
Fungal Biotransformation of 2′-Methylflavanone and 2′-Methylflavone as a Method to Obtain Glycosylated Derivatives


Abstract
:1. Introduction
2. Results
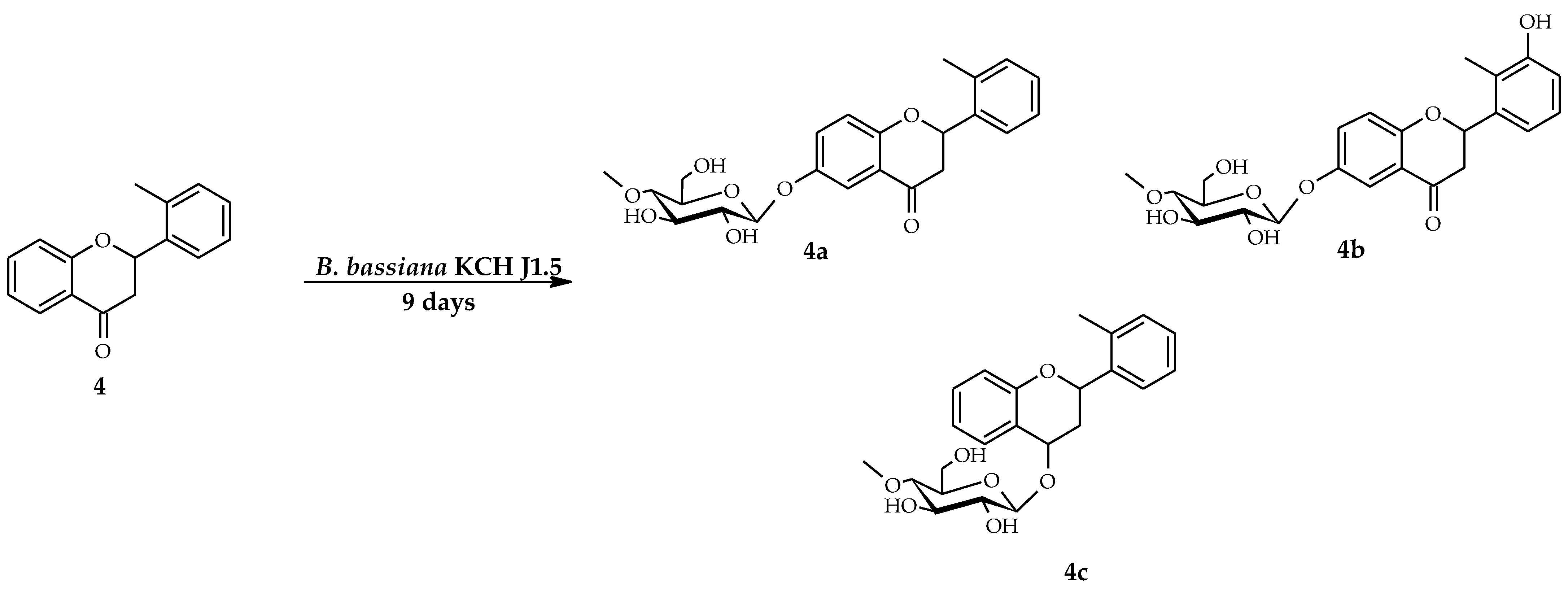
2.1. Biotransformations of 2′-Methylflavanone (4) in the Culture of B. bassiana KCH J1.5
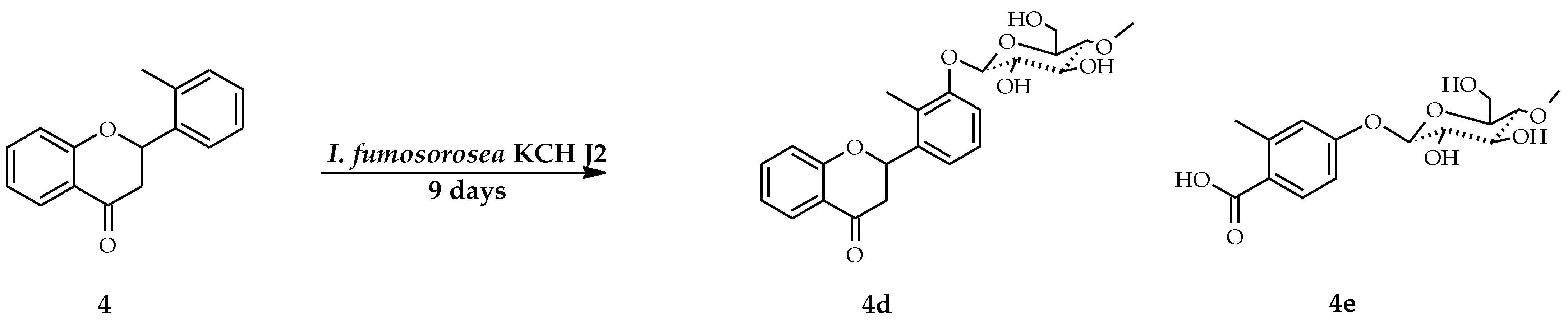
2.2. Biotransformations of 2′-Methylflavanone (4) in the Culture of I. fumosorosea KCH J2
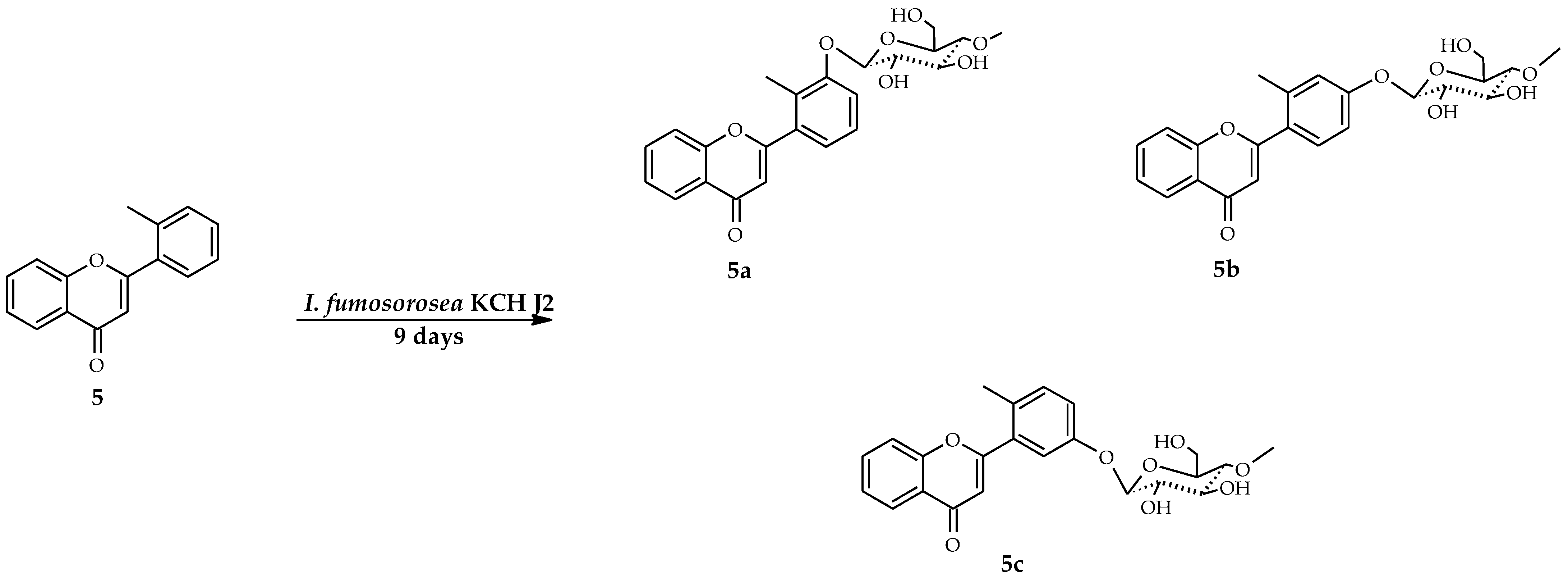
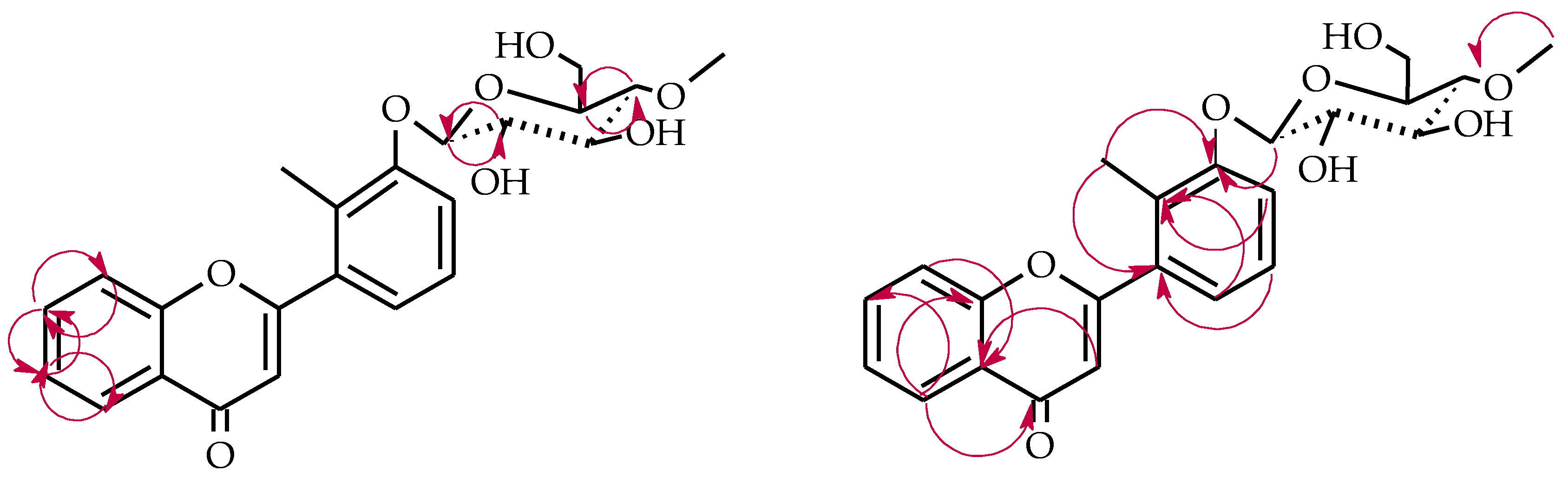
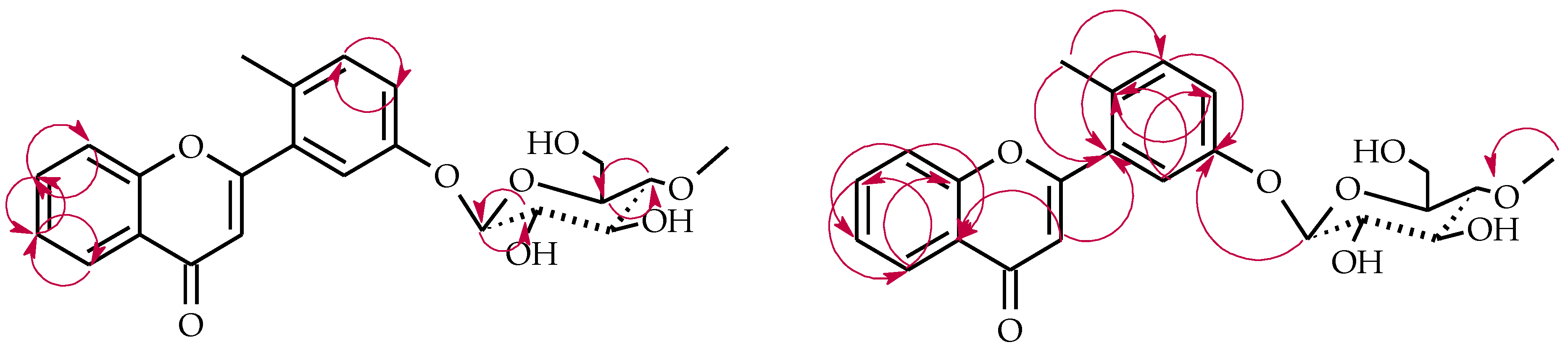
2.3. Biotransformations of 2′-Methylflavone (5) in the Culture of I. fumosorosea KCH J2
3. Discussion
4. Materials and Methods
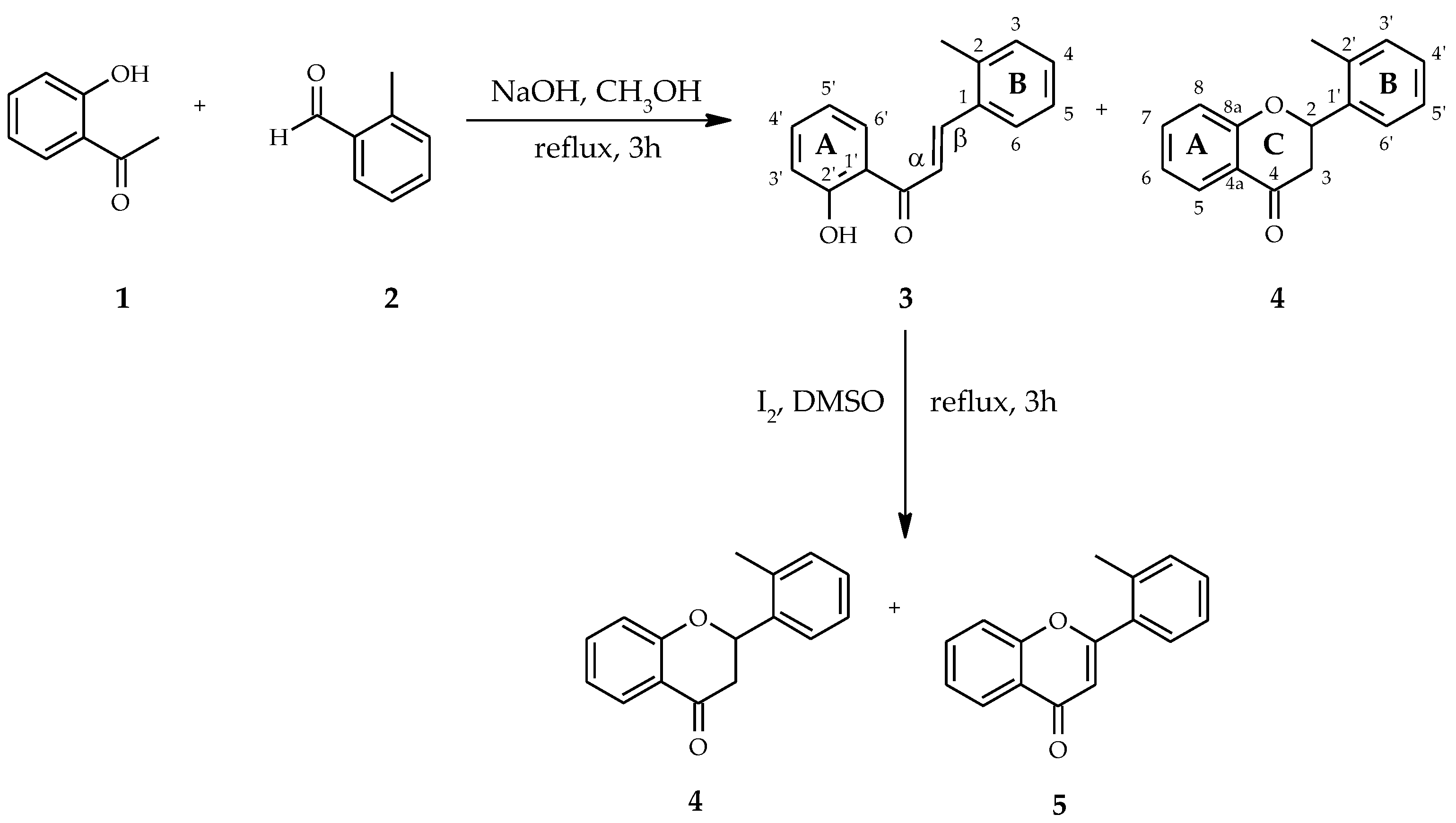
4.1. Substrates
4.1.1. 2′-Methylflavanone (4)
4.1.2. 2′-Methylflavone (5)
4.2. Microorganisms
4.3. Analysis
4.4. Screening Procedure
4.5. The Semipreparative Biotransformations
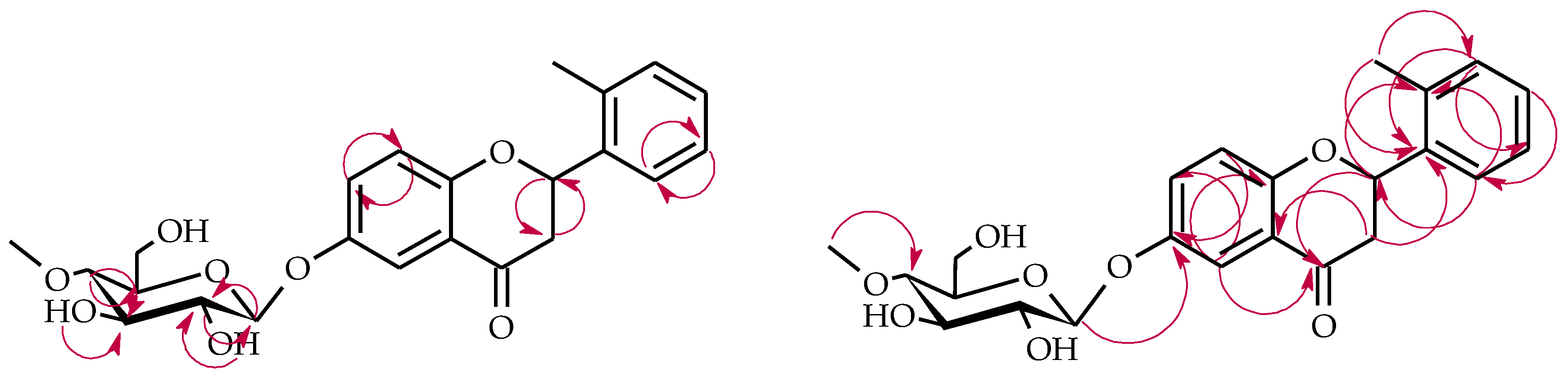
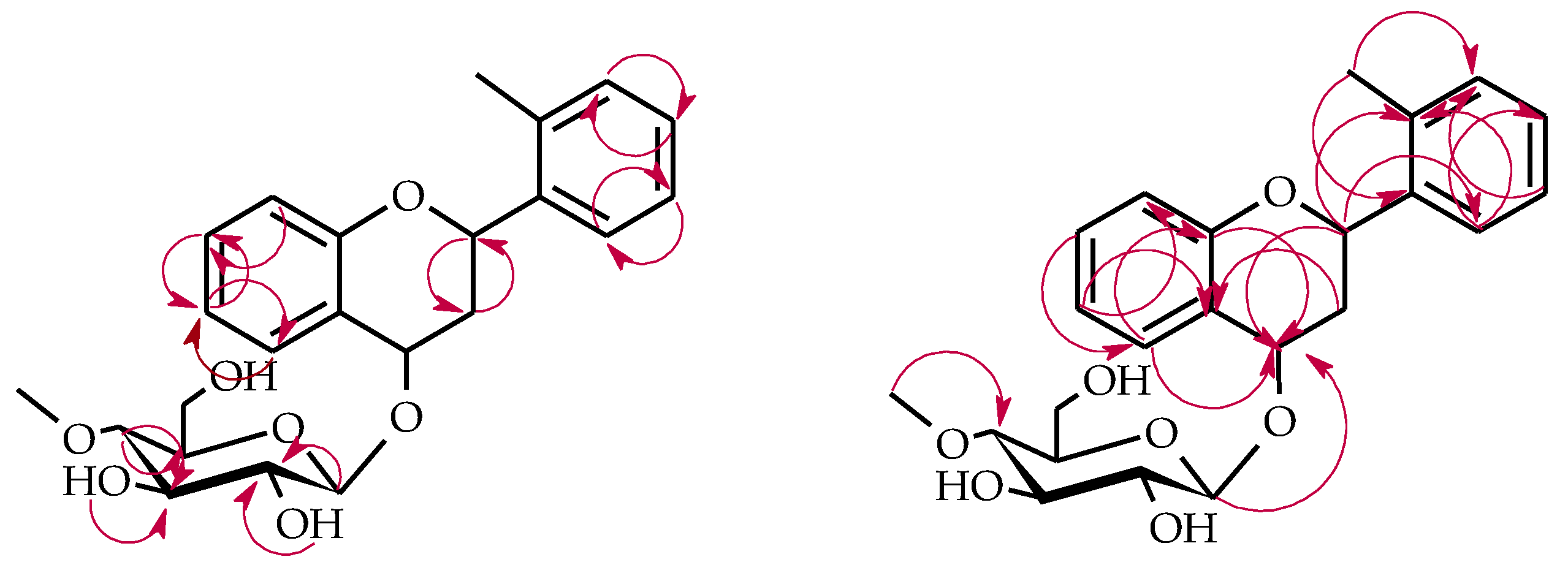
4.5.1. 2′-Methylflavanone 6-O-β-d-(4″-O-Methyl)-glucopyranoside (4a)
4.5.2. 3′-Hydroxy-2′-Methylflavanone 6-O-β-d-(4″-O-Methyl)-glucopyranoside (4b)
4.5.3. 2-(2′-Methylphenyl)-chromane 4-O-β-d-(4″-O-Methyl)-glucopyranoside (4c)
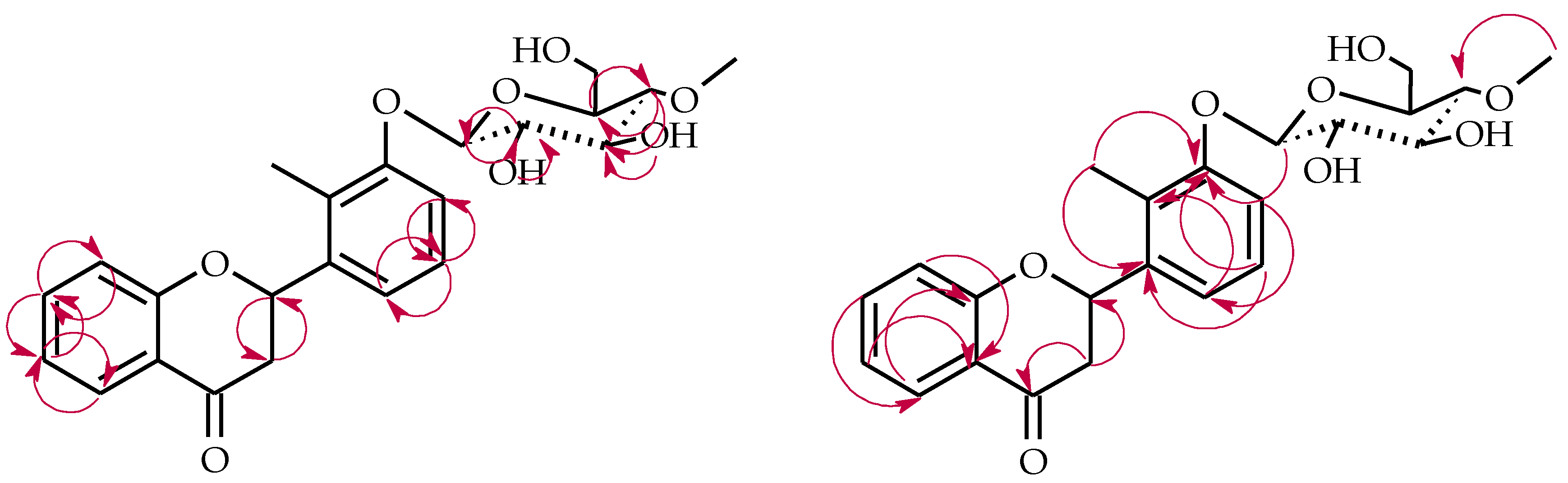
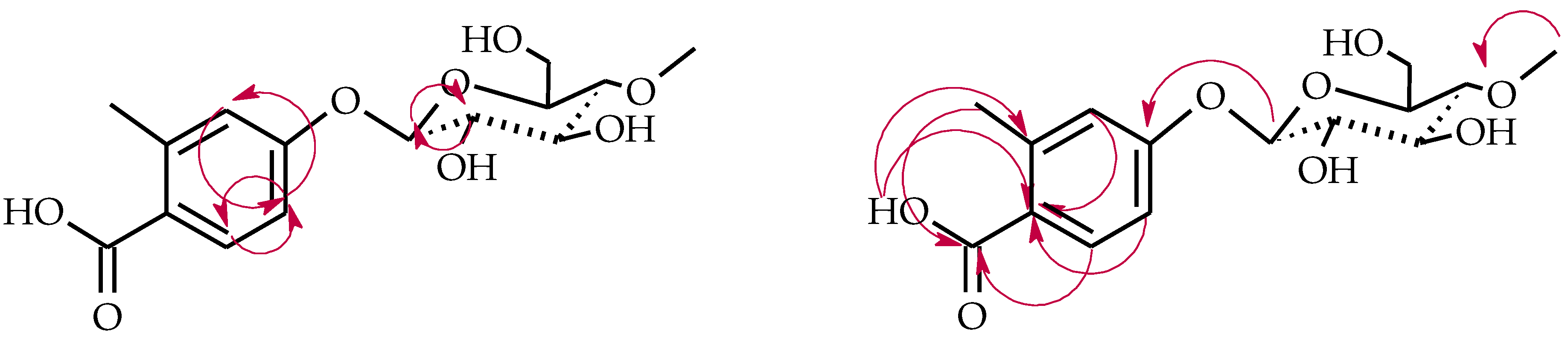
4.5.4. 2′-Methylflavanone 3′-O-β-d-(4″-O-Methyl)-glucopyranoside (4d)
4.5.5. 2-Methylbenzoic Acid 4-O-β-d-(4″-O-Methyl)-glucopyranoside (4e)
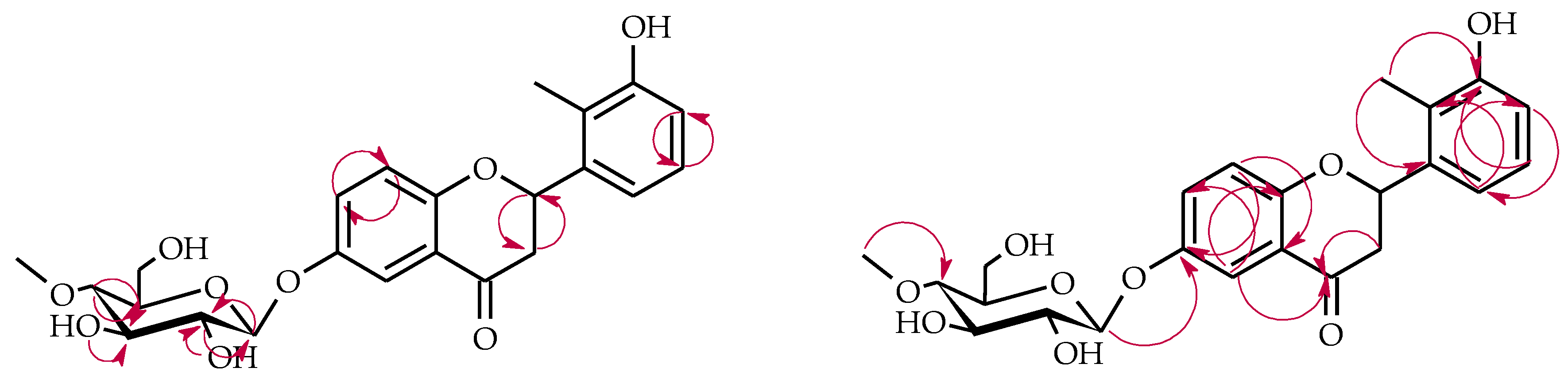
4.5.6. 2′-Methylflavone 3′-O-β-d-(4″-O-Methyl)-glucopyranoside (5a)
4.5.7. 2′-Methylflavone 4′-O-β-d-(4″-O-Methyl)-glucopyranoside (5b)
4.5.8. 2′-Methylflavone 5′-O-β-d-(4″-O-Methyl)-glucopyranoside (5c)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Raffa, D.; Maggio, B.; Raimondi, M.V.; Plescia, F.; Daidone, G. Recent discoveries of anticancer flavonoids. Eur. J. Med. Chem. 2017, 142, 213–228. [Google Scholar] [CrossRef]
- Wang, T.Y.; Li, Q.; Bi, K. Bioactive flavonoids in medicinal plants: Structure, activity and biological fate. Asian J. Pharm. Sci. 2018, 13, 12–23. [Google Scholar] [CrossRef]
- Xiao, J.; Muzashvili, T.S.; Georgiev, M.I. Advances in the biotechnological glycosylation of valuable flavonoids. Biotechnol. Adv. 2014, 32, 1145–1156. [Google Scholar] [CrossRef]
- Koirala, N.; Thuan, N.H.; Ghimire, G.P.; Thang, D.V.; Sohng, J.K. Methylation of flavonoids: Chemical structures, bioactivities, progress and perspectives for biotechnological production. Enzym. Microb. Technol. 2016, 86, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Blunder, M.; Orthaber, A.; Bauer, R.; Bucar, F.; Kunert, O. Efficient identification of flavones, flavanones and their glycosides in routine analysis via off-line combination of sensitive NMR and HPLC experiments. Food Chem. 2017, 218, 600–609. [Google Scholar] [CrossRef]
- Wen, X.; Walle, T. Methylated flavonoids have greatly improved intestinal absorption and metabolic stability. Drug Metab. Dispos. 2006, 34, 1786–1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thilakarathna, S.H.; Rupasinghe, V.H.P. Flavonoid bioavailability and attempts for bioavailability enhancement. Nutrients 2013, 5, 3367–3387. [Google Scholar] [CrossRef]
- Walle, T.; Ta, N.; Kawamori, T.; Wen, X.; Tsuji, P.A.; Walle, U.K. Cancer chemopreventive properties of orally bioavailable flavonoids-methylated versus unmethylated flavones. Biochem. Pharmacol. 2007, 73, 1288–1296. [Google Scholar] [CrossRef] [Green Version]
- Xiao, J. Dietary flavonoid aglycones and their glycosides: Which show better biological significance? Crit. Rev. Food Sci. Nutr. 2017, 57, 1874–1905. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, A.; Minihane, A.M. The role of metabolism (and the microbiome) in defining the clinical efficacy of dietary flavonoids. Am. J. Clin. Nutr. 2017, 105, 10–22. [Google Scholar] [CrossRef] [Green Version]
- Hollman, P. Absorption, bioavailability, and metabolism of flavonoids. Pharm. Biol. 2004, 42, 74–83. [Google Scholar] [CrossRef]
- Kawabata, K.; Yoshioka, Y.; Terao, J. Role of intestinal microbiota in the bioavailability and physiological functions of dietary polyphenols. Molecules 2019, 24, 370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oteiza, P.I.; Fraga, C.G.; Mills, D.A.; Taft, D.H. Flavonoids and the gastrointestinal tract: Local and systemic effects. Mol. Asp. Med. 2018, 61, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Pei, R.; Liu, X.; Bolling, B. Flavonoids and gut health. Curr. Opin. Biotechnol. 2020, 61, 153–159. [Google Scholar] [CrossRef]
- Hostetler, G.L.; Ralston, R.A.; Schwartz, S.J. Flavones: Food sources, bioavailability, metabolism, and bioactivity. Adv. Nutr. 2017, 8, 423–435. [Google Scholar] [CrossRef] [Green Version]
- Dymarska, M.; Janeczko, T.; Kostrzewa-Susłow, E. Biotransformations of flavones and an isoflavone (daidzein) in cultures of entomopathogenic filamentous fungi. Molecules 2018, 23, 1356. [Google Scholar] [CrossRef] [Green Version]
- Dou, F.; Wang, Z.; Li, G.; Dun, B. Microbial transformation of flavonoids by Isaria fumosorosea ACCC 37814. Molecules 2019, 24, 1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dymarska, M.; Janeczko, T.; Kostrzewa-Susłow, E. Glycosylation of methoxylated flavonoids in the cultures of Isaria fumosorosea KCH J2. Molecules 2018, 23, 2578. [Google Scholar] [CrossRef] [Green Version]
- Dymarska, M.; Grzeszczuk, J.; Urbaniak, M.; Janeczko, T.; Pląskowska, E.; Stępień, Ł.; Kostrzewa-Susłow, E. Glycosylation of 6-methylflavone by the strain Isaria fumosorosea KCH J2. PLoS ONE 2017, 12, e0184885. [Google Scholar] [CrossRef] [PubMed]
- Dymarska, M.; Janeczko, T.; Kostrzewa-Susłow, E. Glycosylation of 3-hydroxyflavone, 3-methoxyflavone, quercetin and baicalein in fungal cultures of the genus Isaria. Molecules 2018, 23, 2477. [Google Scholar] [CrossRef] [Green Version]
- Krawczyk-Łebek, A.; Dymarska, M.; Janeczko, T.; Kostrzewa-Susłow, E. Entomopathogenic filamentous fungi as biocatalysts in glycosylation of methylflavonoids. Catalysts 2020, 10, 1148. [Google Scholar] [CrossRef]
- Sordon, S.; Popłoński, J.; Tronina, T.; Huszcza, E. Regioselective O-glycosylation of flavonoids by fungi Beauveria bassiana, Absidia coerulea and Absidia glauca. Bioorganic Chem. 2019, 93, 102750. [Google Scholar] [CrossRef] [PubMed]
- Sordon, S.; Popłoński, J.; Tronina, T.; Huszcza, E. Microbial glycosylation of daidzein, genistein and biochanin a: Two new glucosides of biochanin A. Molecules 2017, 22, 81. [Google Scholar] [CrossRef] [Green Version]
- Strugała, P.; Tronina, T.; Huszcza, E.; Gabrielska, J. Bioactivity in vitro of quercetin glycoside obtained in Beauveria bassiana culture and its interaction with liposome membranes. Molecules 2017, 22, 1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tronina, T.; Strugała, P.; Popłoński, J.; Włoch, A.; Sordon, S.; Bartmańska, A.; Huszcza, E. The Influence of Glycosylation of Natural and Synthetic Prenylated Flavonoids on Binding to Human Serum Albumin and Inhibition of Cyclooxygenases COX-1 and COX-2. Molecules 2017, 22, 1230. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Zhang, L.; Wang, C.; Wang, X.; Xu, Y.; Yu, H.; Wu, P.; Li, S.; Han, L.; Gunatilaka, A.A.L.; et al. Methylglucosylation of aromatic amino and phenolic moieties of drug-like biosynthons by combinatorial biosynthesis. Proc. Natl. Acad. Sci. USA 2018, 115, E4980–E4989. [Google Scholar] [CrossRef] [Green Version]
- Kozłowska, E.; Urbaniak, M.; Hoc, N.; Grzeszczuk, J.; Dymarska, M.; Stępień, Ł.; Pląskowska, E.; Kostrzewa-Susłow, E.; Janeczko, T. Cascade biotransformation of dehydroepiandrosterone (DHEA) by Beauveria species. Sci. Rep. 2018, 8, 13449. [Google Scholar] [CrossRef]
- Łużny, M.; Tronina, T.; Kozłowska, E.; Dymarska, M.; Popłoński, J.; Łyczko, J.; Kostrzewa-Susłow, E.; Janeczko, T. Biotransformation of methoxyflavones by selected entomopathogenic filamentous fungi. Int. J. Mol. Sci. 2020, 21, 6121. [Google Scholar] [CrossRef] [PubMed]
- Włoch, A.; Strugała-Danak, P.; Pruchnik, H.; Krawczyk-Łebek, A.; Szczecka, K.; Janeczko, T.; Kostrzewa-Susłow, E. Interaction of 4′-methylflavonoids with biological membranes, liposomes, and human albumin. Sci. Rep. 2021, 11, 16003. [Google Scholar] [CrossRef]
- Sordon, S.; Popłoński, J.; Huszcza, E. Microbial glycosylation of flavonoids. Pol. J. Microbiol. 2016, 65, 137–151. [Google Scholar] [CrossRef] [Green Version]
- Zhan, J.; Leslie Gunatilaka, A.A. Selective 4′-O-methylglycosylation of the pentahydroxy-flavonoid quercetin by Beauveria bassiana ATCC 7159. Biocatal. Biotransformation 2006, 24, 396–399. [Google Scholar] [CrossRef]
- Dao, T.T.; Tung, B.T.; Nguyen, P.H.; Thuong, P.T.; Yoo, S.S.; Kim, E.H.; Kim, S.K.; Oh, W.K. C-methylated flavonoids from Cleistocalyx operculatus and their inhibitory effects on novel influenza A (H1N1) neuraminidase. J. Nat. Prod. 2010, 73, 1636–1642. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.Q.; Song, Z.J.; Xu, H.H. A new antifungal and cytotoxic C-methylated flavone glycoside from Picea neoveitchii. Bioorgranic Med. Chem. Lett. 2012, 22, 5819–5822. [Google Scholar] [CrossRef] [PubMed]
- Nobakht, M.; Trueman, S.J.; Wallace, H.M.; Brooks, P.R.; Streeter, K.J.; Katouli, M. Antibacterial properties of flavonoids from kino of the eucalypt tree, Corymbia torelliana. Plants 2017, 6, 39. [Google Scholar] [CrossRef] [Green Version]
- Ye, C.L.; Liu, Y.; Wei, D.Z. Antioxidant and anticancer activity of 3′-formyl-4′, 6′-dihydroxy-2′-methoxy-5′-methylchalcone and (2S)-8-formyl-5-hydroxy-7-methoxy-6-methylflavanone. J. Pharm. Pharmacol. 2007, 59, 553–559. [Google Scholar] [CrossRef]
- Salmazzo, G.R.; Verdan, M.H.; Silva, F.; Cicarelli, R.M.; da Silva Mota, J.; Salvador, M.J.; de Carvalho, J.E.; Cardoso, C.A.L. Chemical composition and antiproliferative, antioxidant and trypanocidal activities of the fruits from Campomanesia xanthocarpa (Mart.) O. Berg (Myrtaceae). Nat. Prod. Res. 2019, 35, 853–857. [Google Scholar] [CrossRef]
- Hall, B.J.; Chebib, M.; Hanrahan, J.R.; Johnston, G.A.R. Flumazenil-independent positive modulation of gamma-aminobutyric acid action by 6-methylflavone at human recombinant α1β2γ2L and α1β2 GABAA receptors. Eur. J. Pharmacol. 2006, 491, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.J.; Chebib, M.; Hanrahan, J.R.; Johnston, G.A.R. 6-Methylflavanone, a more efficacious positive allosteric modulator of γ-aminobutyric acid (GABA) action at human recombinant α2β2γ2L than at α1β2γ2L and α1β2 GABAA receptors expressed in Xenopus oocytes. Eur. J. Pharmacol. 2006, 512, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.M.S.; Tavares, H.R.; Barros, A.I.N.R.A.; Cavaleiro, J.A.S. NMR and structural and conformational features of 2′-hydroxychalcones and flavones. Spectrosc. Lett. 1997, 30, 1655–1667. [Google Scholar] [CrossRef]
- Yadav, N.; Dixit, S.K.; Bhattacharya, A.; Mishra, L.C.; Sharma, M.; Awasthi, S.K.; Bhasin, V.K. Antimalarial activity of newly synthesized chalcone derivatives in vitro. Chem. Biol. Drug Des. 2012, 80, 340–347. [Google Scholar] [CrossRef]
- Li, C.; Zhang, X.; Xue, X.; Zhang, F.; Xu, Q.; Liang, X. Structural characterization of iridoid glucosides by ultra-performance liquid chromatography/electrospray ionization quadrupole time-of-flight tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2008, 22, 1941–1954. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
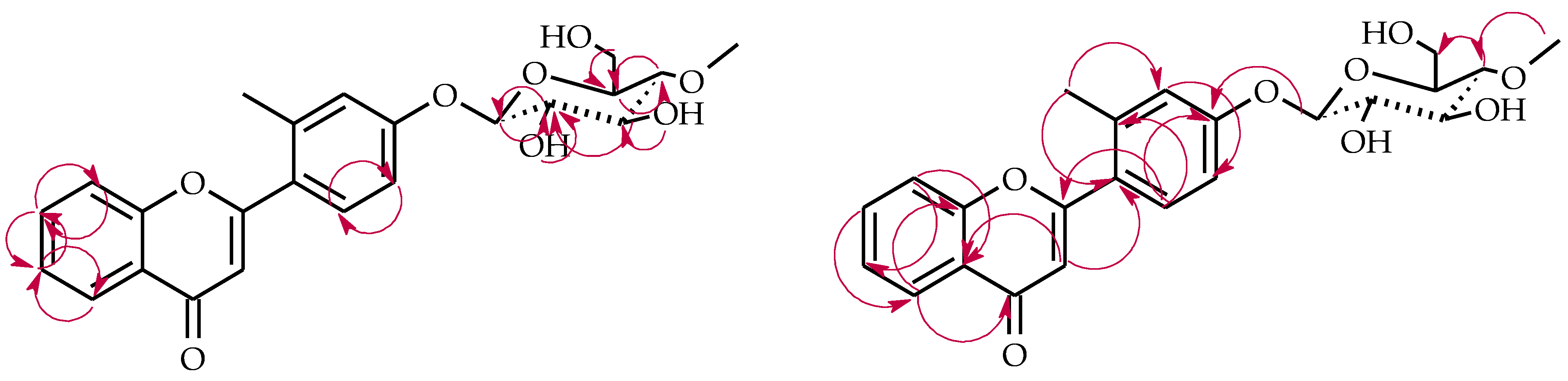{kind=link}
{kind=link}
| Proton | Compound | |||||
|---|---|---|---|---|---|---|
| 4 | 4a | 4b | 4c | 4d | 4e | |
| H-2 | 5.84 (dd) J = 13.5, J = 2.6 | 5.78 (dd) J = 13.6, J = 2.6 | 5.77 (dd) J = 13.4, J = 2.7 | 5.47 (dd) J = 12.1, J = 1.7 | 5.86 (dd) J = 13.5, J = 2.6 | - |
| H-3ax | 3.15 (dd) J = 16.8, J = 13.5 | 3.12 (ddd) J = 16.8, J = 13.5, J = 3.3 | 3.08 (dd) J = 16.9, J = 13.4 | 2.45 (dt) J = 14.8, J = 2.0 | 3.12 (dd) J = 16.8, J = 13.4 | - |
| H-3eq | 2.81 (dd) J = 16.8, J = 2.6 | 2.80 (dd) J = 16.9, J = 2.7 | 2.79 (dd) J = 17.1 J = 2.7 | 1.96 (m) | 2.81 (dd) J = 16.9, J = 2.7 | - |
| H-4 | - | - | - | 4.97 (m) | - | - |
| H-5 | 7.87 (dd) J = 7.8, J = 1.7 | 7.49 (d) J = 3.1 | 7.48 (d) J = 3.1 | 7.43 (dd) J = 7.7, J = 1.6 | 7.86 (dd) J = 7.9, J = 1.7 | - |
| H-6 | 7.10 (ddd) J = 7.9, J = 7.2, J = 0.9 | - | - | 6.93 (td) J = 7.4, J = 1.1 | 7.10 (td) J = 7.7, J = 1.0 | - |
| H-7 | 7.58 (m) | 7.33 (d) J = 8.9, J = 3.1 | 7.33 (dd) J = 8.9, J = 3.1 | 7.23 (m) | 7.58 (ddd) J = 8.5, J = 7.2, J = 1.7 | - |
| H-8 | 7.07 (dd) J = 8.5, J = 0.8 | 7.02 (dd) J = 9.0, J = 1.8 | 7.01 (dd) J = 8.9, J = 2.0 | 6.87 (m) | 7.07 (m) | - |
| H-3′ | 7.30 (m) | 7.28 (m) | - | 7.23 (m) | - | (3) 6.86 (d) J = 9.0 |
| H-4′ | 7.27 (m) | 7.28 (m) | 6.89 (dd) J = 6.6, J = 2.5 | 7.23 (m) | 7.18 (d) J = 7.5 | - |
| H-5′ | 7.30 (m) | 7.28 (m) | 7.11 (m) | 7.23 (m) | 7.24 (t) J = 8.0 | (5) 7.29 (d) J = 9.0, J = 3.0 |
| H-6′ | 7.64 (dd) J = 7.0, J = 2.0 | 7.62 (m) | 7.11 (m) | 7.54 (m) | 7.33 (d) J = 7.7 | (6) 7.62 (d) J = 2.9 |
| H-1″ | - | 4.88 (d) J = 7.8 | 4.88 (d) J = 7.8 | 4.63 (d) J = 7.8 | 4.94 (d) J = 7.8 | (1′) 4.86 (d) J = 7.8 |
| H-2″ | - | 3.46 (ddd) J = 10.8, J = 6.6, J = 3.2 | 3.46 (m) | 3.22 (m) | 3.54 (m) | (2′) 3.50 (ddd) J = 9.6, J = 5.7, J = 1.7 |
| H-3″ | - | 3.63 (m) | 3.63 (m) | 3.57 (dd) J = 8.9, J = 3.4 | 3.64 (td) J = 9.0, J = 4.0 | (3′) 3.61 (m) |
| H-4″ | - | 3.24 (m) | 3.24 (m) | 3.14 (m) | 3.23 (m) | (4′) 3.15 (m) |
| H-5″ | - | 3.46 (ddd) J = 10.8, J = 6.6, J = 3.2 | 3.46 (m) | 3.36 (dd) J = 9.7, J = 5.0 | 3.47 (ddd) J = 9.8, J = 4.8, J = 2.2 | (5′) 3.45 (tt) J = 5.3, J = 3.6 |
| H-6″ | - | 3.83 (ddd) J = 8.0, J = 4.1, J = 1.9 3.70 (dd) J = 7.2, J = 4.4 | 3.83 (ddd) J = 12.7, J = 8.2, J = 4.1 3.70 (dd) J = 7.1, J = 4.5 | 3.87 (m) 3.72 (dd) J = 14.0, J = 3.9 | 3.84 (ddd) J = 11.6, J = 5.1, J = 2.2 3.70 (ddd) J = 11.8, J = 7.1, J = 4.8 | (6′) 3.87 (qd) J = 5.7, J = 3.0 3.68 (m) |
| C4″-OCH3 | - | 3.57 (s) | 3.57 (s) | 3.54 (s) | 3.57 (s) | (4′-OCH3) 3.55 (s) |
| C2′-CH3 | 2.44 (s) | 2.43 (s) | 2.27 (s) | 2.43 (s) | 2.34 (s) | (2-CH3) 2.65 (s) |
| 2″-OH | - | 4.69 (d) J = 3.9 | 4.69 (d) J = 3.3 | 4.44 (d) J = 4.0 | 4.67 (d) J = 4.6 | (2′-OH) 4.64 (d) J = 3.6 |
| 3″-OH | - | 4.40 (d) J = 3.9 | 4.39 (d) J = 3.7 | 4.23 (d) J = 3.7 | 4.44(d) J = 4.1 | (3′-OH) 4.41 (d) J = 4.0 |
| 6″-OH | - | 3.72 (t) J = 3.1 | 3.72 (m) | 3.72 (dd) J = 14.0, J = 3.9 | 3.76 (m) | - |
| -COOH | - | - | - | 11.92 (s) | ||
| Carbon | Compound | |||||
|---|---|---|---|---|---|---|
| 4 | 4a | 4b | 4c | 4d | 4e | |
| C- 2 | 77.6 | 77.7 | 77.9 | 71.1 | 77.6 | - |
| C-3 | 43.8 | 43.8 | 43.9 | 34.5 | 44.0 | - |
| C-4 | 192.1 | 191.9 | 192.0 | 69.2 | 192.0 | (C=O) 205.9 |
| C-4a | 122.0 | 122.1 | 123.1 | 122.8 | 122.0 | - |
| C-5 | 127.4 | 114.0 | 113.9 | 132.9 | 127.4 | - |
| C-6 | 122.3 | 153.2 | 153.2 | 121.1 | 122.3 | - |
| C-7 | 136.8 | 126.7 | 126.7 | 130.3 | 136.8 | - |
| C-8 | 118.9 | 119.8 | 119.9 | 117.5 | 118.9 | - |
| C-8a | 162.7 | 158.1 | 158.1 | 156.5 | 162.7 | - |
| C-1′ | 138.1 | 138.2 | 122.1 | 140.4 | 139.3 | (1) 158.4 |
| C-2′ | 136.5 | 136.5 | 139.5 | 136.2 | 126.2 | (2) 120.1 |
| C-3′ | 129.3 | 131.5 | 156.3 | 128.4 | 156.9 | (3) 119.3 |
| C-4′ | 131.5 | 129.3 | 115.6 | 131.2 | 116.1 | (4) 150.8 |
| C-5′ | 127.1 | 127.1 | 127.3 | 126.9 | 127.5 | (5) 127.6 |
| C-6′ | 126.9 | 126.9 | 118.1 | 126.7 | 120.5 | (6) 118.9 |
| C-1″ | - | 102.7 | 102.7 | 100.7 | 102.2 | (1′) 102.8 |
| C-2″ | - | 75.0 | 75.0 | 75.0 | 75.1 | (2′) 77.3 |
| C-3″ | - | 77.9 | 77.9 | 78.2 | 78.1 | (3′) 78.1 |
| C-4″ | - | 80.0 | 80.0 | 80.7 | 80.1 | (4′) 80.5 |
| C-5″ | - | 77.0 | 77.0 | 77.0 | 77.0 | (5′) 74.9 |
| C-6″ | - | 62.0 | 62.0 | 62.6 | 62.1 | (6′) 62.3 |
| 4″-OCH3 | - | 60.5 | 60.5 | 60.5 | 60.5 | (4′-OCH3) 60.6 |
| 2′-CH3 | 19.1 | 19.1 | 11.1 | 19.3 | 11.4 | (2-CH3) 27.0 |
| Proton | Compound | |||
|---|---|---|---|---|
| 5 | 5a | 5b | 5c | |
| H-3 | 6.42 (s) | 6.39 (s) | 6.39 (s) | 6.44 (s) |
| H-5 | 8.15 (dd) J = 7.9, J = 1.7 | 8.15 (m) | 8.14 (dd) J = 8.0, J = 1.6 | 8.14 (dd) J = 8.0, J = 1.7 |
| H-6 | 7.50 (m) | 7.51 (t) J = 7.4, | 7.50 (m) | 7.51 (m) |
| H-7 | 7.82 (ddd) J = 8.7, J = 7.2, J = 1.7 | 7.82 (m) | 7.80 (ddd) J = 8.7, J = 7.2, J = 1.7 | 7.82 (ddd) J = 8.7, J = 7.2, J = 1.7 |
| H-8 | 7.64 (m) | 7.63 (d) J = 8.4 | 7.63 (d) J = 8.4 | 7.66 (dd) J = 8.4 J = 0.8 |
| H-3′ | 7.40 (m) | - | 7.06 (m) | 7.31 (d) J = 8.4 |
| H-4′ | 7.50 (m) | 7.36 (d) J = 8.1 | - | 7.17 (dd) J = 8.4, J = 2.7 |
| H-5′ | 7.40 (m) | 7.32 (t) J = 7.9 | 7.06 (m) | - |
| H-6′ | 7.64 (m) | 7.28 (d) J = 6.9 | 7.59 (d) J = 8.4 | 7.35 (d) J = 2.6 |
| H-1″ | - | 5.00 (d) J = 7.7 | 5.06 (d) J = 7.8 | 5.02 (d) J = 7.8 |
| H-2″ | - | 3.55 (d) J = 6.6 | 3.49 (m) | 3.47 (dd) J = 8.4, J = 3.2 |
| H-3″ | - | 3.65 (m) | 3.65 (tt) J = 6.7, J = 3.4 | 3.63 (m) |
| H-4″ | - | 3.25 (t) J = 9.3 | 3.23 (m) | 3.20 (m) |
| H-5″ | - | 3.51 (ddd) J = 9.6, J = 4.7, J = 2.0 | 3.53 (ddd) J = 9.8, J = 5.0, J = 2.1 | 3.50 (ddd) J = 9.8, J = 5.0, J = 2.1 |
| H-6″ | - | 3.86 (m) 3.71 (ddd) J = 10.8, J = 7.8, J = 4.5 | 3.86 (ddd) J = 11.4, J = 5.2, J = 2.0 3.70 (ddd) J = 11.5, J = 6.5, J = 5.1 | 3.84 (ddd) J = 11.6, J = 5.1, J = 2.1 3.68 (m) |
| C4″-OCH3 | - | 3.57 (s) | 3.57 (s) | 3.55 (s) |
| C2′-CH3 | 2.51 (s) | 2.37 (s) | 2.49 (s) | 2.44 (s) |
| 2″-OH | - | 4.72 (d) J = 4.3 | 4.72 (d) J = 4.2 | 4.67 (d) J = 3.8 |
| 3″-OH | - | 4.46 (d) J = 4.0 | 4.46 (d) J = 4.1 | 4.41 (d) J = 3.8 |
| 6″-OH | - | 3.80 (dd) J = 14.3, J = 7.8 | 3.82 (m) | 3.78 (dd) J = 10.2, J = 4.3 |
| Carbon | Compound | |||
|---|---|---|---|---|
| 5 | 5a | 5b | 5c | |
| C-2 | 166.7 | 166.5 | 166.5 | 166.2 |
| C-3 | 112.4 | 112.7 | 111.9 | 112.4 |
| C-4 | 177.8 | 177.8 | 177.8 | 177.8 |
| C-4a | 124.7 | 124.7 | 124.7 | 124.7 |
| C-5 | 126.0 | 126.1 | 126.0 | 126.0 |
| C-6 | 126.2 | 126.2 | 126.1 | 126.2 |
| C-7 | 134.9 | 134.9 | 134.8 | 134.9 |
| C-8 | 119.2 | 119.2 | 119.2 | 119.3 |
| C-8a | 157.4 | 157.4 | 157.3 | 157.4 |
| C-1′ | 133.7 | 135.1 | 127.4 | 134.3 |
| C-2′ | 137.7 | 127.4 | 139.6 | 131.0 |
| C-3′ | 132.1 | 157.2 | 119.9 | 133.1 |
| C-4′ | 131.6 | 118.2 | 160.4 | 119.9 |
| C-5′ | 127.1 | 127.8 | 114.9 | 156.9 |
| C-6′ | 130.2 | 123.8 | 131.7 | 118.1 |
| C-1″ | - | 102.2 | 101.3 | 101.8 |
| C-2″ | - | 75.0 | 74.9 | 75.0 |
| C-3″ | - | 78.1 | 78.0 | 78.0 |
| C-4″ | - | 80.1 | 80.1 | 80.2 |
| C-5″ | - | 77.1 | 77.2 | 77.1 |
| C-6″ | - | 62.1 | 62.1 | 62.2 |
| 4″-OCH3 | - | 60.6 | 60.6 | 60.5 |
| 2′-CH3 | 20.5 | 13.5 | 21.0 | 19.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krawczyk-Łebek, A.; Dymarska, M.; Janeczko, T.; Kostrzewa-Susłow, E. Fungal Biotransformation of 2′-Methylflavanone and 2′-Methylflavone as a Method to Obtain Glycosylated Derivatives. Int. J. Mol. Sci. 2021, 22, 9617. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179617
Krawczyk-Łebek A, Dymarska M, Janeczko T, Kostrzewa-Susłow E. Fungal Biotransformation of 2′-Methylflavanone and 2′-Methylflavone as a Method to Obtain Glycosylated Derivatives. International Journal of Molecular Sciences. 2021; 22(17):9617. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179617
Chicago/Turabian StyleKrawczyk-Łebek, Agnieszka, Monika Dymarska, Tomasz Janeczko, and Edyta Kostrzewa-Susłow. 2021. "Fungal Biotransformation of 2′-Methylflavanone and 2′-Methylflavone as a Method to Obtain Glycosylated Derivatives" International Journal of Molecular Sciences 22, no. 17: 9617. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179617