
Heat Shock Protein 90 Chaperones E1A Early Protein of Adenovirus 5 and Is Essential for Replication of the Virus


Abstract
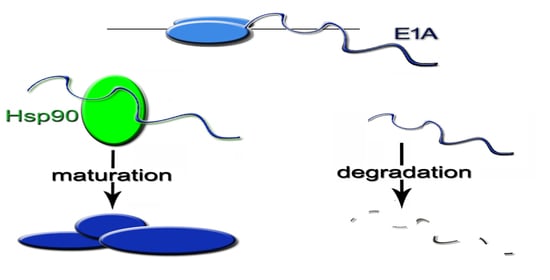
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
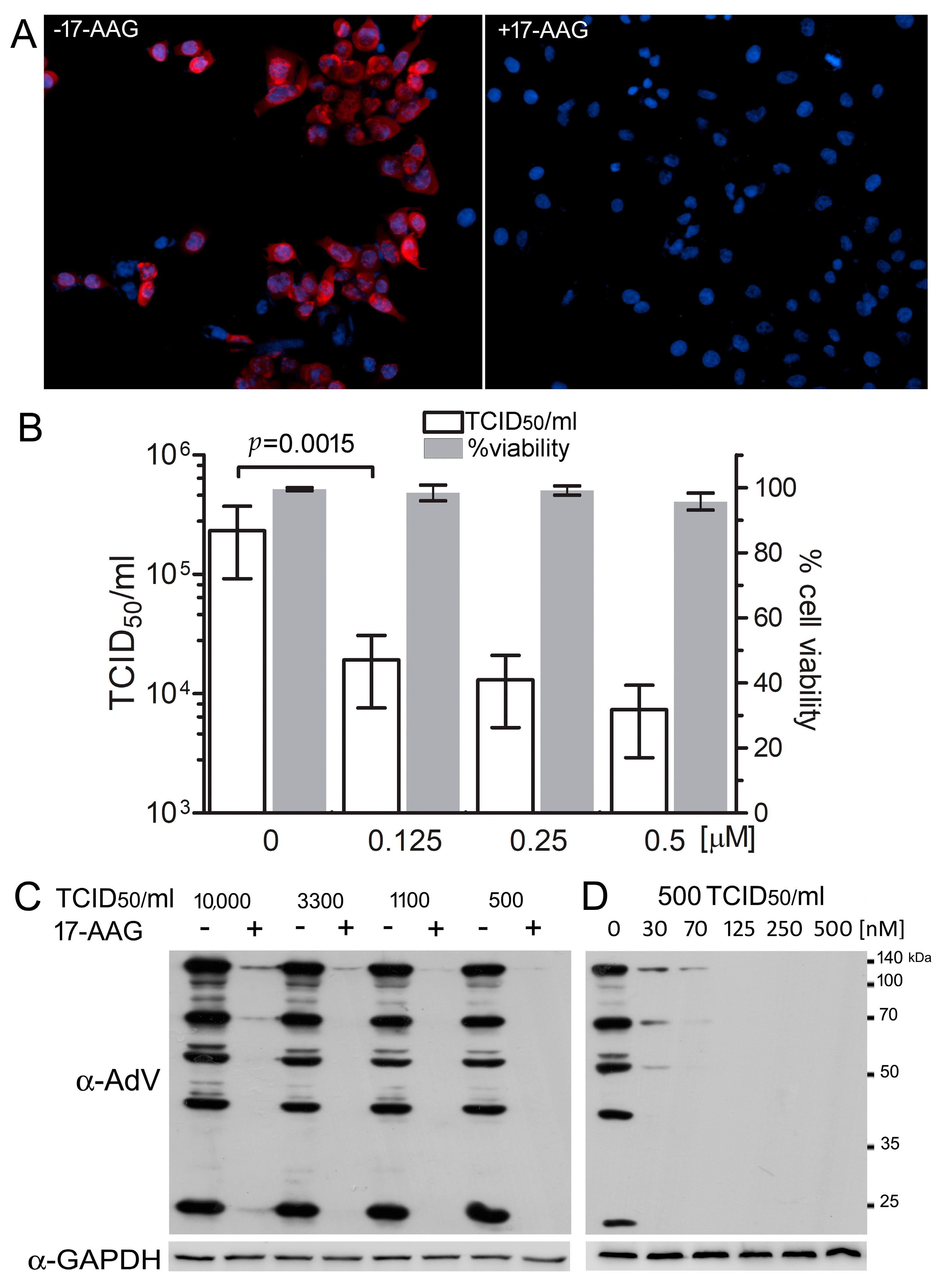
2.1. Hsp90 Is Necessary for Efficient HAdV-5 Replication
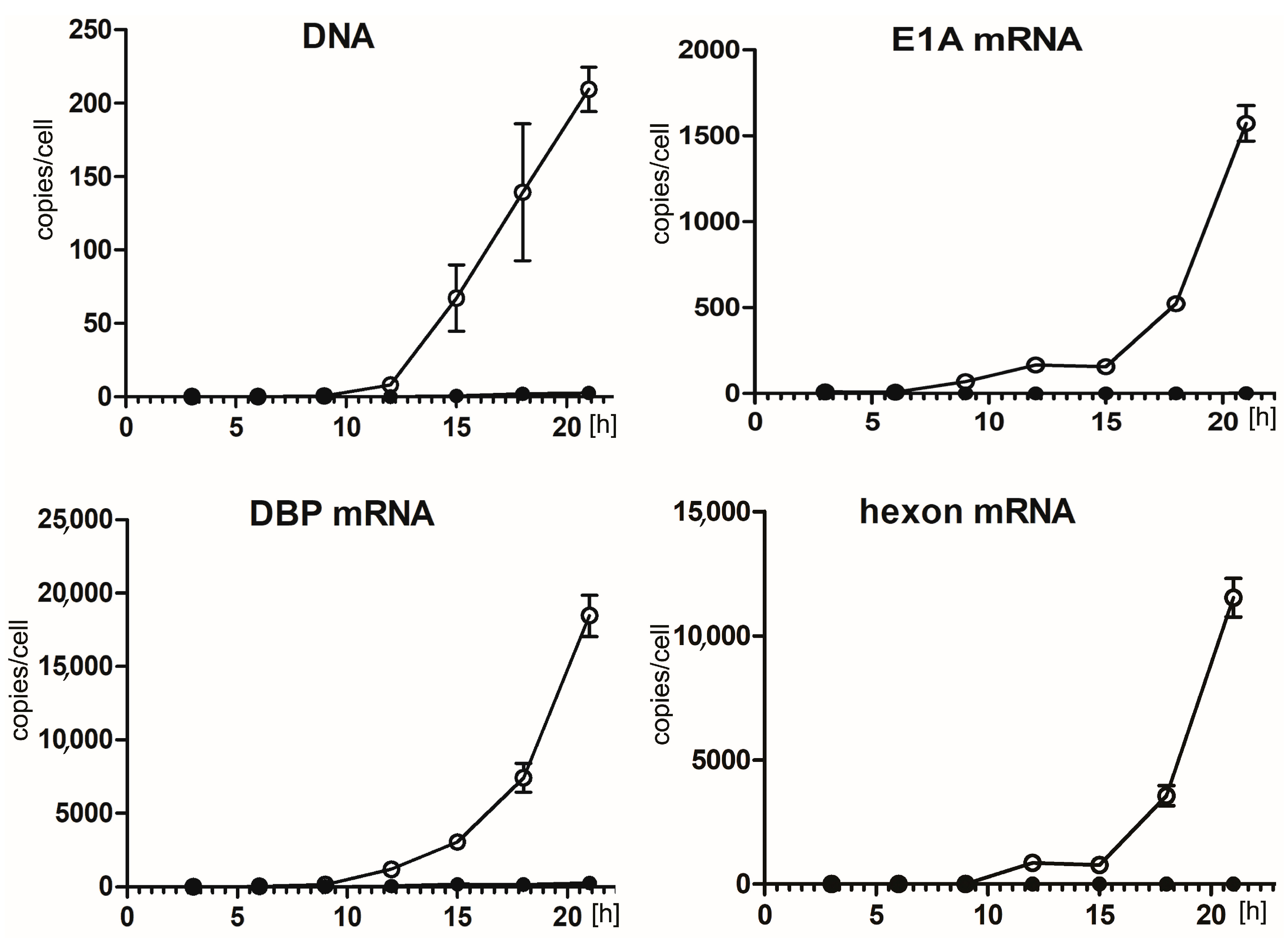
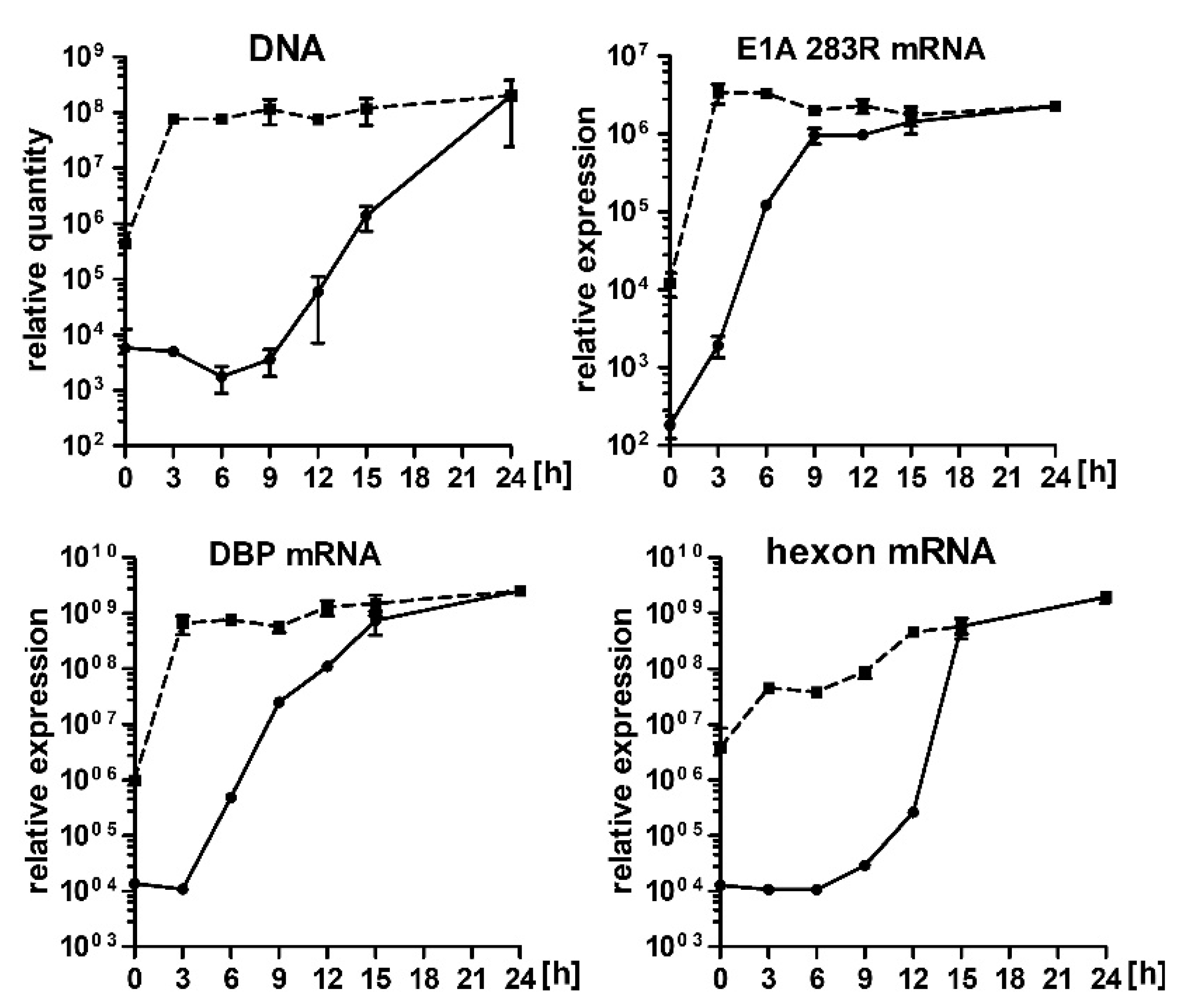
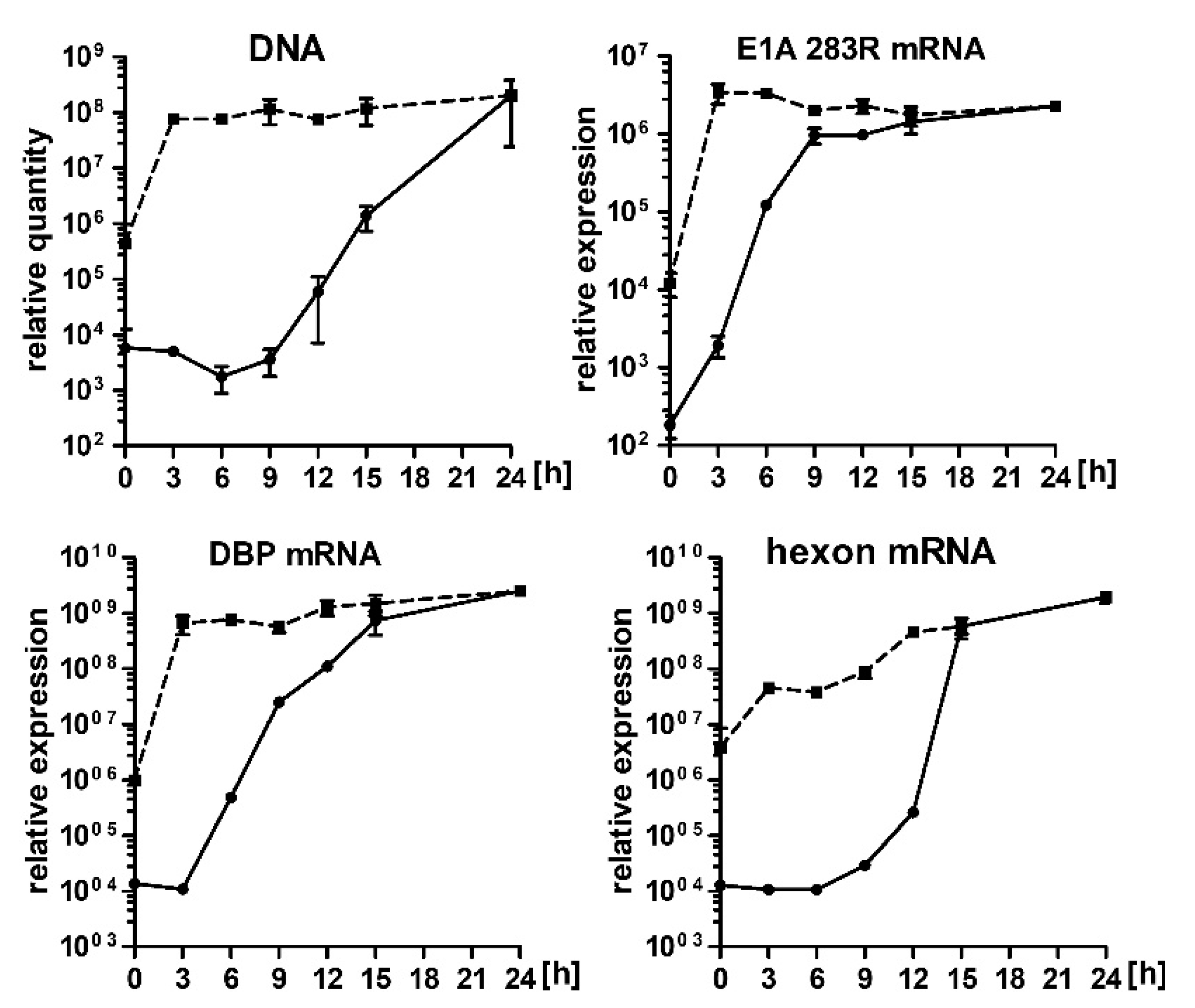
2.2. Hsp90 Inhibition Decreases Transcription of HAdV-5 Early and Late Genes and Replication of HAdV-5 Genome
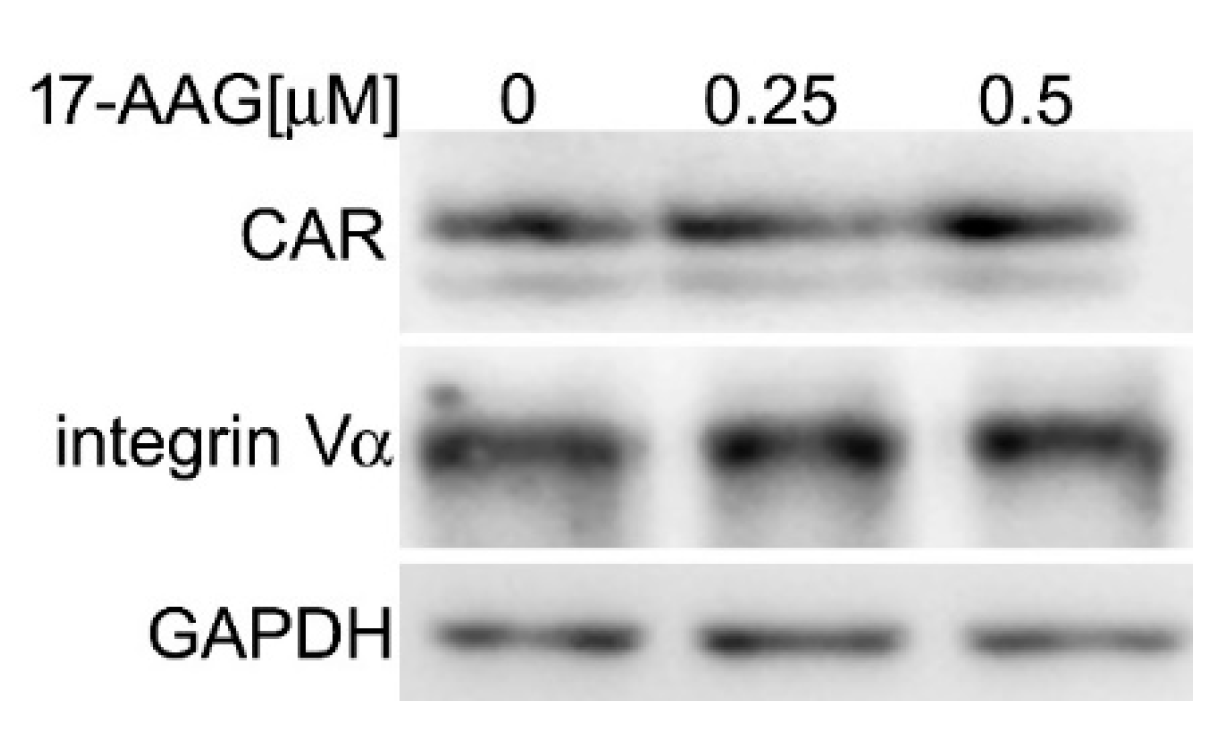
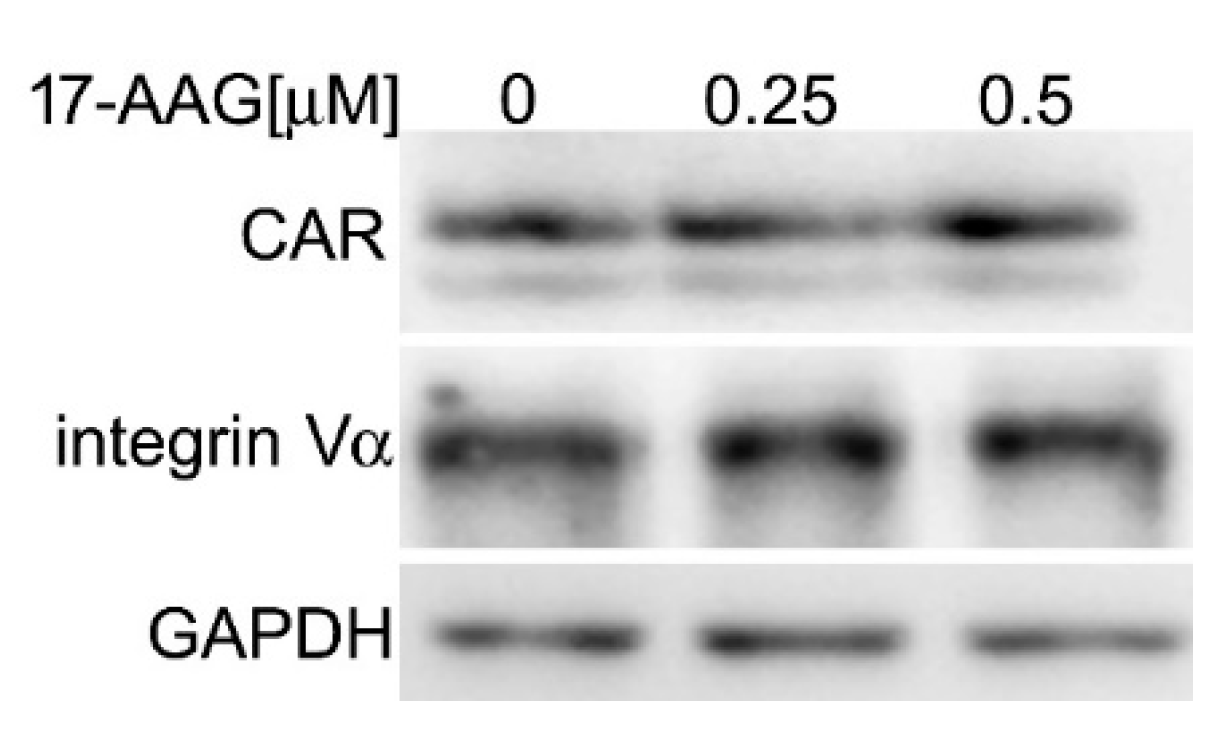
2.3. 17-AAG Does Not Decrease the Level of Cellular Receptors for HAdV-5
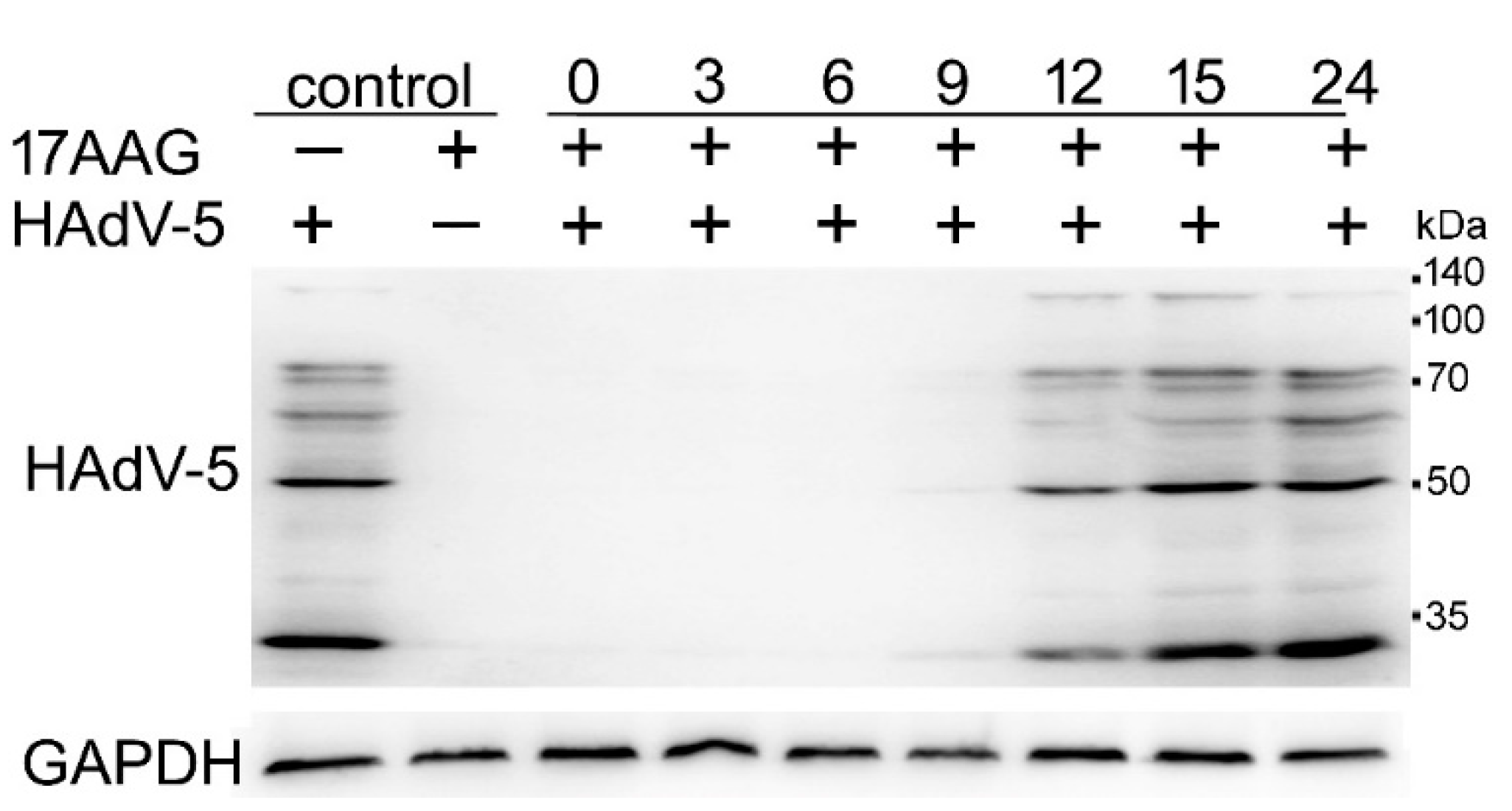
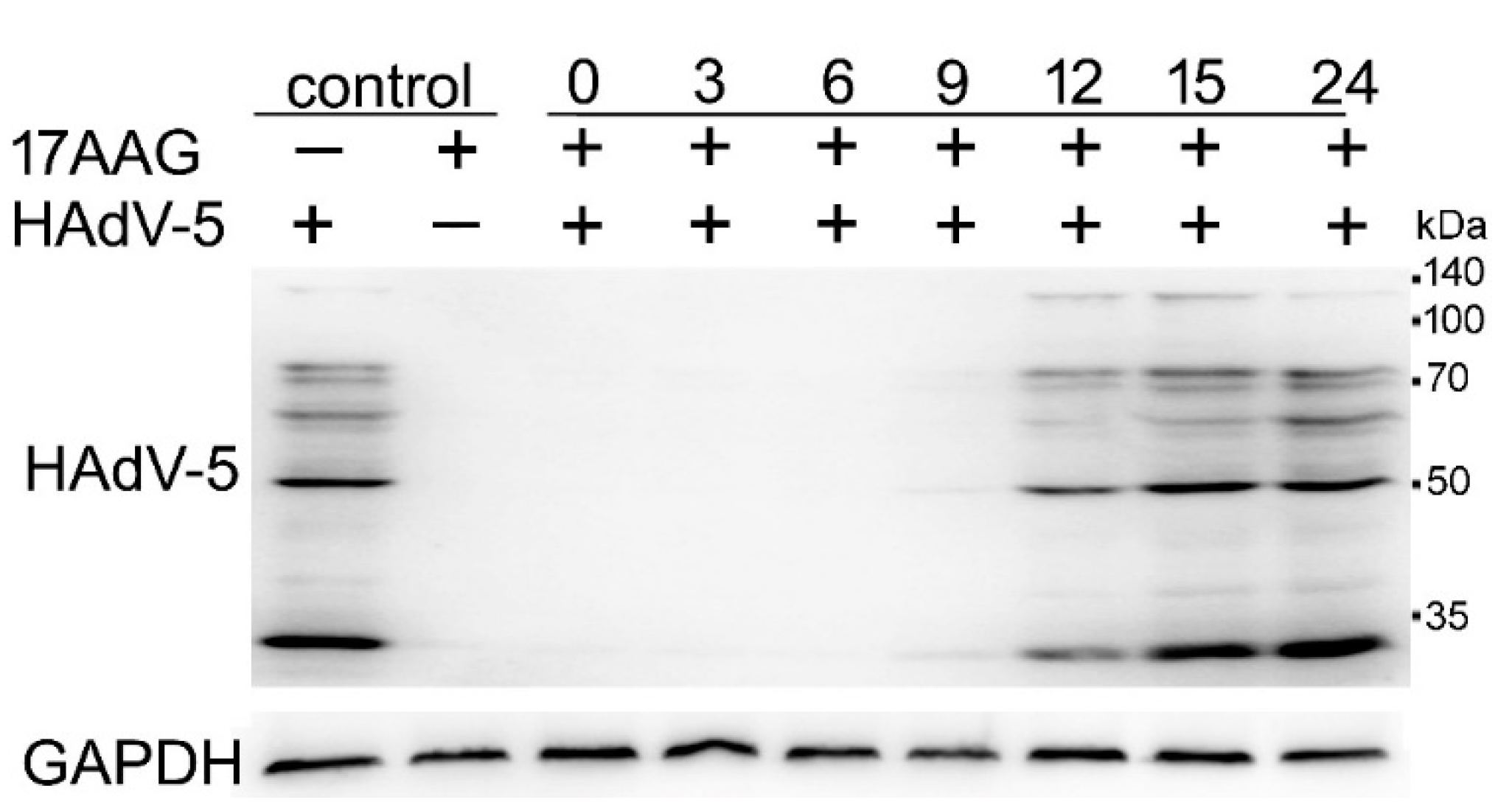
2.4. Expression of HAdV-5 Structural Proteins Is Sensitive to Hsp90 Inhibition Several Hours after Infection
2.5. 17-AAG Inhibits Transcription of HAdV-5 Genes at the Early Steps after Infection
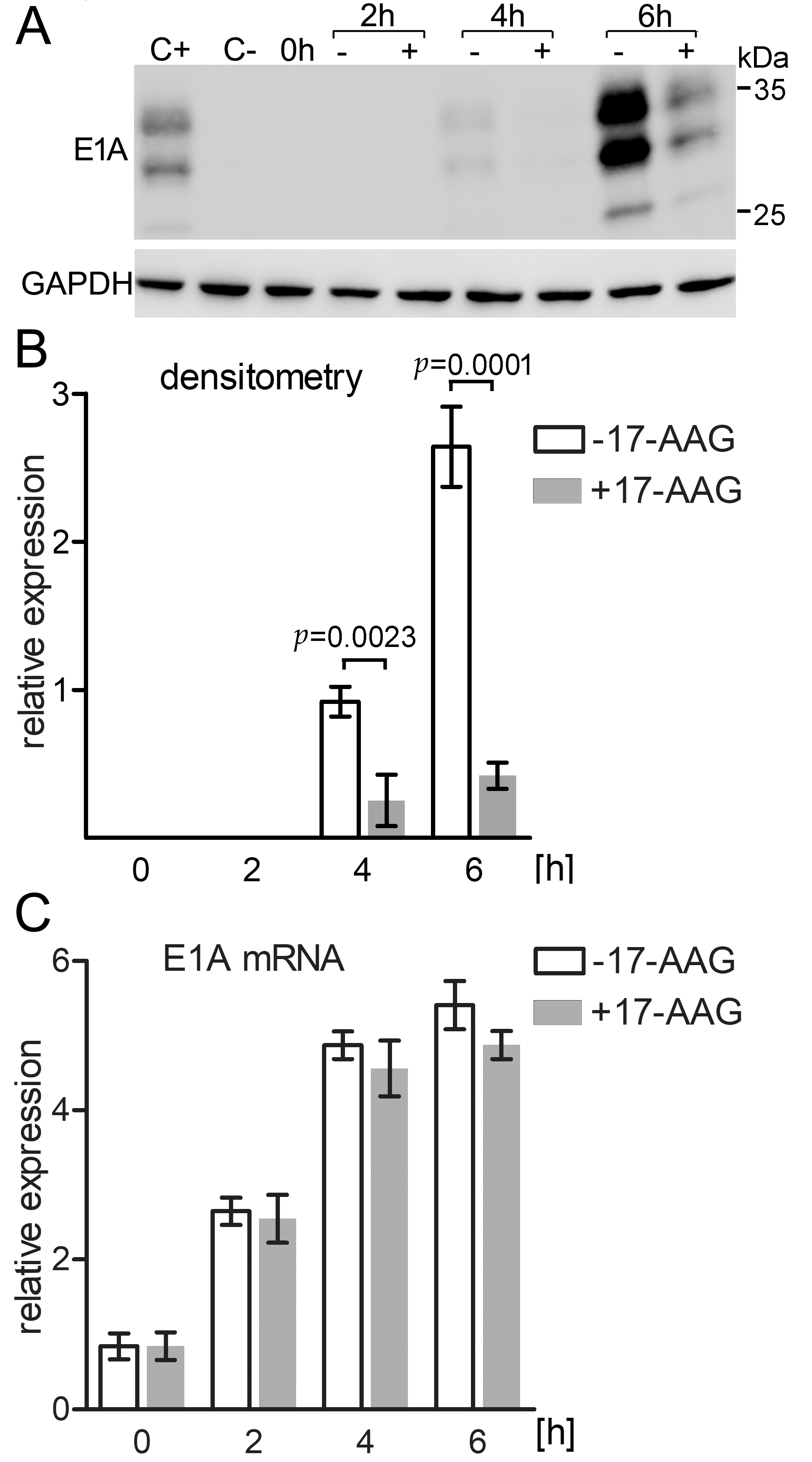
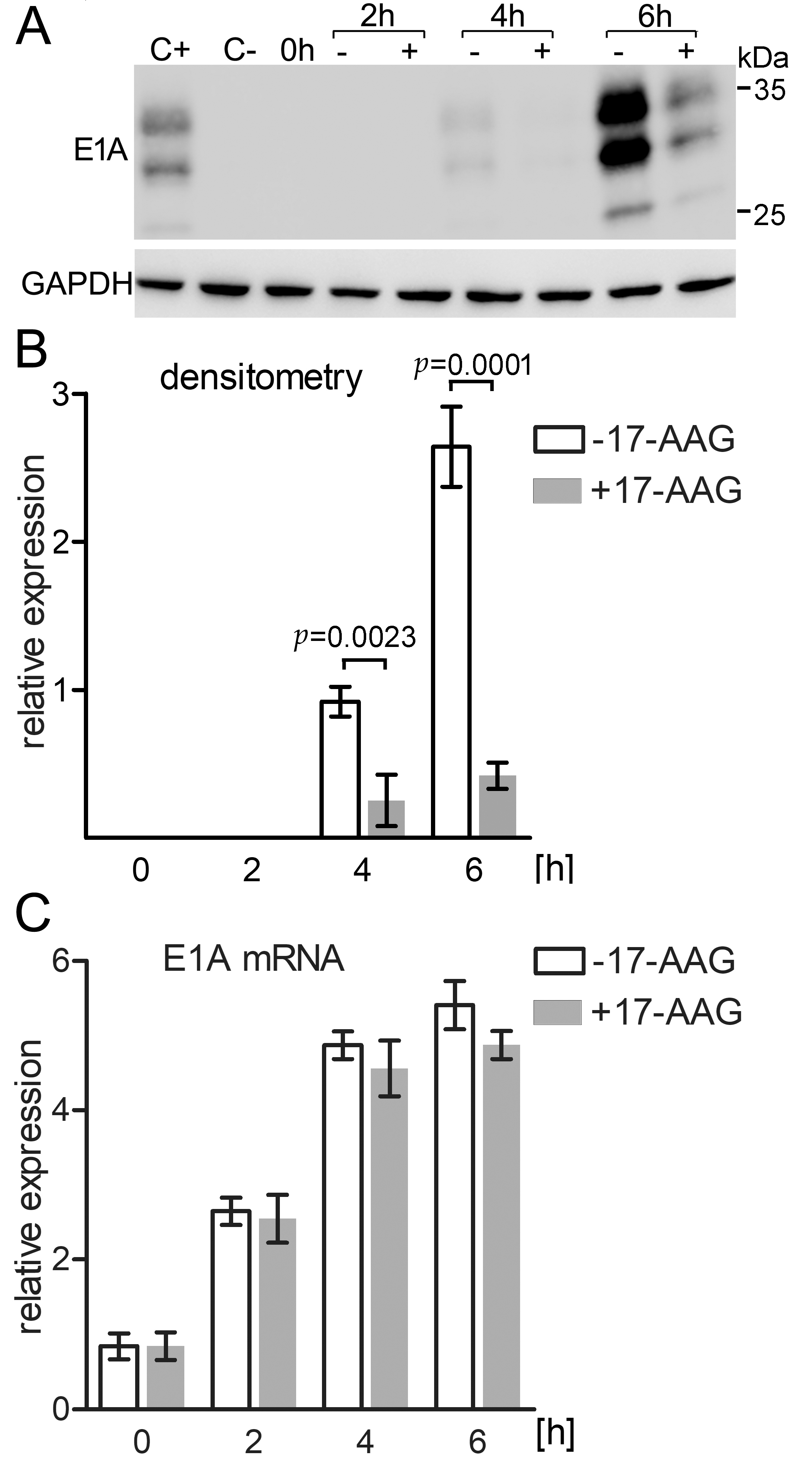
2.6. Immediately after Infection 17-AAG Inhibits E1A Protein but Not E1A mRNA Expression
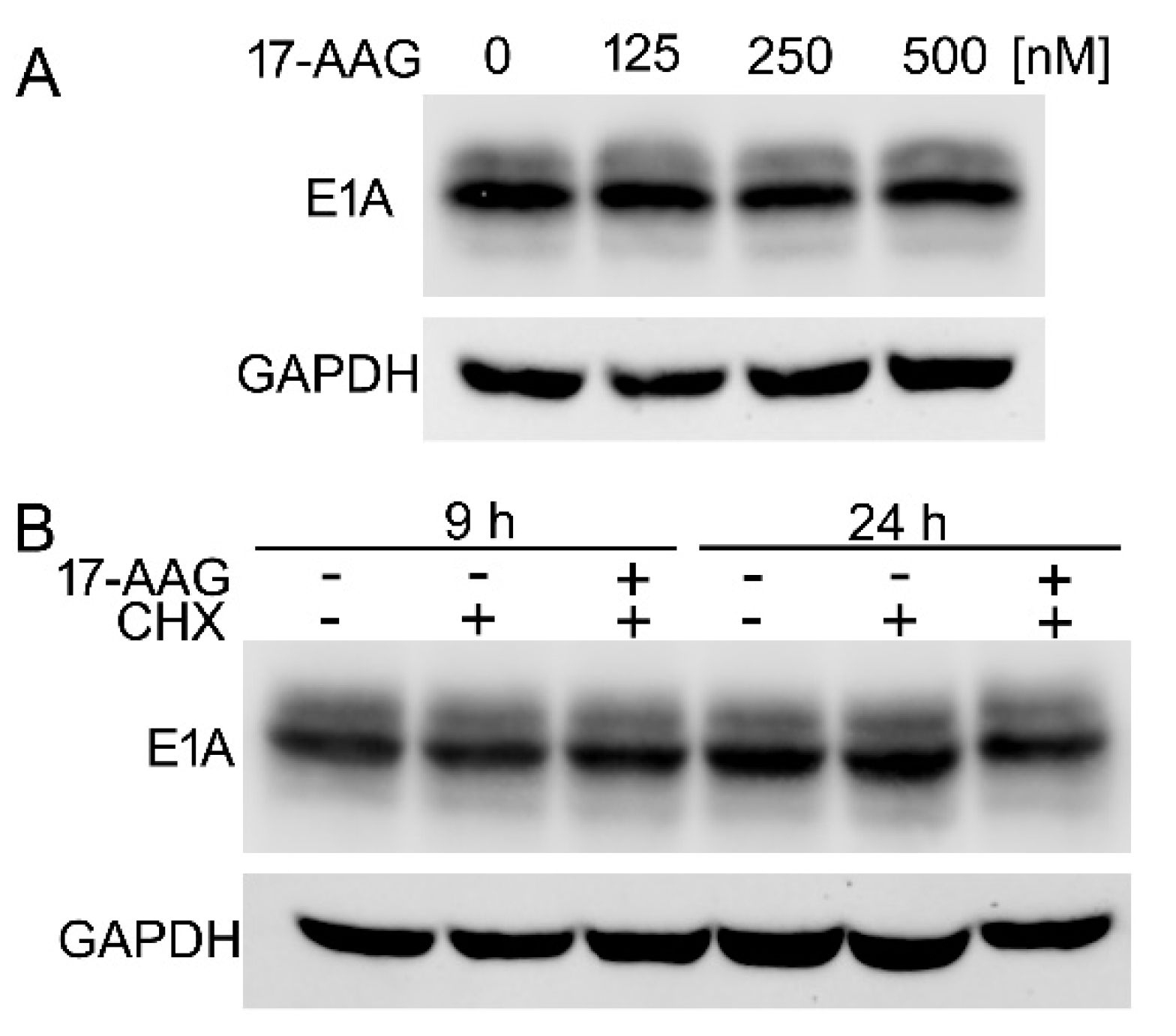
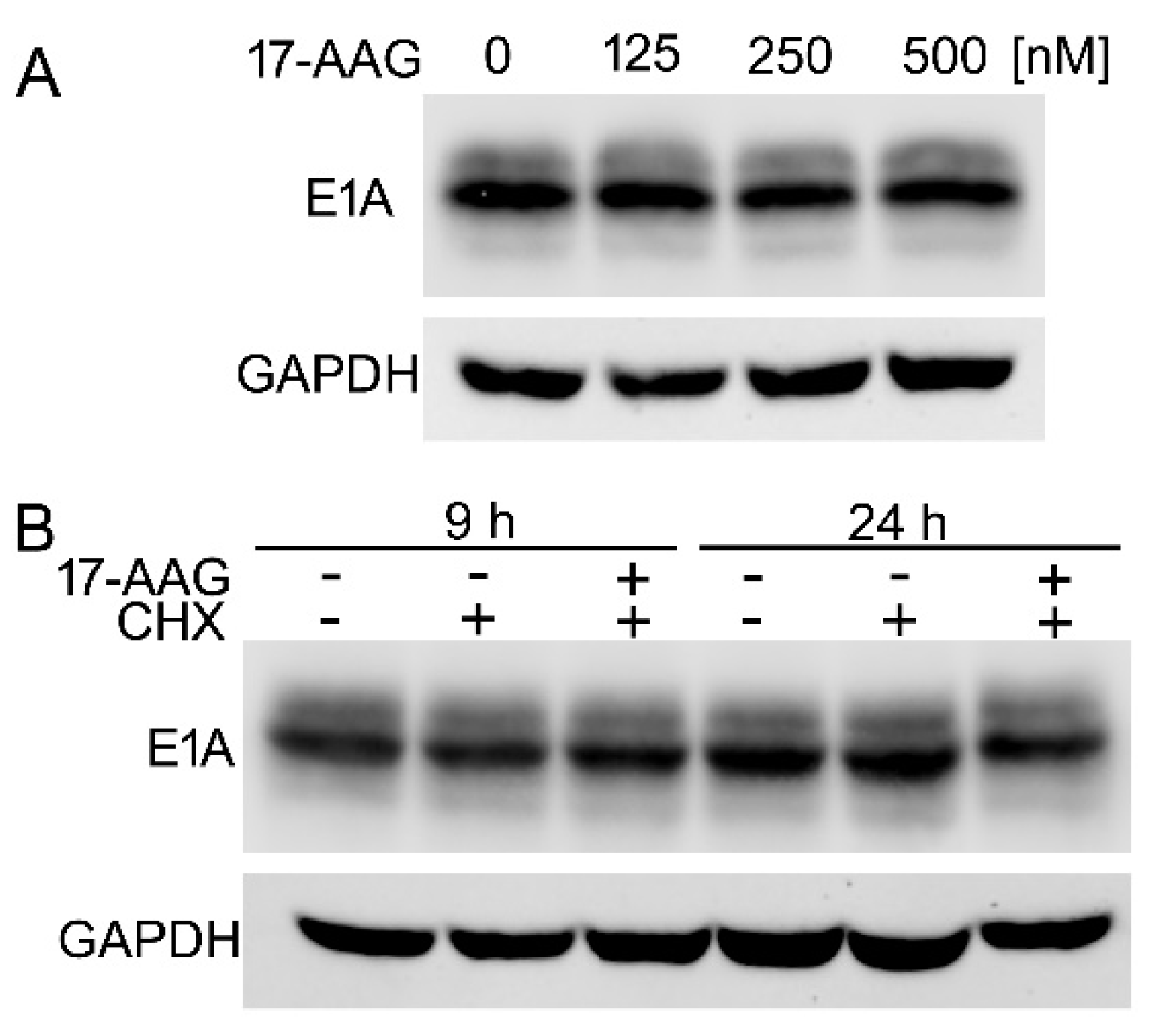
2.7. The Inhibition of Hsp90 Increases Degradation Rate of the Newly Translated E1A
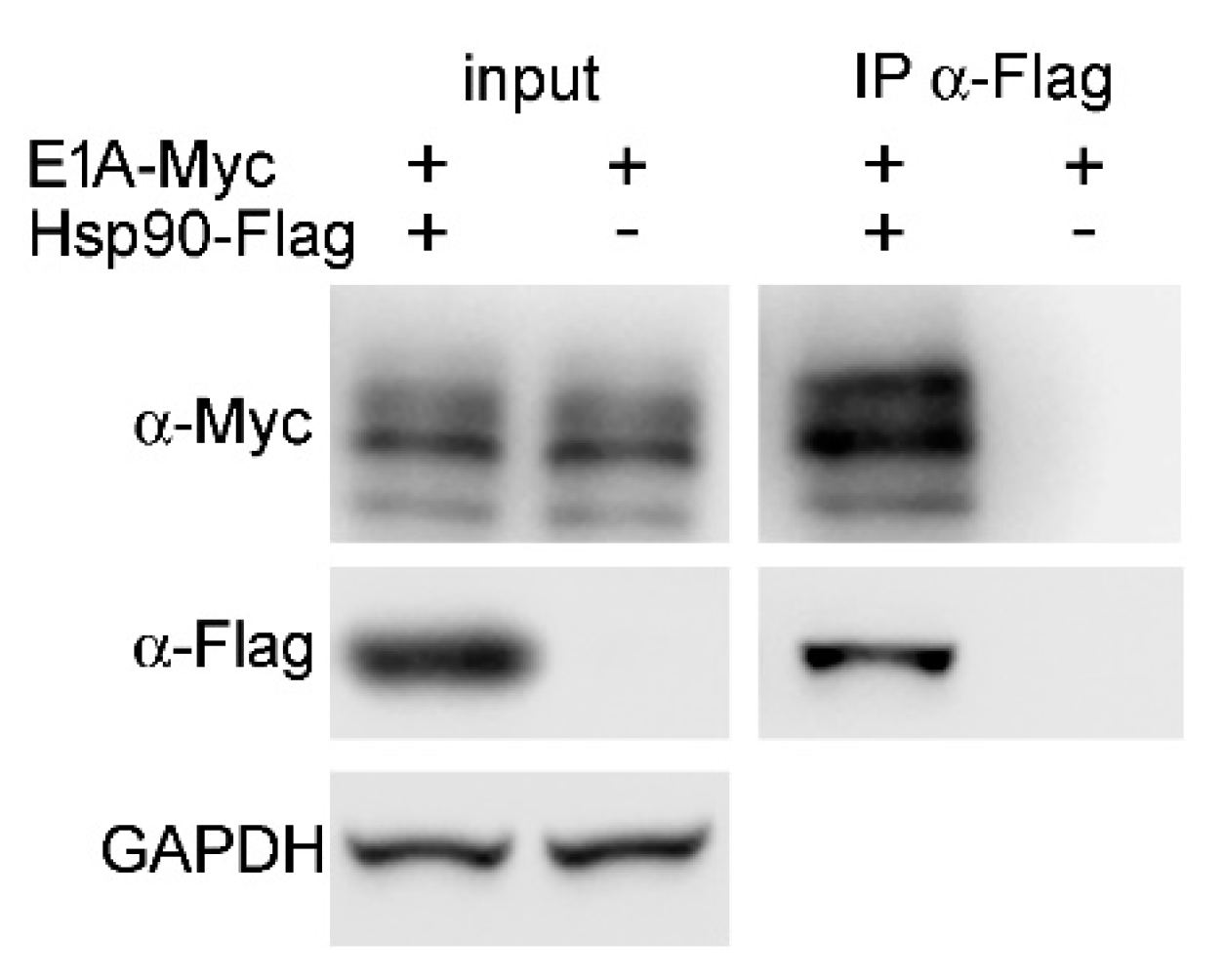
2.8. Hsp90α Interacts with E1A Protein
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Virus Infection
4.2. Cell Viability Assay
4.3. Plasmid Construction
4.4. Immunofluorescence Microscopy
4.5. Co-Immunoprecipitation Assay (Co-IP)
4.6. Western Blot Analysis
4.7. qPCR
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hartson, S.D.; Matts, R.L. Approaches for defining the Hsp90-dependent proteome. Biochim. Biophys. Acta 2012, 1823, 656–667. [Google Scholar] [CrossRef] [Green Version]
- Lai, B.T.; Chin, N.W.; Stanek, A.E.; Keh, W.; Lanks, K.W. Quantitation and intracellular localization of the 85K heat shock protein by using monoclonal and polyclonal antibodies. Mol. Cell Biol. 1984, 4, 2802–2810. [Google Scholar] [CrossRef] [Green Version]
- Echeverria, P.C.; Bernthaler, A.; Dupuis, P.; Mayer, B.; Picard, D. An interaction network predicted from public data as a discovery tool: Application to the Hsp90 molecular chaperone machine. PLoS ONE 2011, 6, e26044. [Google Scholar] [CrossRef]
- Schulte, T.W.; Neckers, L.M. The benzoquinone ansamycin 17-allylamino-17-demethoxygeldanamycin binds to HSP90 and shares important biologic activities with geldanamycin. Cancer Chemother Pharm. 1998, 42, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Tatokoro, M.; Koga, F.; Yoshida, S.; Kihara, K. Heat shock protein 90 targeting therapy: State of the art and future perspective. EXCLI J. 2015, 14, 48–58. [Google Scholar] [PubMed]
- Sanchez, J.; Carter, T.R.; Cohen, M.S.; Blagg, B.S.J. Old and New Approaches to Target the Hsp90 Chaperone. Curr. Cancer Drug Targets 2020, 20, 253–270. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jin, F.; Wang, R.; Li, F.; Wu, Y.; Kitazato, K. HSP90: A promising broad-spectrum antiviral drug target. Arch. Virol. 2017, 162, 3269–3282. [Google Scholar] [CrossRef]
- Geller, R.; Taguwa, S.; Frydman, J. Broad action of Hsp90 as a host chaperone required for viral replication. Biochim. Biophys. Acta 2012, 1823, 698–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Flores, D.; Toft, D.; Wang, X.; Nguyen, D. Requirement of heat shock protein 90 for human hepatitis B virus reverse transcriptase function. J. Virol. 2004, 78, 13122–13131. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Bristol, J.A.; Iwahori, S.; Hagemeier, S.R.; Meng, Q.; Barlow, E.A.; Fingeroth, J.D.; Tarakanova, V.L.; Kalejta, R.F.; Kenney, S.C. Hsp90 inhibitor 17-DMAG decreases expression of conserved herpesvirus protein kinases and reduces virus production in Epstein-Barr virus-infected cells. J. Virol. 2013, 87, 10126–10138. [Google Scholar] [CrossRef] [Green Version]
- Tsou, Y.L.; Lin, Y.W.; Chang, H.W.; Lin, H.Y.; Shao, H.Y.; Yu, S.L.; Liu, C.C.; Chitra, E.; Sia, C.; Chow, Y.H. Heat shock protein 90: Role in enterovirus 71 entry and assembly and potential target for therapy. PLoS ONE 2013, 8, e77133. [Google Scholar] [CrossRef] [PubMed]
- Bukong, T.N.; Momen-Heravi, F.; Kodys, K.; Bala, S.; Szabo, G. Exosomes from hepatitis C infected patients transmit HCV infection and contain replication competent viral RNA in complex with Ago2-miR122-HSP90. PLoS Pathog. 2014, 10, e1004424. [Google Scholar] [CrossRef] [Green Version]
- Seo, H.W.; Seo, J.P.; Jung, G. Heat shock protein 70 and heat shock protein 90 synergistically increase hepatitis B viral capsid assembly. Biochem. Biophys. Res. Commun. 2018, 503, 2892–2898. [Google Scholar] [CrossRef] [PubMed]
- Burch, A.D.; Weller, S.K. Herpes simplex virus type 1 DNA polymerase requires the mammalian chaperone hsp90 for proper localization to the nucleus. J. Virol. 2005, 79, 10740–10749. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Harhaj, E.W. HSP90 protects the human T-cell leukemia virus type 1 (HTLV-1) tax oncoprotein from proteasomal degradation to support NF-kappaB activation and HTLV-1 replication. J. Virol. 2013, 87, 13640–13654. [Google Scholar] [CrossRef] [Green Version]
- Gilmore, R.; Coffey, M.C.; Lee, P.W. Active participation of Hsp90 in the biogenesis of the trimeric reovirus cell attachment protein sigma1. J. Biol. Chem. 1998, 273, 15227–15233. [Google Scholar] [CrossRef] [Green Version]
- Dhingra, A.; Hage, E.; Ganzenmueller, T.; Bottcher, S.; Hofmann, J.; Hamprecht, K.; Obermeier, P.; Rath, B.; Hausmann, F.; Dobner, T.; et al. Molecular Evolution of Human Adenovirus (HAdV) Species C. Sci. Rep. 2019, 9, 1039. [Google Scholar] [CrossRef]
- Smith, J.G.; Wiethoff, C.M.; Stewart, P.L.; Nemerow, G.R. Adenovirus. Curr. Top Microbiol. Immunol. 2010, 343, 195–224. [Google Scholar]
- Robinson, C.M.; Singh, G.; Lee, J.Y.; Dehghan, S.; Rajaiya, J.; Liu, E.B.; Yousuf, M.A.; Betensky, R.A.; Jones, M.S.; Dyer, D.W.; et al. Molecular evolution of human adenoviruses. Sci. Rep. 2013, 3, 1812. [Google Scholar] [CrossRef] [Green Version]
- Echavarria, M. Adenoviruses in immunocompromised hosts. Clin. Microbiol. Rev. 2008, 21, 704–715. [Google Scholar] [CrossRef] [Green Version]
- Lion, T. Adenovirus infections in immunocompetent and immunocompromised patients. Clin. Microbiol. Rev. 2014, 27, 441–462. [Google Scholar] [CrossRef] [Green Version]
- Arnberg, N.; Pring-Akerblom, P.; Wadell, G. Adenovirus type 37 uses sialic acid as a cellular receptor on Chang C cells. J. Virol. 2002, 76, 8834–8841. [Google Scholar] [CrossRef] [Green Version]
- Roelvink, P.W.; Lizonova, A.; Lee, J.G.; Li, Y.; Bergelson, J.M.; Finberg, R.W.; Brough, D.E.; Kovesdi, I.; Wickham, T.J. The coxsackievirus-adenovirus receptor protein can function as a cellular attachment protein for adenovirus serotypes from subgroups A, C, D, E, and F. J. Virol. 1998, 72, 7909–7915. [Google Scholar] [CrossRef] [Green Version]
- Bergelson, J.M.; Cunningham, J.A.; Droguett, G.; Kurt-Jones, E.A.; Krithivas, A.; Hong, J.S.; Horwitz, M.S.; Crowell, R.L.; Finberg, R.W. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science 1997, 275, 1320–1323. [Google Scholar] [CrossRef]
- Gaggar, A.; Shayakhmetov, D.M.; Lieber, A. CD46 is a cellular receptor for group B adenoviruses. Nat. Med. 2003, 9, 1408–1412. [Google Scholar] [CrossRef]
- Wickham, T.J.; Mathias, P.; Cheresh, D.A.; Nemerow, G.R. Integrins alpha v beta 3 and alpha v beta 5 promote adenovirus internalization but not virus attachment. Cell 1993, 73, 309–319. [Google Scholar] [CrossRef]
- Svensson, U. Role of vesicles during adenovirus 2 internalization into HeLa cells. J. Virol. 1985, 55, 442–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greber, U.F.; Willetts, M.; Webster, P.; Helenius, A. Stepwise dismantling of adenovirus 2 during entry into cells. Cell 1993, 75, 477–486. [Google Scholar] [CrossRef] [Green Version]
- Spector, D.J.; McGrogan, M.; Raskas, H.J. Regulation of the appearance of cytoplasmic RNAs from region 1 of the adenovirus 2 genome. J. Mol. Biol. 1978, 126, 395–414. [Google Scholar] [CrossRef]
- Nevins, J.R.; Raychaudhuri, P.; Yee, A.S.; Rooney, R.J.; Kovesdi, I.; Reichel, R. Transactivation by the adenovirus E1A gene. Biochem. Cell Biol. 1988, 66, 578–583. [Google Scholar] [CrossRef]
- de Jong, R.N.; van der Vliet, P.C.; Brenkman, A.B. Adenovirus DNA replication: Protein priming, jumping back and the role of the DNA binding protein DBP. Curr. Top Microbiol. Immunol. 2003, 272, 187–211. [Google Scholar]
- Alestrom, P.; Akusjarvi, G.; Pettersson, M.; Pettersson, U. DNA sequence analysis of the region encoding the terminal protein and the hypothetical N-gene product of adenovirus type 2. J. Biol. Chem. 1982, 257, 13492–13498. [Google Scholar] [CrossRef]
- Sohn, S.Y.; Hearing, P. The adenovirus E4-ORF3 protein functions as a SUMO E3 ligase for TIF-1gamma sumoylation and poly-SUMO chain elongation. Proc. Natl. Acad. Sci. USA 2016, 113, 6725–6730. [Google Scholar] [CrossRef] [Green Version]
- Weitzman, M.D. Functions of the adenovirus E4 proteins and their impact on viral vectors. Front. Biosci. 2005, 10, 1106–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, E. Regulation of the cell cycle and apoptosis by the oncogenes of adenovirus. Oncogene 2001, 20, 7836–7846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lichtenstein, D.L.; Toth, K.; Doronin, K.; Tollefson, A.E.; Wold, W.S. Functions and mechanisms of action of the adenovirus E3 proteins. Int. Rev. Immunol. 2004, 23, 75–111. [Google Scholar] [CrossRef]
- Morris, S.J.; Scott, G.E.; Leppard, K.N. Adenovirus late-phase infection is controlled by a novel L4 promoter. J. Virol. 2010, 84, 7096–7104. [Google Scholar] [CrossRef] [Green Version]
- Akusjarvi, G. Temporal regulation of adenovirus major late alternative RNA splicing. Front. Biosci. 2008, 13, 5006–5015. [Google Scholar] [CrossRef]
- Murali, V.K.; Ornelles, D.A.; Gooding, L.R.; Wilms, H.T.; Huang, W.; Tollefson, A.E.; Wold, W.S.; Garnett-Benson, C. Adenovirus death protein (ADP) is required for lytic infection of human lymphocytes. J. Virol. 2014, 88, 903–912. [Google Scholar] [CrossRef] [Green Version]
- Tollefson, A.E.; Scaria, A.; Hermiston, T.W.; Ryerse, J.S.; Wold, L.J.; Wold, W.S. The adenovirus death protein (E3-11.6K) is required at very late stages of infection for efficient cell lysis and release of adenovirus from infected cells. J. Virol. 1996, 70, 2296–2306. [Google Scholar] [CrossRef] [Green Version]
- Simon, M.C.; Kitchener, K.; Kao, H.T.; Hickey, E.; Weber, L.; Voellmy, R.; Heintz, N.; Nevins, J.R. Selective induction of human heat shock gene transcription by the adenovirus E1A gene products, including the 12S E1A product. Mol. Cell. Biol. 1987, 7, 2884–2890. [Google Scholar] [CrossRef] [Green Version]
- Macejak, D.G.; Luftig, R.B. Association of HSP70 with the adenovirus type 5 fiber protein in infected HEp-2 cells. Virology 1991, 180, 120–125. [Google Scholar] [CrossRef]
- Niewiarowska, J.; D’Halluin, J.C.; Belin, M.T. Adenovirus capsid proteins interact with HSP70 proteins after penetration in human or rodent cells. Exp. Cell. Res. 1992, 201, 408–416. [Google Scholar] [CrossRef]
- Louis, N.; Evelegh, C.; Graham, F.L. Cloning and sequencing of the cellular-viral junctions from the human adenovirus type 5 transformed 293 cell line. Virology 1997, 233, 423–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephens, C.; Harlow, E. Differential splicing yields novel adenovirus 5 E1A mRNAs that encode 30 kd and 35 kd proteins. EMBO J. 1987, 6, 2027–2035. [Google Scholar] [CrossRef]
- Srisutthisamphan, K.; Jirakanwisal, K.; Ramphan, S.; Tongluan, N.; Kuadkitkan, A.; Smith, D.R. Hsp90 interacts with multiple dengue virus 2 proteins. Sci. Rep. 2018, 8, 4308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Zhang, X.; Ma, C.; You, J.; Dong, M.; Yun, S.; Jiang, P. Heat shock protein 90 is essential for replication of porcine circovirus type 2 in PK-15 cells. Virus Res. 2016, 224, 29–37. [Google Scholar] [CrossRef]
- Mohl, B.P.; Roy, P. Hsp90 Chaperones Bluetongue Virus Proteins and Prevents Proteasomal Degradation. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Liu, F.; Liu, J.; Wang, D.; Yan, Y.; Ji, S.; Zan, J.; Zhou, J. The co-chaperone Cdc37 regulates the rabies virus phosphoprotein stability by targeting to Hsp90AA1 machinery. Sci. Rep. 2016, 6, 27123. [Google Scholar] [CrossRef]
- Shim, H.Y.; Quan, X.; Yi, Y.S.; Jung, G. Heat shock protein 90 facilitates formation of the HBV capsid via interacting with the HBV core protein dimers. Virology 2011, 410, 161–169. [Google Scholar] [CrossRef] [Green Version]
- Chai, K.; Ning, X.; Nguyen, T.T.T.; Zhong, B.; Morinaga, T.; Li, Z.; Shingyoji, M.; Tada, Y.; Tatsumi, K.; Shimada, H.; et al. Heat shock protein 90 inhibitors augment endogenous wild-type p53 expression but down-regulate the adenovirally-induced expression by inhibiting a proteasome activity. Oncotarget 2018, 9, 26130–26143. [Google Scholar] [CrossRef] [Green Version]
- Berk, A.J.; Lee, F.; Harrison, T.; Williams, J.; Sharp, P.A. Pre-early adenovirus 5 gene product regulates synthesis of early viral messenger RNAs. Cell 1979, 17, 935–944. [Google Scholar] [CrossRef]
- Lewis, J.B.; Mathews, M.B. Control of adenovirus early gene expression: A class of immediate early products. Cell 1980, 21, 303–313. [Google Scholar] [CrossRef]
- Marrugal-Lorenzo, J.A.; Serna-Gallego, A.; Gonzalez-Gonzalez, L.; Bunuales, M.; Poutou, J.; Pachon, J.; Gonzalez-Aparicio, M.; Hernandez-Alcoceba, R.; Sanchez-Cespedes, J. Inhibition of adenovirus infection by mifepristone. Antiviral. Res. 2018, 159, 77–83. [Google Scholar] [CrossRef]
- Marrugal-Lorenzo, J.A.; Serna-Gallego, A.; Berastegui-Cabrera, J.; Pachon, J.; Sanchez-Cespedes, J. Repositioning salicylanilide anthelmintic drugs to treat adenovirus infections. Sci. Rep. 2019, 9, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Kiefer, J.; Whalen, D.; Flint, S.J. DNA synthesis-dependent relief of repression of transcription from the adenovirus type 2 IVa(2) promoter by a cellular protein. Virology 2003, 314, 394–402. [Google Scholar] [CrossRef] [Green Version]
- Farley, D.C.; Brown, J.L.; Leppard, K.N. Activation of the early-late switch in adenovirus type 5 major late transcription unit expression by L4 gene products. J. Virol. 2004, 78, 1782–1791. [Google Scholar] [CrossRef] [Green Version]
- Morris, S.J.; Leppard, K.N. Adenovirus serotype 5 L4-22K and L4-33K proteins have distinct functions in regulating late gene expression. J. Virol. 2009, 83, 3049–3058. [Google Scholar] [CrossRef] [Green Version]
- Berk, A.J. Adenovirus promoters and E1A transactivation. Annu. Rev. Genet. 1986, 20, 45–79. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, V.; Salzman, N.P. Cis and trans activation of adenovirus IVa2 gene transcription. Nucleic. Acids. Res. 1985, 13, 4067–4083. [Google Scholar] [CrossRef] [Green Version]
- King, C.R.; Tessier, T.M.; Dodge, M.J.; Weinberg, J.B.; Mymryk, J.S. Inhibition of Human Adenovirus Replication by the Importin alpha/beta1 Nuclear Import Inhibitor Ivermectin. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Basha, W.; Kitagawa, R.; Uhara, M.; Imazu, H.; Uechi, K.; Tanaka, J. Geldanamycin, a potent and specific inhibitor of Hsp90, inhibits gene expression and replication of human cytomegalovirus. Antivir. Chem. Chemother. 2005, 16, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Katsuma, S. Hsp90 function is required for stable transcription of the baculovirus transactivator ie-1 gene. Virus Res. 2021, 291, 198200. [Google Scholar] [CrossRef]
- Ujino, S.; Yamaguchi, S.; Shimotohno, K.; Takaku, H. Combination therapy for hepatitis C virus with heat-shock protein 90 inhibitor 17-AAG and proteasome inhibitor MG132. Antivir. Chem. Chemother. 2010, 20, 161–167. [Google Scholar] [CrossRef] [Green Version]
- Munday, D.C.; Wu, W.; Smith, N.; Fix, J.; Noton, S.L.; Galloux, M.; Touzelet, O.; Armstrong, S.D.; Dawson, J.M.; Aljabr, W.; et al. Interactome analysis of the human respiratory syncytial virus RNA polymerase complex identifies protein chaperones as important cofactors that promote L-protein stability and RNA synthesis. J. Virol. 2015, 89, 917–930. [Google Scholar] [CrossRef] [Green Version]
- Chase, G.; Deng, T.; Fodor, E.; Leung, B.W.; Mayer, D.; Schwemmle, M.; Brownlee, G. Hsp90 inhibitors reduce influenza virus replication in cell culture. Virology 2008, 377, 431–439. [Google Scholar] [CrossRef] [Green Version]
- Pacey, S.; Gore, M.; Chao, D.; Banerji, U.; Larkin, J.; Sarker, S.; Owen, K.; Asad, Y.; Raynaud, F.; Walton, M.; et al. A Phase II trial of 17-allylamino, 17-demethoxygeldanamycin (17-AAG, tanespimycin) in patients with metastatic melanoma. Invest New Drugs 2012, 30, 341–349. [Google Scholar] [CrossRef]
- Solit, D.B.; Ivy, S.P.; Kopil, C.; Sikorski, R.; Morris, M.J.; Slovin, S.F.; Kelly, W.K.; DeLaCruz, A.; Curley, T.; Heller, G.; et al. Phase I trial of 17-allylamino-17-demethoxygeldanamycin in patients with advanced cancer. Clin. Cancer Res. 2007, 13, 1775–1782. [Google Scholar] [CrossRef] [Green Version]
- Zurawska, A.; Urbanski, J.; Matuliene, J.; Baraniak, J.; Klejman, M.P.; Filipek, S.; Matulis, D.; Bieganowski, P. Mutations that increase both Hsp90 ATPase activity in vitro and Hsp90 drug resistance in vivo. Biochim. Biophys. Acta 2010, 1803, 575–583. [Google Scholar] [CrossRef] [Green Version]
- Baker, A.T.; Aguirre-Hernandez, C.; Hallden, G.; Parker, A.L. Designer Oncolytic Adenovirus: Coming of Age. Cancers (Basel) 2018, 10, 201. [Google Scholar] [CrossRef] [Green Version]
- Goradel, N.H.; Mohajel, N.; Malekshahi, Z.V.; Jahangiri, S.; Najafi, M.; Farhood, B.; Mortezaee, K.; Negahdari, B.; Arashkia, A. Oncolytic adenovirus: A tool for cancer therapy in combination with other therapeutic approaches. J. Cell Physiol. 2019, 234, 8636–8646. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Satoh, M.; Abe, H.; Sunamura, M.; Moriya, T.; Ishidoya, S.; Saito, S.; Hamada, H.; Arai, Y. Oncolytic viral therapy by bladder instillation using an E1A, E1B double-restricted adenovirus in an orthotopic bladder cancer model. Urology 2006, 68, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Lion, T. Adenovirus persistence, reactivation, and clinical management. FEBS Lett. 2019, 593, 3571–3582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pennington, M.R.; Saha, A.; Painter, D.F.; Gavazzi, C.; Ismail, A.M.; Zhou, X.; Chodosh, J.; Rajaiya, J. Disparate Entry of Adenoviruses Dictates Differential Innate Immune Responses on the Ocular Surface. Microorganisms 2019, 7, 351. [Google Scholar] [CrossRef] [Green Version]
- Kosulin, K. Intestinal HAdV Infection: Tissue Specificity, Persistence, and Implications for Antiviral Therapy. Viruses 2019, 11, 804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, L.J.; Muench, H. A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 1938, 27, 493–497. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dalidowska, I.; Gazi, O.; Sulejczak, D.; Przybylski, M.; Bieganowski, P. Heat Shock Protein 90 Chaperones E1A Early Protein of Adenovirus 5 and Is Essential for Replication of the Virus. Int. J. Mol. Sci. 2021, 22, 2020. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042020
Dalidowska I, Gazi O, Sulejczak D, Przybylski M, Bieganowski P. Heat Shock Protein 90 Chaperones E1A Early Protein of Adenovirus 5 and Is Essential for Replication of the Virus. International Journal of Molecular Sciences. 2021; 22(4):2020. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042020
Chicago/Turabian StyleDalidowska, Iga, Olga Gazi, Dorota Sulejczak, Maciej Przybylski, and Pawel Bieganowski. 2021. "Heat Shock Protein 90 Chaperones E1A Early Protein of Adenovirus 5 and Is Essential for Replication of the Virus" International Journal of Molecular Sciences 22, no. 4: 2020. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042020