zenno, S.; Saigo, K.; Murphy, M.; Tanokura, M. Structure and site-directed mutagenesis of a flavoprotein from Escherichia coli that reduces nitrocompounds: Alteration of pyridine nucleotide binding by a single amino acid substitution. J. Biol. Chem. 2001, 276, 2816–2823. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Zhan, J.; Bai, J.; Liu, P.; Xue, Y.; Yang, Q. Residue Phe42 is critical for the catalytic activity of Escherichia coli major nitroreductase NfsA. Biotechnol. Lett. 2013, 35, 1693–1700. [Google Scholar] [CrossRef]
- Williams, E.M. Development of Bacterial Nitroreductase Enzymes for Noninvasive Imaging in Cancer Gene Therapy. Ph.D. Thesis, Victoria University, Wellington, Australia, 2013. [Google Scholar]
- Rich, M.H.; Sharrock, A.V.; Hall, K.R.; Ackerley, D.F.; MacKichan, J.K. Evaluation of NfsA-like nitroreductases from Neisseria meningitidis and Bartonella henselae for enzyme-prodrug therapy, targeted cellular ablation, and dinitrotoluene bioremediation. Biotechnol. Lett. 2018, 40, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Manina, G.; Bellinzoni, M.; Pasca, M.R.; Neres, J.; Milano, A.; Ribeiro, A.L.J.L.; Buroni, S.; Skovierova, H.; Dianiskova, P.; Mikusova, K.; et al. Biological and structural characterization of the Mycobacterium smegmatis nitroreductase NfnB, and its role in benzothiazinone resistance. Mol. Microbiol. 2010, 77, 1172–1185. [Google Scholar] [CrossRef]
- Olekhnovich, I.N.; Goodwin, A.; Hoffman, P.S. Characterization of the NAD(P)H oxidase and metronidazole reductase activities of the RdxA nitroreductase of Helicobacter pylori. FEBS J. 2009, 276, 3354–3364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Júlvez, M.; Rojas, A.L.; Olekhnovich, I.; Angarica, V.E.; Hoffman, P.S.; Sancho, J. Structure of RdxA—An oxygen-intensitive nitroreductase essential for metronidazole activation in Helicobacter pylori. FEBS J. 2012, 279, 4306–4317. [Google Scholar] [CrossRef] [Green Version]
- Sisson, G.; Goodwin, A.; Raudonikiene, A.; Hughes, N.J.; Mukhopadhyay, A.K.; Berg, D.E.; Hoffman, P.S. Enzymes associated with reductive activation and action of nitazoxanide, nitrofurans, and metronidazole in Helicobacter pylori. Antimicrob. Agents Chemother. 2002, 46, 2116–2123. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, S.R.; Bot, C.; Kelly, J.M.; Hall, B.S. Trypanocidal activity of nitroaromatic prodrugs: Current treatments and future perspectives. Curr. Top. Med. Chem. 2011, 11, 2072–2084. [Google Scholar] [CrossRef]
- Hall, B.S.; Bot, C.; Wilkinson, S.R. Nifurtimox activation by trypanosomal type I nitroreductase generates cytotoxic nitrile metabolites. J. Biol. Chem. 2011, 286, 130088–130095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, B.S.; Meredith, E.L.; Wilkinson, S.R. Targeting the substrate preference of a tupe 1 nitroreductaseto develop antitrypanosomal quinone-based prodrugs. Antimicrob. Agents Chemother. 2011, 56, 5821–5830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voak, A.A.; Gobalakrishnapillai, V.; Seifert, K.; Balczo, E.; Hu, L.; Hall, B.S.; Wilkinson, S.R. An essential type I nitroreductase from Leishmania major can be used to activate leishmanicidal prodrugs. J. Biol. Chem. 2013, 288, 28466–28476. [Google Scholar] [CrossRef] [Green Version]
- Wyllie, S.; Patterson, S.; Stojanovski, L.; Simeons, F.R.C.; Norval, S.; Kime, R.; Read, K.D.; Fairlamb, A.H. The anti-trypanosome drug fexinidazole shows potential for treating visceral leishmaniasis. Sci. Transl. Med. 2012, 4, 119re1. [Google Scholar] [CrossRef] [Green Version]
- Wyllie, S.; Roberts, A.J.; Norval, S.; Patterson, S.; Foth, B.J.; Berriman, M.; Read, K.D.; Fairlamb, A.H. Activation of bicyclic nitro-drugs by a novel nitroreductase (NTR2) in Leishmania. PloS Pathog. 2016, 12, e1005971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaignon, P.; Cortial, S.; Ventura, A.P.; Lopes, P.; Halgand, F.; Laprevote, O.; Ouazzani, J. Purification and identification of a Bacillus nitroreductase: Potential use in 3,5-DNBTF biosensoring system. Enzyme Microb. Technol. 2006, 39, 1499–1506. [Google Scholar] [CrossRef]
- Crofts, T.S.; Sontha, P.; King, A.O.; Wang, B.; Biddy, B.A.; Zanolli, N.; Gaumnitz, J.; Dantas, G. Discovery and characterization of a nitroreductase capable of conferring bacterial resistance to chloramphenicol. Cell Chem. Biol. 2019, 26, 559–570. [Google Scholar] [CrossRef]
- Gurumurthy, M.; Mukherjee, T.; Dowd, C.; Singh, R.; Niyomrattanakit, P.; Tay, J.A.; Nayar, A.; Lee, Y.S.; Cherian, S.; Boshoff, H.I.; et al. Substrate specificity of the deazaflavin-dependent nitroreductase from Mycobacterium tuberculosis responsible for the bioactivation of bicyclic nitroimidazoles. FEBS J. 2012, 279, 113–125. [Google Scholar] [CrossRef]
- Guise, C.P.; Abbattista, M.R.; Singleton, R.S.; Holford, S.D.; Connolly, J.; Dachs, G.U.; Fox, S.B.; Pollock, R.; Harvey, J.; Guilford, P.; et al. The bioreductive prodrug PR-104A is activated under aerobic conditions by human aldo-keto reductase 1C3. Cancer Res. 2010, 70, 1573–1584. [Google Scholar] [CrossRef] [Green Version]
- Williams, C.H., Jr. Lipoamide dehydrogenase, glutathione reductase, thioredoxin reductase, and mercuric ion reductase—A family of flavoenzyme transhydrogenases. In Chemistry and Biochemistry of Flavoenzymes, 2nd ed.; Miiller, F., Ed.; CRC Press: Boca Raton, FL, USA, 1992; Volume 3, pp. 121–211. [Google Scholar]
- Argyrou, A.; Blanchard, J.S. Flavoprotein disulfide reductases: Advances in chemistry and function. Prog. Nucleic Acid Res. Mol. Biol. 2004, 78, 89–142. [Google Scholar]
- Nauser, T.; Dockheer, S.; Kissner, R.; Koppenol, W.H. Catalysis of electron transfer by selenocysteine. Biochemistry 2006, 45, 6038–6043. [Google Scholar] [CrossRef]
- Karplus, P.; Pai, E.F.; Schulz, G.E. A crystallographic study of the glutathione binding site of glutathione reductase at 0.3-nm resolution. Eur. J. Biochem. 1989, 178, 693–703. [Google Scholar] [CrossRef]
- Kuriyan, J.; Kong, X.; Krishna, T.; Sweet, R.; Murgolo, N.; Field, H.; Cerami, A.; Henderson, G. X-ray structure of trypanothione reductase from Crithidia fasciculata at 2.4-Å resolution. Proc. Natl. Acad. Sci. USA 1991, 88, 8764–8768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarma, G.N.; Savvides, S.N.; Becker, K.; Schirmer, M.; Schirmer, R.H.; Karplus, P.A. Glutathione reductase of the malarial parasite Plasmodium falciparum: Crystal structure and inhibitor development. J. Mol. Biol. 2003, 328, 893–907. [Google Scholar] [CrossRef]
- Sullivan, F.X.; Sobolov, S.B.; Bradley, M.; Walsh, C.T. Mutational analysis of parasite tripanothione reductase: Acquisition of glutathione reductase activity in triple mutant. Biochemistry 1991, 30, 2761–2767. [Google Scholar] [CrossRef] [PubMed]
- Krauth-Siegel, R.L.; Ender, S.B.; Henderson, G.B.; Fairamb, A.H.; Schirmer, R.H. Trypanothione reductase from Trypanosoma cruzi. Purification and characterization of the crystalline enzyme. Eur. J. Biochem. 1987, 164, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Čėnas, N.K.; Arscott, D.; Williams, C.H., Jr.; Blanchard, J.S. Mechanism of reduction of quinones by Trypanosoma congolense trypanothione reductase. Biochemistry 1994, 33, 2509–2515. [Google Scholar] [CrossRef]
- Veine, D.; Arscott, L.; Williams, C.H., Jr. Redox potentials for yeast, Escherichia coli and human glutathione reductase relative to the NAD+/NADH redox couple: Enzyme forms active in catalysis. Biochemistry 1998, 37, 15575–15582. [Google Scholar] [CrossRef]
- Salmon-Chemin, L.; Buisine, E.; Yardley, V.; Kohler, S.; Debreu, M.A.; Ladry, V.; Sergheraert, C.; Croft, S.L.; Krauth-Siegel, R.L.; Davioud-Charvet, E. 2- And 3-substituted 1,4-naphthoquinone derivatives as subversive substrates of trypanothione reductase and lipoamide dehydrogenase fron Trypanosoma cruzi: Synthesis and correlation between redox cycling activities and in vitro cytotoxicity. J. Med. Chem. 2001, 44, 548–565. [Google Scholar] [CrossRef] [PubMed]
- Marozienė, A.; Lesanavičius, M.; Davioud-Charvet, E.; Aliverti, A.; Grellier, P.; Šarlauskas, J.; Čėnas, N. Antiplasmodial activity of nitroaromatic compounds: Correlation with their reduction potential and inhibitory action on Plasmodium falciparum glutathione reductase. Molecules 2019, 24, 4509. [Google Scholar] [CrossRef] [Green Version]
- Bulger, J.E.; Brandt, K.G. Yeast glutathione reductase. II. Interaction of dinucleotide phosphate. J. Biol. Chem. 1971, 246, 5578–5587. [Google Scholar] [CrossRef]
- Böhme, C.C.; Arscott, L.D.; Becker, K.; Schirmer, R.H.; Williams, C.H., Jr. Kinetic characterization of gliutathione reductase from malarial parasite Plasmodium falciparum. Comparison with the human enzyme. J. Biol. Chem. 2000, 275, 37317–37323. [Google Scholar] [CrossRef] [Green Version]
- Zheng, R.J.; Čėnas, N.; Blanchard, J.S. Catalytic and potentiometric characterization of E201 and E201Q mutants of Trypanosoma congolense trypanothione reductase. Biochemistry 1995, 34, 12697–12703. [Google Scholar] [CrossRef]
- Henderson, G.B.; Ulrich, P.; Fairlamb, A.H.; Rosenberg, I.; Pereira, M.; Sela, M.; Cerami, A. “Subversive” substrates of the enzyme trypanothione disulfide reductase: Alternative approach to chemotherapy of Chagas disease. Proc. Natl. Acad. Sci. USA 1988, 85, 5374–5378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Čėnas, N.; Bironaitė, D.; Dičkancaitė, E.; Anusevičius, Ž.; Šarlauskas, J.; Blanchard, J.S. Chinifur, a selective inhibitor and subversive substrate for Trypanosoma congolense trypanothione reductase. Biochem. Biophys. Res. Commun. 1994, 204, 224–229. [Google Scholar] [CrossRef]
- Čėnas, N.K.; Bironaitė, D.A.; Kulys, J.J.; Sukhova, N.M. Interaction of nitrofurans with glutathione reductase. Biochim Biophys. Acta 1991, 1073, 195–199. [Google Scholar]
- Bauer, H.; Fritz-Wolf, K.; Winzer, A.; Kühner, S.; Little, S.; Yardley, V.; Vezin, H.; Palfey, B.; Schirmer, R.H.; Davioud-Charvet, E. A fluoro analogue of the menadione derivative 6-[2′-(3′-methyl)-1′,4′-naphthoquinolyl]hexanoic acid is a suicide substrate of glutathione reductase. Crystal structure of the alkylated human enzyme. J. Am. Chem. Soc. 2006, 23, 10784–10794. [Google Scholar] [CrossRef]
- Blumenstiel, K.; Schöneck, R.; Yardley, V.; Croft, S.L.; Krauth-Siegel, R.L. Nitrofuran drugs as common subversive substrates of Trypanosoma cruzi lipoamide dehydrogenase and lipoamide dehydrogenase. Biochem. Pharmacol. 1999, 58, 1791–1799. [Google Scholar] [CrossRef]
- Taylor, M.C.; Kelly, J.M.; Chapman, C.J.; Fairlamb, A.H.; Miles, M.A. The structure, organization, and expression of the Leishmania donovani gene encoding trypanothione reductase. Mol. Biochem. Parasitol. 1994, 64, 293–301. [Google Scholar] [CrossRef]
- Belorgey, D.; Lanfranchi, D.A.; Davioud-Charvet, E. 1,4-Naphthoquinones and other NADPH-dependent glutathione reductase-catalyzed redox cyclers as antimalarial agents. Curr. Pharm. Des. 2013, 19, 2512–2528. [Google Scholar] [CrossRef]
- Vienožinskis, J.; Butkus, A.; Čėnas, N.; Kulys, J. The mechanism of quinone reductase reaction of pig heart lipoamide dehydrogenase. Biochem. J. 1990, 269, 101–105. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.S. Nitroreductase activity of heart lipoamide dehydrogenase. Biochem. J. 1987, 242, 447–452. [Google Scholar] [CrossRef] [Green Version]
- Argyrou, A.; Sun, G.; Palfey, B.A.; Blanchard, J.S. Catalysis of diaphorase reactions by Mycobacterium tuberculosis lipoamide dehydrogenase occurs at the EH4 level. Biochemistry 2003, 42, 2218–2228. [Google Scholar] [CrossRef]
- Arner, E.S.; Holmgren, A. Physiological function of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 2000, 267, 6102–6109. [Google Scholar] [CrossRef]
- Florencio, F.; Yee, B.; Johnson, T.; Buchanan, B.B. An NADP/thioredoxin system in leaves: Purification and characterization of NADP-thioredoxin reductase and thioredoxin h from spinach. Arch. Biochem. Biophys. 1988, 266, 496–507. [Google Scholar] [CrossRef]
- Meyer, Y.; Buchanan, B.B.; Vignols, F.; Reichheld, J.P. Thioredoxins and glutaredoxins: Unifying elements in redox biology. Annu. Rev. Genet. 2009, 43, 335–367. [Google Scholar] [CrossRef]
- Lincoln, D.T.; Ali Emadi, E.M.; Tonissen, K.F.; Clarke, F.M. The thioredoxin-thioredoxin reductase system: Over-expression in human cancer. Anticancer Res. 2003, 23, 2425–2433. [Google Scholar]
- Arscott, L.; Gromer, S.; Schirmer, R.; Becker, K.; Williams, C.H., Jr. The mechanism of thioredoxin reductase from human placenta is similar to the mechanisms of lipoamide dehydrogenase and glutathione reductase and is distinct from the mechanism of thioredoxin reductase from Escherichia coli. Proc. Natl. Acad. Sci. USA 1997, 94, 3621–3626. [Google Scholar] [CrossRef] [Green Version]
- Sandalova, T.; Zhong, L.; Lindqvist, Y.; Holmgren, A.; Schneider, G. Three-dimensional structure of a mammalian thioredoxin reductase: Implications of mechanism and evolution of a selenocysteine-dependent enzyme. Proc. Natl. Acad. Sci. USA 2001, 98, 9533–9538. [Google Scholar] [CrossRef] [Green Version]
- Čėnas, N.; Nivinskas, H.; Anusevičius, Ž.; Šarlauskas, J.; Lederer, F.; Arner, E.S.J. Interactions of quinones with thioredoxin reductase: A challenge to the antioxidant role of the mammalian selenoprotein. J. Biol. Chem. 2004, 279, 2583–2592. [Google Scholar] [CrossRef] [Green Version]
- Čėnas, N.; Prast, S.; Nivinskas, H.; Šarlauskas, J.; Arner, E.S.J. Interactions of nitroaromatic compounds with the mammalian selenoprotein thioredoxin reductase and the relation to induction of apoptosis in human cancer cells. J. Biol. Chem. 2006, 281, 5593–5603. [Google Scholar] [CrossRef] [Green Version]
- Millet, R.; Urig, S.; Jacob, J.; Amtmann, E.; Moulinoux, J.P.; Gromer, S.; Becker, K.; Davioud-Charvet, E. Synthesis of 5-nitro-2-furancarbohydrazides and their cis-diamminedichloroplatinum complexes as bitopic and irreversible human thioredoxin reductase inhibitors. J. Med. Chem. 2005, 48, 7024–7039. [Google Scholar] [CrossRef]
- Nordberg, J.; Arnér, E.S. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free. Radic. Biol. Med. 2001, 31, 1287–1312. [Google Scholar] [CrossRef]
- Stafford, W.C.; Peng, X.; Olofsson, M.H.; Zhang, X.; Luci, D.K.; Lu, L.; Cheng, Q.; Tresauges, L.; Dexheimer, T.S.; Coussens, N.P.; et al. Irreversible inhibition of cytosolic thioredoxin reductase 1 as a mechanistic basis for anticancer therapy. Sci. Transl. Med. 2018, 10, eaaf7444. [Google Scholar] [CrossRef] [Green Version]
- Gencheva, R.; Cheng, Q.; Arner, E.S.J. Efficient selenocysteine-dependent reduction of toxoflavin by mammalian thioredoxin reductase. Biochim. Biophys. Acta 2018, 1862, 2511–2517. [Google Scholar] [CrossRef]
- Wang, P.F.; Arscott, L.D.; Gilberger, T.W.; Müller, S.; Williams, C.H., Jr. Thioredoxin reductase from Plasmodium falciparum: Evidence for interaction between the C-terminal cysteine residues and the active site disulfide-dithiol. Biochemistry 1999, 38, 3187–3196. [Google Scholar] [CrossRef] [PubMed]
- Morin, C.; Besset, T.; Moutet, J.C.; Fayolle, M.; Brückner, M.; Limosin, D.; Becker, K.; Davioud-Charvet, E. The aza-analogues of 1,4-naphthoquinones are potent substrates and inhibitors of plasmodial thioredoxin and glutathione reductase and of human erythrocyte glutathione reductase. Org. Biomol. Chem. 2008, 6, 2731–2742. [Google Scholar] [CrossRef]
- O’Donnell, M.E.; Williams, C.H., Jr. Graphical analysis of interactions between oxidation-reduction sites in two site oxidation-reduction proteins. Anal. Biochem. 1984, 136, 235–246. [Google Scholar] [CrossRef] [Green Version]
- Williams, C.H., Jr. Mechanism and structure of thioredoxin reductase from Escherichia coli. FASEB J. 1995, 13, 1267–1276. [Google Scholar] [CrossRef] [Green Version]
- Lennon, B.W.; Williams, C.H., Jr. Reductive half-reaction of thioredoxin reductase from Escherichia coli. Biochemistry 1997, 36, 9464–9477. [Google Scholar] [CrossRef]
- Mulrooney, S.; Williams, C.H., Jr. Evidence for two conformational states of thioredoxin reductase from Escherichia coli: Use of intrinsic and extrinsic quenchers of flavin fluorescence as probes to observe domain rotation. Protein Sci. 1997, 6, 2188–2195. [Google Scholar] [CrossRef] [Green Version]
- Zanetti, G.; Williams, C.H., Jr.; Massey, V. Influence of photoirradiation on the oxidation-reduction state of thioredoxin reductase. J. Biol. Chem. 1968, 243, 4013–4019. [Google Scholar] [CrossRef]
- Dai, S.; Saarinen, M.; Ramaswami, S.; Meyer, Y.; Jacquot, J.-P.; Eklund, H. Crystal structure of Arabidopsis thaliana NADPH dependent thioredoxin reductase at 2.5 Å resolution. J. Mol. Biol. 1996, 264, 1044–1057. [Google Scholar] [CrossRef] [PubMed]
- Bironaitė, D.; Anusevičius, Ž.; Jacquot, J.-P.; Čėnas, N. Interaction of quinones with Arabidopsis thaliana thioredoxin reductase. Biochim. Biophys. Acta 1998, 1383, 82–92. [Google Scholar] [CrossRef]
- Miškinienė, V.; Šarlauskas, J.; Jacquot, J.-P.; Čėnas, N. Nitroreductase reactions of Arabidopsis thaliana thioredoxin reductase. Biochim. Biophys. Acta 1998, 1366, 275–283. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Ma, K. Characterization of a thioredoxin-thioredoxin reductase system from the hyperthermophilic bacterium Thermotoga maritima. J. Bacteriol. 2010, 192, 1370–1376. [Google Scholar] [CrossRef] [Green Version]
- Valiauga, B.; Rouhier, N.; Jacquot, J.-P.; Čėnas, N. Quinone- and nitroreductase reactions of Thermotoga maritima thioredoxin reductase. Acta Biochim. Pol. 2015, 62, 303–309. [Google Scholar] [CrossRef]
- Valiauga, B.; Rouhier, N.; Jacquot, J.-P.; Čėnas, N. Characterization of redox properties of FAD cofactor of Thermotoga maritima thioredoxin reductase. Chemija 2020, 31, 191–195. [Google Scholar] [CrossRef]
- Leitsch, D.; Kolarich, D.; Binder, M.; Stadlmann, J.; Altmann, F.; Duchêne, M. Trichomonas vaginalis: Metronidazole and other nitroimidazole drugs are reduced by the flavin enzyme thioredoxin reductase and disrupt the cellular redox system. Implication for nitroimidazole toxicity and resistance. Mol. Microbiol. 2009, 72, 518–536. [Google Scholar] [CrossRef]
- Leitsch, D.; Müller, J.; Müller, N. Evaluation of Giardia lamblia thioredoxin reductase as drug activating enzyme and as drug target. Int. J. Parasitol. Drug Resist. 2016, 6, 148–153. [Google Scholar] [CrossRef] [Green Version]
- Jönsson, T.J.; Ellis, H.R.; Poole, L.B. Cysteine reactivity and thiol-disulfide interchange pathways in AhpF and AhpC of the bacterial alkyl hydroperoxide reductase system. Biochemistry 2007, 46, 5709–5721. [Google Scholar] [CrossRef] [Green Version]
- Le, V.V.H.; Davies, I.G.; Moon, C.D.; Wheeler, D.; Biggs, P.J.; Rakonjac, J. Novel 5-nitrofuran-activating reductase of Escherichia coli. Antimicrob. Agents Chemother. 2019, 63, e00868-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, G.E.; Clarke, E.D.; Jacobs, R.S.; Stratford, I.J.; Wallace, R.G.; Wardman, P.; Watts, M.E. Mammalian cell toxicity of nitro compounds: Dependence upon reduction potential. Biochem. Biophys. Res. Commun. 1976, 72, 824–829. [Google Scholar] [CrossRef]
- Adams, G.E.; Clarke, E.D.; Gray, P.; Jacobs, R.S.; Stratford, I.J.; Wardman, P.; Watts, M.E.; Parrick, J.; Wallace, R.G.; Smithen, C.E. Structure-activity relationships in the development of hypoxic cell radiosensitizers. II. Cytotoxicity and therapeutic ratio. Int. J. Radiat. Biol. 1979, 35, 151–160. [Google Scholar]
- Miškinienė, V.; Sergedienė, E.; Nemeikaitė, A.; Segura-Aguilar, J.; Čėnas, N. Role of redox cycling and activation by DT-diaphorase in cytotoxicity of 5-(aziridin-1-yl)-2,4-dinitrobenzamide (CB-1954) and its analogs. Cancer Lett. 1999, 146, 217–222. [Google Scholar] [CrossRef]
- Miliukienė, V.; Čėnas, N. Cytotoxicity of nitroaromatic explosives and their biodegradation products in mice splenocytes: Implications for their immunotoxicity. Z. Naturforsch. C 2008, 63, 519–525. [Google Scholar] [CrossRef] [Green Version]
- Wilson, W.R.; Anderson, R.F.; Denny, W.A. Hypoxia-selective antitumor agents. 1. Relationships between structure, redox properties and hypoxia-selective cytotoxicity for 4-substituted derivatives of nitracrine. J. Med. Chem. 1989, 32, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Čėnas, N.; Nemeikaitė-Čėnienė, A.; Sergedienė, E.; Nivinskas, H.; Anusevičius, Ž.; Šarlauskas, J. Quantitative structure-activity relationships in enzymatic single-electron reduction of nitroaromatic explosives: Implications for their cytotoxicity. Biochim. Biophys. Acta 2001, 1528, 31–38. [Google Scholar] [CrossRef]
- Šarlauskas, J.; Nemeikaitė-Čėnienė, A.; Anusevičius, Ž.; Misevičienė, L.; Marozienė, A.; Markevičius, A.; Čėnas, N. Enzymatic redox properties of novel nitrotriazole explosives: Implications for their toxicity. Z. Naturforsch. C 2004, 59, 399–404. [Google Scholar] [CrossRef]
- Šarlauskas, J.; Nemeikaitė-Čėnienė, A.; Anusevičius, Ž.; Misevičienė, L.; Martines-Julvez, M.M.; Medina, M.; Gomez-Moreno, C.; Čėnas, N. Flavoenzyme-catalyzed redox cycling of hydroxylamino and amino metabolites of 2,4,6-trinitrotoluene for their cytotoxicity. Arch. Biochem. Biophys. 2004, 425, 184–192. [Google Scholar] [CrossRef]
- Nemeikaitė-Čėnienė, A.; Šarlauskas, J.; Misevičienė, L.; Anusevičius, Ž.; Marozienė, A.; Čėnas, N. Enzymatic redox reactions of explosive 4,6-dinitrobenzofuran (DNBF): Implications for its toxic action. Acta Biochim. Pol. 2004, 51, 1081–1086. [Google Scholar]
- Cabanillas Stanchi, K.M.; Bruchelt, G.; Handgretinger, R.; Holzer, U. Nifurtimox reduces N-Myc expression and aerobic glycolysis in neuroblastoma. Cancer Biol. Ther. 2015, 16, 1353–1363. [Google Scholar] [CrossRef] [Green Version]
- Du, M.; Zhang, L.; Scorsone, K.A.; Woodfield, S.E.; Zage, P.E. Nifurtimox is effective against tumour cells and is synergistic with buthionine sulfoximine. Sci. Rep. 2016, 6, 27458. [Google Scholar] [CrossRef] [Green Version]
- Siim, B.G.; Atwell, G.J.; Anderson, R.F.; Wardman, P.; Pullen, S.M.; Wilson, W.R.; Denny, W.A. Hypoxia-selective antitumor agents. 15. Modification of rate of nitroreduction and extent of lysosomal uptake by polysubstitution of 4-(alkylamino)-5-nitroquinoline bioreductive drugs. J. Med. Chem. 1997, 40, 1381–1390. [Google Scholar] [CrossRef]
- Tercel, M.; Wilson, W.R.; Anderson, R.F.; Denny, W.A. Hypoxia-selective antitumor agents. 12. Nitrobenzyl quaternary salts as bioreductive prodrug of the alkylating agent mechlorethamine. J. Med. Chem. 1996, 39, 1084–1094. [Google Scholar] [CrossRef]
- Naylor, M.A.; Stephens, M.A.; Cole, S.; Threadgill, M.D.; Stratford, I.J.; O’Neill, P.; Fielden, E.M.; Adams, G.E. Synthesis and evaluation of novel electrophilic nitrofuran carboxamides and carboxylates as radiosensitizers and bioreductively activated cytotoxins. J. Med. Chem. 1990, 33, 2508–2513. [Google Scholar] [CrossRef]
- Naylor, M.A.; Stephens, M.A.; Stratford, I.J.; Keohane, A.; O’Neill, P.; Threadgill, M.D.; Webb, P.; Fielden, E.M.; Adams, G.E. Aziridinyl nitropyrroles and nitropyrazoles as hypoxia-selective cytotoxins and radiosensitizers. Anticancer Drug Des. 1991, 6, 151–167. [Google Scholar]
- Threadgill, M.D.; Webb, P.; O’Neill, P.; Naylor, M.A.; Stephens, M.A.; Stratford, I.J.; Cole, S.; Adams, G.E.; Fielden, E.M. Synthesis of a series of nitrothiophenes with basic or electrophilic substituents and evaluation as radiosensitizers and as bioreductively activated cytotoxins. J. Med. Chem. 1991, 34, 2112–2120. [Google Scholar] [CrossRef]
- Adams, G.E.; Ahmed, I.; Clarke, E.D.; O’Neill, P.; Parrick, J.; Stratford, I.J.; Wallace, R.G.; Wardman, P.; Watts, M.E. Structure-activity relationships in the development of hypoxic cell radiosensitizers. Int. J. Radiat. Biol. 1980, 38, 613–626. [Google Scholar] [CrossRef]
- Wang, J.; Guise, C.P.; Dachs, G.U.; Phung, Y.; Lambie, N.K.; Patterson, A.V.; Wilson, W.R. Identification of one-electron reductases that activate both the hypoxia prodrug SN30000 and diagnostic probe EF5. Biochem. Pharmacol. 2014, 91, 436–446. [Google Scholar] [CrossRef]
- Hunter, F.W.; Wouters, B.G.; Wilson, W.R. Hypoxia-activated prodrugs: Paths forward in the era of personalized medicine. Br. J. Cancer 2016, 114, 1071–1077. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Basran, J.; Roitel, O.; Wolf, C.R.; Munro, A.W.; Paine, M.J.I.; Scrutton, N.S. Determination of the redox potentials and electron transfer properties of the FAD- and FMN-binding domains of the human oxidoreductase NR1. Eur. J. Biochem. 2003, 270, 1164–1175. [Google Scholar] [CrossRef]
- Wolthers, K.R.; Basran, J.; Munro, A.W.; Scrutton, N.S. Molecular dissection of human methionine synthase-reductase: Determination of the flavin redox potentials in full-length enzyme and isolated flavin-binding domains. Biochemistry 2003, 42, 3911–3920. [Google Scholar] [CrossRef] [PubMed]
- Hunter, W.F.; Devaux, J.B.L.; Meng, F.; Hong, C.R.; Khan, A.; Tsai, P.; Ketela, T.W.; Sharma, I.; Kakadia, P.M.; Marastoni, S.; et al. Functional CRISPR and shRNA screens identify involvement of mitochondrial electron transport in the activation of evofosfamide. Mol. Pharmacol. 2019, 95, 638–651. [Google Scholar] [CrossRef] [PubMed]
- Helsby, N.A.; Wheeler, S.J.; Pruijn, F.B.; Palmer, B.D.; Yang, S.; Denny, W.A.; Wilson, W.R. Effect of nitroreduction on the alkylating reactivity and cytotoxicity of the 2,4-dinitrobenzamide-5-aziridine CB 1954 and the corresponding nitrogen mustard SN 23862: Distinct mechanisms of bioreductive activation. Chem. Res. Toxicol. 2003, 16, 469–478. [Google Scholar] [CrossRef]
- Wilson, W.R.; Stribbling, S.M.; Pruijn, F.B.; Syddall, S.P.; Petterson, A.V.; Liyanage, H.D.S.; Smith, E.; Botting, K.J.; Tercel, M. Nitro-chloromethylbenzindolines: Hypoxia-activated prodrugs of potent adenine N3 DNA minor groove alkylators. Mol. Cancer Ther. 2009, 8, 2903–2913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabbani, G.; Ahmad, E.; Zaidi, N.; Fatima, S.; Khan, R.H. pH-Induced molten globule state of Rhizopus niveus lipase is more resistant against thermal and chemical denaturation than its native state. Cell Biochem. Biophys. 2012, 62, 487–499. [Google Scholar] [CrossRef]
- Copp, J.N.; Mowday, A.M.; Williams, E.M.; Guise, C.P.; Ashoorzadeh, A.; Sharrock, A.V.; Flanagan, J.U.; Smaill, J.B.; Patterson, A.V.; Ackerley, D.F. Engineering a multifunctional nitroreductase for improved activation of prodrugs and PET probes for cancer gene therapy. Cell Chem. Biol. 2017, 24, 391–403. [Google Scholar] [CrossRef] [Green Version]
- Williams, E.M.; Rich, M.H.; Mowday, A.M.; Ashoorzadeh, A.; Copp, J.N.; Guise, C.P.; Anderson, R.F.; Flanagan, J.H.; Smaill, J.B.; Patterson, A.V.; et al. Engineering Escherichia coli NfsB to activate a hypoxia-resistant analogue of the PET probe EF5 to enable non-invasive imaging during enzyme prodrug therapy. Biochemistry 2019, 58, 3700–3710. [Google Scholar] [CrossRef]
- Dai, C.; Li, D.; Gong, L.; Xiao, X.; Tang, S. Curcumin ameliorates furazolidone-induced DNA damage and apoptosis in human hepatocyte LO2 cells by inhibiting ROS production and mitochondrial pathway. Molecules 2016, 21, 1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Liu, K.; Yao, K.; Reddy, K.; Fu, Y.; Yang, G.; Zykova, T.A.; Shin, S.H.; Li, H.; Ryu, J.; et al. HOI-O2 induces apoptosis and G2-M arrest in esophageal cancer mediated by ROS. Cell Death Dis. 2015, 6, e1912. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Tang, S.; Zhang, S.; Zhang, C.; Wang, C.; Zhou, Y.; Dai, C.; Xiao, X. Furazolidone induces apoptosis through activating reactive oxygen species-dependent mitochondrial signaling pathway and suppressing P13K/Akt signaling pathay in HepG2 cells. Food Chem. Toxicol. 2015, 75, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Gwenin, C.D. The role of nitroreductases in resistance to nitroimidazoles. Biology 2021, 10, 388. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Zhang, X.; Leong, H.; Shin, Y.M.; Park, D.H.; You, S.; Kim, D.-K. A novel bispidinone analog induces S-phase cell cycle arrest and apoptosis in HeLa human cervical carcinoma cells. Oncol. Rep. 2015, 33, 1526–1532. [Google Scholar] [CrossRef]
- Zhang, S.; Zhou, L.; Hong, B.; van den Heuvel, P.J.; Prabhu, V.V.; Warfel, N.A.; Kline, C.L.B.; Dicker, D.T.; Kopelovich, L.; El-Deiry, W. Small-molecule NSC59984 restores p53 pathway signaling and antitumor effects against colorectal cancer via p73 activation and degradation of mutant p53. Cancer Res. 2015, 75, 3842–3852. [Google Scholar] [CrossRef] [Green Version]
- Su, J.-G.J.; Liao, P.-J.; Huang, M.-C.; Chu, M.-C.; Lin, S.-C.; Chang, Y.-J. Aldo-keto reductase 1C2 is essential for 1-nitropyrene’s but not for benzo[a]pyrene’s induction of p53 phosphorylation and apoptosis. Toxicology 2008, 244, 257–270. [Google Scholar] [CrossRef]
- Lee, Y.S.; Yoon, H.J.; Oh, J.H.; Park, H.J.; Lee, E.H.; Song, C.W.; Yoon, S. 1,3-Dinitrobenzene induces apoptosis in TM4 mous Sertoli cells: Involvement of the c-Jun N-terminal kinase (JNK) MAPK pathway. Toxicol. Lett. 2009, 189, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Nelson, E.A.; Walker, S.R.; Kepich, A.; Gashin, L.B.; Hideshima, T.; Ikeda, H.; Chauhan, D.; Anderson, K.C.; Frank, D.A. Nifuroxazide inhibits survival of multiple myeloma cells by directly inhibiting STAT3. Blood 2008, 112, 5095–5102. [Google Scholar] [CrossRef] [Green Version]
- Andrade, J.K.E.; Souza, M.I.F.; Gomes Filho, M.A.; Silva, D.M.F.; Barros, A.L.S.; Rodrigues, M.D.; Silva, P.B.N.; Nascimento, S.C.; Aguiar, J.S.; Brondani, D.J.; et al. N-pentyl-nitrofurantoin induces apoptosis in HL-60 leukemia cell line by upregulating BAX and downregulating BCL-xL gene expression. Pharmacol. Rep. 2016, 68, 1046–1053. [Google Scholar] [CrossRef]
- Tseng, C.-H.; Tzeng, C.-C.; Chio, C.-C.; Hsu, C.-Y.; Chou, C.-K.; Chen, Y.-L. Discovery of 2-[2-(5-nitrofuran-2-yl)vinyl] quinoline derivatives as a novel type of antimetastatic agents. Bioorg. Med. Chem. 2015, 23, 141–148. [Google Scholar] [CrossRef]
- Yang, F.; Hu, M.; Lei, Q.; Xia, Y.; Zhu, Y.; Song, X.; Li, Y.; Lie, H.; Liu, C.; Xiong, Y.; et al. Nifuroxazide induces apoptosis and impairs pulmonary metastasis in breast cancer model. Cell Death Dis. 2015, 6, e1701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, D.H.; Milner, A.E.; Kerr, D.J.; Young, L.S. Mechanism of cell death induced by the novel enzyme-prodrug combination, nitroreductase/CB1954, and identification of synergism with 5-fluorouracil. Br. J. Cancer 2003, 89, 944–950. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Wang, Y.; Wang, J.; Yang, F.; Li, X.; Wu, Y. Trinitrotoluene induces endoplasmic reticulum atress and apoptosis in HePG2 cells. Med. Sci. Monit. 2015, 21, 3434–3441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, M.; Duan, Y.; Wang, P.; Gao, R.; Chen, X.; Ou, Y.; Liang, M.; Wang, L.; Zhu, H. DYT-40, a novel synthetic 2-styryl-5-nitroimidazole derivative, blocks malignant glioblastoma growth and invasion by inhibiting AEG-1 and NF-κB signaling pathway. Sci. Rep. 2016, 6, 27331. [Google Scholar] [CrossRef] [Green Version]
- Saulnier Sholler, G.L.; Brand, L.; Straub, J.A.; Dorf, L.; Illeyne, S.; Koto, K.; Kalkunte, S.; Bosenberg, M.; Ashikaga, T.; Nishi, R. Nifurtimox induces apoptosis of neuroblastoma cells in vitro and in vivo. J. Pediatr. Hematol. Oncol. 2009, 31, 187–193. [Google Scholar] [CrossRef] [Green Version]
- Hof, H.; Ströder, J.; Buisson, J.P.; Royer, R. Effect of different nitroheterocyclic compounds on aerobic, microaerophilic, and anaerobic bacteria. Antimicrob. Agents Chemother. 1986, 30, 679–683. [Google Scholar] [CrossRef] [Green Version]
- Hof, H.; Chakraborty, T.; Royer, R.; Buisson, J.P. Mode of action of nitro-heterocyclic compounds on Escherichia coli. Drugs Exp. Clin. Res. 1987, 13, 635–639. [Google Scholar] [PubMed]
- Olive, P.L. Correlation between metabolic reduction rates and electron affinity of nitroheterocycles. Cancer Res. 1979, 39, 4512–4515. [Google Scholar]
- Reynolds, A.V. The activity of nitro-compounds against Bacteroides fragilis is related to their electron affinity. J. Antimicrob. Chemother. 1981, 8, 91–99. [Google Scholar] [CrossRef]
- Rabbani, G.; Ahmad, E.; Khan, M.V.; Ashraf, M.T.; Bhat, R.; Khan, R.H. Impact of structural stability of cold adapted Candida antarctica lipase B (CaLB): In relation to pH, chemical and thermal denaturation. RSC Adv. 2015, 5, 20115–20131. [Google Scholar] [CrossRef]
- Müller, M. Reductive activation of nitroimidazoles in anaerobic microorganisms. Biochem. Pharmacol. 1986, 35, 37–41. [Google Scholar] [CrossRef]
- Olekhnovich, I.N.; Vitko, S.; Valliere, M.; Hoffman, P.S. Response to metronidazole and oxidative stress is mediated through homeostatic regulator HsrA (HP 1043) in Helicobacter pylori. J. Bacteriol. 2014, 196, 729–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trend, M.A.; Jorgensen, M.A.; Hazell, S.L.; Mendz, G.L. Oxidases and reductases are involved in metronidazole sensitivity in Helicobacter pylori. Int. J. Biochem. Cell Biol. 2001, 33, 143–153. [Google Scholar] [CrossRef]
- Salillas, S.; Alias, M.; Michel, V.; Mahia, A.; Lucia, A.; Rodriguez, L.; Bueno, J.; Galano-Frutos, J.J.; De Reuse, H.; Velazquez-Campoy, A.; et al. Design, synthesis, and efficacy testing of nitroethylene- and 7-nitrobenzoxadiazol-based flavodoxin inhibitors against Helicobacter pylori drug-resistant clinical strains and in Helicobater pylori-infected mice. J. Med. Chem. 2019, 62, 6102–6115. [Google Scholar] [CrossRef]
- Lopes-Oliveira, L.A.P.; Fantinati, M.; Da-Cruz, A.M. In vitro-induction of metronidazole-resistant Giardia duodenalis is not associated with nucleotide alterations in the genes involved in pro-drug activation. Mem. Inst. Oswaldo Cruz 2020, 115, e200303. [Google Scholar] [CrossRef]
- Müller, J.; Heller, M.; Uldry, A.C.; Braga, S.; Müller, N. Nitroreductase activities in Giardia lamblia: ORF 17150 encodes a quinone reductase with nitroreductase activity. Pathogens 2021, 10, 129. [Google Scholar] [CrossRef]
- Papadopoulou, M.V.; Bloomer, W.D.; Rosenzweig, H.S.; Arena, A.; Arrieta, F.; Rebolledo, J.C.J.; Smith, D.K. Nitrotriazole- and imidazole-based amides and sulfonamides as antitubercular agents. Antimicrob. Agents Chemother. 2014, 58, 6828–6836. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, R.; Moraski, G.C.; Krchňák, V.; Miller, P.A.; Colon-Martinez, M.; Herrero, E.; Oliver, A.G.; Miller, M.J. Thiolates chemically induce redox activation of BTZ043 and related potent nitroaromatic anti-tuberculosis agents. J. Am. Chem. Soc. 2013, 135, 3539–3549. [Google Scholar] [CrossRef] [Green Version]
- Hartkoorn, R.C.; Ryabova, O.B.; Chiarelli, L.R.; Riccardi, G.; Makarov, V.; Cole, S.T. Mechanism of action of 5-nitrothiophenes agains Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 2944–2947. [Google Scholar] [CrossRef] [Green Version]
- Cherian, J.; Choi, I.; Nayyar, A.; Manjunatha, U.H.; Mukherjee, T.; Lee, Y.S.; Boshoff, H.I.; Singh, R.; Ha, Y.H.; Goodwin, M.; et al. Structure–activity relationships of antitubercular nitroimidazoles. 3. Exploration of the linker and lipophilic tail of ((S)-2-nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3] oxazin-6-yl)-(4-trifluoromethoxybenzyl)amine (6-amino PA-824). J. Med. Chem. 2011, 54, 5639–5659. [Google Scholar] [CrossRef] [Green Version]
- Patterson, S.; Fairlamb, A.H. Current and future prospects of nitro-compounds as drugs for trypanosomiasis and leishmaniasis. Curr. Med. Chem. 2019, 26, 4454–4475. [Google Scholar] [CrossRef] [Green Version]
- Boiani, M.; Piacenza, L.; Hernandez, P.; Boiani, L.; Cerecetto, H.; Gonzalez, M.; Denicola, A. mode of action of nifurtimox and N-oxide-containing heterocycles against Trypanosoma cruzi: Is oxidative stress involved? Biochem. Pharmacol. 2010, 79, 1736–1745. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, M.V.; Trunz, B.B.; Bloomer, W.D.; McKenzie, C.; Wilkinson, S.R.; Prasittichai, C.; Brun, R.; Kaiser, M.; Torreele, E. Novel 3-nitro-1h-1,2,4-triazole-based aliphatic and aromatic amines as anti-chagasic agents. J. Med. Chem. 2011, 54, 8214–8223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadopoulou, M.V.; Bloomer, W.D.; Rosenzweig, H.S.; Chatelain, E.; Kaiser, M.; Wilkinson, S.R.; McKenzie, C.; Ioset, J.R. Novel 3-nitro-1h-1,2,4-triazole-based amides and sulfonamides as potential antitrypanosomal agents. J. Med. Chem. 2012, 55, 5554–5565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadopoulou, M.V.; Bloomer, W.D.; Rosenzweig, H.S.; Wilkinson, S.R.; Kaiser, M. Novel nitro(triazole/imidazole)- based heteroarylamides/sulfonamides as potential antitrypanosomal agents. Eur. J. Med. Chem. 2014, 87, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, M.V.; Bloomer, W.D.; Rosenzweig, H.S.; Wilkinson, S.R.; Szular, J.; Kaiser, M. Antitrypanosomal activity of 5-nitro-2-aminothiazole-based compounds. Eur. J. Med. Chem. 2016, 117, 179–186. [Google Scholar] [CrossRef] [Green Version]
- Pedron, J.; Boudot, C.; Hutter, S.; Bourgade-Delmas, S.; Stigliani, J.-L.; Sournia-Saquet, A.; Moreau, A.; Boutet-Robinet, E.; Paloque, L.; Mothes, E.; et al. Novel 8-nitroquinolin-2(1H)-ones as NTR-bioactivated antikinetoplasmid molecules: Synthesis, electrochemical and SAR study. Eur. J. Med. Chem. 2018, 155, 135–152. [Google Scholar] [CrossRef] [Green Version]
- Arias, D.G.; Herrera, F.E.; Garay, A.S.; Rodrigues, D.; Forastieri, P.S.; Luna, L.E.; Bürgi, M.D.; Prieto, C.; Iglesias, A.A.; Cravero, R.M.; et al. Rational design of nitrofuran derivatives. Synthesis and valuation as inhibitors of Trypanosoma cruzi trypanothione reductase. Eur. J. Med. Chem. 2017, 125, 1088–1097. [Google Scholar] [CrossRef]
- Benitez, D.; Comini, M.A.; Anusevičius, Ž.; Šarlauskas, J.; Miliukienė, V.; Miliuvienė, E.; Čėnas, N. 5-Vinylquinoline- substituted nitrofurans as inhibitors of trypanothione reductase and antitrypanosomal agents. Chemija 2020, 31, 111–117. [Google Scholar] [CrossRef] [Green Version]
- Wiesner, J.; Fucik, K.; Kettler, K.; Sakowski, J.; Ortmann, R.; Jamaa, H.; Schlitzer, M. Structure-activity relationships of novel anti-malarial agents. Part 6: N-(4-arylpropionylamino-3-benzoylphenyl)-[5-(4-nitrophenyl)-2-furyl]acrylic acid amides. Bioorg. Med. Chem. Lett. 2003, 13, 1539–1541. [Google Scholar] [CrossRef]
- Tukulula, M.; Sharma, R.-K.; Meurillon, M.; Mahajan, A.; Naran, K.; Warner, D.; Huang, J.; Mekonen, B.; Chibale, K. Synthesis and antiplasmodial and antimycobacterial evaluation of new nitroimidazole and nitroimidazooxazine derivatives. ACS Med. Chem. Lett. 2013, 4, 128–131. [Google Scholar] [CrossRef] [Green Version]
- Becker, K.; Tilley, L.; Vennerstrom, J.L.; Roberts, D.; Rogerson, S.; Ginsburg, H. Oxidative stress in malaria parasite-infected erythrocytes. Host-parasite interactions. Int. J. Parasitol. 2004, 34, 163–189. [Google Scholar] [CrossRef] [PubMed]








| Enzyme | Redox Potential vs. NHE | Rate Constants of Electron (Hydride) Transfer, pH 7.0 |
|---|---|---|
| NADPH: cytochrome P-450 reductase (rat liver) | −0.325 V (FAD/FADH·), −0.372 V (FADH·/FADH2), −0.068 V (FMN/FMNH·), −0.246 V (FMNH·/FMNH2), pH 7.4 [60] | 30 s−1 (NADPH to cytochrome c via FAD and FMN, steady-state) [61], kcat/Km = 6.8 × 102–5.9 × 106 M−1s−1 (ArNO2, steady-state) [13] |
| Nitric oxide synthase (rat neurons) | −0.250 V (FAD/FADH·), −0.260 V (FADH·/FADH2), −0.120 V (FMN/FMNH·), −0.220 V (FMNH·/FMNH2), −0.290 V (Fe3+/Fe2+), H 7.0 [62] | 242 s−1 (NADPH to flavins, fast phase), 46 s−1 (FMNH2 to heme), pH 7.6 [63]; kcat/Km = 1.2 × 102–2.8 × 105 M−1s−1, (steady-state, ArNO2, calmodulin is absent) [64] |
| Flavohemoglobin (bacteria, fungi) | ≤−0.150 V (FAD/FADH−), −0.120 V (heme) (E. coli FHb, pH 8.0) [65]; −0.190 V (FAD/FADH−), −0.170 V (heme) (S. aureus FHb, pH 7.6) [66] | 130 s−1 (NADH to heme via FAD), kcat = 4–25 s−1, kcat/Km = 6.2 × 102–1.1 × 105 M−1s−1 (steady-state, ArNO2, S. aureus FHb) [67] |
| Enzyme | Redox Potential vs. NHE, pH 7.0 | Rate Constants of Electron (Hydride) Transfer, pH 7.0 |
|---|---|---|
| Bovine adrenal cortex NADPH: adrenodoxin reductase | −0.250–−0.274 V (FAD/FADH2), −0.320 V (FAD/FADH·), −0.260 V (ADX, free), −0.360 V (ADX, ADR-bound) [86,87,88] | 28 s−1 (NADPH to FAD), 10 e−/s (FADH2 to ADXox,steady-state) [86,87,88] |
| Ferredoxin: NADP(H) oxidoreductase, Anabaena PCC7119 [89,90,91] | −0.296 V (FAD/FADH−), −0.280 V (FAD/FADH·), −0.384 V(Fd), pH 7.5 | 6200 e−/s (Fdred to FAD), 250 e−/s (Fdred to FADH), >600 s−1 (FADH− to NADP+), pH 8.0; 140 s−1 (NADPH to FAD), >600 s−1 (FADH− to Fdox) |
| Ferredoxin: NADP(H) oxidoreductase, Plasmodium falciparum [92,93] | −0.280 V (FAD/FADH−), −0.260 V (Fd) | 125–148 s−1 (NADPH to FAD) 13–15 e−/s (FADH− to Fdox, steady-state) |
| Bovine heart mitochondrial NADH:ubiquinone reductase (complex I) | −0.345 V (FMN/FMNH2), −0.382 V (FMN/FMNH·) [94]; ≤−0.380 V (N1a), −0.240–−0.270 V (N1b, N3, N4, N5), −0.050–−0.120 V (N2) [95] | 3500 e−/s (NADH to FMN (ferricyanide), steady-state) [96,97]; 150–380 s−1 (NADH to ubiquinone, steady-state, proteoliposomes) [98] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Čėnas, N.; Nemeikaitė-Čėnienė, A.; Kosychova, L. Single- and Two-Electron Reduction of Nitroaromatic Compounds by Flavoenzymes: Mechanisms and Implications for Cytotoxicity. Int. J. Mol. Sci. 2021, 22, 8534. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168534
Čėnas N, Nemeikaitė-Čėnienė A, Kosychova L. Single- and Two-Electron Reduction of Nitroaromatic Compounds by Flavoenzymes: Mechanisms and Implications for Cytotoxicity. International Journal of Molecular Sciences. 2021; 22(16):8534. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168534
Chicago/Turabian StyleČėnas, Narimantas, Aušra Nemeikaitė-Čėnienė, and Lidija Kosychova. 2021. "Single- and Two-Electron Reduction of Nitroaromatic Compounds by Flavoenzymes: Mechanisms and Implications for Cytotoxicity" International Journal of Molecular Sciences 22, no. 16: 8534. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168534