Mitochondrial Dysfunction and Heart Disease: Critical Appraisal of an Overlooked Association

 , , , and
, , , and 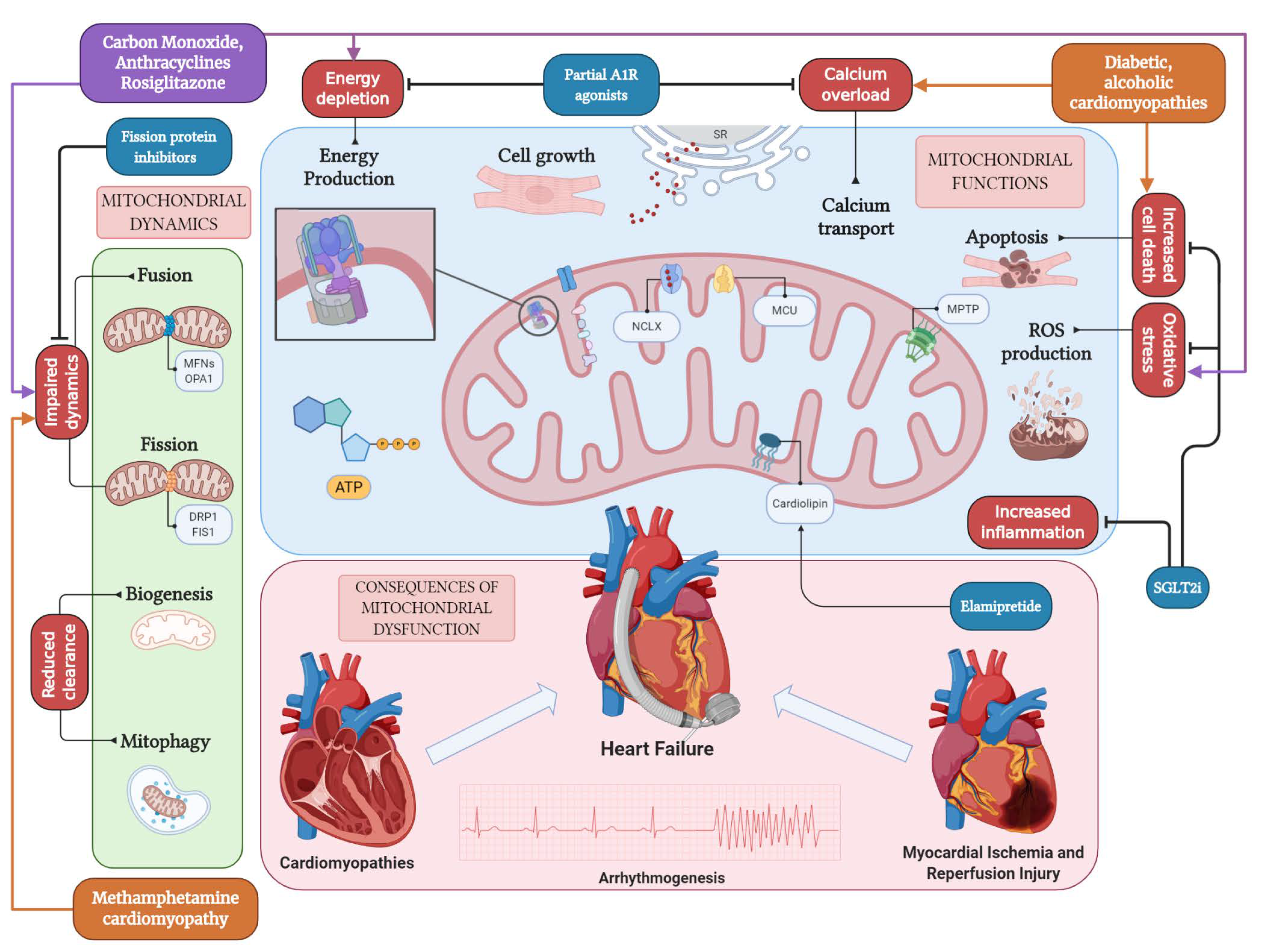{kind=link}
Abstract
:1. Introduction
2. Mitochondrial Structure and Function in the Normal Heart
2.1. Origin and Morphology of Mitochondria
2.2. Mitochondrial Networks in the Heart: Biogenesis, Mitophagy, and Mitochondrial Dynamics
2.3. Mitochondrial Bioenergetics and Ion Handling in the Heart
3. Mitochondrial Dysfunction and Cardiovascular Disorders
3.1. Myocardial Infarction and Ischemia/Reperfusion Injury
3.2. Drug-Induced and Toxic Cardiomyopathies
3.2.1. Alcohol
3.2.2. Methamphetamine
3.2.3. Anticancer Drugs
3.2.4. Carbon Monoxide
3.2.5. Other Drugs
3.3. Metabolic Cardiomyopathies: The Case for Diabetes and the Metabolic Syndrome
3.4. Conduction Disorders
4. Mitochondrial Involvement in Heart Failure
5. Targeting the Mitochondrion in Heart Failure
5.1. Mitochondrial Antioxidants: Elamipretide, mitoTEMPO, and mitoQ
5.2. Partial Adenosine A1 Receptor Agonists
5.3. SGLT2 Inhibitors
5.4. Mitochondria-Targeting Natural Compounds
6. Materials and Methods
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| A1R | Adenosine A1 receptor |
| AIDS | Acquired immune deficiency syndrome |
| ANTs | Anthracyclines |
| ATP | Adenosine triphosphate |
| CANVAS | Canagliflozin Cardiovascular Assessment Study |
| CVD | Cardiovascular disease |
| CMP | Cardiomyopathy |
| CO | Carbon monoxide |
| CREDENCE | Canagliflozin and Renal Endpoints in Diabetes with Established Nephropathy Clinical Evaluation |
| DAPA-HF | Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction |
| DECLARE-TIMI | Dapagliflozin Effect on Cardiovascular Events |
| DRP1 | Dynamin-related protein 1 |
| EMA | European Medicines Agency |
| EMPA-REG | Empagliflozin Cardiovascular Outcome Event Trial in Type 2 Diabetes Mellitus Patients |
| FDA | U.S. Food and Drug Administration |
| FIS1 | Mitochondrial fission 1 protein |
| HF | Heart failure |
| HFpEF | Heart failure with preserved ejection fraction |
| HFrEF | Heart failure with reduced ejection fraction |
| IRI | Ischemia/reperfusion injury |
| LCAC | Long-chain acylcarnitine |
| LV | Left ventricle/left ventricular |
| MCU | Mitochondrial calcium uniporter |
| METH | Methamphetamine |
| MFN1 | Mitofusin 1 |
| MFN2 | Mitofusin 2 |
| MI | Myocardial infarction |
| MPTP | Mitochondrial permeability transition pore |
| NAFLD | Nonalcoholic fatty liver disease |
| NCLX | Li+-permeable Na+–Ca2+ exchanger |
| OPA1 | Optic atrophy 1, mitochondrial dynamin like GTPase |
| OXPHOS | Oxidative phosphorylation |
| PGC-1α | Peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| RIRR | ROS-induced ROS release |
| ROS | Reactive oxygen species |
| SGLT | Sodium glucose cotransporter |
| SigmaR1 | Sigma-1 receptor |
| SR | Sarcoplasmic reticulum |
References
- Weiss, R.G.; Gerstenblith, G.; Bottomley, P.A. ATP flux through creatine kinase in the normal, stressed, and failing human heart. Proc. Natl. Acad. Sci. USA 2005, 102, 808–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, Y.S.; Turnbull, D.M. Mitochondrial disease: Genetics and management. J. Neurol. 2016, 263, 179–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Hattab, A.W.; Scaglia, F. Mitochondrial Cardiomyopathies. Front. Cardiovasc. Med. 2016, 3, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filardi, T.; Ghinassi, B.; Di Baldassarre, A.; Tanzilli, G.; Morano, S.; Lenzi, A.; Basili, S.; Crescioli, C. Cardiomyopathy Associated with Diabetes: The Central Role of the Cardiomyocyte. Int. J. Mol. Sci. 2019, 20, 3299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabbah, H.N. Targeting the Mitochondria in Heart Failure: A Translational Perspective. JACC Basic Transl. Sci. 2020, 5, 88–106. [Google Scholar] [CrossRef]
- Chan, D.C. Mitochondrial Dynamics and Its Involvement in Disease. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 235–259. [Google Scholar] [CrossRef] [Green Version]
- Taanman, J.W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Biophys. Acta 1999, 1410, 103–123. [Google Scholar] [CrossRef] [Green Version]
- Marín-García, J.; Goldenthal, M.J. Mitochondria and the Heart; Springer: New York, NY, USA, 2005; 400p. [Google Scholar]
- Giacomello, M.; Pyakurel, A.; Glytsou, C.; Scorrano, L. The cell biology of mitochondrial membrane dynamics. Nat. Rev. Mol. Cell Biol. 2020, 21, 204–224. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; De Benedictis, V.; Ruggiero, F.M.; Petrosillo, G. Functional role of cardiolipin in mitochondrial bioenergetics. Biochim. Biophys. Acta 2014, 1837, 408–417. [Google Scholar] [CrossRef] [Green Version]
- Hollander, J.M.; Thapa, D.; Shepherd, D.L. Physiological and structural differences in spatially distinct subpopulations of cardiac mitochondria: Influence of cardiac pathologies. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1–H14. [Google Scholar] [CrossRef] [Green Version]
- Dorn, G.W., 2nd; Vega, R.B.; Kelly, D.P. Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev. 2015, 29, 1981–1991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Knowlton, A.A. Mitochondrial dynamics in heart failure. Congest. Heart Fail. 2011, 17, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Olichon, A.; Baricault, L.; Gas, N.; Guillou, E.; Valette, A.; Belenguer, P.; Lenaers, G. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J. Biol. Chem. 2003, 278, 7743–7746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar] [PubMed] [Green Version]
- Yang, Y.; Li, T.; Li, Z.; Liu, N.; Yan, Y.; Liu, B. Role of Mitophagy in Cardiovascular Disease. Aging Dis. 2020, 11, 419–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Mitochondria and the Autophagy–Inflammation–Cell Death Axis in Organismal Aging. Science 2011, 333, 1109–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taneike, M.; Yamaguchi, O.; Nakai, A.; Hikoso, S.; Takeda, T.; Mizote, I.; Oka, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; et al. Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy 2010, 6, 600–606. [Google Scholar] [CrossRef]
- Shires, S.E.; Gustafsson, Å.B. Mitophagy and heart failure. J. Mol. Med. (Berl. Ger.) 2015, 93, 253–262. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Z.; Wei, Y.; Song, Q.; Du, B.; Wang, H.; Chu, Y.; Hu, Y. The Role of Myocardial Mitochondrial Quality Control in Heart Failure. Front. Pharmacol. 2019, 10. [Google Scholar] [CrossRef]
- Picca, A.; Mankowski, R.T.; Burman, J.L.; Donisi, L.; Kim, J.-S.; Marzetti, E.; Leeuwenburgh, C. Mitochondrial quality control mechanisms as molecular targets in cardiac ageing. Nat. Rev. Cardiol. 2018, 15, 543–554. [Google Scholar] [CrossRef]
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz-Juarez, J.; Suarez, J.A.; Dillmann, W.H.; Suarez, J. Mitochondrial calcium handling and heart disease in diabetes mellitus. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2021, 1867, 165984. [Google Scholar] [CrossRef] [PubMed]
- Duchen, M.R. Mitochondria and calcium: From cell signalling to cell death. J. Physiol. 2000, 529, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Feno, S.; Rizzuto, R.; Raffaello, A.; Vecellio Reane, D. The molecular complexity of the Mitochondrial Calcium Uniporter. Cell Calcium 2021, 93, 102322. [Google Scholar] [CrossRef]
- Boyman, L.; Lederer, W.J. How the mitochondrial calcium uniporter complex (MCUcx) works. Proc. Natl. Acad. Sci. USA 2020, 117, 22634. [Google Scholar] [PubMed]
- Wang, Y.; Han, Y.; She, J.; Nguyen, N.X.; Mootha, V.K.; Bai, X.-C.; Jiang, Y. Structural insights into the Ca2+-dependent gating of the human mitochondrial calcium uniporter. eLife 2020, 9, e60513. [Google Scholar] [CrossRef]
- Pan, X.; Liu, J.; Nguyen, T.; Liu, C.; Sun, J.; Teng, Y.; Fergusson, M.M.; Rovira, I.I.; Allen, M.; Springer, D.A.; et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol. 2013, 15, 1464–1472. [Google Scholar] [CrossRef] [Green Version]
- Luongo, T.; Lambert, J.; Yuan, A.; Zhang, X.; Gross, P.; Song, J.; Santhanam, S.; Gao, E.; Jain, M.; Houser, S.; et al. The Mitochondrial Calcium Uniporter Matches Energetic Supply with Cardiac Workload during Stress and Modulates Permeability Transition. Cell Rep. 2015, 12, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Holmström, K.M.; Pan, X.; Liu, J.C.; Menazza, S.; Liu, J.; Nguyen, T.T.; Pan, H.; Parks, R.J.; Anderson, S.; Noguchi, A.; et al. Assessment of cardiac function in mice lacking the mitochondrial calcium uniporter. J. Mol. Cell. Cardiol. 2015, 85, 178–182. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Rasmussen, T.P.; Koval, O.M.; Joiner, M.-l.A.; Hall, D.D.; Chen, B.; Luczak, E.D.; Wang, Q.; Rokita, A.G.; Wehrens, X.H.T.; et al. The mitochondrial uniporter controls fight or flight heart rate increases. Nat. Commun. 2015, 6, 6081. [Google Scholar] [CrossRef] [Green Version]
- Ferdinandy, P.; Hausenloy, D.J.; Heusch, G.; Baxter, G.F.; Schulz, R. Interaction of Risk Factors, Comorbidities, and Comedications with Ischemia/Reperfusion Injury and Cardioprotection by Preconditioning, Postconditioning, and Remote Conditioning. Pharmacol. Rev. 2014, 66, 1142–1174. [Google Scholar] [CrossRef]
- Honda, H.M.; Korge, P.; Weiss, J.N. Mitochondria and ischemia/reperfusion injury. Ann. N. Y. Acad. Sci. 2005, 1047, 248–258. [Google Scholar] [CrossRef]
- Ong, S.B.; Subrayan, S.; Lim, S.Y.; Yellon, D.M.; Davidson, S.M.; Hausenloy, D.J. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 2010, 121, 2012–2022. [Google Scholar] [CrossRef] [Green Version]
- Szteyn, K.; Singh, H. BKCa Channels as Targets for Cardioprotection. Antioxidants 2020, 9, 760. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial ROS-induced ROS release: An update and review. Biochim. Biophys. Acta (BBA) Bioenerg. 2006, 1757, 509–517. [Google Scholar] [CrossRef] [Green Version]
- Ramachandra, C.J.A.; Hernandez-Resendiz, S.; Crespo-Avilan, G.E.; Lin, Y.-H.; Hausenloy, D.J. Mitochondria in acute myocardial infarction and cardioprotection. EBioMedicine 2020, 57, 102884. [Google Scholar] [CrossRef]
- Hernandez-Resendiz, S.; Prunier, F.; Girao, H.; Dorn, G.; Hausenloy, D.J. Targeting mitochondrial fusion and fission proteins for cardioprotection. J. Cell. Mol. Med. 2020, 24, 6571–6585. [Google Scholar] [CrossRef]
- Aishwarya, R.; Alam, S.; Abdullah, C.S.; Morshed, M.; Nitu, S.S.; Panchatcharam, M.; Miriyala, S.; Kevil, C.G.; Bhuiyan, M.S. Pleiotropic effects of mdivi-1 in altering mitochondrial dynamics, respiration, and autophagy in cardiomyocytes. Redox Biol. 2020, 36, 101660. [Google Scholar] [CrossRef]
- Goswami, S.K.; Ponnalagu, D.; Hussain, A.T.; Shah, K.; Karekar, P.; Gururaja Rao, S.; Meredith, A.L.; Khan, M.; Singh, H. Expression and Activation of BKCa Channels in Mice Protects Against Ischemia-Reperfusion Injury of Isolated Hearts by Modulating Mitochondrial Function. Front. Cardiovasc. Med. 2019, 5, 194. [Google Scholar] [CrossRef] [Green Version]
- Seferović, P.M.; Polovina, M.; Bauersachs, J.; Arad, M.; Gal, T.B.; Lund, L.H.; Felix, S.B.; Arbustini, E.; Caforio, A.L.P.; Farmakis, D.; et al. Heart failure in cardiomyopathies: A position paper from the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2019, 21, 553–576. [Google Scholar] [CrossRef] [Green Version]
- Maisch, B. Alcoholic cardiomyopathy: The result of dosage and individual predisposition. Herz 2016, 41, 484–493. [Google Scholar] [CrossRef] [Green Version]
- Steiner, J.L.; Lang, C.H. Etiology of alcoholic cardiomyopathy: Mitochondria, oxidative stress and apoptosis. Int. J. Biochem. Cell Biol. 2017, 89, 125–135. [Google Scholar] [CrossRef]
- Sivakumar, A.; Shanmugarajan, S.; Subbiah, R.; Balakrishnan, R. Cardiac Mitochondrial PTEN-L determines cell fate between apoptosis and survival during chronic alcohol consumption. Apoptosis 2020, 25, 590–604. [Google Scholar] [CrossRef]
- Kevil, C.G.; Goeders, N.E.; Woolard, M.D.; Bhuiyan, M.S.; Dominic, P.; Kolluru, G.K.; Arnold, C.L.; Traylor, J.G.; Orr, A.W. Methamphetamine Use and Cardiovascular Disease. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1739–1746. [Google Scholar] [CrossRef]
- Abdullah, C.S.; Aishwarya, R.; Alam, S.; Morshed, M.; Remex, N.S.; Nitu, S.; Kolluru, G.K.; Traylor, J.; Miriyala, S.; Panchatcharam, M.; et al. Methamphetamine induces cardiomyopathy by Sigmar1 inhibition-dependent impairment of mitochondrial dynamics and function. Commun. Biol. 2020, 3, 682. [Google Scholar] [CrossRef]
- Murabito, A.; Hirsch, E.; Ghigo, A. Mechanisms of Anthracycline-Induced Cardiotoxicity: Is Mitochondrial Dysfunction the Answer? Front. Cardiovasc. Med. 2020, 7, 35. [Google Scholar] [CrossRef] [Green Version]
- Kerkelä, R.; Grazette, L.; Yacobi, R.; Iliescu, C.; Patten, R.; Beahm, C.; Walters, B.; Shevtsov, S.; Pesant, S.; Clubb, F.J.; et al. Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat. Med. 2006, 12, 908–916. [Google Scholar] [CrossRef] [Green Version]
- D’Amico, M.A.; Ghinassi, B.; Izzicupo, P.; Di Ruscio, A.; Di Baldassarre, A. IL-6 Activates PI3K and PKCzeta Signaling and Determines Cardiac Differentiation in Rat Embryonic H9c2 Cells. J. Cell. Physiol. 2016, 231, 576–586. [Google Scholar] [CrossRef]
- Bouitbir, J.; Alshaikhali, A.; Panajatovic, M.V.; Abegg, V.F.; Paech, F.; Krähenbühl, S. Mitochondrial oxidative stress plays a critical role in the cardiotoxicity of sunitinib: Running title: Sunitinib and oxidative stress in hearts. Toxicology 2019, 426, 152281. [Google Scholar] [CrossRef]
- Mohan, N.; Jiang, J.; Dokmanovic, M.; Wu, W.J. Trastuzumab-mediated cardiotoxicity: Current understanding, challenges, and frontiers. Antib. Ther. 2018, 1, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Bouitbir, J.; Panajatovic, M.V.; Frechard, T.; Roos, N.J.; Krähenbühl, S. Imatinib and Dasatinib Provoke Mitochondrial Dysfunction Leading to Oxidative Stress in C2C12 Myotubes and Human RD Cells. Front. Pharmacol. 2020, 11, 1106. [Google Scholar] [CrossRef]
- Pecoraro, M.; Pinto, A.; Popolo, A. Trastuzumab-induced cardiotoxicity and role of mitochondrial connexin43 in the adaptive response. Toxicol. In Vitro 2020, 67, 104926. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.S.; Lee, J.S.; Min, Y.G.; Park, J.S.; Jeon, W.C.; Park, E.J.; Shin, J.H.; Oh, S.; Choi, S.C. Carbon monoxide-induced cardiomyopathy. Circ. J. Off. J. Jpn. Circ. Soc. 2014, 78, 1437–1444. [Google Scholar] [CrossRef] [Green Version]
- Cha, Y.S.; Kim, H.; Hwang, S.O.; Kim, J.Y.; Kim, Y.K.; Choi, E.H.; Kim, O.H.; Kim, H.I.; Cha, K.C.; Lee, K.H. Incidence and patterns of cardiomyopathy in carbon monoxide-poisoned patients with myocardial injury. Clin. Toxicol. 2016, 54, 481–487. [Google Scholar] [CrossRef]
- Lippi, G.; Rastelli, G.; Meschi, T.; Borghi, L.; Cervellin, G. Pathophysiology, clinics, diagnosis and treatment of heart involvement in carbon monoxide poisoning. Clin. Biochem. 2012, 45, 1278–1285. [Google Scholar] [CrossRef]
- Wallach, J.D.; Wang, K.; Zhang, A.D.; Cheng, D.; Grossetta Nardini, H.K.; Lin, H.; Bracken, M.B.; Desai, M.; Krumholz, H.M.; Ross, J.S. Updating insights into rosiglitazone and cardiovascular risk through shared data: Individual patient and summary level meta-analyses. bmj 2020, 368, l7078. [Google Scholar] [CrossRef] [Green Version]
- Nissen, S.E. The rise and fall of rosiglitazone. Eur. Heart J. 2010, 31, 773–776. [Google Scholar] [CrossRef] [Green Version]
- Mishra, P.; Singh, S.V.; Verma, A.K.; Srivastava, P.; Sultana, S.; Rath, S.K. Rosiglitazone induces cardiotoxicity by accelerated apoptosis. Cardiovasc. Toxicol. 2014, 14, 99–119. [Google Scholar] [CrossRef]
- He, H.; Tao, H.; Xiong, H.; Duan, S.Z.; McGowan, F.X., Jr.; Mortensen, R.M.; Balschi, J.A. Rosiglitazone causes cardiotoxicity via peroxisome proliferator-activated receptor γ-independent mitochondrial oxidative stress in mouse hearts. Toxicol. Sci. 2014, 138, 468–481. [Google Scholar] [CrossRef] [Green Version]
- D’Andrea, G.; Brisdelli, F.; Bozzi, A. AZT: An old drug with new perspectives. Curr. Clin. Pharmacol. 2008, 3, 20–37. [Google Scholar]
- McKee, E.E.; Bentley, A.T.; Hatch, M.; Gingerich, J.; Susan-Resiga, D. Phosphorylation of thymidine and AZT in heart mitochondria: Elucidation of a novel mechanism of AZT cardiotoxicity. Cardiovasc. Toxicol. 2004, 4, 155–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, R.Y.; Mukhopadhyay, P.; Mohanraj, R.; Wang, H.; Horváth, B.; Yin, S.; Pacher, P. Resveratrol attenuates azidothymidine-induced cardiotoxicity by decreasing mitochondrial reactive oxygen species generation in human cardiomyocytes. Mol. Med. Rep. 2011, 4, 151–155. [Google Scholar] [PubMed] [Green Version]
- Varga, Z.V.; Ferdinandy, P.; Liaudet, L.; Pacher, P. Drug-induced mitochondrial dysfunction and cardiotoxicity. American J. Physiol. Heart Circ. Physiol. 2015, 309, H1453–H1467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guertl, B.; Noehammer, C.; Hoefler, G. Metabolic cardiomyopathies. Int. J. Exp. Pathol. 2000, 81, 349–372. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.L.; Zipes, D.; Libby, P.; Bonow, R. Braunwald’s Heart Disease: A Textbook of Cardiovascular Medicine. BMH Med. J. 2015, 5. [Google Scholar]
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef]
- Boudina, S.; Abel, E.D. Diabetic cardiomyopathy revisited. Circulation 2007, 115, 3213–3223. [Google Scholar] [CrossRef]
- Bugger, H.; Riehle, C.; Jaishy, B.; Wende, A.R.; Tuinei, J.; Chen, D.; Soto, J.; Pires, K.M.; Boudina, S.; Theobald, H.A.; et al. Genetic loss of insulin receptors worsens cardiac efficiency in diabetes. J. Mol. Cell. Cardiol. 2012, 52, 1019–1026. [Google Scholar] [CrossRef] [Green Version]
- Schooneman, M.G.; Vaz, F.M.; Houten, S.M.; Soeters, M.R. Acylcarnitines: Reflecting or inflicting insulin resistance? Diabetes 2013, 62, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Boudina, S.; Sena, S.; O’Neill, B.T.; Tathireddy, P.; Young, M.E.; Abel, E.D. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation 2005, 112, 2686–2695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rainer, P.P.; Primessnig, U.; Harenkamp, S.; Doleschal, B.; Wallner, M.; Fauler, G.; Stojakovic, T.; Wachter, R.; Yates, A.; Groschner, K.; et al. Bile acids induce arrhythmias in human atrial myocardium--implications for altered serum bile acid composition in patients with atrial fibrillation. Heart 2013, 99, 1685–1692. [Google Scholar] [CrossRef] [PubMed]
- Vasavan, T.; Ferraro, E.; Ibrahim, E.; Dixon, P.; Gorelik, J.; Williamson, C. Heart and bile acids—Clinical consequences of altered bile acid metabolism. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2018, 1864, 1345–1355. [Google Scholar] [CrossRef] [PubMed]
- Gambardella, J.; Sorriento, D.; Ciccarelli, M.; Del Giudice, C.; Fiordelisi, A.; Napolitano, L.; Trimarco, B.; Iaccarino, G.; Santulli, G. Functional Role of Mitochondria in Arrhythmogenesis. Adv. Exp. Med. Biol. 2017, 982, 191–202. [Google Scholar] [PubMed]
- O’Rourke, B.; Cortassa, S.; Akar, F.; Aon, M. Mitochondrial ion channels in cardiac function and dysfunction. Novartis Found. Symp. 2007, 287, 140–156. [Google Scholar] [PubMed] [Green Version]
- Kistamás, K.; Veress, R.; Horváth, B.; Bányász, T.; Nánási, P.P.; Eisner, D.A. Calcium Handling Defects and Cardiac Arrhythmia Syndromes. Front. Pharmacol. 2020, 11, 72. [Google Scholar] [CrossRef]
- Xie, W.; Santulli, G.; Reiken, S.R.; Yuan, Q.; Osborne, B.W.; Chen, B.-X.; Marks, A.R. Mitochondrial oxidative stress promotes atrial fibrillation. Sci. Rep. 2015, 5, 11427. [Google Scholar] [CrossRef] [Green Version]
- Neubauer, S. The Failing Heart—An Engine Out of Fuel. New Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef] [Green Version]
- Lindemayer, G.E.; Sordahl, L.A.; Schwartz, A. Reevaluation of Oxidative Phosphorylation in Cardiac Mitochondria from Normal Animals and Animals in Heart Failure. Circ. Res. 1968, 23, 439–450. [Google Scholar] [CrossRef] [Green Version]
- Sabbah, H.N.; Sharov, V.; Riddle, J.M.; Kono, T.; Lesch, M.; Goldstein, S. Mitochondrial abnormalities in myocardium of dogs with chronic heart failure. J. Mol. Cell. Cardiol. 1992, 24, 1333–1347. [Google Scholar] [CrossRef]
- Sharov, V.G.; Goussev, A.; Lesch, M.; Goldstein, S.; Sabbah, H.N. Abnormal Mitochondrial Function in Myocardium of Dogs with Chronic Heart Failure. J. Mol. Cell. Cardiol. 1998, 30, 1757–1762. [Google Scholar] [CrossRef] [PubMed]
- Sharov, V.G.; Todor, A.V.; Silverman, N.; Goldstein, S.; Sabbah, H.N. Abnormal Mitochondrial Respiration in Failed Human Myocardium. J. Mol. Cell. Cardiol. 2000, 32, 2361–2367. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, S.; Horn, M.; Cramer, M.; Harre, K.; Newell, J.B.; Peters, W.; Pabst, T.; Ertl, G.; Hahn, D.; Ingwall, J.S.; et al. Myocardial Phosphocreatine-to-ATP Ratio Is a Predictor of Mortality in Patients With Dilated Cardiomyopathy. Circulation 1997, 96, 2190–2196. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.T.; Abozguia, K.; Shivu, G.N.; Mahadevan, G.; Ahmed, I.; Williams, L.; Dwivedi, G.; Patel, K.; Steendijk, P.; Ashrafian, H.; et al. Heart failure with preserved ejection fraction is characterized by dynamic impairment of active relaxation and contraction of the left ventricle on exercise and associated with myocardial energy deficiency. J. Am. Coll. Cardiol. 2009, 54, 402–409. [Google Scholar] [CrossRef]
- Hunter, W.G.; Kelly, J.P.; McGarrah, R.W., 3rd; Khouri, M.G.; Craig, D.; Haynes, C.; Ilkayeva, O.; Stevens, R.D.; Bain, J.R.; Muehlbauer, M.J.; et al. Metabolomic Profiling Identifies Novel Circulating Biomarkers of Mitochondrial Dysfunction Differentially Elevated in Heart Failure With Preserved Versus Reduced Ejection Fraction: Evidence for Shared Metabolic Impairments in Clinical Heart Failure. J. Am. Heart Assoc. 2016, 5, e003190. [Google Scholar] [CrossRef] [Green Version]
- Wirth, C.; Brandt, U.; Hunte, C.; Zickermann, V. Structure and function of mitochondrial complex I. Biochim. Biophys. Acta (BBA) Bioenerg. 2016, 1857, 902–914. [Google Scholar] [CrossRef]
- Forte, M.; Palmerio, S.; Bianchi, F.; Volpe, M.; Rubattu, S. Mitochondrial complex I deficiency and cardiovascular diseases: Current evidence and future directions. J. Mol. Med. 2019, 97, 579–591. [Google Scholar] [CrossRef]
- Ke, B.-X.; Pepe, S.; Grubb, D.R.; Komen, J.C.; Laskowski, A.; Rodda, F.A.; Hardman, B.M.; Pitt, J.J.; Ryan, M.T.; Lazarou, M.; et al. Tissue-specific splicing of an Ndufs6 gene-trap insertion generates a mitochondrial complex I deficiency-specific cardiomyopathy. Proc. Natl. Acad. Sci. USA 2012, 109, 6165. [Google Scholar] [CrossRef] [Green Version]
- Karamanlidis, G.; Lee, C.F.; Garcia-Menendez, L.; Kolwicz Jr, S.C.; Suthammarak, W.; Gong, G.; Sedensky, M.M.; Morgan, P.G.; Wang, W.; Tian, R. Mitochondrial Complex I Deficiency Increases Protein Acetylation and Accelerates Heart Failure. Cell Metab. 2013, 18, 239–250. [Google Scholar] [CrossRef] [Green Version]
- Rineau, E.; Gaillard, T.; Gueguen, N.; Procaccio, V.; Henrion, D.; Prunier, F.; Lasocki, S. Iron deficiency without anemia is responsible for decreased left ventricular function and reduced mitochondrial complex I activity in a mouse model. Int. J. Cardiol. 2018, 266, 206–212. [Google Scholar] [CrossRef]
- Pisano, A.; Cerbelli, B.; Perli, E.; Pelullo, M.; Bargelli, V.; Preziuso, C.; Mancini, M.; He, L.; Bates, M.G.D.; Lucena, J.R.; et al. Impaired mitochondrial biogenesis is a common feature to myocardial hypertrophy and end-stage ischemic heart failure. Cardiovasc. Pathol. 2016, 25, 103–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowen, T.S.; Rolim, N.P.; Fischer, T.; Baekkerud, F.H.; Medeiros, A.; Werner, S.; Brønstad, E.; Rognmo, O.; Mangner, N.; Linke, A.; et al. Heart failure with preserved ejection fraction induces molecular, mitochondrial, histological, and functional alterations in rat respiratory and limb skeletal muscle. Eur. J. Heart Fail. 2015, 17, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.; Parashar, A.; Kumbhani, D.; Agarwal, S.; Garg, J.; Kitzman, D.; Levine, B.; Drazner, M.; Berry, J. Exercise training in patients with heart failure and preserved ejection fraction: Meta-analysis of randomized control trials. Circ. Heart Fail. 2015, 8, 33–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.A.; Kelly, D.P.; Chirinos, J.A. Mitochondrial Dysfunction in Heart Failure With Preserved Ejection Fraction. Circulation 2019, 139, 1435–1450. [Google Scholar] [CrossRef]
- Calafiore, A.M.; Lorusso, R.; Kheirallah, H.; Alsaied, M.M.; Alfonso, J.J.; Di Baldassare, A.; Gallina, S.; Gaudino, M.; Di Mauro, M. Late tricuspid regurgitation and right ventricular remodeling after tricuspid annuloplasty. J. Card. Surg. 2020, 35, 1891–1900. [Google Scholar] [CrossRef]
- Di Mauro, M.; Bezante, G.P.; Di Baldassarre, A.; Clemente, D.; Cardinali, A.; Acitelli, A.; Salerni, S.; Penco, M.; Calafiore, A.M.; Gallina, S.; et al. Functional tricuspid regurgitation: An underestimated issue. Int. J. Cardiol. 2013, 168, 707–715. [Google Scholar] [CrossRef]
- Di Mauro, M.; Gallina, S.; D’Amico, M.A.; Izzicupo, P.; Lanuti, P.; Bascelli, A.; Di Fonso, A.; Bartoloni, G.; Calafiore, A.M.; Di Baldassarre, A.; et al. Functional mitral regurgitation: From normal to pathological anatomy of mitral valve. Int. J. Cardiol. 2013, 163, 242–248. [Google Scholar] [CrossRef]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; González-Juanatey, J.R.; Harjola, V.-P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar]
- Wolfel, E.E. Exploring the Mechanisms of Exercise Intolerance in Patients With HFpEF. JACC Heart Fail. 2016, 4, 646–648. [Google Scholar] [CrossRef]
- Szeto, H.H.; Birk, A.V. Serendipity and the discovery of novel compounds that restore mitochondrial plasticity. Clin. Pharmacol. Ther. 2014, 96, 672–683. [Google Scholar] [CrossRef] [Green Version]
- Dudek, J.; Hartmann, M.; Rehling, P. The role of mitochondrial cardiolipin in heart function and its implication in cardiac disease. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2019, 1865, 810–821. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, H.N.; Gupta, R.C.; Kohli, S.; Wang, M.; Hachem, S.; Zhang, K. Chronic Therapy With Elamipretide (MTP-131), a Novel Mitochondria-Targeting Peptide, Improves Left Ventricular and Mitochondrial Function in Dogs With Advanced Heart Failure. Circ. Heart Fail. 2016, 9, e002206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatfield, K.C.; Sparagna, G.C.; Chau, S.; Phillips, E.K.; Ambardekar, A.V.; Aftab, M.; Mitchell, M.B.; Sucharov, C.C.; Miyamoto, S.D.; Stauffer, B.L. Elamipretide Improves Mitochondrial Function in the Failing Human Heart. JACC Basic Transl. Sci. 2019, 4, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Daubert, M.A.; Yow, E.; Dunn, G.; Marchev, S.; Barnhart, H.; Douglas, P.S.; O’Connor, C.; Goldstein, S.; Udelson, J.E.; Sabbah, H.N. Novel Mitochondria-Targeting Peptide in Heart Failure Treatment. Circ. Heart Fail. 2017, 10, e004389. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.; Khan, M.S.; Anker, S.D.; Fonarow, G.C.; Kim, R.J.; Nodari, S.; O’Connor, C.M.; Pieske, B.; Pieske-Kraigher, E.; Sabbah, H.N.; et al. Effects of Elamipretide on Left Ventricular Function in Patients With Heart Failure With Reduced Ejection Fraction: The PROGRESS-HF Phase 2 Trial. J. Card. Fail. 2020, 26, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Dey, S.; DeMazumder, D.; Sidor, A.; Foster, D.B.; O’Rourke, B. Mitochondrial ROS Drive Sudden Cardiac Death and Chronic Proteome Remodeling in Heart Failure. Circ. Res. 2018, 123, 356–371. [Google Scholar] [CrossRef]
- Ni, R.; Cao, T.; Xiong, S.; Ma, J.; Fan, G.C.; Lacefield, J.C.; Lu, Y.; Le Tissier, S.; Peng, T. Therapeutic inhibition of mitochondrial reactive oxygen species with mito-TEMPO reduces diabetic cardiomyopathy. Free Radic. Biol. Med. 2016, 90, 12–23. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.A.; Murphy, M.P. Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann. N. Y. Acad. Sci. 2010, 1201, 96–103. [Google Scholar] [CrossRef]
- Adlam, V.J.; Harrison, J.C.; Porteous, C.M.; James, A.M.; Smith, R.A.; Murphy, M.P.; Sammut, I.A. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2005, 19, 1088–1095. [Google Scholar] [CrossRef]
- Junior, R.F.; Dabkowski, E.R.; Shekar, K.C.; Hecker, P.A.; Murphy, M.P. MitoQ improves mitochondrial dysfunction in heart failure induced by pressure overload. Free Radic. Biol. Med. 2018, 117, 18–29. [Google Scholar] [CrossRef]
- Kim, S.; Song, J.; Ernst, P.; Latimer, M.N.; Ha, C.-M.; Goh, K.Y.; Ma, W.; Rajasekaran, N.-S.; Zhang, J.; Liu, X.; et al. MitoQ regulates redox-related noncoding RNAs to preserve mitochondrial network integrity in pressure-overload heart failure. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H682–H695. [Google Scholar] [CrossRef] [PubMed]
- Headrick, J.P.; Peart, J.N.; Reichelt, M.E.; Haseler, L.J. Adenosine and its receptors in the heart: Regulation, retaliation and adaptation. Biochim. Biophys. Acta (BBA) Biomembr. 2011, 1808, 1413–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meibom, D.; Albrecht-Küpper, B.; Diedrichs, N.; Hübsch, W.; Kast, R.; Krämer, T.; Krenz, U.; Lerchen, H.G.; Mittendorf, J.; Nell, P.G.; et al. Neladenoson Bialanate Hydrochloride: A Prodrug of a Partial Adenosine A(1) Receptor Agonist for the Chronic Treatment of Heart Diseases. ChemMedChem 2017, 12, 728–737. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, H.N.; Gupta, R.C.; Kohli, S.; Wang, M.; Rastogi, S.; Zhang, K.; Zimmermann, K.; Diedrichs, N.; Albrecht-Küpper, B.E. Chronic therapy with a partial adenosine A1-receptor agonist improves left ventricular function and remodeling in dogs with advanced heart failure. Circ. Heart Fail. 2013, 6, 563–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.C.; Szekely, K.; Wang, M.; Zhang, K.; Rastogi, S.; Albrecht-Küpper, B.; Sabbah, H.N. Long-term therapy with the partial adenosine A1-Receptor agonist capadenoson, improves peroxisome proliferator-activated receptor coactivator-1α phosphorylation and protein expression in left ventricular myocardium of dogs with chronic heart failure. J. Am. Coll. Cardiol. 2013, 61 (Suppl. 10), E702. [Google Scholar] [CrossRef] [Green Version]
- Tendera, M.; Gaszewska-Żurek, E.; Parma, Z.; Ponikowski, P.; Jankowska, E.; Kawecka-Jaszcz, K.; Czarnecka, D.; Krzemińska-Pakuła, M.; Bednarkiewicz, Z.; Sosnowski, M.; et al. The new oral adenosine A1 receptor agonist capadenoson in male patients with stable angina. Clin. Res. Cardiol. 2012, 101, 585–591. [Google Scholar] [CrossRef]
- Voors, A.A.; Bax, J.J.; Hernandez, A.F.; Wirtz, A.B.; Pap, A.F.; Ferreira, A.C.; Senni, M.; van der Laan, M.; Butler, J. Safety and efficacy of the partial adenosine A1 receptor agonist neladenoson bialanate in patients with chronic heart failure with reduced ejection fraction: A phase IIb, randomized, double-blind, placebo-controlled trial. Eur. J. Heart Fail. 2019, 21, 1426–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, S.J.; Voors, A.A.; McMurray, J.J.; Kitzman, D.W.; Viethen, T.; Wirtz, A.B.; Huang, E.; Pap, A.F.; Solomon, S.D. Effect of Neladenoson Bialanate on Exercise Capacity Among Patients With Heart Failure With Preserved Ejection Fraction: A Randomized Clinical Trial. JAMA 2019, 321, 2101–2112. [Google Scholar] [CrossRef]
- Vrhovac, I.; Eror, D.B.; Klessen, D.; Burger, C.; Breljak, D.; Kraus, O.; Radović, N.; Jadrijević, S.; Aleksic, I.; Walles, T.; et al. Localizations of Na+-d-glucose cotransporters SGLT1 and SGLT2 in human kidney and of SGLT1 in human small intestine, liver, lung, and heart. Pflügers Arch. Eur. J. Physiol. 2015, 467, 1881–1898. [Google Scholar] [CrossRef]
- Ghezzi, C.; Loo, D.D.F.; Wright, E.M. Physiology of renal glucose handling via SGLT1, SGLT2 and GLUT2. Diabetologia 2018, 61, 2087–2097. [Google Scholar] [CrossRef] [Green Version]
- Chao, E.C.; Henry, R.R. SGLT2 inhibition—a novel strategy for diabetes treatment. Nat. Rev. Drug Discov. 2010, 9, 551–559. [Google Scholar] [CrossRef] [PubMed]
- FDA. FDA approves new treatment for a type of heart failure. FDA NEWS RELEASE 2020. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-new-treatment-type-heart-failure (accessed on 14 December 2020).
- Toyama, T.; Neuen, B.L.; Jun, M.; Ohkuma, T.; Neal, B.; Jardine, M.J.; Heerspink, H.L.; Wong, M.G.; Ninomiya, T.; Wada, T.; et al. Effect of SGLT2 inhibitors on cardiovascular, renal and safety outcomes in patients with type 2 diabetes mellitus and chronic kidney disease: A systematic review and meta-analysis. Diabetes Obes. Metab. 2019, 21, 1237–1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, K.L.; Ofori-Asenso, R.; Hopper, I.; von Lueder, T.G.; Reid, C.M.; Zoungas, S.; Wang, B.H.; Liew, D. Potential mechanisms underlying the cardiovascular benefits of sodium glucose cotransporter 2 inhibitors: A systematic review of data from preclinical studies. Cardiovasc. Res. 2019, 115, 266–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallina, S.; Di Francescomarino, S.; Di Mauro, M.; Izzicupo, P.; D’Angelo, E.; D’Amico, M.A.; Pennelli, A.; Amicarelli, F.; Di Baldassarre, A. NAD(P)H oxidase p22(phox) polymorphism and cardiovascular function in amateur runners. Acta Physiol. (Oxf) 2012, 206, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Di Mauro, M.; Izzicupo, P.; Santarelli, F.; Falone, S.; Pennelli, A.; Amicarelli, F.; Calafiore, A.M.; Di Baldassarre, A.; Gallina, S. ACE and AGTR1 polymorphisms and left ventricular hypertrophy in endurance athletes. Med. Sci. Sports Exerc. 2010, 42, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Uthman, L.; Baartscheer, A.; Bleijlevens, B.; Schumacher, C.A.; Fiolet, J.W.T.; Koeman, A.; Jancev, M.; Hollmann, M.W.; Weber, N.C.; Coronel, R.; et al. Class effects of SGLT2 inhibitors in mouse cardiomyocytes and hearts: Inhibition of Na(+)/H(+) exchanger, lowering of cytosolic Na(+) and vasodilation. Diabetologia 2018, 61, 722–726. [Google Scholar] [CrossRef] [Green Version]
- Cingolani, H.E.; Ennis, I.L. Sodium-Hydrogen Exchanger, Cardiac Overload, and Myocardial Hypertrophy. Circulation 2007, 115, 1090–1100. [Google Scholar] [CrossRef]
- Packer, M. Activation and Inhibition of Sodium-Hydrogen Exchanger Is a Mechanism That Links the Pathophysiology and Treatment of Diabetes Mellitus With That of Heart Failure. Circulation 2017, 136, 1548–1559. [Google Scholar] [CrossRef]
- Izzicupo, P.; Di Valerio, V.; MA, D.A.; Di Mauro, M.; Pennelli, A.; Falone, S.; Alberti, G.; Amicarelli, F.; Miscia, S.; Gallina, S.; et al. NAD(P)H oxidase and pro-inflammatory response during maximal exercise: Role of C242T polymorphism of the P22PHOX subunit. Int. J. Immunopathol. Pharmacol. 2010, 23, 203–211. [Google Scholar] [CrossRef]
- Arauna, D.; Furrianca, M.; Espinosa-Parrilla, Y.; Fuentes, E.; Alarcón, M.; Palomo, I. Natural Bioactive Compounds As Protectors Of Mitochondrial Dysfunction In Cardiovascular Diseases And Aging. Molecules 2019, 24, 4259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, C.-M.; Ryu, D.; Cho, S.; Jang, Y. Mitochondrial Quality Control in the Heart: New Drug Targets for Cardiovascular Disease. Korean Circ. J. 2020, 50, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Liobikas, J.; Skemiene, K.; Trumbeckaite, S.; Borutaite, V. Anthocyanins in cardioprotection: A path through mitochondria. Pharmacol. Res. 2016, 113, 808–815. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.-Q.; Zhou, H.-Z.; Teerlink, J.R.; Karliner, J.S. Pyrroloquinoline Quinone (PQQ) Decreases Myocardial Infarct Size and Improves Cardiac Function in Rat Models of Ischemia and Ischemia/Reperfusion. Cardiovasc. Drugs Ther. 2004, 18, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Zhu, D.; Chen, D.; Zhang, Q.; Dong, H.; Wu, W.; Lu, H.; Wu, G. Sulforaphane, a Natural Isothiocyanate Compound, Improves Cardiac Function and Remodeling by Inhibiting Oxidative Stress and Inflammation in a Rabbit Model of Chronic Heart Failure. Med Sci. Monit. Int. Med J. Exp. Clin. Res. 2018, 24, 1473–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siti, H.N.; Jalil, J.; Asmadi, A.Y.; Kamisah, Y. Effects of Quercetin on Cardiac Function in Pressure Overload and Postischemic Cardiac Injury in Rodents: A Systematic Review and Meta-Analysis. Cardiovasc. Drugs Ther. 2020, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Savi, M.; Bocchi, L.; Mena, P.; Dall’Asta, M.; Crozier, A.; Brighenti, F.; Stilli, D.; Del Rio, D. In vivo administration of urolithin A and B prevents the occurrence of cardiac dysfunction in streptozotocin-induced diabetic rats. Cardiovasc. Diabetol. 2017, 16, 80. [Google Scholar] [CrossRef]
- Wu, X.; Zhu, X.; Zhou, Y. Abstract 531: Urolithin a Suppress Cardiac Fibrosis via Autophagy Pathway in the Diabetic Cardiomyopathy. Circ. Res. 2019, 125, A531-A531. [Google Scholar] [CrossRef]
- Zheng, D.; Liu, Z.; Zhou, Y.; Hou, N.; Yan, W.; Qin, Y.; Ye, Q.; Cheng, X.; Xiao, Q.; Bao, Y.; et al. Urolithin B, a gut microbiota metabolite, protects against myocardial ischemia/reperfusion injury via p62/Keap1/Nrf2 signaling pathway. Pharmacol. Res. 2020, 153, 104655. [Google Scholar] [CrossRef]
- de Cabo, R.; Navas, P. Spermidine to the rescue for an aging heart. Nat. Med. 2016, 22, 1389–1390. [Google Scholar] [CrossRef] [Green Version]
- Eisenberg, T.; Abdellatif, M.; Schroeder, S.; Primessnig, U.; Stekovic, S.; Pendl, T.; Harger, A.; Schipke, J.; Zimmermann, A.; Schmidt, A.; et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat. Med. 2016, 22, 1428–1438. [Google Scholar] [CrossRef] [PubMed]
- de Cabo, R.; Carmona-Gutierrez, D.; Bernier, M.; Hall, M.N.; Madeo, F. The search for antiaging interventions: From elixirs to fasting regimens. Cell 2014, 157, 1515–1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bisaccia, G.; Ricci, F.; Gallina, S.; Di Baldassarre, A.; Ghinassi, B. Mitochondrial Dysfunction and Heart Disease: Critical Appraisal of an Overlooked Association. Int. J. Mol. Sci. 2021, 22, 614. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020614
Bisaccia G, Ricci F, Gallina S, Di Baldassarre A, Ghinassi B. Mitochondrial Dysfunction and Heart Disease: Critical Appraisal of an Overlooked Association. International Journal of Molecular Sciences. 2021; 22(2):614. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020614
Chicago/Turabian StyleBisaccia, Giandomenico, Fabrizio Ricci, Sabina Gallina, Angela Di Baldassarre, and Barbara Ghinassi. 2021. "Mitochondrial Dysfunction and Heart Disease: Critical Appraisal of an Overlooked Association" International Journal of Molecular Sciences 22, no. 2: 614. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020614