
Macrophage Motility in Wound Healing Is Regulated by HIF-1α via S1P Signaling

 , , , , and
, , , , and 
Abstract
:1. Introduction
2. Biological Regulation of HIF-1α in Macrophage
2.1. The Cellular Regulation of HIF-1α
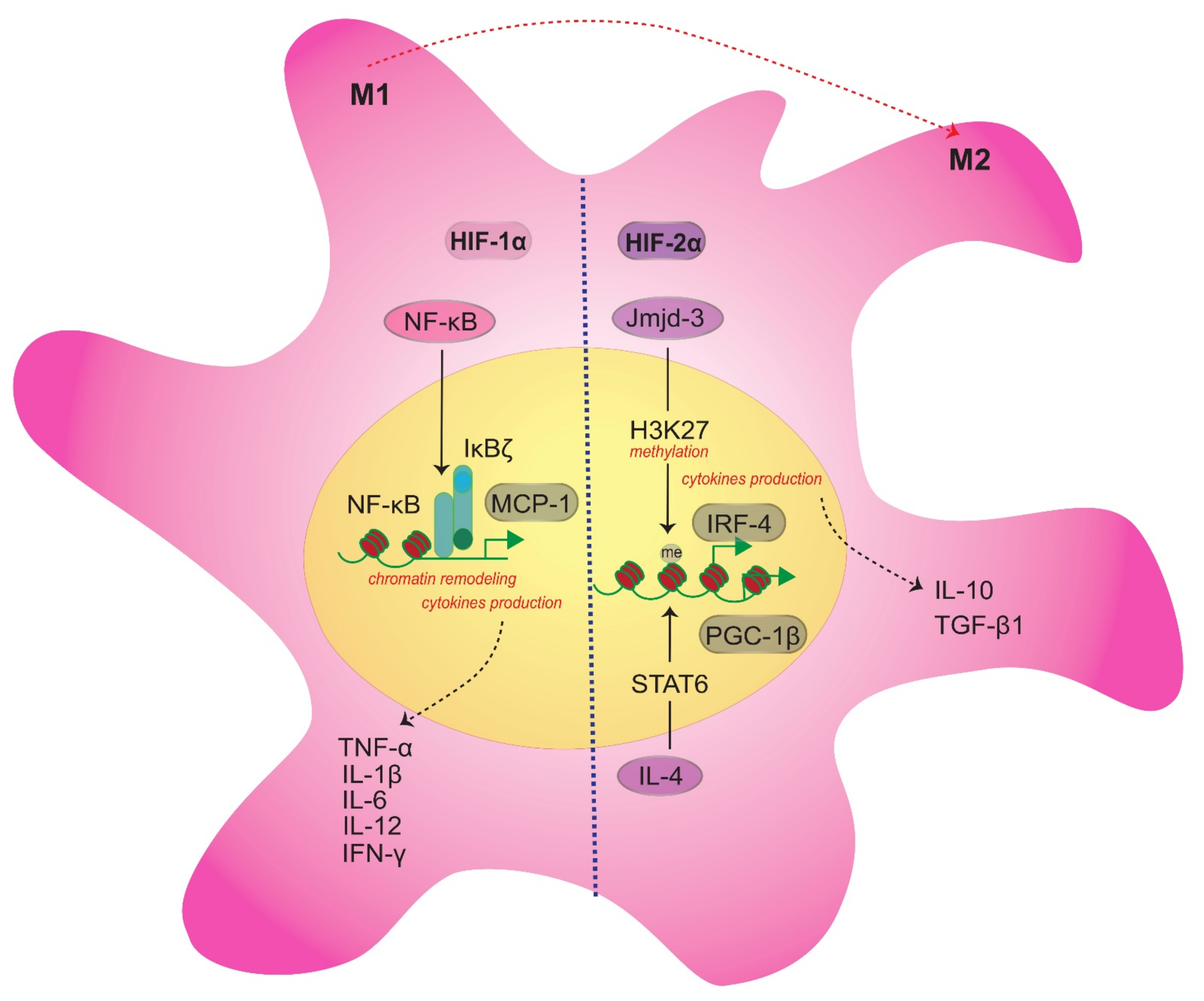
2.2. Role of HIF-1α in Inflammation and Immune System Regulation Involving Macrophages
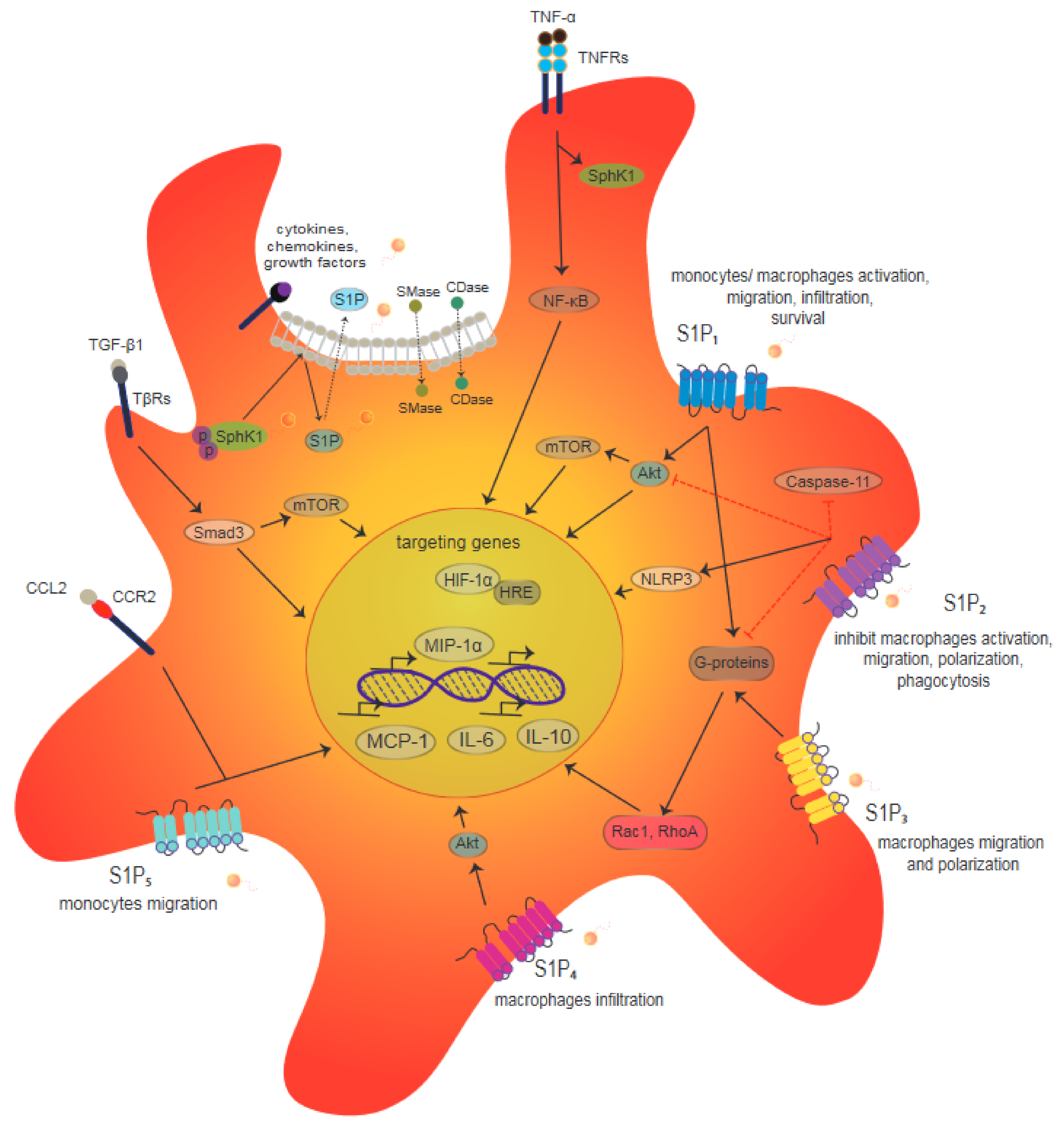
3. The Role of S1P/S1PR in Macrophages during Inflammation
4. Cellular Regulation of HIF-1α through S1P/S1P1 Signaling in Macrophages during Wound Healing
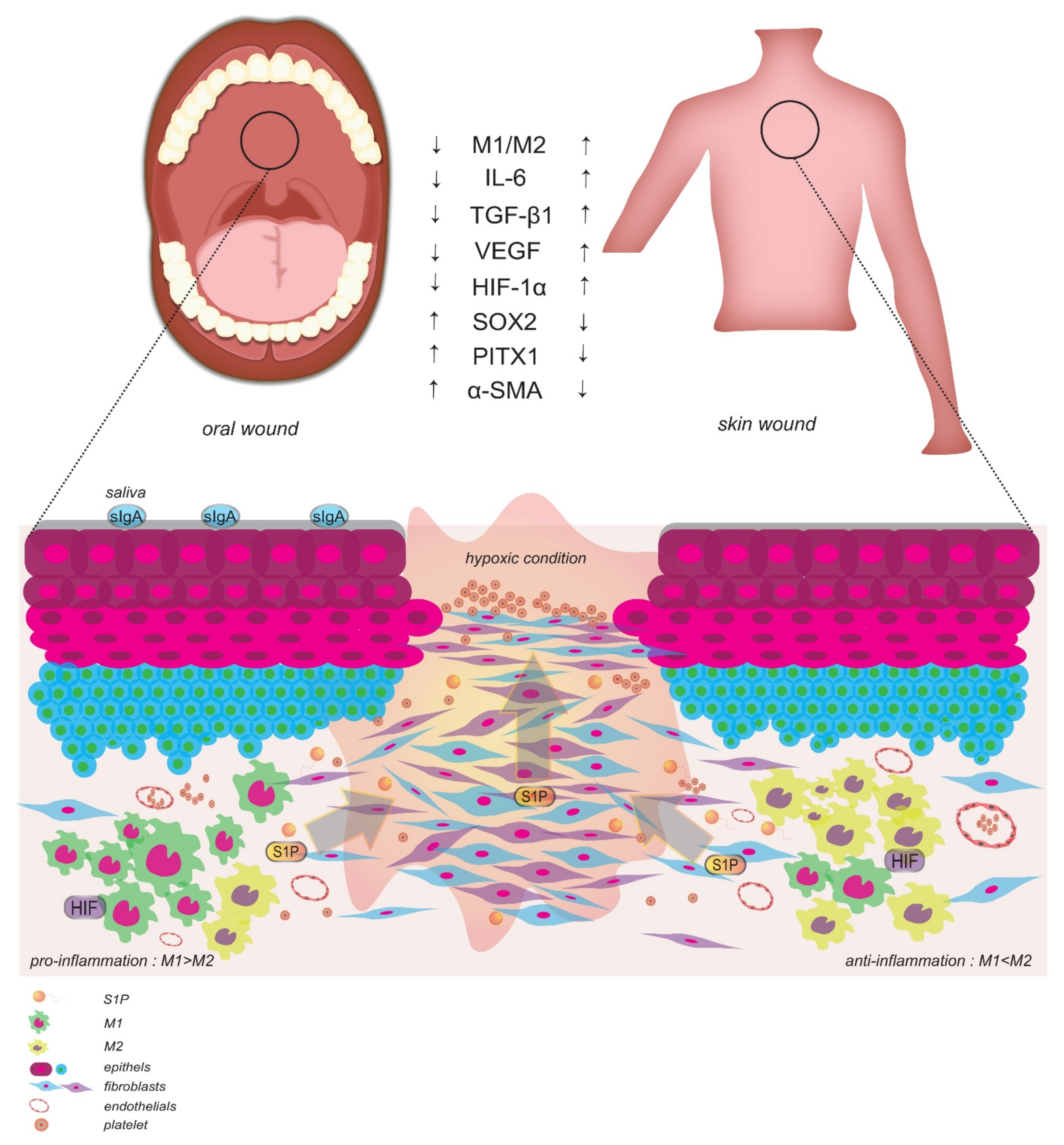
4.1. The Roles of Macrophage Profiles and Hypoxia on Wound Healing
4.2. Differential Wound Healing Is Accelerated at Skin and Mucosal Sites of Injury
4.3. Cross Signaling among HIF-1α/S1P/S1PRs in Macrophages
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wang, G.L.; Jiang, B.-H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weidemann, A.; Johnson, R.S. Biology of HIF-1α. Cell Death Differ. 2008, 15, 621–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ema, M.; Hirota, K.; Mimura, J.; Abe, H.; Yodoi, J.; Sogawa, K.; Poellinger, L.; Fujii-Kuriyama, Y. Molecular mechanisms of transcription activation by HLF and HIF1α in response to hypoxia: Their stabilization and redox signal-induced interaction with CBP/p300. EMBO J. 1999, 18, 1905–1914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.L.; Jiang, B.H.; Semenza, G.L. Effect of protein kinase and phosphatase inhibitors on expression of hypoxia-inducible factor 1. Biochem. Biophys. Res. Commun. 1995, 216, 669–675. [Google Scholar] [CrossRef]
- Li, H.; Ko, H.P.; Whitlock, J.P. Induction of phosphoglycerate kinase 1 gene expression by hypoxia. Roles of Arnt and HIF1α. J. Biol. Chem. 1996, 271, 21262–21267. [Google Scholar] [CrossRef] [Green Version]
- Wiesener, M.S.; Jürgensen, J.S.; Rosenberger, C.; Scholze, C.K.; Hörstrup, J.H.; Warnecke, C.; Mandriota, S.J.; Bechmann, I.; Frei, U.A.; Pugh, C.W.; et al. Widespread, hypoxia-inducible expression of HIF-2α in distinct cell populations of different organs. FASEB J. 2002, 17, 271–273. [Google Scholar] [CrossRef] [Green Version]
- Gothie, E.; Richard, D.E.; Berra, E.; Pages, G.; Pouyssegur, J. Identification of alternative spliced variants of human hypoxia-inducible factor-1α. J. Biol. Chem. 2000, 275, 6922–6927. [Google Scholar] [CrossRef] [Green Version]
- Chun, Y.S.; Choi, E.; Yeo, E.J.; Lee, J.H.; Kim, M.S.; Park, J.W. A new HIF-1α variant induced by zinc ion suppresses HIF-1-mediated hypoxic responses. J. Cell. Sci. 2001, 114, 4051–4061. [Google Scholar] [CrossRef]
- Chun, Y.S.; Choi, E.; Kim, T.Y.; Kim, M.S.; Park, J.W. A dominant-negative isoform lacking exons 11 and 12 of the human hypoxia-inducible factor-1α gene. Biochem. J. 2002, 362, 71–79. [Google Scholar] [CrossRef]
- Chun, Y.S.; Lee, K.H.; Choi, E.; Bae, S.Y.; Yeo, E.J.; Huang, L.E.; Kim, M.S.; Park, J.W. Phorbol ester stimulates the nonhypoxic induction of a novel hypoxia-inducible factor 1α isoform: Implications for tumor promotion. Cancer Res. 2003, 63, 8700–8707. [Google Scholar]
- Tian, H.; McKnight, S.L.; Russell, D.W. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 1997, 11, 72–82. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.W.; Bae, S.H.; Jeong, J.W.; Kim, S.H.; Kim, K.W. Hypoxia-inducible factor (HIF-1)α: Its protein stability and biological functions. Exp. Mol. Med. 2004, 36, 1–12. [Google Scholar] [CrossRef]
- Makino, Y.; Cao, R.; Svensson, K.; Bertilsson, G.; Asman, M.; Tanaka, H.; Cao, Y.; Berkenstam, A.; Poellinger, L. Inhibitory PAS domain protein is a negative regulator of hypoxia-inducible gene expression. Nature 2001, 414, 550–554. [Google Scholar] [CrossRef]
- Murdoch, C.; Muthana, M.; Lewis, C.E. Hypoxia regulates macrophage functions in inflammation. J. Immunol. 2005, 175, 6257–6263. [Google Scholar] [CrossRef] [Green Version]
- Nizet, V.; Johnson, R.S. Interdependence of hypoxic and innate immune responses. Nat. Rev. Immunol. 2009, 9, 609–617. [Google Scholar] [CrossRef] [Green Version]
- Corrado, C.; Fontana, S. Hypoxia and HIF signaling: One axis with divergent effects. Int. J. Mol. Sci. 2020, 21, 5611. [Google Scholar] [CrossRef] [PubMed]
- Takeda, N.; O’Dea, E.L.; Doedens, A.; Kim, J.W.; Weidemann, A.; Stockmann, C.; Asagiri, M.; Simon, M.C.; Hoffmann, A.; Johnson, R.S. Differential activation and antagonistic function of HIF-{α} isoforms in macrophages are essential for NO homeostasis. Genes Dev. 2010, 24, 491–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wacker, B.K.; Park, T.S.; Gidday, J.M. Hypoxic preconditioning-induced cerebral ischemic tolerance: Role of microvascular sphingosine kinase 2. Stroke 2009, 40, 3342–3348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wacker, B.K.; Perfater, J.L.; Gidday, J.M. Hypoxic preconditioning induces stroke tolerance in mice via a cascading HIF, sphingosine kinase, and CCL2 signaling pathway. J. Neurochem. 2012, 123, 954–962. [Google Scholar] [CrossRef] [Green Version]
- Yung, L.M.; Wei, Y.; Qin, T.; Wang, Y.; Smith, C.D.; Waeber, C. Sphingosine kinase 2 mediates cerebral preconditioning and protects the mouse brain against ischemic injury. Stroke 2012, 43, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Weigert, A.; Olesch, C.; Brüne, B. Sphingosine-1-phosphate and macrophage biology—How the sphinx tames the big eater. Front. Immunol. 2019, 10, 1706. [Google Scholar] [CrossRef]
- Gude, D.R.; Alvarez, S.E.; Paugh, S.W.; Mitra, P.; Yu, J.; Griffiths, R.; Barbour, S.E.; Milstien, S.; Spiegel, S. Apoptosis induces expression of sphingosine kinase 1 to release sphingosine-1-phosphate as a "come-and-get-me" signal. FASEB J. 2008, 22, 2629–2638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutami, I.R.; Izawa, T.; Mino-Oka, A.; Shinohara, T.; Mori, H.; Iwasa, A.; Tanaka, E. Fas/S1P1 crosstalk via NF-κB activation in osteoclasts controls subchondral bone remodeling in murine TMJ arthritis. Biochem. Biophys. Res. Commun. 2017, 490, 1274–1281. [Google Scholar] [CrossRef] [PubMed]
- Roggeri, A.; Schepers, M.; Tiane, A.; Rombaut, B.; Van Veggel, L.; Hellings, N.; Prickaerts, J.; Pittaluga, A.; Vanmierlo, T. Sphingosine-1-phosphate receptor modulators and oligodendroglial cells: Beyond immunomodulation. Int. J. Mol. Sci. 2020, 21, 7537. [Google Scholar] [CrossRef] [PubMed]
- Baeyens, A.; Bracero, S.; Chaluvadi, V.S.; Khodadadi-Jamayran, A.; Cammer, M.; Schwab, S.R. Monocyte-derived S1P in the lymph node regulates immune responses. Nature 2021, 592, 290–295. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Feagan, B.G.; Wolf, D.C.; D’Haens, G.; Vermeire, S.; Hanauer, S.B.; Ghosh, S.; Smith, H.; Cravets, M.; Frohna, P.A.; et al. Ozanimod induction and maintenance treatment for ulcerative colitis. N. Engl. J. Med. 2016, 374, 1754–1762. [Google Scholar] [CrossRef]
- Ebenezer, D.L.; Fu, P.; Natarajan, V. Targeting sphingosine-1-phosphate signaling in lung diseases. Pharmacol. Ther. 2016, 168, 143–157. [Google Scholar] [CrossRef] [Green Version]
- Barnawi, J.; Tran, H.; Jersmann, H.; Pitson, S.; Roscioli, E.; Hodge, G.; Meech, R.; Haberberger, R.V.; Hodge, S. Potential link between the sphingosine-1-phosphate (S1P) system and defective alveolar macrophage phagocytic function in chronic obstructive pulmonary disease (COPD). PLoS ONE 2015, 10, e0122771. [Google Scholar] [CrossRef]
- Hou, L.; Yang, L.; Chang, N.; Zhao, X.; Zhou, X.; Dong, C.; Liu, F.; Yang, L.; Li, L. Macrophage sphingosine 1-phosphate receptor 2 blockade attenuates liver inflammation and fibrogenesis triggered by NLRP3 inflammasome. Front. Immunol. 2020, 11, 1149. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Zhang, Z.; Yang, L.; Chang, N.; Zhao, X.; Zhou, X.; Yang, L.; Li, L. NLRP3 inflammasome priming and activation in cholestatic liver injury via the sphingosine 1-phosphate/S1P receptor 2/Gα(12/13)/MAPK signaling pathway. J. Mol. Med. (Berl.) 2021, 99, 273–288. [Google Scholar] [CrossRef]
- Grimm, M.; Tischner, D.; Troidl, K.; Albarran Juarez, J.; Sivaraj, K.K.; Ferreiros Bouzas, N.; Geisslinger, G.; Binder, C.J.; Wettschureck, N. S1P2/G12/13 signaling negatively regulates macrophage activation and indirectly shapes the atheroprotective B1-cell population. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 37–48. [Google Scholar] [CrossRef] [Green Version]
- Keul, P.; Lucke, S.; Lipinski, K.V.W.; Bode, C.; Gräler, M.; Heusch, G.; Levkau, B. Sphingosine-1-phosphate receptor 3 promotes recruitment of monocyte/macrophages in inflammation and atherosclerosis. Circ. Res. 2011, 108, 314–323. [Google Scholar] [CrossRef] [Green Version]
- Potì, F.; Gualtieri, F.; Sacchi, S.; Weißen-Plenz, G.; Varga, G.; Brodde, M.; Weber, C.; Simoni, M.; Nofer, J.-R. KRP-203, sphingosine 1-phosphate receptor type 1 agonist, ameliorates atherosclerosis in LDL-R−/−mice. Arter. Thromb. Vasc. Biol. 2013, 33, 1505–1512. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Yemisci, M.; Kim, H.-H.; Yung, L.M.; Shin, H.K.; Hwang, S.-K.; Guo, S.; Qin, T.; Alsharif, N.; Brinkmann, V.; et al. Fingolimod provides long-term protection in rodent models of cerebral ischemia. Ann. Neurol. 2010, 69, 119–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grassi, S.; Mauri, L.; Prioni, S.; Cabitta, L.; Sonnino, S.; Prinetti, A.; Giussani, P. Sphingosine 1-phosphate receptors and metabolic enzymes as druggable targets for brain diseases. Front. Pharmacol. 2019, 10, 807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Liu, D.; Ding, L.-H.; Ma, K.-L.; Wu, M.; Lv, L.-L.; Wen, Y.; Liu, H.; Tang, R.-N.; Liu, B.-C. FTY720 inhibits tubulointerstitial inflammation in albumin overload-induced nephropathy of rats via the Sphk1 pathway. Acta Pharmacol. Sin. 2014, 35, 1537–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oskeritzian, C.A.; Hait, N.C.; Wedman, P.; Chumanevich, A.; Kolawole, E.M.; Price, M.M.; Falanga, Y.T.; Harikumar, K.B.; Ryan, J.J.; Milstien, S.; et al. The sphingosine-1-phosphate/sphingosine-1-phosphate receptor 2 axis regulates early airway T-cell infiltration in murine mast cell-dependent acute allergic responses. J. Allergy Clin. Immunol. 2015, 135, 1008–1018e1. [Google Scholar] [CrossRef] [Green Version]
- McGowan, E.M.; Haddadi, N.; Nassif, N.T.; Lin, Y. Targeting the SphK-S1P-SIPR pathway as a potential therapeutic approach for COVID-19. Int. J. Mol. Sci. 2020, 21, 7189. [Google Scholar] [CrossRef]
- Wenderfer, S.E.; Stepkowski, S.M.; Braun, M.C. Increased survival and reduced renal injury in MRL/lpr mice treated with a novel sphingosine-1-phosphate receptor agonist. Kidney Int. 2008, 74, 1319–1326. [Google Scholar] [CrossRef] [Green Version]
- Kono, M.; Allende, M.L.; Proia, R.L. Sphingosine-1-phosphate regulation of mammalian development. Biochim. Biophys. Acta 2008, 1781, 435–441. [Google Scholar] [CrossRef] [Green Version]
- Tsai, H.C.; Nguyen, K.; Hashemi, E.; Engleman, E.; Hla, T.; Han, M.H. Myeloid sphingosine-1-phosphate receptor 1 is important for CNS autoimmunity and neuroinflammation. J. Autoimmun. 2019, 105, 102290. [Google Scholar] [CrossRef] [PubMed]
- Golfier, S.; Kondo, S.; Schulze, T.; Takeuchi, T.; Vassileva, G.; Achtman, A.H.; Graler, M.H.; Abbondanzo, S.J.; Wiekowski, M.; Kremmer, E.; et al. Shaping of terminal megakaryocyte differentiation and proplatelet development by sphingosine-1-phosphate receptor S1P4. FASEB J. 2010, 24, 4701–4710. [Google Scholar] [CrossRef]
- Jaillard, C.; Harrison, S.; Stankoff, B.; Aigrot, M.S.; Calver, A.R.; Duddy, G.; Walsh, F.S.; Pangalos, M.N.; Arimura, N.; Kaibuchi, K.; et al. Edg8/S1P5: An oligodendroglial receptor with dual function on process retraction and cell survival. J. Neurosci. 2005, 25, 1459–1469. [Google Scholar] [CrossRef]
- Ruf, W.; Furlan-Freguia, C.; Niessen, F. Vascular and dendritic cell coagulation signaling in sepsis progression. J. Thromb. Haemost. 2009, 7 (Suppl. 1), S118–S121. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, S.; Milstien, S. The outs and the ins of sphingosine-1-phosphate in immunity. Nat. Rev. Immunol. 2011, 11, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Bryan, A.M.; Del Poeta, M. Sphingosine-1-phosphate receptors and innate immunity. Cell. Microbiol. 2018, 20, e12836. [Google Scholar] [CrossRef] [Green Version]
- Al-Jarallah, A.; Chen, X.; Gonzalez, L.; Trigatti, B.L. High density lipoprotein stimulated migration of macrophages depends on the scavenger receptor class B, type I, PDZK1 and Akt1 and is blocked by sphingosine 1 phosphate receptor antagonists. PLoS ONE 2014, 9, e106487. [Google Scholar] [CrossRef] [Green Version]
- Michaud, J.; Im, D.S.; Hla, T. Inhibitory role of sphingosine 1-phosphate receptor 2 in macrophage recruitment during inflammation. J. Immunol. 2010, 184, 1475–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaire, B.P.; Song, M.R.; Choi, J.W. Sphingosine 1-phosphate receptor subtype 3 (S1P3) contributes to brain injury after transient focal cerebral ischemia via modulating microglial activation and their M1 polarization. J. Neuroinflamm. 2018, 15, 284. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.Y.; Im, D.S. Pro-inflammatory role of S1P3 in macrophages. Biomol Ther. (Seoul) 2019, 27, 373–380. [Google Scholar] [CrossRef]
- Wang, W.; Graeler, M.H.; Goetzl, E.J. Type 4 sphingosine 1-phosphate G protein-coupled receptor (S1P 4) transduces S1P effects on T cell proliferation and cytokine secretion without signaling migration. FASEB J. 2005, 19, 1731–1733. [Google Scholar] [CrossRef]
- Duong, C.Q.; Bared, S.M.; Abu-Khader, A.; Buechler, C.; Schmitz, A.; Schmitz, G. Expression of the lysophospholipid receptor family and investigation of lysophospholipid-mediated responses in human macrophages. Biochim. Biophys. Acta 2004, 1682, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Muller, J.; von Bernstorff, W.; Heidecke, C.D.; Schulze, T. Differential S1P receptor profiles on M1- and M2-polarized macrophages affect macrophage cytokine production and migration. Biomed. Res. Int. 2017, 2017, 7584621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuster, C.; Huard, A.; Sirait-Fischer, E.; Dillmann, C.; Brüne, B.; Weigert, A. S1PR4-dependent CCL2 production promotes macrophage recruitment in a murine psoriasis model. Eur. J. Immunol. 2020, 50, 839–845. [Google Scholar] [CrossRef] [Green Version]
- Debien, E.; Mayol, K.; Biajoux, V.; Daussy, C.; De Agüero, M.G.; Taillardet, M.; Dagany, N.; Brinza, L.; Henry, T.; Dubois, B.; et al. S1PR5 is pivotal for the homeostasis of patrolling monocytes. Eur. J. Immunol. 2013, 43, 1667–1675. [Google Scholar] [CrossRef] [Green Version]
- Kalhori, V.; Kemppainen, K.; Asghar, M.Y.; Bergelin, N.; Jaakkola, P.; Törnquist, K. Sphingosine-1-phosphate as a regulator of hypoxia-induced factor-1α in thyroid follicular carcinoma cells. PLoS ONE 2013, 8, e66189. [Google Scholar] [CrossRef] [Green Version]
- Hutami, I.R.; Izawa, T.; Khurel-Ochir, T.; Sakamaki, T.; Iwasa, A.; Tomita, S.; Tanaka, E. HIF-1α controls palatal wound healing by regulating macrophage motility via S1P/S1P1 signaling axis. Oral. Dis. 2021. [Google Scholar] [CrossRef]
- Sanagawa, A.; Iwaki, S.; Asai, M.; Sakakibara, D.; Norimoto, H.; Sobel, B.E.; Fujii, S. Sphingosine 1phosphate induced by hypoxia increases the expression of PAI1 in HepG2 cells via HIF1α. Mol. Med. Rep. 2016, 14, 1841–1848. [Google Scholar] [CrossRef]
- Jewell, U.R.; Kvietikova, I.; Scheid, A.; Bauer, C.; Wenger, R.H.; Gassmann, M. Induction of HIF-1α in response to hypoxia is instantaneous. FASEB J. 2001, 15, 1312–1314. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.H.; Semenza, G.L.; Bauer, C.; Marti, H.H. Hypoxia-inducible factor 1 levels vary exponentially over a physiologically relevant range of O2 tension. Am. J. Physiol. 1996, 271, C1172–C1180. [Google Scholar] [CrossRef] [Green Version]
- Cockman, M.E.; Masson, N.; Mole, D.R.; Jaakkola, P.; Chang, G.W.; Clifford, S.C.; Maher, E.R.; Pugh, C.W.; Ratcliffe, P.J.; Maxwell, P.H. Hypoxia inducible factor-α binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein. J. Biol. Chem. 2000, 275, 25733–25741. [Google Scholar] [CrossRef] [Green Version]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFα targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Jaakkola, P.; Mole, D.R.; Tian, Y.-M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.; et al. Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factor 1 and cancer pathogenesis. IUBMB Life 2008, 60, 591–597. [Google Scholar] [CrossRef]
- Maxwell, P.; Wiesener, M.S.; Chang, G.-W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.; Wykoff, C.C.; Pugh, C.; Maher, E.; Ratcliffe, P. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Ohh, M.; Park, C.W.; Ivan, M.; Hoffman, M.A.; Kim, T.Y.; Huang, L.E.; Pavletich, N.; Chau, V.; Kaelin, W.G. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat. Cell Biol 2000, 2, 423–427. [Google Scholar] [CrossRef]
- Bai, S.; Datta, J.; Jacob, S.T.; Ghoshal, K. Treatment of PC12 cells with nerve growth factor induces proteasomal degradation of T-cadherin that requires tyrosine phosphorylation of its cadherin domain. J. Biol. Chem. 2018, 282, 27171–27180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamura, T.; Sato, S.; Iwai, K.; Czyzyk-Krzeska, M.; Conaway, R.C.; Conaway, J.W. Activation of HIF1α ubiquitination by a reconstituted von Hippel-Lindau (VHL) tumor suppressor complex. Proc. Natl. Acad. Sci. USA 2000, 97, 10430–10435. [Google Scholar] [CrossRef] [Green Version]
- Tanimoto, K.; Makino, Y.; Pereira, T.; Poellinger, L. Mechanism of regulation of the hypoxia-inducible factor-1 α by the von Hippel-Lindau tumor suppressor protein. EMBO J. 2000, 19, 4298–4309. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Regulation of cancer cell metabolism by hypoxia-inducible factor 1. Semin. Cancer Biol. 2009, 19, 12–16. [Google Scholar] [CrossRef]
- Metzen, E.; Zhou, J.; Jelkmann, W.; Fandrey, J.; Brune, B. Nitric oxide impairs normoxic degradation of HIF-1α by inhibition of prolyl hydroxylases. Mol. Biol. Cell 2003, 14, 3470–3481. [Google Scholar] [CrossRef] [Green Version]
- Ravi, R.; Mookerjee, B.; Bhujwalla, Z.M.; Sutter, C.H.; Artemov, D.; Zeng, Q.; Dillehay, L.E.; Madan, A.; Semenza, G.L.; Bedi, A. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1α. Genes Dev. 2000, 14, 34–44. [Google Scholar]
- Tang, T.T.-L.; Lasky, L.A. The forkhead transcription factor FOXO4 induces the down-regulation of hypoxia-inducible factor 1 α by a von Hippel-Lindau protein-independent mechanism. J. Biol. Chem. 2003, 278, 30125–30135. [Google Scholar] [CrossRef] [Green Version]
- Lee, P.J.; Jiang, B.H.; Chin, B.Y.; Iyer, N.V.; Alam, J.; Semenza, G.L.; Choi, A.M. Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase-1 gene in response to hypoxia. J. Biol. Chem. 1997, 272, P5375–P5381. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L.; Agani, F.; Booth, G.; Forsythe, J.; Iyer, N.; Jiang, B.H.; Leung, S.; Roe, R.; Wiener, C.; Yu, A. Structural and functional analysis of hypoxia-inducible factor 1. Kidney Int. 1997, 51, 553–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, R.; Zhang, H.; Kim, J.W.; Shimoda, L.; Dang, C.V.; Semenza, G.L. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007, 129, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Forsythe, J.A.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 1996, 16, 4604–4613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, N.S.; Chung, S.; Furneaux, H.; Levy, A.P. Hypoxic stabilization of vascular endothelial growth factor mRNA by the RNA-binding protein HuR. J. Biol. Chem. 1998, 273, 6417–6423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, N.; Wang, L.; Esko, J.; Giordano, F.J.; Huang, Y.; Gerber, H.P.; Ferrara, N.; Johnson, R.S. Loss of HIF-1α in endothelial cells disrupts a hypoxia-driven VEGF autocrine loop necessary for tumorigenesis. Cancer Cell 2004, 6, 485–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, N.; Ding, M.; Zheng, J.Z.; Zhang, Z.; Leonard, S.S.; Liu, K.J.; Shi, X.; Jiang, B.H. Vanadate-induced expression of hypoxia-inducible factor 1 α and vascular endothelial growth factor through phosphatidylinositol 3-kinase/Akt pathway and reactive oxygen species. J. Biol. Chem. 2002, 277, 31963–31971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herb, M.; Schramm, M. Functions of ROS in macrophages and antimicrobial immunity. Antioxidants 2021, 10, 313. [Google Scholar] [CrossRef]
- Klimova, T.; Chandel, N.S. Mitochondrial complex III regulates hypoxic activation of HIF. Cell. Death Differ. 2008, 15, 660–666. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.N.; Yang, W.K.; Kim, J.; Kim, H.S.; Kim, E.J.; Yun, H.; Park, H.; Kim, S.S.; Choe, W.; Kang, I.; et al. Reactive oxygen species stabilize hypoxia-inducible factor-1 α protein and stimulate transcriptional activity via AMP-activated protein kinase in DU145 human prostate cancer cells. Carcinogenesis 2008, 29, 713–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1α during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [Green Version]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Kim, H.E.; Cho, H.; Shi, S.; Kim, B.; Kim, O. Red light-emitting diode irradiation regulates oxidative stress and inflammation through SPHK1/NF-κB activation in human keratinocytes. J. Photochem. Photobiol. B 2018, 186, 31–40. [Google Scholar] [CrossRef]
- Li, Q.; Qian, J.; Li, Y.; Huang, P.; Liang, H.; Sun, H.; Liu, C.; Peng, J.; Lin, X.; Chen, X.; et al. Generation of sphingosine-1-phosphate by sphingosine kinase 1 protects nonalcoholic fatty liver from ischemia/reperfusion injury through alleviating reactive oxygen species production in hepatocytes. Free Radic. Biol. Med. 2020, 159, 136–149. [Google Scholar] [CrossRef]
- Xia, C.; Meng, Q.; Liu, L.-Z.; Rojanasakul, Y.; Wang, X.-R.; Jiang, B.-H. Reactive oxygen species regulate angiogenesis and tumor growth through vascular endothelial growth factor. Cancer Res. 2007, 67, 10823–10830. [Google Scholar] [CrossRef] [Green Version]
- Ader, I.; Brizuela, L.; Bouquerel, P.; Malavaud, B.; Cuvillier, O. Sphingosine kinase 1: A new modulator of hypoxia inducible factor 1α during hypoxia in human cancer cells. Cancer Res. 2008, 68, 8635–8642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.-Y.; Lee, H.-J.; Jeong, S.-J.; Kim, H.-S.; Chen, C.Y.; Lee, E.-O.; Kim, S.-H. Sphingosine kinase 1 pathway is involved in melatonin-induced HIF-1α inactivation in hypoxic PC-3 prostate cancer cells. J. Pineal Res. 2011, 51, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.L.; Klimova, T.A.; Eisenbart, J.; Moraes, C.T.; Murphy, M.P.; Budinger, G.R.; Chandel, N.S. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J. Cell Biol. 2007, 177, 1029–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef] [Green Version]
- Kaelin, W.G., Jr.; Ratcliffe, P.J. Oxygen sensing by metazoans: The central role of the HIF hydroxylase pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Xu, R.; Hu, Z.; Tian, Y.; Zhu, Y.; Gu, L.; Zhou, L. PI3K and ERK-induced Rac1 activation mediates hypoxia-induced HIF-1α expression in MCF-7 breast cancer cells. PLoS ONE 2011, 6, e25213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mylonis, I.; Chachami, G.; Samiotaki, M.; Panayotou, G.; Paraskeva, E.; Kalousi, A.; Georgatsou, E.; Bonanou, S.; Simos, G. Identification of MAPK phosphorylation sites and their role in the localization and activity of hypoxia-inducible factor-1α. J. Biol. Chem. 2006, 281, 33095–33106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koshikawa, N.; Hayashi, J.; Nakagawara, A.; Takenaga, K. Reactive oxygen species-generating mitochondrial DNA mutation up-regulates hypoxia-inducible factor-1α gene transcription via phosphatidylinositol 3-kinase-Akt/protein kinase C/histone deacetylase pathway. J. Biol. Chem. 2009, 284, 33185–33194. [Google Scholar] [CrossRef] [Green Version]
- Flügel, D.; Görlach, A.; Michiels, C.; Kietzmann, T. Glycogen synthase kinase 3 phosphorylates hypoxia-inducible factor 1α and mediates its destabilization in a VHL-independent manner. Mol. Cell. Biol. 2007, 27, 3253–3265. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Yao, L.; Yang, J.; Wang, Z.; Du, G. PI3K/Akt and HIF1 signaling pathway in hypoxiaischemia (Review). Mol. Med. Rep. 2018, 18, 3547–3554. [Google Scholar]
- Liu, M.-H.; Li, G.-H.; Peng, L.-J.; Qu, S.-L.; Zhang, Y.; Peng, J.; Luo, X.-Y.; Hu, H.-J.; Ren, Z.; Liu, Y.; et al. PI3K/Akt/FoxO3a signaling mediates cardioprotection of FGF-2 against hydrogen peroxide-induced apoptosis in H9c2 cells. Mol. Cell. Biochem. 2016, 414, 57–66. [Google Scholar] [CrossRef]
- Liu, J.; Wu, P.; Xu, Z.; Zhang, J.; Liu, J.; Yang, Z. Ginkgolide B inhibits hydrogen peroxide-induced apoptosis and attenuates cytotoxicity via activating the PI3K/Akt/mTOR signaling pathway in H9c2 cells. Mol. Med. Rep. 2020, 22, 310–316. [Google Scholar] [CrossRef]
- Galanis, A.; Pappa, A.; Giannakakis, A.; Lanitis, E.; Dangaj, D.; Sandaltzopoulos, R. Reactive oxygen species and HIF-1 signalling in cancer. Cancer Lett. 2008, 266, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, A.; Montaner, S.; Miyazaki, H.; Gutkind, J.S. MAPK and Akt act cooperatively but independently on hypoxia inducible factor-1α in rasV12 upregulation of VEGF. Biochem. Biophys. Res. Commun. 2001, 287, 292–300. [Google Scholar] [CrossRef]
- Mottet, D.; Dumont, V.; Deccache, Y.; Demazy, C.; Ninane, N.; Raes, M.; Michiels, C. Regulation of hypoxia-inducible factor-1α protein level during hypoxic conditions by the phosphatidylinositol 3-kinase/Akt/glycogen synthase kinase 3beta pathway in HepG2 cells. J. Biol. Chem. 2003, 278, 31277–31285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimova, E.Y.; Michiels, C.; Kietzmann, T. Kinases as upstream regulators of the HIF system: Their emerging potential as anti-cancer drug targets. Curr. Pharm. Des. 2009, 15, 3867–3877. [Google Scholar] [CrossRef] [PubMed]
- Karar, J.; Cerniglia, G.J.; Lindsten, T.; Koumenis, C.; Maity, A. Dual PI3K/mTOR inhibitor NVP-BEZ235 suppresses hypoxia-inducible factor (HIF)-1α expression by blocking protein translation and increases cell death under hypoxia. Cancer Biol. Ther. 2012, 13, 1102–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, H.; Chiles, K.; Feldser, D.; Laughner, E.; Hanrahan, C.; Georgescu, M.M.; Simons, J.W.; Semenza, G.L. Modulation of hypoxia-inducible factor 1α expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: Implications for tumor angiogenesis and therapeutics. Cancer Res. 2000, 60, 1541–1545. [Google Scholar]
- Yang, X.-M.; Wang, Y.-S.; Zhang, J.; Li, Y.; Xu, J.-F.; Zhu, J.; Zhao, W.; Chu, D.-K.; Wiedemann, P. Role of PI3K/Akt and MEK/ERK in mediating hypoxia-induced expression of HIF-1 and VEGF in laser-induced rat choroidal neovascularization. Investig. Opthalmol. Vis. Sci. 2009, 50, 1873–1879. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Qu, Y.; Mao, M.; Xiong, Y.; Mu, D. The involvement of phosphoinositid 3-kinase/Akt pathway in the activation of hypoxia-inducible factor-1α in the developing rat brain after hypoxia–ischemia. Brain Res. 2008, 1197, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Kilic-Eren, M.; Boylu, T.; Tabor, V. Targeting PI3K/Akt represses hypoxia inducible factor-1α activation and sensitizes Rhabdomyosarcoma and Ewing’s sarcoma cells for apoptosis. Cancer Cell Int. 2013, 13, 1–36. [Google Scholar] [CrossRef] [Green Version]
- Jung, S. Macrophages and monocytes in 2017: Macrophages and monocytes: Of tortoises and hares. Nat. Rev. Immunol. 2018, 18, 85–86. [Google Scholar] [CrossRef] [PubMed]
- Epelman, S.; Lavine, K.J.; Randolph, G.J. Origin and functions of tissue macrophages. Immunity 2014, 41, 21–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, L.C.; Jenkins, S.J.; Allen, J.E.; Taylor, P.R. Tissue-resident macrophages. Nat. Immunol. 2013, 14, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, G.; Chen, C.; Portnoy, D.A. Strategies used by bacteria to grow in macrophages. Microbiol. Spectr. 2016, 4, 701–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, A. The phagosome: Compartment with a license to kill. Traffic 2007, 8, 311–330. [Google Scholar] [CrossRef]
- Djaldetti, M.; Salman, H.; Bergman, M.; Djaldetti, R.; Bessler, H. Phagocytosis--the mighty weapon of the silent warriors. Microsc. Res. Tech. 2002, 57, 421–431. [Google Scholar] [CrossRef]
- Lemke, G. How macrophages deal with death. Nat. Rev. Immunol. 2019, 19, 539–549. [Google Scholar] [CrossRef]
- Weiss, G.; Schaible, U.E. Macrophage defense mechanisms against intracellular bacteria. Immunol. Rev. 2015, 264, 182–203. [Google Scholar] [CrossRef] [Green Version]
- Del Cerro-Vadillo, E.; Madrazo-Toca, F.; Marin, E.C.; Fernandez-Prieto, L.; Beck, C.; Leyva-Cobián, F.; Saftig, P.; Alvarez-Dominguez, C. Cutting edge: A novel nonoxidative phagosomal mechanism exerted by cathepsin-D controls listeria monocytogenes intracellular growth. J. Immunol. 2006, 176, 1321–1325. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Dominguez, C.; Carrasco-Marin, E.; Lopez-Mato, P.; Leyva-Cobian, F. The contribution of both oxygen and nitrogen intermediates to the intracellular killing mechanisms of C1q-opsonized Listeria monocytogenes by the macrophage-like IC-21 cell line. Immunology 2000, 101, 83–89. [Google Scholar] [CrossRef]
- Arango Duque, G.; Descoteaux, A. Macrophage cytokines: Involvement in immunity and infectious diseases. Front. Immunol. 2014, 5, 491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emerling, B.M.; Platanias, L.C.; Black, E.; Nebreda, A.R.; Davis, R.J.; Chandel, N.S. Mitochondrial reactive oxygen species activation of p38 mitogen-activated protein kinase is required for hypoxia signaling. Mol. Cell. Biol. 2005, 25, 4853–4862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmeen, A.; Barford, D. Functions and mechanisms of redox regulation of cysteine-based phosphatases. Antioxid. Redox Signal 2005, 7, 560–577. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.T.; Colgan, S.P. Regulation of immunity and inflammation by hypoxia in immunological niches. Nat. Rev. Immunol. 2017, 17, 774–785. [Google Scholar] [CrossRef]
- Palazon, A.; Goldrath, A.W.; Nizet, V.; Johnson, R.S. HIF transcription factors, inflammation, and immunity. Immunity 2014, 41, 518–528. [Google Scholar] [CrossRef] [Green Version]
- Peyssonnaux, C.; Datta, V.; Cramer, T.; Doedens, A.; Theodorakis, E.A.; Gallo, R.L.; Hurtado-Ziola, N.; Nizet, V.; Johnson, R.S. HIF-1α expression regulates the bactericidal capacity of phagocytes. J. Clin. Investig. 2005, 115, 1806–1815. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Pascual, A.; Lorente-Cebrian, S.; Moreno-Aliaga, M.J.; Martinez, J.A.; Gonzalez-Muniesa, P. Inflammation stimulates hypoxia-inducible factor-1α regulatory activity in 3T3-L1 adipocytes with conditioned medium from lipopolysaccharide-activated RAW 264.7 macrophages. J. Cell. Physiol. 2018, 234, 550–560. [Google Scholar] [CrossRef] [Green Version]
- Aarup, A.; Pedersen, T.X.; Junker, N.; Christoffersen, C.; Bartels, E.D.; Madsen, M.; Nielsen, C.H.; Nielsen, L.B. Hypoxia-inducible factor-1α expression in macrophages promotes development of atherosclerosis. Arter. Thromb. Vasc. Biol. 2016, 36, 1782–1790. [Google Scholar] [CrossRef] [Green Version]
- Watts, E.R.; Walmsley, S.R. Inflammation and hypoxia: HIF and PHD isoform selectivity. Trends Mol. Med. 2019, 25, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Liu, H.; Lian, G.; Zhang, S.-Y.; Wang, X.; Jiang, C. HIF1α-induced glycolysis metabolism is essential to the activation of inflammatory macrophages. Mediat. Inflamm. 2017, 2017, 9029327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrante, C.J.; Leibovich, S.J. Regulation of macrophage polarization and wound healing. Adv. Wound Care (N. Rochelle) 2012, 1, 10–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Allen, J.; Biswas, S.K.; Fisher, E.; Gilroy, D.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannella, K.M.; Wynn, T.A. Mechanisms of organ injury and repair by macrophages. Annu. Rev. Physiol. 2017, 79, 593–617. [Google Scholar] [CrossRef]
- Krzyszczyk, P.; Schloss, R.; Palmer, A.; Berthiaume, F. The role of macrophages in acute and chronic wound healing and interventions to promote pro-wound healing phenotypes. Front. Physiol. 2018, 9, 419. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Hildebrand, D.G.; Alexander, E.; Horber, S.; Lehle, S.; Obermayer, K.; Munck, N.A.; Rothfuss, O.; Frick, J.S.; Morimatsu, M.; Schmitz, I.; et al. IκBzeta is a transcriptional key regulator of CCL2/MCP-1. J. Immunol. 2013, 190, 4812–4820. [Google Scholar] [CrossRef]
- Oliver, K.M.; Taylor, C.T.; Cummins, E.P. Hypoxia. Regulation of NFκB signalling during inflammation: The role of hydroxylases. Arthritis Res. Ther. 2009, 11, 215. [Google Scholar] [CrossRef] [Green Version]
- Fang, H.-Y.; Hughes, R.; Murdoch, C.; Coffelt, S.; Biswas, S.K.; Harris, A.; Johnson, R.; Imityaz, H.Z.; Simon, M.C.; Fredlund, E.; et al. Hypoxia-inducible factors 1 and 2 are important transcriptional effectors in primary macrophages experiencing hypoxia. Blood 2009, 114, 844–859. [Google Scholar] [CrossRef] [Green Version]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef]
- Hesketh, M.; Sahin, K.B.; West, Z.E.; Murray, R.Z. Macrophage phenotypes regulate scar formation and chronic wound healing. Int. J. Mol. Sci. 2017, 18, 1545. [Google Scholar] [CrossRef] [Green Version]
- St-Pierre, J.; Lin, J.; Krauss, S.; Tarr, P.T.; Yang, R.; Newgard, C.B.; Spiegelman, B.M. Bioenergetic analysis of peroxisome proliferator-activated receptor gamma coactivators 1α and 1β (PGC-1α and PGC-1β) in muscle cells. J. Biol. Chem. 2003, 278, 26597–26603. [Google Scholar] [CrossRef] [Green Version]
- Shao, D.; Liu, Y.; Liu, X.; Zhu, L.; Cui, Y.; Cui, A.; Qiao, A.; Kong, X.; Liu, Y.; Chen, Q.; et al. PGC-1β-regulated mitochondrial biogenesis and function in myotubes is mediated by NRF-1 and ERR α. Mitochondrion 2010, 10, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Vats, D.; Mukundan, L.; Odegaard, J.I.; Zhang, L.; Smith, K.L.; Morel, C.R.; Wagner, R.A.; Greaves, D.R.; Murray, P.J.; Chawla, A. Oxidative metabolism and PGC-1β attenuate macrophage-mediated inflammation. Cell Metab. 2006, 4, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Desanta, F.; Totaro, M.G.; Prosperini, E.; Notarbartolo, S.; Testa, G.; Natoli, G. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell 2007, 130, 1083–1094. [Google Scholar] [CrossRef] [Green Version]
- Satoh, T.; Takeuchi, O.; Vandenbon, A.; Yasuda, K.; Tanaka, Y.; Kumagai, Y.; Miyake, T.; Matsushita, K.; Okazaki, T.; Saitoh, T.; et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat. Immunol. 2010, 11, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Cyster, J.G.; Schwab, S. Sphingosine-1-phosphate and lymphocyte egress from lymphoid organs. Annu. Rev. Immunol. 2012, 30, 69–94. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, S.; Milstien, S. Sphingosine-1-phosphate: An enigmatic signalling lipid. Nat. Rev. Mol. Cell. Biol. 2003, 4, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Bruce, C.R.; Risis, S.; Babb, J.R.; Yang, C.; Kowalski, G.M.; Selathurai, A.; Lee-Young, R.S.; Weir, J.M.; Yoshioka, K.; Takuwa, Y.; et al. Overexpression of sphingosine kinase 1 prevents ceramide accumulation and ameliorates muscle insulin resistance in high-fat diet-fed mice. Diabetes 2012, 61, 3148–3155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiegel, S.; Milstien, S. Functions of the multifaceted family of sphingosine kinases and some close relatives. J. Biol. Chem. 2007, 282, 2125–2129. [Google Scholar] [CrossRef] [Green Version]
- Hannun, Y.A.; Obeid, L. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Merrill, A.H., Jr.; Stokes, T.H.; Momin, A.; Park, H.; Portz, B.J.; Kelly, S.; Wang, E.; Sullards, M.C.; Wang, M.D. Sphingolipidomics: A valuable tool for understanding the roles of sphingolipids in biology and disease. J. Lipid Res. 2009, 50, S97–S102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitman, M.; Costabile, M.; Pitson, S.M. Recent advances in the development of sphingosine kinase inhibitors. Cell. Signal. 2016, 28, 1349–1363. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.R.; Pyne, S.; Pyne, N.J. Sphingosine kinases: Emerging structure-function insights. Trends Biochem. Sci. 2016, 41, 395–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiegel, S.; English, D.; Milstien, S. Sphingosine 1-phosphate signaling: Providing cells with a sense of direction. Trends Cell Biol. 2002, 12, 236–242. [Google Scholar] [CrossRef]
- Rosen, H.; Stevens, R.; Hanson, M.; Roberts, E.; Oldstone, M.B. Sphingosine-1-phosphate and its receptors: Structure, signaling, and influence. Annu. Rev. Biochem. 2013, 82, 637–662. [Google Scholar] [CrossRef]
- Blaho, V.; Hla, T. An update on the biology of sphingosine 1-phosphate receptors. J. Lipid Res. 2014, 55, 1596–1608. [Google Scholar] [CrossRef] [Green Version]
- Lattin, J.; Zidar, D.A.; Schroder, K.; Kellie, S.; Hume, D.A.; Sweet, M.J. G-protein-coupled receptor expression, function, and signaling in macrophages. J. Leukoc. Biol. 2007, 82, 16–32. [Google Scholar] [CrossRef] [Green Version]
- Worzfeld, T.; Wettschureck, N.; Offermanns, S. G12/G13-mediated signalling in mammalian physiology and disease. Trends Pharmacol. Sci. 2008, 29, 582–589. [Google Scholar] [CrossRef]
- Siehler, S. Regulation of RhoGEF proteins by G12/13-coupled receptors. Br. J. Pharmacol. 2009, 158, 41–49. [Google Scholar] [CrossRef] [Green Version]
- Di, A.; Kawamura, T.; Gao, X.P.; Tang, H.; Berdyshev, E.; Vogel, S.M.; Zhao, Y.Y.; Sharma, T.; Bachmaier, K.; Xu, J.; et al. A novel function of sphingosine kinase 1 suppression of JNK activity in preventing inflammation and injury. J. Biol. Chem. 2010, 285, 15848–15857. [Google Scholar] [CrossRef] [Green Version]
- Nishi, T.; Kobayashi, N.; Hisano, Y.; Kawahara, A.; Yamaguchi, A. Molecular and physiological functions of sphingosine 1-phosphate transporters. Biochim. Biophys. Acta 2014, 1841, 759–765. [Google Scholar] [CrossRef]
- Osborne, N.; Brand-Arzamendi, K.; Ober, E.A.; Jin, S.-W.; Verkade, H.; Holtzman, N.G.; Yelon, D.; Stainier, D.Y. The spinster homolog, two of hearts, is required for sphingosine 1-phosphate signaling in zebrafish. Curr. Biol. 2008, 18, 1882–1888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawahara, A.; Nishi, T.; Hisano, Y.; Fukui, H.; Yamaguchi, A.; Mochizuki, N. The sphingolipid transporter spns2 functions in migration of zebrafish myocardial precursors. Science 2009, 323, 524–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pappu, R.; Schwab, S.R.; Cornelissen, I.; Pereira, J.P.; Regard, J.B.; Xu, Y.; Camerer, E.; Zheng, Y.W.; Huang, Y.; Cyster, J.G.; et al. Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science 2007, 316, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Hait, N.C.; Allegood, J.; Maceyka, M.; Strub, G.M.; Harikumar, K.B.; Singh, S.K.; Luo, C.; Marmorstein, R.; Kordula, T.; Milstien, S.; et al. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 2009, 325, 1254–1257. [Google Scholar] [CrossRef] [Green Version]
- Mori, H.; Izawa, T.; Tanaka, E. Smad3 deficiency leads to mandibular condyle degradation via the sphingosine 1-phosphate (S1P)/S1P3 signaling axis. Am. J. Pathol. 2015, 185, 2742–2756. [Google Scholar] [CrossRef]
- Alvarez, S.E.; Harikumar, K.; Hait, N.C.; Allegood, J.; Strub, G.M.; Kim, E.Y.; Maceyka, M.; Jiang, H.; Luo, C.; Kordula, T.; et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nat. Cell Biol. 2010, 465, 1084–1088. [Google Scholar] [CrossRef] [Green Version]
- Strub, G.M.; Paillard, M.; Liang, J.; Gomez, L.; Allegood, J.C.; Hait, N.C.; Maceyka, M.; Price, M.M.; Chen, Q.; Simpson, D.; et al. Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. FASEB J. 2010, 25, 600–612. [Google Scholar] [CrossRef] [Green Version]
- Maceyka, M.; Alvarez, S.E.; Milstien, S.; Spiegel, S. Filamin A links sphingosine kinase 1 and sphingosine-1-phosphate receptor 1 at lamellipodia to orchestrate cell migration. Mol. Cell Biol. 2008, 28, 5687–5697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, M.; Egen, J.G.; Klauschen, F.; Meier-Schellersheim, M.; Saeki, Y.; Vacher, J.; Proia, R.L.; Germain, R.N. Sphingosine-1-phosphate mobilizes osteoclast precursors and regulates bone homeostasis. Nature 2009, 458, 524–528. [Google Scholar] [CrossRef]
- Yu, H. Sphingosine-1-phosphate receptor 2 regulates proinflammatory cytokine production and osteoclastogenesis. PLoS ONE 2016, 11, e0156303. [Google Scholar] [CrossRef] [PubMed]
- Song, F.; Hou, J.; Chen, Z.; Cheng, B.; Lei, R.; Cui, P.; Sun, Y.; Wang, H.; Fang, X. Sphingosine-1-phosphate receptor 2 signaling promotes caspase-11-dependent macrophage pyroptosis and worsens escherichia coli sepsis outcome. Anesthesiology 2018, 129, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Brecht, K.; Weigert, A.; Hu, J.; Popp, R.; Fisslthaler, B.; Korff, T.; Fleming, I.; Geisslinger, G.; Brune, B. Macrophages programmed by apoptotic cells promote angiogenesis via prostaglandin E2. FASEB J. 2011, 25, 2408–2417. [Google Scholar] [CrossRef]
- Hutami, I.R.; Tanaka, E.; Izawa, T. Crosstalk between Fas and S1P1 signaling via NF-kB in osteoclasts controls bone destruction in the TMJ due to rheumatoid arthritis. Jpn. Dent. Sci. Rev. 2019, 55, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Segar, C.E.; Hughley, B.B.; Bowers, D.T.; Botchwey, E.A. The promotion of mandibular defect healing by the targeting of S1P receptors and the recruitment of alternatively activated macrophages. Biomaterials 2013, 34, 9853–9862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, J.; Chen, Q.; Wu, X.; Zhao, D.; Reuveni, H.; Licht, T.; Xu, M.; Hu, H.; Hoeft, A.; Ben-Sasson, S.A.; et al. S1PR3 signaling drives bacterial killing and is required for survival in bacterial sepsis. Am. J. Respir. Crit. Care Med. 2017, 196, 1559–1570. [Google Scholar] [CrossRef] [PubMed]
- Heilmann, A.; Schinke, T.; Bindl, R.; Wehner, T.; Rapp, A.; Haffner-Luntzer, M.; Liedert, A.; Amling, M.; Ignatius, A. Systemic treatment with the sphingosine-1-phosphate analog FTY720 does not improve fracture healing in mice. J. Orthop. Res. 2013, 31, 1845–1850. [Google Scholar] [CrossRef]
- Yang, J.; Yang, L.; Tian, L.; Ji, X.; Yang, L.; Li, L. Sphingosine 1-phosphate (S1P)/S1P receptor2/3 axis promotes inflammatory M1 polarization of bone marrow-derived monocyte/macrophage via G(α)i/o/PI3K/JNK pathway. Cell Physiol. Biochem. 2018, 49, 1677–1693. [Google Scholar] [CrossRef] [PubMed]
- Hymel, L.A.; Ogle, M.E.; Anderson, S.E.; Emeterio, C.L.S.; Turner, T.C.; York, W.Y.; Liu, A.Y.; Olingy, C.E.; Sridhar, S.; Lim, H.S.; et al. Modulating local S1P receptor signaling as a regenerative immunotherapy after volumetric muscle loss injury. J. Biomed. Mater. Res. Part A 2020, 109, 695–712. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Sun, C.; Argraves, K.M. Periodontal inflammation and alveolar bone loss induced by Aggregatibacter actinomycetemcomitans is attenuated in sphingosine kinase 1-deficient mice. J. Periodontal Res. 2016, 51, 38–49. [Google Scholar] [CrossRef] [Green Version]
- Hoeffel, G.; Wang, Y.; Greter, M.; See, P.; Teo, P.; Malleret, B.; Leboeuf, M.; Low, D.; Oller, G.; de Almeida, F.F.; et al. Adult Langerhans cells derive predominantly from embryonic fetal liver monocytes with a minor contribution of yolk sac–derived macrophages. J. Exp. Med. 2012, 209, 1167–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minutti, C.; Knipper, J.; Allen, J.E.; Zaiss, D.M. Tissue-specific contribution of macrophages to wound healing. Semin. Cell Dev. Biol. 2017, 61, 3–11. [Google Scholar] [CrossRef] [Green Version]
- DiPietro, L. Wound healing: The role of the macrophage and other immune cells. Shock 1995, 4, 233–240. [Google Scholar] [CrossRef]
- Lin, N.; Simon, M.C. Hypoxia-inducible factors: Key regulators of myeloid cells during inflammation. J. Clin. Investig. 2016, 126, 3661–3671. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Gajendrareddy, P.; DiPietro, L.A. Differential expression of HIF-1α in skin and mucosal wounds. J. Dent. Res. 2012, 91, 871–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, A.D.; Basi, D.L.; Abubaker, A.O. Wound healing: Findings of the 2005 AAOMS research summit. J. Oral Maxillofac. Surg. 2005, 63, 1426–1435. [Google Scholar] [CrossRef]
- Martin, P.; Leibovich, S.J. Inflammatory cells during wound repair: The good, the bad and the ugly. Trends Cell Biol. 2005, 15, 599–607. [Google Scholar] [CrossRef]
- Reinke, J.M.; Sorg, H. Wound repair and regeneration. Eur. Surg. Res. 2012, 49, 35–43. [Google Scholar] [CrossRef]
- Brancato, S.K.; Albina, J.E. Wound macrophages as key regulators of repair: Origin, phenotype, and function. Am. J. Pathol. 2011, 178, 19–25. [Google Scholar] [CrossRef]
- Sindrilaru, A.; Peters, T.; Wieschalka, S.; Baican, C.; Baican, A.; Peter, H.; Hainzl, A.; Schatz, S.; Qi, Y.; Schlecht, A.; et al. An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J. Clin. Investig. 2011, 121, 985–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucas, T.; Waisman, A.; Ranjan, R.; Roes, J.; Krieg, T.; Müller, W.; Roers, A.; Eming, S.A. Differential roles of macrophages in diverse phases of skin repair. J. Immunol. 2010, 184, 3964–3977. [Google Scholar] [CrossRef] [Green Version]
- Szpaderska, A.M.; Zuckerman, J.D.; DiPietro, L.A. Differential injury responses in oral mucosal and cutaneous wounds. J. Dent. Res. 2003, 82, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, F.; Yoshida, A.; Nakajima, A.; Wada-Takahashi, -S.; Takahashi, S.S.-I.; Lee, M.C. Alteration of the redox state with reactive oxygen species for 5-fluorouracil-induced oral mucositis in hamsters. PLoS ONE 2013, 8, e82834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glim, J.E.; Beelen, R.H.; Niessen, F.B.; Everts, V.; Ulrich, M.M. The number of immune cells is lower in healthy oral mucosa compared to skin and does not increase after scarring. Arch. Oral Biol. 2015, 60, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Szpaderska, A.M.; Walsh, C.G.; Steinberg, M.J.; DiPietro, L.A. Distinct patterns of angiogenesis in oral and skin wounds. J. Dent. Res. 2005, 84, 309–314. [Google Scholar] [CrossRef]
- Wong, J.W.; Gallant-Behm, C.; Wiebe, C.; Mak, K.; Hart, D.A.; Larjava, H.; Häkkinen, L. Wound healing in oral mucosa results in reduced scar formation as compared with skin: Evidence from the red Duroc pig model and humans. Wound Repair Regen. 2009, 17, 717–729. [Google Scholar] [CrossRef]
- Iglesias-Bartolome, R.; Uchiyama, A.; Molinolo, A.A.; Abusleme, L.; Brooks, S.R.; Callejas-Valera, J.L.; Edwards, D.; Doci, C.; Asselin-Labat, M.-L.; Onaitis, M.W.; et al. Transcriptional signature primes human oral mucosa for rapid wound healing. Sci. Transl. Med. 2018, 10, eaap8798. [Google Scholar] [CrossRef] [Green Version]
- Mak, K.; Manji, A.; Gallant-Behm, C.; Wiebe, C.; Hart, D.A.; Larjava, H.; Häkkinen, L. Scarless healing of oral mucosa is characterized by faster resolution of inflammation and control of myofibroblast action compared to skin wounds in the red Duroc pig model. J. Dermatol. Sci. 2009, 56, 168–180. [Google Scholar] [CrossRef]
- O’Kane, S.; Ferguson, M.W. Transforming growth factor beta s and wound healing. Int. J. Biochem. Cell Biol. 1997, 29, 63–78. [Google Scholar] [CrossRef]
- Jinno, K.; Takahashi, T.; Tsuchida, K.; Tanaka, E.; Moriyama, K. Acceleration of palatal wound healing in Smad3-deficient mice. J. Dent. Res. 2009, 88, 757–761. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, N.; Yasue, A.; Watanabe, T.; Tanaka, E. Down-regulation of Smad3 accelerates palatal wound repair. J. Dent. Res. 2013, 92, 716–720. [Google Scholar] [CrossRef]
- Wilgus, T.A. Immune cells in the healing skin wound: Influential players at each stage of repair. Pharmacol. Res. 2008, 58, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Zelles, T.; Purushotham, K.R.; Macauley, S.P.; Oxford, G.E.; Humphreys-Beher, M.G. Saliva and growth factors: The fountain of youth resides in us all. J. Dent. Res. 1995, 74, 1826–1832. [Google Scholar] [CrossRef]
- Ashcroft, G.S.; Lei, K.; Jin, W.; Longenecker, G.; Kulkarni, A.B.; Greenwell-Wild, T.; Hale-Donze, H.; McGrady, G.; Song, X.-Y.; Wahl, S.M. Secretory leukocyte protease inhibitor mediates non-redundant functions necessary for normal wound healing. Nat. Med. 2000, 6, 1147–1153. [Google Scholar] [CrossRef]
- Tamaki, N.; Campos, R.C.O.; Fukui, M.; Ito, H.-O. Hydrogen-rich water intake accelerates oral palatal wound healing via activation of the Nrf2/antioxidant defense pathways in a rat model. Oxidative Med. Cell. Longev. 2015, 2016, 5679040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, H.; Li, X.; Hu, S.; Liu, T.; Yuan, B.; Gu, H.; Ni, Q.; Zhang, X.; Zheng, F. IL-33 accelerates cutaneous wound healing involved in upregulation of alternatively activated macrophages. Mol. Immunol. 2013, 56, 347–353. [Google Scholar] [CrossRef]
- Yin, H.; Li, X.Y.; Yuan, B.H.; Zhang, B.B.; Hu, S.L.; Gu, H.B.; Jin, X.B.; Zhu, J.Y. Adenovirus-mediated overexpression of soluble ST2 provides a protective effect on lipopolysaccharide-induced acute lung injury in mice. Clin. Exp. Immunol. 2011, 164, 248–255. [Google Scholar] [CrossRef]
- Duan, L.; Chen, J.; Zhang, H.; Yang, H.; Zhu, P.; Xiong, A.; Xia, Q.; Zheng, F.; Tan, Z.; Gong, F.; et al. Interleukin-33 ameliorates experimental colitis through promoting Th2/Foxp3(+) regulatory T-cell responses in mice. Mol. Med. 2012, 18, 753–761. [Google Scholar] [CrossRef]
- Cramer, T.; Yamanishi, Y.; Clausen, B.E.; Forster, I.; Pawlinski, R.; Mackman, N.; Haase, V.H.; Jaenisch, R.; Corr, M.; Nizet, V.; et al. HIF-1α is essential for myeloid cell-mediated inflammation. Cell 2003, 112, 645–657. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Li, Y.; Li, Z.; Huang, C.; Yang, Y.; Lang, M.; Cao, J.; Jiang, W.; Xu, Y.; Dong, J.; et al. Hypoxia inducible factor 1 (HIF-1) recruits macrophage to activate pancreatic stellate cells in pancreatic ductal adenocarcinoma. Int. J. Mol. Sci. 2016, 17, 799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prabhakar, N.R. Sensing hypoxia: Physiology, genetics and epigenetics. J. Physiol. 2013, 591, 2245–2257. [Google Scholar] [CrossRef]
- Ratcliffe, P.J. Oxygen sensing and hypoxia signalling pathways in animals: The implications of physiology for cancer. J. Physiol. 2013, 591, 2027–2042. [Google Scholar] [CrossRef]
- Loboda, A.; Jozkowicz, A.; Dulak, J. HIF-1 and HIF-2 transcription factors—Similar but not identical. Mol. Cells 2010, 29, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Monick, M.M.; Mallampalli, R.K.; Bradford, M.; McCoy, D.; Gross, T.J.; Flaherty, D.M.; Powers, L.S.; Cameron, K.; Kelly, S.; Merrill, A.H.; et al. Cooperative prosurvival activity by ERK and Akt in human alveolar macrophages is dependent on high levels of acid ceramidase activity. J. Immunol. 2004, 173, 123–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Ponti, C.; Carini, R.; Alchera, E.; Nitti, M.P.; Locati, M.; Albano, E.; Cairo, G.; Tacchini, L. Adenosine A2a receptor-mediated, normoxic induction of HIF-1 through PKC and PI-3K-dependent pathways in macrophages. J. Leukoc. Biol. 2007, 82, 392–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blouin, C.C.; Page, E.L.; Soucy, G.M.; Richard, D.E. Hypoxic gene activation by lipopolysaccharide in macrophages: Implication of hypoxia-inducible factor 1α. Blood 2004, 103, 1124–1130. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Jeong, E.; Joung, S.M.; Lee, J.Y. PI3K/Akt contributes to increased expression of Toll-like receptor 4 in macrophages exposed to hypoxic stress. Biochem. Biophys. Res. Commun. 2012, 419, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Semba, H.; Takeda, N.; Isagawa, T.; Sugiura, Y.; Honda, K.; Wake, M.; Miyazawa, H.; Yamaguchi, Y.; Miura, M.; Jenkins, D.M.R.; et al. HIF-1α-PDK1 axis-induced active glycolysis plays an essential role in macrophage migratory capacity. Nat. Commun. 2016, 7, 11635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Real-Hohn, A.; Zancan, P.; Da Silva, D.; Martins, E.R.; Salgado, L.T.; Mermelstein, C.S.; Gomes, A.M.; Sola-Penna, M. Filamentous actin and its associated binding proteins are the stimulatory site for 6-phosphofructo-1-kinase association within the membrane of human erythrocytes. Biochimie 2010, 92, 538–544. [Google Scholar] [CrossRef]
- Shiraishi, T.; Verdone, J.E.; Huang, J.; Kahlert, U.D.; Hernandez, J.R.; Torga, G.; Zarif, J.C.; Epstein, T.; Gatenby, R.; McCartney, A.; et al. Glycolysis is the primary bioenergetic pathway for cell motility and cytoskeletal remodeling in human prostate and breast cancer cells. Oncotarget 2015, 6, 130–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.; Zhang, D.-X.; Shi, X.; Zhang, Q.; Huang, Y.-S. Activation of the prolyl-hydroxylase oxygen-sensing signal cascade leads to AMPK activation in cardiomyocytes. J. Cell. Mol. Med. 2011, 16, 2049–2059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takaku, M.; Tomita, S.; Kurobe, H.; Kihira, Y.; Morimoto, A.; Higashida, M.; Ikeda, Y.; Ushiyama, A.; Hashimoto, I.; Nakanishi, H.; et al. Systemic preconditioning by a prolyl hydroxylase inhibitor promotes prevention of skin flap necrosis via HIF-1-induced bone marrow-derived cells. PLoS ONE 2012, 7, e42964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhdanov, A.V.; Okkelman, I.A.; Collins, F.W.; Melgar, S.; Papkovsky, D.B. A novel effect of DMOG on cell metabolism: Direct inhibition of mitochondrial function precedes HIF target gene expression. Biochim. Biophys. Acta 2015, 1847, 1254–1266. [Google Scholar] [CrossRef] [Green Version]
- Reznick, R.M.; Shulman, G.I. The role of AMP-activated protein kinase in mitochondrial biogenesis. J. Physiol. 2006, 574, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Jager, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef] [Green Version]
- Chae, S.S.; Paik, J.H.; Allende, M.L.; Proia, R.L.; Hla, T. Regulation of limb development by the sphingosine 1-phosphate receptor S1p1/EDG-1 occurs via the hypoxia/VEGF axis. Dev. Biol. 2004, 268, 441–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.H.; Kim, C.; Aref, A.R.; Kamm, R.D.; Raghunath, M. Complementary effects of ciclopirox olamine, a prolyl hydroxylase inhibitor and sphingosine 1-phosphate on fibroblasts and endothelial cells in driving capillary sprouting. Integr. Biol. (Camb) 2013, 5, 1474–1484. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Inflammatory Conditions | Signaling Pathways | S1P Receptors | Roles |
|---|---|---|---|
| Palatal wound healing [57] | MCP-1, HIF-1α, MIP-1α, iNOS, TNF-α, Akt, p38 | S1P1 | Infiltration of M1/M2 macrophages |
| CNS autoimmunity and neuroinflammation [41] | STAT-3, JAK, IL-6 | S1P1 | Pronounced activation of monocytes |
| Inflammation and atherosclerosis [21,24,25,31,32,33] | TNF-α, MCP-1, IL-6, Akt, PI3K, PKC, ERK1/2, p38, Rho kinases | S1P1 | Activation and migration of macrophages |
| Gα12/13, NF-κB, RhoA | S1P2 | Inhibits macrophage migration (negative effect) and inhibits M1 activation | |
| MCP-1, TNF-α, Sphk1 | S1P3 | Migration of monocytes/macrophages | |
| Tubulointerstitial inflammation [36] | MCP-1, SphK1, TNF-α, arginase-1, IL-6, IL-10 | S1P1 S1P3 | Macrophage infiltration |
| Acute allergic [37] | MCP-1/CCL2, MIP-1α, RANTES/CCL5 | S1P2 | Macrophage infiltration |
| Inflammation and liver injury [29,30,179] | Gαi/o, PI3K, JNK, Rac1 | S1P2 S1P3 | Migration of BMMs, M1 polarization |
| NLRP3, IL-1β, IL-18, p38, ERK, JNK, Gα12/13 | S1P2 | Blockade BMMs activation and M1 polarization | |
| Rheumatoid arthritis [23,175] | NF-κB, Fas, Akt, p38, ERK1/2, Rac, Rho | S1P1 | Motility and number of BMMs induced osteoclast |
| Cerebral ischemia [49] | NF-κB p65, ERK1/2, p38, Akt | S1P3 | M1 polarization |
| Bacterial sepsis [50,173,177] | Caspase-11 | S1P2 | Macrophage pyroptosis |
| MCP-1, Sphk1, TNF-α, arginase-1, IL-6, IL-10 | S1P1 S1P3 | Macrophage infiltration | |
| Psoriasis [54] | IL-6, CCL2, CXCL1 | S1P4 | M2 macrophage infiltration |
| Bone marrow sinusoidal inflammation [55] | CCR2 | S1P5 | Monocyte migration |
| Pulmonary disease [27,28] | CCR2 | S1P5 | Monocyte migration |
| Encephalomyelitis [25] | CCR2 | S1P5 | Inflammatory monocytes supply the concentration of lymph node S1P |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hutami, I.R.; Izawa, T.; Khurel-Ochir, T.; Sakamaki, T.; Iwasa, A.; Tanaka, E. Macrophage Motility in Wound Healing Is Regulated by HIF-1α via S1P Signaling. Int. J. Mol. Sci. 2021, 22, 8992. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168992
Hutami IR, Izawa T, Khurel-Ochir T, Sakamaki T, Iwasa A, Tanaka E. Macrophage Motility in Wound Healing Is Regulated by HIF-1α via S1P Signaling. International Journal of Molecular Sciences. 2021; 22(16):8992. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168992
Chicago/Turabian StyleHutami, Islamy Rahma, Takashi Izawa, Tsendsuren Khurel-Ochir, Takuma Sakamaki, Akihiko Iwasa, and Eiji Tanaka. 2021. "Macrophage Motility in Wound Healing Is Regulated by HIF-1α via S1P Signaling" International Journal of Molecular Sciences 22, no. 16: 8992. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168992