
The GRKs Reactome: Role in Cell Biology and Pathology

Laboratory of Veterinary Pathology and Platelet Signaling, College of Veterinary Medicine, Chungbuk National University, Cheongju 28644, Korea
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2021, 22(7), 3375; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073375
Submission received: 28 February 2021
/
Revised: 21 March 2021
/
Accepted: 22 March 2021
/
Published: 25 March 2021
(This article belongs to the Special Issue G Protein-Coupled Receptor and Their Kinases in Cell Biology and Disease)
Abstract
:G protein-coupled receptor kinases (GRKs) are protein kinases that function in concert with arrestins in the regulation of a diverse class of G protein-coupled receptors (GPCRs) signaling. Although GRKs and arrestins are key participants in the regulation of GPCR cascades, the complex regulatory mechanisms of GRK expression, its alternation, and their function are not thoroughly understood. Several studies together with the work from our lab in recent years have revealed the critical role of these kinases in various physiological and pathophysiological processes, including cardiovascular biology, inflammation and immunity, neurodegeneration, thrombosis, and hemostasis. A comprehensive understanding of the mechanisms underlying functional interactions with multiple receptor proteins and how these interactions take part in the development of various pathobiological processes may give rise to novel diagnostic and therapeutic strategies. In this review, we summarize the current research linking the role of GRKs to various aspects of cell biology, pathology, and therapeutics, with a particular focus on thrombosis and hemostasis.
1. Introduction
G protein-coupled receptors (GPCRs) are the largest and most diverse class of signaling proteins that affect an incredible array of physiological processes throughout the human body. Therefore, one-third to half of all marketed drugs act by targeting GPCRs today. However, humans alone have nearly 1000 various GPCRs, and single ligand–receptor interaction leads to the divergent downstream signaling pathways in cells [1]. The continuous activation of these receptors may cause alteration in the physiological properties of the normal cell. Moreover, “druggable GPCRs” may develop drug resistance due to long-term exposure. These factors suggest that the long-held concept regarding GPCR signaling will no longer be adequate to design the next generation of therapeutic drugs.
Several mechanisms prevent the hyperactivation of GPCR signaling, among which GRKs and arrestins are the most important mechanism of terminating the GPCR activation, and these are therefore gaining popularity among researchers. GRKs initiate the kinase-dependent homologous desensitization of GPCRs by phosphorylating the activated GPCRs and allowing arrestin recruitment in a canonical pathway, thereby allowing cells to adapt to changing extracellular signals and prevent excessive signaling. There is a rapid loss of receptor responsiveness. GRKs also interact with other protein substrates and regulate various cellular interactions in a non-canonical manner (the interactions of GRKs with various proteins are depicted in Figure 1). Nevertheless, the profound data implicating GRKs in various cell biologies and pathologies suggest that GRK regulation can be considered as an important target of investigation in multiple aspects of diseases and their comorbidities, new diagnostic and novel therapeutics that would uncover improved treatments for diseases. Therefore, completely understanding the link between agonist-induced GPCR phosphorylation and the associated physiological effects is critical for the efficient targeting of cell signaling pathways in cell biology and diseases of high clinical importance.
Here, we will review the current research linking the role of GRKs to several aspects of cell biology and discuss the therapeutic strategies of targeting specific GRKs in different diseases, with a particular focus on thrombosis and hemostasis.
2. GRKs and Arrestins in GPCRs Family
2.1. GPCR Signaling Pathway; General Overview
GPCRs are seven-transmembrane (7-TM) receptors constituting a large protein family that connect intracellular and extracellular environments, and activate inside signal transduction pathways, and ultimately cellular response. There are five subfamilies of GPCRs: rhodopsin-like, secretin receptor, metabotropic glutamate, adhesion, and frizzled [2]. In terms of structure, each receptor is characterized by an extracellular region consisting of an N-terminus and three extracellular loops followed by a 7-TM α-helices region, and finally the intracellular region which includes three intracellular loops, an intracellular amphipathic helix, and the C-terminus [3]. Ligands such as light, proteins, ions, or small chemicals induce conformational changes in GPCRs and activate these receptors by coupling to heterotrimeric G protein complexes that consist of three subunits (Gα, β, and γ) [4,5]. Upon receptor binding of the ligand, the Gα subunit is activated and exchanges the active GTP-bound form in place of a GDP-bound form, which in turn triggers the dissociation of the Gα subunit from the Gβγ dimer. These dissociated subunits individually interact with the other specific intracellular effectors to continue the downstream GPCR signaling cascade. The βγ complex also interacts with downstream effectors including phosphoinositide 3-kinase γ (PI3Kγ) to activate GPCR-mediated signaling [6]. Further signaling depends on the type of G protein (Gq/G11, G12/G13, Gi/Go/Gz, and Gs) each GPCRs encounters, followed by unique physiological responses [7].
2.2. The GRK and Arrestin Family
Although several proteins have been shown to interact directly with the cell surface 7-TM GPCRs [8], only two protein families besides heterotrimeric G proteins have the potential to interact with the activated conformation of 7-TM receptors: the GRKs and arrestins [9]. They are considered as the key modulators of important intracellular GPCR signaling cascades, as well as GPCR phosphorylation, desensitization, intracellular trafficking and resensitization [10,11,12,13]. GRKs belong to a subfamily of AGC-like kinases and thus share a conserved AGC sequence necessary for kinase activity [14]. There are three main types of GRKs based on the sequence homology: rhodopsin/visual kinases (GRK1 and GRK7), the β-adrenergic receptor kinases (GRK2 and GRK3), and the GRK4 subfamily (GRK4, GRK5, and GRK6). GRK2, 3, 5, and 6 are ubiquitously expressed in mammalian tissues, whereas GRK1 and 7 are exclusively expressed in the rod and cone cells, respectively, and GRK4 is expressed in testis, cerebellum, and kidney [15,16].
These seven different genes share a similar basic protein structure, with an N-terminal, a central catalytic domain, and a unique variable-length C-terminal domain [17]. Each GRK has a highly conserved N-terminus and shares a considerable homology, which is believed to be critical for GPCR recognition, which renders it an important structure for the selective targeting of GPCRs. Apart from bearing homology, the N-terminal region displays several other motifs leading to an increase in its kinase catalytic activity. The C-terminal region contains phosphorylation sites, and key determinants of the cellular location and interaction with the lipids and/or membrane proteins, such as binding phosphatidylinositol 4,5-bisphosphate (PIP2) and Gβγ [18,19]. GRKs are also substrates for different kinases that take part in the modulation of kinase activity, localization, and stability [20]. The structure of GRKs is depicted in Figure 2.
The arrestins are a small family of soluble proteins that specifically bind with the GRK-phosphorylated GPCRs and turn off intracellular signaling [22,23,24]. There are four members of the arrestin family. The expression of visual arrestins, arrestin 1 and arrestin 4, is restricted to retinal rod and cone cells, respectively. By contrast, non-visual arrestins, arrestin 2 and arrestin 3 (known as β-arrestin1 and β-arrestin 2), are expressed ubiquitously [25]. The amino acid sequences of the arrestin isoforms are 78% identical with most of the coding differences at the C terminus of the protein [26]. Mice lacking either arrestin 2 or arrestin 3 are shown to be viable [27,28], but arrestin 2/arrestin 3 double knockout mice die in utero [29]. Although it is not clear, arrestin isoforms might substitute each other functionally to some degree.
2.3. Cell Biology of GRKs and Arrestins in Signaling, Desensitization, Internalization, and Sorting of GPCRs
Upon the ligand-binding activation of GPCR, G protein coupling, and signal transduction, GRK comes and phosphorylates the cytoplasmic serine and threonine residues of activated GPCR, enabling arrestin recruitment. The recruited arrestin then binds to the phosphorylated GPCR, inhibits further G-protein coupling and terminates the downstream signaling, a process termed desensitization [30]. These arrestin-bound receptor complexes are then targeted to clathrin-coated pits, where arrestin forms a multicomponent complex with the key elements of internalization machinery, such as clathrin [31] and adaptor protein-2 (AP-2) [32], and phosphoinositides, promoting endocytosis by budding inwardly from the membrane, thus resulting in receptor internalization. Finally, internalized GPCRs are sorted either into degradation or recycling compartments (Figure 3). This phenomenon was first demonstrated by the Kuhn group by showing that the phosphorylation of rhodopsin was necessary to stop its signaling [33]. Later, the Lefkowitz group reported that the β2-adrenergic receptor (β2-AR) is phosphorylated and desensitized by a cAMP-independent kinase [34]. It was verified that there is a general mode of two-step homologous GPCR desensitization where the active receptor is phosphorylated by one or more GRKs, and further arrestin binding stops the receptor signaling by direct competition with the G protein [35,36,37,38]. Various studies have been carried out showing that the GRKs’ kinase activity is regulated by interacting with multiple proteins, such as caveolin, RKIP, calmodulin, clathrin, actin, PI3K, Akt, etc.
There is a distribution of potential and established phosphorylation sites of GRKs in the selected GPCRs. These phosphorylation sites may be localized at the cytoplasmic region of the receptor. However, the potential GRK phosphorylation sites, their number, positions, and arrestin-binding capacity may vary among the same as well as various receptor subtypes. As the substitute of receptor desensitization, receptor internalization is often used as a readout for arrestin recruitment. However, these two processes are separate and have differential mechanistic requirements. In some cases, the phosphorylation of the receptor may not be necessary or have an insignificant role in binding arrestins to the receptor, as several arrestin-associated receptors do not require coated pits for internalization, while other arrestins might appear to only mediate receptor internalization, not desensitization. Usually, arrestin binding depends on the receptor’s C-cluster, which includes Thr307-Ser311 phosphorylation sites, and internalization is promoted by the phosphorylation of either the N-cluster that includes Ser286-Ser290 residues, or the C-cluster [40,41]. It has also been shown that some receptors do not recycle following internalization [42,43]. Therefore, there are multiple modes of not only the GRK-mediated phosphorylation of receptor subtypes, but also the succeeding arrestin recruitment, binding, and further downstream signaling, and thus the classical model does not necessarily apply to overall GPCR signaling.
Moreover, there are substantially more types of GPCRs compared to GRKs that might play a role in the interaction and phosphorylation of multiple GPCRs [44]. GRKs might exhibit substrate specificity with selective GPCRs, despite their domains’ structural similarity. Therefore, the biological function of each GRK isoform may differ remarkably [45]. Interestingly, GRKs can also substitute phosphorytable regions of their targeted GPCR when the interacting domain is mutated [46]. It has been shown that agonist-induced specific receptor conformation should be favorable for GRK binding and/or activation for a receptor to be phosphorylated by GRKs [47]. Among other things, GPCRs have plenty of promising serine and threonine phosphorylation sites. This suggests that these sites are not likely to be targeted all at once. It has been suggested that the order of phosphorylation may be either barcoded (where receptors responding to specific agonists will be phosphorylated by various GRKs at distinct sites, thus establishing a “barcode”), sequential (where a higher number of serine/threonine residues will be phosphorylated first) or hierarchical (where a particular sequence of serine and threonine will be preferentially targeted) [48,49,50,51]. However, the role of GRKs during GPCR activation and how GRKs recognize conformational changes in GPCR are still undetermined [52].
The cell type-dependent mechanisms of these GRKs on GPCRs, and how the specific phosphorylation of protein residues would control the consequent physiopathology of cell-biology and diseases, have just started to be untangled. The recent research in this area is discussed below.
3. The Role of GRKs and Arrestins in Regulation of GPCRs Family
3.1. The Role of GRKs in Immune Cells and Inflammation
Chemotaxis plays a significant role in inflammatory signaling by enabling the immune cells to appear at the site of inflammation. Chemokines-producing cells initiate the chemotaxis via chemokine receptor-mediated signaling (mainly GPCRs) through the integrated modulation of different steps, including receptor sensing, cell polarization, membrane protrusion, adhesion, or de-adhesion [53,54]. It has been demonstrated that chemokine GPCRs undergo desensitization upon constant stimulation of the receptors. Immune cells express high levels of GRK2, 3, 5, and 6, and are known to play a critical role in the modulation of immune cell response by involvement in the regulation of various cellular responses, such as scavenging, arrestins recruitment, signaling, and the desensitization of various chemokine receptors. The detailed function of GRKs in immune cells and inflammation is listed in Table 1.
Among various GRKs, GRK2 has been extensively studied in terms of chemotaxis and has been shown to negatively regulate chemotactic responses via canonical negative GPCR signaling [55,56,57]. It was shown to be involved in the regulation of cell type and stimulus-dependent immune cell migration [58,59]. Myeloid-specific GRK2 KO exhibited increased circulating neutrophils and macrophages mobilization to inflammatory sites, suggesting the desensitizing effect of GRK3 on chemokine receptors [60]. In malaria and sepsis, neutrophils showed an increased expression of GRK2 levels associated with decreased CXCR2 expression and reduced responses to IL-8 [61,62]. In contrast, it was shown that chemotactic responses in few other cell types are positively mediated by GRK2 [63]. GRK2 also plays a non-canonical role in cell motility, as it was shown to phosphorylate proteins such as ezrin, radixin and ERK1/2 in a kinase-independent manner [64,65]. Similarly, although in very few cell types, GRK5 has also been reported to regulate GPCRs such as CCR2 and CXCR4, which are critical in chemotaxis in both canonical and non-canonical manners [66,67,68,69]. Several chemokine receptors such as CXCR2, CXCR4, and LTB4 were found to be regulated by GRK6 in a canonical manner [70,71,72]. GRK6 was shown to modulate neutrophil and lymphocyte recruitment in vivo in various disease models [73,74,75]. The non-canonical role of GRK6 in immune cell chemotaxis is yet to be understood.
GPCRs have also been known to play a major role in the pathophysiological events in sepsis, a severe inflammatory response, via cardiovascular, immune, and coagulatory responses. It was shown that GRK2 and GRK5 play a notable role in the pathogenesis of human sepsis by regulating neutrophil chemotaxis, and modulate the outcomes of septic shock via the NF-κB1p105-TPL2-MEK-ERK pathway [61,76,77,78,79]. However, these GRKs have no role in immune cell infiltration, the presence of bacteria, or patient survival. It was shown that GRK5 deficiency leads to decreased cytokine levels, decreased thymocyte apoptosis and immune suppression, and reduced plasma corticosterone levels, leading to sepsis-induced mortality even in the presence of antibiotics in both endotoxemia and polymicrobial sepsis models [77,80].
All these results indicate the important roles of GRKs in immune cell chemotaxis and inflammatory signaling, and indicate that the identification of the small-molecule compounds that regulate GRK isoforms could be a plausible therapeutic approach in inflammatory disease management.
3.2. The Role of GRKs in Cardiovascular Diseases
Although significant improvements have been made with the drugs targeting GPCRs to treat cardiovascular patients, the treatments remain insufficient. The disease-causing effect of GPCRs can initially be mitigated by negative feedback via GRKs, as GRKs have widely established themselves as having a primary role in modulating GPCR signaling. GRKs have been thoroughly studied as possible diagnostic or therapeutic targets in the cardiovascular system. GRK activity and expression were shown to be altered in various cardiovascular system diseases, including congestive heart failure, hypertension, myocardial infarction, and cardiac hypertrophy [95]. GRK2, GRK3, and GRK5 play a well-established role in the progress of diseases of the cardiovascular system [96,97]. GRK2 and GRK5 have been associated with heart failure in humans and their increased expression/activity was shown to induce β-adrenergic receptor desensitization [98,99]. GRK2 expression was shown to be enhanced in hypertension, cardiac hypertrophy, and myocardial infarction, and the inhibition of GRK2 was observed to have beneficial effects on these diseases [100,101,102,103]. GRK5 has been found to regulate the vascular endothelial growth factor (VEGF) receptor in the endothelial cells of the coronary artery [104]. The overexpression of GRK2 and GRK5 in vivo has been shown to decrease adrenergic receptor-induced myocardial contractility and cardiac output, which were counteracted by GRK2, GRK3, and GRK5 inhibition [96,98,103,105]. It was shown that hypertensive patients display enhanced GRK2 activity and protein expression with no any changes in GRK5, GRK6, PKA or arrestins, suggesting disease-dependent variation in GRK2 [101]. GRK2 and GRK5 have also been associated with the development of atherosclerosis [60,66].
Overall, studies indicate that GRK inhibition has direct advantageous effects in cardiovascular disease conditions by promoting survival signals, but the pathophysiological role of GRKs in cardiovascular diseases is still incompletely understood. Therefore, increasing the understanding of GRKs’ molecular and cellular processes is of critical importance in order to improve therapeutic strategies.
3.3. The Role of GRKs in Neurodegeneration and Autoimmune Diseases
GRKs have been shown to play a role in the pathogenesis of neurodegenerative diseases, including Alzheimer’s disease (AD), multiple sclerosis (MS), and Parkinson’s disease [106,107,108]. GRK2 levels were increased in the human brain and this was reported to serve as a marker for early hypoperfusion-induced brain damage in AD patients [109]. During hypoxic-ischemic injury, GRK2 was shown to exacerbate brain damage via p38-dependent TNFα production [83], regulate the metabotropic glutamate receptor function and expression implicated in the pathogenesis of AD and MS, and cause neurodegeneration via the over-activation of group I mGLuTs [110,111]. GRK2 was also shown to reduce inflammatory hyperalgesia by inhibiting microglial activation via the inhibiting of p38-dependent TNFα production and PGE2-mediated pathways [112]. Nociceptor function leading to chronic pain was shown to ause a long-lasting neuroplastic change with the inhibition of GRK2 levels [113]. Similarly, GRK5 was shown to be involved in the regulation of the desensitization of muscarinic receptors, specifically M2 and M4 [114]. GRK5-deficient mice have been shown to increase the incidence of AD-like pathology. Mice-deficient in GRK6 were shown to develop an autoimmune disease, such as arthritis and colitis, by modulating the infiltration of immune cells [73,75,115].
Together, these studies reveal that GRKs do play a critical role in the pathogenesis of neurodegenerative as well as autoimmune disease, and thus can be targeted for therapeutic development.
3.4. The Role of GRKs in Cancer
The evolving evidence shows that GPCRs are being used as an early diagnosis biomarker, as GPCR signaling plays integral roles in regulating various aspects of cancer biology, including vascular remodeling, invasion, cell proliferation, apoptosis and migration, by regulating cancer-associated signaling pathways [116,117]. Developing drugs targeting GPCR and its signaling pathway has potential as a new therapeutic strategy for the treatment of cancers. As GRKs have established themselves as a negative regulator of GPCR activity, several studies have displayed the function of GRKs in cancer progression in a cell type-dependent manner [118,119]. An overview of the established roles of GRKs in different types of cancer is summarized in Table 2.
Briefly discussing the role of GRKs in cancer, GRK1 and GRK7 were shown to play a role in embryogenesis, while interacting indirectly with Rho GDP-dissociation inhibitor (RhoGDI) and phosphodiesterase γ (PDEγ), both of which are shown to be abnormally regulated in cancer [153,154]. GRK1/7 might play a role in the development of cancer-related retinopathy in lung cancer patients by interacting with a calcium-binding protein called recoverin [152]. GRK2, but not GRK5, is more highly expressed in differentiated thyroid carcinoma than normal thyroid tissue, and is involved in the rapid desensitization of thyroid-stimulating hormone receptor (TSHR), which desensitization can be further inhibited by GRK5. A study found out that cancer cell proliferation was increased due to the activation of TSHR in thyroid carcinoma [120]. GRK2 has been shown to negatively regulate the insulin-like growth factor-1 receptor (IGF1-R) signaling involved in proliferation and migration by decreasing cyclin in human hepatocellular carcinoma HepG2 cells [121]. GRK2 also plays an important role in suppressing cell cycle progression, regulating early growth response 1 (EGR1) expression, and the progression of breast, gastric, and skin cancer by modulating GPCR signaling [122]. Furthermore, clinical studies have revealed a correlation between high GRK2 expression and a high tumor (T) stage, as well as the poor survival rates of patients suffering from pancreatic cancer. Interestingly, nerve growth factor (NGF) in combination with GRK2 promotes opioid receptors phosphorylation whilst amplifying the pain, and when treated with anti-NGF therapies, there was significant relief in cancer bone pain by mediating the outcome of GRK2 and arrestins. GRK2 acted as a negative regulator of the chemokine receptor CXCR4 (C-X-C chemokine receptor type 4), which is responsible for mediating metastasis and is generally applied as a patient prognosis indicator [155]. Several studies showed that GRK3 inhibited breast cancer metastasis by regulating CXCR4 signaling [44,139]. GRK3 was abnormally expressed in oral squamous carcinoma cells probably via the activation of the β2-adrenergic receptor [128]. Arterial angiotensin type 1 (AT1) and dopamine D receptor signaling have been shown to be modulated by GRK4 [156,157]. Several studies found that GRK4α/β desensitizes the follicle-stimulating hormone receptor (FSHR) and its expression was significantly lower in malignant ovarian granulosa cells compared to benign granulosa cells, suggesting the crucial function of GRK4α/β in the course of ovarian granulosa cell transformation [135]. GRK5 expression is associated with a worse prognosis in patients that are suffering from stage II-IV glioblastoma. GRK2 and GRK5 are known to work oppositely in thyroid cancer [120]. GRK5 could be responsible for the direct phosphorylation of P53 tumor suppression in U2OS and Saos-2 osteosarcoma cells degradation promotion, consequently inhibiting tumor cell apoptosis [145]. A positive association was found between GRK6 and Ki-67 expression, pathological disease stage, metastasis, and survival rate in the patients with hepatocellular carcinoma, and GRK6 was hypothesized as a biomarker for the early diagnosis of hepatocellular carcinoma [148].
3.5. The Role of GRKs in Thrombosis and Hemostasis
Platelets develop from bone marrow, and are anucleated discoid cells of approximately 2 to 4 µm in diameter [158]. The resting platelet consists of an organelle zone formed by alpha granules, dense granules, lysosomal granules, and it glycogen granules, and contains more than 800 different proteins involved in major platelet functions [159,160]. Platelets are primarily responsible for the aggregation process and contribute to coagulation. During a vascular lesion, platelets are activated, and their granules release factors that are involved in the coagulation.
The activation of platelets is important in hemostasis and thrombosis [161]. Initial platelet activation signals are greatly amplified by a series of rapid positive feedback loops, enabling robust platelet recruitment and thrombus stabilization. The detailed mechanism of platelet activation has been reviewed elsewhere [162]. In brief, upon the vascular injury or high shear stress of the blood flow, circulating platelets come into contact with the exposed sub-endothelium, causing the release, generation, or exposure of agonists, which in turn can activate platelets, resulting in the onset of the thrombus formation process. Platelets are activated by adhesion to adhesive proteins (von Willebrand factor (vWF), collagen) or soluble agonists (ADP, thrombin, thromboxane A2 (TxA2), serotonin, epinephrine) via their respective adhesion receptors or GPCRs, respectively [163,164,165].
The key initiators of the platelet activation at the sites of endothelial damage are the adhesion receptors, while GPCRs play a critical and central role in platelet activation and thrombus formation [165]. Interestingly, despite significant variations in functions and downstream signaling pathways, important platelet receptors share a lot of similarities in their signal transduction mechanisms. For example, the complex of adhesion receptors GP Ib–IX–V, glycoprotein VI (GPVI), and integrins all involve Src family kinases (SFKs), PI3Ks, and the immunoreceptor tyrosine-based activation motif (ITAM) signaling pathway, while GPCRs involve phospholipase C (PLC), Ca2+, diacyl gylcerol (DAG), protein kinase C (PKC), and the PI3Ks signaling pathway. These receptor-specific platelet activations signal pathway cross-talk and merge into common signaling events that trigger platelet shape change, granule secretion, and TxA2 generation, activating integrin αIIbβ3. Ligand binding to integrin αIibβ3 causes platelet adhesion and aggregation, leading to the activation of “inside-out” signaling. Inside-out signaling triggers the “outside-in” signaling, leading to the spreading of platelets, additional granule content secretion and clot retraction, finally stabilizing the platelet adhesion and aggregation.
It has been reported that human platelets can become refractory to activation after major surgery, possibly leading to an increased risk of post-surgical bleeding [166]. This clearly demonstrates that, like platelet activation and aggregation, the desensitization mechanism plays an equally critical role in regulating platelet responsiveness. There are several mechanisms by which GPCR- and G-protein-dependent signaling is turned off in various cells, including GRKs- and arrestin-induced desensitization, RGS protein-regulated turning off, and the internalization of receptors by forming endosomes. Platelets express GRKs and arrestins, and it is well known that the signaling of platelets largely occurs through GPCRs. Platelet GPCRs regulate platelet function by coupling to their respective G-protein, such as Gq-coupled PARs, thromboxane A2 receptor (TP), P2Y1, 5HT; Gi-coupled P2Y12; Gz-coupled adrenergic receptors; and Gs-coupled IP, upon the stimulation of platelets with various agonists, including ADP, TxA2, thrombin, serotonin, epinephrine, and prostacyclin.
Platelet GPCRs are clinically relevant GPCRs, which are targeted by multitudinous drugs of therapeutical importance for bleeding disorders or anticoagulation. Considering the critical roles of GRKs in GPCR functions in other cells, very little is known about the regulation and mechanisms of GPCR signaling and GPCR desensitization by GRKs in platelets. Very recently, we and others have unveiled the interesting features of the GRK system in platelet GPCR signaling. Using GRK6 KO mouse platelets, we showed that the platelet aggregation and dense granule secretion induced by GPCR agonists, including 2-MeSADP, U46619 (TxA2 analog), AYPGKF, and thrombin, is significantly potentiated compared to WT platelets [167]. However, GPVI agonist collagen-related peptide (CRP)-induced platelet aggregation and dense granule secretion are not affected in the GRK6-deficient platelets, indicating that GRK6 does play a role in the regulation of P2Y1, P2Y12, TPα, and PARs-mediated signaling in platelets, and does not regulate non-GPCR-mediated platelet activation. We found that the U46619-induced aggregation response curve shifts left, but the extent of potentiation was not as significant as it was for the other GPCR agonists. This may be due to the shorter C-terminus of the platelet TxA2 receptor TPα that has lesser phosphorylatable serine residues, leading to the decreased affinity of GRK6 towards it compared to other GPCRs in platelets. Similar to our study, it was demonstrated that the GRK phosphorylation sites for β1AR and β2AR are in their C-termini, whereas β3AR lacks GRK targets and it has a very short C-terminus [168]. GRK6 also affects GPCR-mediated integrin αIIbβ3 activation and P-selectin expression in platelets [167]. Most recently, Chen et al. showed that GRK6 has a role in ADP-induced P2Y12 receptor desensitization, but not P2Y1 receptor desensitization in platelets [169]. Hardy et al. also reported that GRK2 and GRK6 mediate P2Y12, but not P2Y1, receptor desensitization in astrocytoma cells [170]. In contrast to their study, we observed that GRK6 is critical in regulating both ADP-induced P2Y1 and P2Y12 receptor desensitization. GRK’s kinase activity has been shown to vary according to cell type, and the same GRK isoform may have a role in desensitization and activation, or show no response at all to a specific receptor.
Interestingly, in contrast to ADP-induced aggregation, GRK6 is not involved in serotonin-induced Gq-coupled 5HT2A receptor and epinephrine-induced Gz-coupled α2A adrenergic receptor-mediated platelet activation, although the co-stimulation of serotonin and epinephrine mimics the ADP-induced P2Y1 and P2Y12 receptor-mediated platelet activation pathway, further suggesting that the kinase activity of GRKs varies with varying ligands, receptors and proteins. Since platelet GPCRs have been the topmost target for anti-thrombotic drug development, research should be done as such, instead of following the “model” concept and predicting the functional mechanism. We also found that the re-stimulation of platelets with ADP and AYPGKF could restore platelet aggregation in GRK6-deficient platelets, suggesting the role of GRK6 in the desensitization of ADP and PAR4 receptors [167]. Moreover, Gq- and Gi-mediated signaling events were regulated by GRK6 in platelets. GRK6 -/- mice are more susceptible to thrombosis, and thus, GRK6 plays a critical role in platelet function in vivo [167].
It remains unknown whether other GRK isoforms except GRK6 phosphorylate the platelet GPCRs when stimulated with various agonists, and whether or how this phosphorylation will change the behavior of the receptor. It is important to know the potential phosphorylation sites, and evidently the number of absolutely phosphorylated sites, in order to establish the receptor’s behavior in platelets. Neither the putative phosphorylation sites nor their specific function is determined in platelet GPCRs. Which GRK isoform is involved in each phosphorylation event during platelet signaling remains unknown. It would be fascinating to see whether or not the platelet agonists promote the receptor conformation in the same way for GRK binding and activation, subsequent receptor phosphorylation, and arrestin recruitment/binding, or whether there is the involvement of some other mechanisms.
Arrestins have been shown to play crucial roles in terminating GPCR-mediated signaling in various cells. Platelets also express arrestin 2 and arrestin 3. Despite the importance of arrestins in GPCR-mediated signaling, the mechanism of GPCR desensitization by arrestins in platelets has not been clearly elucidated yet. Li et al. first showed that the PAR4 and ADP receptor signaling in platelets is differentially regulated by arrestin 2, as only thrombin-, but not ADP-, induced PI3K-Akt phosphorylation and fibrinogen binding was arrestin 2-dependent [171]. Later, Schaff et al. reported that neither arrestin 2 nor arrestin 3 deficiency altered platelet activation, suggesting that arrestins are not involved in platelet GPCR desensitization [172]. They further reported that the deletion of arrestin 2 in mice, but not arrestin 3, is critical for thrombus formation. In contrast, very recently, a study demonstrated the negative regulatory role of arrestin 3 downstream of PAR4- and P2Y12-mediated signaling pathways in mouse platelets [173]. It is not clear which arrestin plays what functional role in platelets, and further study is required to determine the functional differences of arrestin 3 versus arrestin 2 in the regulation of GPCR signaling and the molecular basis of GPCR desensitization in platelets.
Evaluating the functional significance of the individual GRK and β-arrestin isoforms in platelets, and characterizing the novel mechanisms involved in GPCR desensitization and trafficking in platelets using knockout mice for each isoform, will help us to seek a novel approach to developing drugs in the area of vascular pathobiology.
Contribution of Platelet Beyond Thrombosis and Hemostasis
Platelets were believed to have only hemostatic activity. In recent years, scientific research and technology have come up with a new perspective on platelets and their functions due to their abundant granular content of growth factors (GFs), cytokines, and other biological modulators that, upon release, can affect wound healing, inflammation, angiogenesis, stem cell migration, and cell proliferation. These factors also have a paracrine effect in different cell populations, including mesenchymal cells, osteoblasts, fibroblasts, and endothelial cells, which are all involved in various pathophysiological processes [174,175].
Platelets have been shown to play a significant role in inflammation and immunity. Activated, adherent platelets at the vascular injury site can recruit leukocytes to the injury or inflammatory sites, and largely mediate the signaling involved in initial and sustained platelet–leukocyte interactions by regulating receptors including the epithelial neutrophil-activating peptide, glycoproteins, growth-related oncogenes, interleukins, intercellular adhesion molecules, junctional adhesion molecules, platelet-activating factor, and P-selectin glycoprotein ligands. Platelets are capable of changing their surface expression and release their granule content through their TLRs, P-selectin, RANTES, TGF-β, etc., thereby engaging different endothelial and immune cells, including neutrophils, monocytes, eosinophils, B cells and T cells, dendritic cells, natural killer cells, etc., and potentially leading to the activation of the innate and adaptive immune responses. Similarly, platelets can increase the endothelial permeability and mediate leukocyte trafficking to the inflamed endothelium by activating endothelial cells [176]. Besides this, platelet-originating TxA2 form a positive feedback loop upon platelet activation that facilitates the further release of their substantial repertoire of stored cytokines, including IL-1α, IL-1β, Macrophage Inflammatory Protein (MIP)-1α, CD40 ligand (CD40L), and polyphosphate (polyp), which have been shown to contribute to multiple inflammation-related diseases. Importantly, it was shown that platelets play a major role in netosis.
Platelets express diverse mediators, such as PDGF, TGF, and VEGF, that integrate various cascades governed by multiple cytokines, and are released after activated platelets become entrapped within the fibrin matrix. These mediators have been shown to play crucial roles in remodeling and wound healing by stimulating the mitogenic responses required for tissue repair [177].
In the early phase of infection, platelets can limit parasite growth by killing plasmodium falciparum through the release of platelet factors [178,179]. Platelets can interact with various strains of bacteria including the staphylococci family, Neisseria gonorrhoeae, Porphyromonas gingivalis, and Helicobacter pylori [180,181,182]. During bacterial infections, platelets actively mediate the host response through interactions with circulating leukocytes. Platelet α-granules contain various antimicrobial compounds such as platelet connective tissue-activating peptide 3 (CTAP-3), platelet basic protein, thymosin β-4 (Tβ-4), and fibrinopeptide (A and B), which are known to target various bacterial organisms [183,184]. Platelets are also known to interact with various types of viruses through TLR2, IL-1β, and VEGF. Recently, platelets were shown to play a major role in the pathological consequences of COVID-19 as well [185]. Therefore, platelets are beneficial in infections, but consistent viral or bacterial infections can cause arterial thrombosis or venous thromboembolism, a process termed immune thrombosis, leading to cardiovascular disease. Recently, it has been suggested that antiplatelet medications may lower mortality rates related to infections and sepsis.
Platelets also participate in neural diseases associated with pathogen-induced and sterile inflammation, including AD, MS, and migraine, suggesting its role in CNS inflammatory and immune response [186,187]. It has been shown that IgE stored in platelet α-granules upon release has the potential to amplify allergic responses [188]. It was shown that there is a significant interconnection between the activation of platelets and eosinophils, platelets and leukocytes in asthma, and lung allergic inflammation, respectively [189,190,191]. Overall, platelets play important roles in diverse inflammatory diseases, and targeting platelet signaling could be a promising approach to modify platelet responses to inflammation.
It is now generally accepted that platelets play a major role in the early stage of endothelial disturbance in the atherosclerotic process and subsequent CVD [192]. As the markers for predicting the clinical fate of CVD, the molecules activating platelets have been suggested, and may be targeted to minimize thrombosis and atherosclerosis. The variability in platelet activation would also affect the formation of atherosclerosis, thereby affecting CVD. Conversely, limiting platelet activation and aggregation, and inhibiting the molecules involved, can regulate platelet interactions, thrombosis, and CVD.
Much of the recent data has indicated that platelets play a serious role in the pathogenesis of malignant cancers. Using animal models, it has been demonstrated that platelets make a major contribution to tumor cell proliferation, metastasis, and tumor angiogenesis in various types of cancers, such as carcinomas of the breast, colon, lung, and ovary, as well as in melanoma [193,194]. It has been shown that circulating platelet properties contribute to some hallmarks of cancer, including resisting cell death, inducing angiogenesis, metastasis, and evading immune detection, assisting cancer stem cells, and sustaining proliferative signals. Besides this, patients with metastatic cancer have higher chances of thrombosis. Platelets interact with tumor cells, resulting in platelet activation, P-selectin expression, and the development of platelet–tumor microthrombi, which may protect tumor cells from the innate immune system [195]. Therefore, some have developed anti-platelet antibodies to recognize the activated platelet and then further fragment the platelet, potentially reducing metastatic potential [196,197]. Aspirin was also shown to be associated with a reduced risk of distant metastasis in patients with cancer, particularly of colorectal origin [198].
In conclusion, since platelets are involved in other disease conditions beyond thrombosis and hemostasis, the identification of GRKs and their functional receptors in platelets will serve to design novel drug targets and receptor blockers, contributing to the improved treatment of patients with platelet-associated diseases. Therefore, current research is focused on possible therapeutic interventions targeting platelet activation, desensitization, or its surface receptors.
4. The Implications of GRKs in Pharmacology
The chronic or acute use of drugs that target GPCR is associated with an increasing level of GRK expression. A study was carried out that investigated GPCR-targeted drug tolerance in the brain, which demonstrated that an increase in GRK expression could be responsible for drug resistance [199,200]. Likewise, there is a correlation between several pathological conditions such as heart failure, depression, Alzheimer’s disease and Parkinson’s disease, and modulated endogenous GRK expression [108,201,202,203]. Due to scientists’ continuous efforts to gain more knowledge about GPCR/GRK signaling biology, new treatment strategies have been developed for diseases by targeting GRKs. Presumably, the development of highly selective GRK inhibitors will either target their specific kinase domains or reduce the expression of GRK by using selective RNA aptamers [204]. So far, none of the effective GRK inhibitors have been permitted for use in clinical practice [205]. GRKs belong to the AGC kinases subfamily whose kinase domains are nearly identical in structure; there is a possibility of cross-reactivity in the nonselective GRK inhibitors with other AGC kinases [206]. Nevertheless, Takeda Compound 103A, which is a highly selective GRK2 inhibitor that has been shown to inhibit GRK2 activity 50-fold compared to other AGC kinases, has been developed by Takeda Pharmaceuticals [207]. Meanwhile, other highly selective GRK-targeted drugs, for instance paroxetine, GSK180736A, balanol, Takeda Compound 101, and sanigivamycin, which target GRK2, GRK3, GRK5, GRK2, and GRK6, respectively, are also being extensively investigated [208]. Even though these drugs are rather selective for GRKs, all of them demonstrate cross-reactivity with other kinases that limit the drug’s therapeutic potential. Each GRK isoform plays a variety of roles in disease progression due to their structural heterogeneity. Investigating the divergent roles of specific isoforms may allow us to develop potential drugs that would guarantee a selective inhibitory effect on specific isoforms for better disease control. The selective knockdown of specific GRKs and understanding the pharmaceutical properties using various cellular ligands would allow us to design and develop drugs more practically and efficiently, and unveil novel concepts in therapy.
5. Conclusions
In conclusion, GRKs along with arrestins play a central role, as well as controlling hundreds of unique GPCRs and their signaling in almost every facet of cell biology and diseases, making it difficult to fully perceive their mechanism of cellular signaling. GRKs act as important regulators to prevent cells from hyper-stimulation, and could directly modulate the physio-pathological functions of cell biology. As such, GRKs help in maintaining homeostasis by mediating inter- and intra-cellular communication in response to the surrounding environment. As several GPCR-targeted disease treatments are becoming more attractive topics, GRKs could be propitious molecular targets for controlling GPCR responsiveness. It is important to identify the full mechanism involved in GPCR–GRK interactions. Which GRK isoform and how it is involved in each phosphorylation event, the specific effects of these phosphorylation events on subsequent arrestin binding, and cell type-dependent GPCR desensitization/internalization, are still aspects that are yet to be understood. It would be interesting to investigate if GPCR agonists fail to uphold the receptor conformation that is suitable for GRK binding or activation, and consequent receptor phosphorylation as well as arrestin recruitment, or whether they function via some other mechanism.
Author Contributions
Conceptualization, P.K.C. and S.K.; validation, S.K.; investigation, P.K.C.; writing―original draft, P.K.C.; writing―review and editing, S.K.; supervision, S.K. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Research Foundation of Korea (NRF) Grant of the Korean government (NRF-2016R1D1A1B01010310), the Korea Institute of Planning and Evaluation for Technology in Food, Agriculture, Forestry and Fisheries (IPET), funded by the Ministry of Agriculture, Food and Rural Affairs (MAFRA; 320005-4), Regional Innovation Strategy (RIS) through the NRF funded by the Ministry of Education (MOE), and the Global Research and Development Center (GRDC) Program through the NRF funded by the Ministry of Education, Science and Technology (2017K1A4A3014959).
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| GRK | G protein-coupled receptor kinase |
| GPCR | G protein-coupled receptor |
| 7-TM | Seven-transmembrane |
| PI3K | Phosphoiositide 3-kinase |
| PIP2 | Phosphatidylinositol 4:5-bisphosphate |
| RGS | Regulator of G protein signaling domain |
| PH | Pleckstrin homology |
| AP-2 | Adaptor protein-2 |
| β2-AR | β2-adrenergic receptor |
| VEGF | Vascular endothelial growth factor |
| AD | Alzheimer’s disease |
| MS | Multiple sclerosis |
| RhoGDI | Rho GDP-dissociation inhibitor |
| PDE | Phosphodiesterase |
| TSHR | Thryoid-stimulating hormone receptor |
| IGF1-R | Insulin-like growth factor-1 receptor |
| EGR1 | Early growth factor 1 |
| NGF | Nerve growth factor |
| CXCR | Chemokine receptor |
| AT1 | Angiotensin type 1 |
| FSHR | Follicle-stimulating hormone receptor |
| vWF | Von Willebrand factor |
| TxA2 | Thromboxane A2 |
| PLC | Phospholipase C |
| DAG | Diacyl glycerol |
| PKC | Protein kinase C |
| TP | Thromboxane A2 receptor |
| GPVI | Glycoprotein VI |
| SFKs | Src family kinases |
| ITAM | Immunoreceptor tyrosine-based activation motif |
| CRP | Collagen-related peptide |
| GF | Growth factor |
| MIP | Macrophage inflammatory protein |
References
- Patel, C.B.; Noor, N.; Rockman, H.A. Functional Selectivity in Adrenergic and Angiotensin Signaling Systems. Mol. Pharmacol. 2010, 78, 983–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katritch, V.; Cherezov, V.; Stevens, R.C. Structure-Function of the G Protein–Coupled Receptor Superfamily. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 531–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatakrishnan, A.J.; Deupi, X.; Lebon, G.; Tate, C.G.; Schertler, G.F.; Babu, M.M. Molecular Signatures of G-Protein-Coupled Receptors. Nat. Cell Biol. 2013, 494, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Pierce, K.L.; Premont, R.T.; Lefkowitz, R.J. Seven-Transmembrane Receptors. Nat. Rev. Mol. Cell Biol. 2002, 3, 639–650. [Google Scholar] [CrossRef]
- Hurowitz, E.H.; Melnyk, J.M.; Chen, Y.J.; Kouros-Mehr, H.; Simon, M.I.; Shizuya, H. Genomic Characterization of the Human Heterotrimeric G Protein Alpha, Beta, and Gamma Subunit Genes. DNA Res. 2000, 7, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Stephens, L.R.; Eguinoa, A.; Erdjument-Bromage, H.; Lui, M.; Cooke, F.; Coadwell, J.; Smrcka, A.S.; Thelen, M.; Cadwallader, K.; Tempst, P.; et al. The G beta Gamma Sensitivity of a PI3K is Dependent upon a Tightly Associated Adaptor, p101. Cell 1997, 89, 105–114. [Google Scholar] [CrossRef] [Green Version]
- Neves, S.R.; Ram, P.T.; Iyengar, R. G Protein Pathways. Science 2002, 296, 1636–1639. [Google Scholar] [CrossRef]
- Bockaert, J.; Fagni, L.; Dumuis, A.; Marin, P. GPCR Interacting Proteins (GIP). Pharmacol. Ther. 2004, 103, 203–221. [Google Scholar] [CrossRef]
- Lefkowitz, R.J.; Shenoy, S.K. Transduction of Receptor Signals by Beta-Arrestins. Science 2005, 308, 512–517. [Google Scholar] [CrossRef]
- Ferguson, S.S. Evolving Concepts in G Protein-Coupled Receptor Endocytosis: The Role in Receptor Desensitization and Signaling. Pharmacol. Rev. 2001, 53, 1–24. [Google Scholar] [PubMed]
- Kohout, T.A.; Lefkowitz, R.J. Regulation of G Protein-Coupled Receptor Kinases and Arrestins during Receptor Desensitization. Mol. Pharmacol. 2003, 63, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Santulli, G.; Iaccarino, G. Adrenergic Signaling in Heart Failure and Cardiovascular Aging. Maturitas 2016, 93, 65–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorriento, D.; Ciccarelli, M.; Cipolletta, E.; Trimarco, B.; Iaccarino, G. “Freeze, Don’t Move”: How to Arrest a Suspect in Heart Failure–a Review on Available GRK2 Inhibitors. Front. Cardiovasc. Med. 2016, 3, 48. [Google Scholar] [CrossRef] [Green Version]
- Beautrait, A.; Michalski, K.R.; Lopez, T.S.; Mannix, K.M.; McDonald, D.J.; Cutter, A.R.; Medina, C.B.; Hebert, A.M.; Francis, C.J.; Bouvier, M.; et al. Mapping the Putative G Protein-Coupled Receptor (GPCR) Docking Site on GPCR Kinase 2: Insights from Intact Cell Phosphor-Ylation and Recruitment Assays. J. Biol. Chem. 2014, 289, 25262–25275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sallese, M.; Mariggiò, S.; Collodel, G.; Moretti, E.; Piomboni, P.; Baccetti, B.; De Blasi, A. G Protein-Coupled Receptor Kinase GRK4: Molecular Analysis of the Four Isoforms and Ultrastructural Localization in Spermatozoa and Germinal Cells. J. Biol. Chem. 1997, 272, 10188–10195. [Google Scholar] [CrossRef] [Green Version]
- Virlon, B.; Firsov, D.; Cheval, L.; Reiter, E.; Troispoux, C.; Guillou, F.; Elalouf, J.-M. Rat G Protein-Coupled Receptor Kinase GRK4: Identification, Functional Expression, and Differential Tissue Distribution of Two Splice Variants. Endocrinology 1998, 139, 2784–2795. [Google Scholar] [CrossRef] [PubMed]
- Willets, J.M.; Challiss, R.; Nahorski, S.R. Non-visual GRKs: Are We Seeing the Whole Picture? Trends Pharmacol. Sci. 2003, 24, 626–633. [Google Scholar] [CrossRef] [Green Version]
- DebBurman, S.K.; Ptasienski, J.; Benovic, J.L.; Hosey, M.M. G Protein-Coupled Receptor Kinase GRK2 Is a Phospholip-ID-Dependent Enzyme That Can Be Conditionally Activated by G Protein Betagamma Subunits. J. Biol. Chem. 1996, 271, 22552–22562. [Google Scholar] [CrossRef] [Green Version]
- Pitcher, J.A.; Freedman, N.J.; Lefkowitz, R.J. G Protein-Coupled Receptor Kinases. Annu. Rev. Biochem. 1998, 67, 653–692. [Google Scholar] [CrossRef] [Green Version]
- Penela, P.; Ribas, C.; Mayor, F., Jr. Mechanisms of Regulation of the Expression and Function of G Protein-Coupled Receptor Kinases. Cell Signal. 2003, 15, 973–981. [Google Scholar] [CrossRef]
- Lodowski, D.T.; Tesmer, V.M.; Benovic, J.L.; Tesmer, J.J. The Structure of G Protein-coupled Receptor Kinase (GRK)-6 Defines a Second Lineage of GRKs. J. Biol. Chem. 2006, 281, 16785–16793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeWire, S.M.; Ahn, S.; Lefkowitz, R.J.; Shenoy, S.K. Beta-Arrestins and Cell Signaling. Annu. Rev. Physiol. 2007, 69, 483–510. [Google Scholar] [CrossRef] [Green Version]
- Krupnick, J.G.; Benovic, J.L. The Role of Receptor Kinases and Arrestins in G Protein–Coupled Receptor Regulation. Annu. Rev. Pharmacol. Toxicol. 1998, 38, 289–319. [Google Scholar] [CrossRef]
- Premont, R.T.; Gainetdinov, R.R. Physiological Roles of G Protein–Coupled Receptor Kinases and Arrestins. Annu. Rev. Physiol. 2007, 69, 511–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shenoy, S.K.; Lefkowitz, R.J. Multifaceted Roles of Beta-Arrestins in the Regulation of Seven-Membrane-Spanning Receptor Trafficking and Signalling. Biochem. J. 2003, 375, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, E.V.; Gurevich, V.V. Arrestins: Ubiquitous Regulators of Cellular Signaling Pathways. Genome Biol. 2006, 7, 236. [Google Scholar] [CrossRef] [Green Version]
- Bohn, L.M.; Lefkowitz, R.J.; Gainetdinov, R.R.; Peppel, K.; Caron, M.G.; Lin, F.T. Enhanced Morphine Analgesia in Mice Lacking Beta-Arrestin 2. Science 1999, 286, 2495–2498. [Google Scholar] [CrossRef] [PubMed]
- Conner, D.A.; Mathier, M.A.; Mortensen, R.M.; Christe, M.; Vatner, S.F.; Seidman, C.E.; Seidman, J.G. Beta-arrestin1 Knockout Mice Appear Normal but Demonstrate Altered Cardiac Responses to Beta-Adrenergic Stimulation. Circ. Res. 1997, 81, 1021–1026. [Google Scholar] [CrossRef]
- Kovacs, J.J.; Hara, M.R.; Davenport, C.L.; Kim, J.; Lefkowitz, R.J. Arrestin Development: Emerging Roles for Beta-Arrestins in Developmental Signaling Pathways. Dev. Cell 2009, 17, 443–458. [Google Scholar] [CrossRef] [Green Version]
- Sorkin, A.; von Zastrow, M. Endocytosis and Signalling: Intertwining Molecular Networks. Nature Reviews. Mol. Cell Biol. 2009, 10, 609–622. [Google Scholar]
- Goodman, O.B.; Krupnick, J.G.; Santini, F.; Gurevich, V.V.; Penn, R.B.; Gagnon, A.W.; Keen, J.H.; Benovic, J.L. β-Arrestin Acts as a Clathrin Adaptor in Endocytosis of the β2-Adrenergic Receptor. Nat. Cell Biol. 1996, 383, 447–450. [Google Scholar] [CrossRef] [PubMed]
- Laporte, S.A.; Oakley, R.H.; Zhang, J.; Holt, J.A.; Ferguson, S.S.; Caron, M.G.; Barak, L.S. The β2-adrenergic Recep-Tor/βArrestin Complex Recruits the Clathrin Adaptor AP-2 during Endocytosis. Proc. Natl. Acad. Sci. USA 1999, 96, 3712–3717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilden, U.; Hall, S.W.; Kuhn, H. Phosphodiesterase Activation by Photoexcited Rhodopsin Is Quenched When Rhodopsin Is Phosphorylated and Binds the Intrinsic 48-kDa Protein of Rod Outer Segments. Proc. Natl. Acad. Sci. USA 1986, 83, 1174–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strasser, R.H.; Sibley, D.R.; Lefkowitz, R.J. A Novel Catecholamine-Activated Adenosine Cyclic 3’, 5’-Phosphate Independent Pathway for. Beta-Adrenergic Receptor Phos-Phorylation in Wild-Type and Mutant s49 Lymphoma Cells: Mechanism of Homologous Desensitization of Adenylate Cyclase. Biochemistry 1986, 25, 1371–1377. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-K.; Burns, M.E.; Spencer, M.; Niemi, G.A.; Chen, J.; Hurley, J.B.; Baylor, D.A.; Simon, M.I. Abnormal Photore-Sponses and Light-Induced Apoptosis in Rods Lacking Rhodopsin Kinase. Proc. Natl. Acad. Sci. USA 1999, 96, 3718–3722. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Dodd, R.L.; Makino, C.L.; Simon, M.I.; Baylor, D.A.; Chen, J. Prolonged Photoresponses in Transgenic Mouse Rods Lacking Arrestin. Nat. Cell Biol. 1997, 389, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Wilden, U. Duration and Amplitude of the Light-Induced cGMP Hydrolysis in Vertebrate Photoreceptors Are Regulated by Mul-Tiple Phos-Phorylation of Rhodopsin and by Arrestin Binding. Biochemistry 1995, 34, 1446–1454. [Google Scholar] [CrossRef]
- Krupnick, J.G.; Gurevich, V.V.; Benovic, J.L. Mechanism of Quenching of Phototransduction: Binding Competition between Arrestin and Transducin for Phosphorhodopsin. J. Biol. Chem. 1997, 272, 18125–18131. [Google Scholar] [CrossRef] [Green Version]
- Mohan, M.L.; Vasudevan, N.T.; Gupta, M.K.; Martelli, E.E.; Naga Prasad, S.V. G-Protein Coupled Receptor Resensitiza-Tion-Appreciating the Balancing Act of Receptor Function. Curr. Mol. Pharmacol. 2012, 5, 350–361. [Google Scholar] [CrossRef]
- Pals-Rylaarsdam, R.; Xu, Y.; Witt-Enderby, P.; Benovic, J.L.; Hosey, M.M. Desensitization and Internalization of the m2 Muscarinic Acetylcholine Receptor Are Directed by Independent Mechanisms. J. Biol. Chem. 1995, 270, 29004–29011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakata, H.; Kameyama, K.; Haga, K.; Haga, T. Location of Agonist-Dependent-Phosphorylation Sites in the Third Intracellular Loop of Muscarinic Acetylcholine Receptors (m2 subtype). JBIC J. Biol. Inorg. Chem. 1994, 220, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, N.; Jojima, E.; Saito, H.; Haga, T. Role of the Third Intracellular Loop in the Subtype-Specific Internalization and Recy-Cling of Muscarinic M2 and M4 Receptors. Biomed. Res. 2014, 35, 185–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, M.; Zhang, W.; Tian, Y.; Xu, C.; Xu, T.; Liu, J.; Zhang, R. Unraveling a Molecular Determinant for Clathrin-Independent Internalization of the M2 Muscarinic Acetylcholine Receptor. Sci. Rep. 2015, 5, 11408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billard, M.J.; Fitzhugh, D.J.; Parker, J.S.; Brozowski, J.M.; McGinnis, M.W.; Timoshchenko, R.G.; Serafin, D.S.; Lininger, R.; Klauber-Demore, N.; Sahagian, G.; et al. G Protein Coupled Receptor Kinase 3 Regulates Breast Cancer Migration, Invasion, and Metastasis. PLoS ONE 2016, 11, e0152856. [Google Scholar] [CrossRef] [PubMed]
- Nobles, K.N.; Xiao, K.; Ahn, S.; Shukla, A.K.; Lam, C.M.; Rajagopal, S.; Strachan, R.T.; Huang, T.Y.; Bressler, E.A.; Hara, M.R.; et al. Distinct Phosphorylation Sites on the β(2)-Adrenergic Receptor Establish a Barcode That Encodes Differential Functions of β-Arrestin. Sci. Sig. 2011, 4, ra51. [Google Scholar] [CrossRef] [Green Version]
- Reiter, E.; Marion, S.; Robert, F.; Troispoux, C.; Boulay, F.; Guillou, F.; Crepieux, P. Kinase-Inactive G-Protein-Coupled Receptor Kinases Are Able to Attenuate Follicle-Stimulating Hormone-Induced Signaling. Biochem. Biophys. Res. Commun. 2001, 282, 71–78. [Google Scholar] [CrossRef]
- Kim, O.J.; Gardner, B.R.; Williams, D.B.; Marinec, P.S.; Cabrera, D.M.; Peters, J.D.; Mak, C.C.; Kim, K.-M.; Sibley, D.R. The Role of Phosphorylation in D1 Dopamine Receptor Desensitization: Evidence for a Novel Mechanism of Arrestin Association. J. Biol. Chem. 2004, 279, 7999–8010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, M.J.; Lee, K.A.; Niemi, G.A.; Craven, K.B.; Garwin, G.G.; Saari, J.C.; Hurley, J.B. Multiple Phosphorylation of Rhodopsin and the in Vivo Chemistry Underlying Rod Photoreceptor Dark Adaptation. Neuron 2001, 31, 87–101. [Google Scholar] [CrossRef] [Green Version]
- Onorato, J.J.; Palczewski, K.; Regan, J.W.; Caron, M.G.; Lefkowitz, R.J.; Benovic, J.L. Role of Acidic Amino Acids in Peptide Substrates of the Beta-Adrenergic Receptor Kinase and Rhodopsin Kinase. Biochemistry 1991, 30, 5118–5125. [Google Scholar] [CrossRef]
- Kim, J.; Ahn, S.; Ren, X.R.; Whalen, E.J.; Reiter, E.; Wei, H.; Lefkowitz, R.J. Functional Antagonism of Different G Protein-Coupled Receptor Kinases for Beta-Arrestin-Mediated Angiotensin II Receptor Signaling. Proc. Natl. Acad. Sci. USA 2005, 102, 1442–1447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, X.R.; Reiter, E.; Ahn, S.; Kim, J.; Chen, W.; Lefkowitz, R.J. Different G Protein-Coupled Receptor Kinases Govern G Protein and Beta-Arrestin-Mediated Signaling of V2 Vasopressin Receptor. Proc. Natl. Acad. Sci. USA 2005, 102, 1448–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homan, K.T.; Tesmer, J.J.G. Structural Insights into G Protein-Coupled Receptor Kinase Function. Curr. Opin. Cell Biol. 2014, 27, 25–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotton, M.; Claing, A. G Protein-Coupled Receptors Stimulation and the Control of Cell Migration. Cell Signal 2009, 21, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Insall, R. The Interaction between Pseudopods and Extracellular Signalling during Chemotaxis and Directed Migration. Curr. Opin. Cell Biol. 2013, 25, 526–531. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Jiang, Y.; Li, Y.; Wang, J.; Fan, L.; Scott, M.J.; Xiao, G.; Li, S.; Billiar, T.R.; Wilson, M.A.; et al. TLR4 Signaling Augments Monocyte Chemotaxis by Regulating G Protein-Coupled Receptor Kinase 2 Translocation. J. Immunol. 2013, 191, 857–864. [Google Scholar] [CrossRef] [Green Version]
- Arnon, T.I.; Xu, Y.; Lo, C.; Pham, T.; An, J.; Coughlin, S.; Dorn, G.W.; Cyster, J.G. GRK2-Dependent S1PR1 Desensitization Is Required for Lymphocytes to Overcome Their Attraction to Blood. Science 2011, 333, 1898–1903. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Ma, B.; Malik, A.B.; Tang, H.; Yang, T.; Sun, B.; Wang, G.; Minshall, R.D.; Li, Y.; Zhao, Y.; et al. Bidirectional Regulation of Neutrophil Migration by Mitogen-Activated Protein Kinases. Nat. Immunol. 2012, 13, 457–464. [Google Scholar] [CrossRef] [Green Version]
- Penela, P.; Nogués, L.; Mayor, F. Role of G Protein-Coupled Receptor Kinases in Cell Migration. Curr. Opin. Cell Biol. 2014, 27, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Lafarga, V.; Mayor, J.F.; Penela, P. The Interplay between G Protein-Coupled Receptor Kinase 2 (GRK2) and Histone Deacetylase 6 (HDAC6) at the Crossroads of Epithelial Cell Motility. Cell Adhes. Migr. 2012, 6, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Otten, J.J.T.; De Jager, S.C.A.; Kavelaars, A.; Seijkens, T.; Bot, I.; Wijnands, E.; Beckers, L.; Westra, M.M.; Bot, M.; Busch, M.; et al. Hematopoietic G-Protein-Coupled Receptor Kinase 2 Deficiency Decreases Atherosclerotic Lesion Formation in LDL Receptor-Knockout Mice. FASEB J. 2013, 27, 265–276. [Google Scholar] [CrossRef] [Green Version]
- Arraes, S.M.A.; Freitas, M.S.; Da Silva, S.V.; Neto, H.A.D.P.; Alves-Filho, J.C.; Martins, M.A.; Basile-Filho, A.; Tavares-Murta, B.M.; Barja-Fidalgo, C.; Cunha, F.Q. Impaired Neutrophil Chemotaxis in Sepsis Associates with GRK Expression and Inhibition of Actin Assembly and Tyrosine Phosphorylation. Blood 2006, 108, 2906–2913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leoratti, F.M.D.S.; Trevelin, S.C.; Cunha, F.Q.; Rocha, B.C.; Costa, P.A.C.; Gravina, H.D.; Tada, M.S.; Pereira, D.B.; Golenbock, D.T.; Antonelli, L.R.D.V.; et al. Neutrophil Paralysis in Plasmodium vivax Malaria. PLoS Negl. Trop. Dis. 2012, 6, e1710. [Google Scholar] [CrossRef] [PubMed]
- Penela, P.; Ribas, C.; Aymerich, I.; Eijkelkamp, N.; Barreiro, O.; Heijnen, C.J.; Kavelaars, A.; Sánchez-Madrid, F.; Mayor, F., Jr. G Protein-Coupled Receptor Kinase 2 Positively Regulates Epithelial Cell Migration. EMBO J. 2008, 27, 1206–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cant, S.H.; Pitcher, J.A. G Protein-Coupled Receptor Kinase 2-Mediated Phosphorylation of Ezrin Is Required for G Protein-Coupled Receptor-Dependent Reorganization of the Actin Cytoskeleton. Mol. Biol. Cell 2005, 16, 3088–3099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahsai, A.W.; Zhu, S.; Fenteany, G. G Protein-Coupled Receptor Kinase 2 Activates Radixin, Regulating Membrane Protrusion and Motility in Epithelial Cells. Biochim. Biophys. Acta (BBA) Bioenerg. 2010, 1803, 300–310. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.-H.; Zhang, L.; Fanaroff, A.C.; Cai, X.; Sharma, K.C.; Brian, L.; Exum, S.T.; Shenoy, S.K.; Peppel, K.; Freedman, N.J. G Protein–Coupled Receptor Kinase-5 Attenuates Atherosclerosis by Regulating Receptor Tyrosine Kinases and 7-Transmembrane Receptors. Arter. Thromb. Vasc. Biol. 2012, 32, 308–316. [Google Scholar] [CrossRef] [Green Version]
- Barker, B.L.; Benovic, J.L. G Protein-Coupled Receptor Kinase 5 Phosphorylation of Hip Regulates Internalization of the Chem-Okine Receptor CXCR4. Biochemistry 2011, 50, 6933–6941. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Wang, F.; Long, H.; Chen, Y.; Wu, Z.; Ma, L. GRK5 Promotes F-Actin Bundling and Targets Bundles to Membrane Structures to Control Neuronal Morphogenesis. J. Cell Biol. 2011, 194, 905–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, P.K.; Zhang, Y.; Coomes, A.S.; Kim, W.-J.; Stupay, R.; Lynch, L.D.; Atkinson, T.; Kim, J.I.; Nie, Z.; Daaka, Y. G Protein–Coupled Receptor Kinase GRK5 Phosphorylates Moesin and Regulates Metastasis in Prostate Cancer. Cancer Res. 2014, 74, 3489–3500. [Google Scholar] [CrossRef] [Green Version]
- Raghuwanshi, S.K.; Su, Y.; Singh, V.; Haynes, K.; Richmond, A.; Richardson, R.M. The Chemokine Receptors CXCR1 and CXCR2 Couple to Distinct G Protein-Coupled Receptor Kinases to Mediate and Regulate Leukocyte Functions. J. Immunol. 2012, 189, 2824–2832. [Google Scholar] [CrossRef] [Green Version]
- Vroon, A.; Heijnen, C.J.; Raatgever, R.; Touw, I.P.; Ploemacher, R.E.; Premont, R.T.; Kavelaars, A. GRK6 Defi-Ciency Is Associated with Enhanced CXCR4-Mediated Neutrophil Chemotaxis in Vitro and Impaired Responsiveness to G-CSF In Vivo. J. Leukoc. Biol. 2004, 75, 698–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kavelaars, A.; Vroon, A.; Raatgever, R.P.; Fong, A.M.; Premont, R.T.; Patel, D.D.; Lefkowitz, R.J.; Heijnen, C.J. Increased Acute Inflammation, Leukotriene B4-Induced Chemotaxis, and Signaling in Mice Deficient for G Protein-Coupled Receptor Kinase 6. J. Immunol. 2003, 171, 6128–6134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarrant, T.K.; Rampersad, R.R.; Esserman, D.; Rothlein, L.R.; Liu, P.; Premont, R.T.; Lefkowitz, R.J.; Lee, D.M.; Patel, D.D. Granulocyte Chemotaxis and Disease Expression Are Differentially Regulated by GRK Subtype in an Acute Inflammatory Arthritis Model (K/BxN). Clin. Immunol. 2008, 129, 115–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fong, A.M.; Premont, R.T.; Richardson, R.M.; Yu, Y.R.; Lefkowitz, R.J.; Patel, D.D. Defective Lymphocyte Chem-Otaxis in Beta-arrestin2-and GRK6-Deficient Mice. Proc. Natl. Acad. Sci. USA 2002, 99, 7478–7483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eijkelkamp, N.; Heijnen, C.J.; Lucas, A.; Premont, R.T.; Elsenbruch, S.; Schedlowski, M.; Kavelaars, A. G Protein-Coupled Receptor Kinase 6 Controls Chronicity and Severity of Dextran Sodium Sulphate-Induced Colitis in Mice. Gut 2007, 56, 847–854. [Google Scholar] [CrossRef] [Green Version]
- Alves-Filho, J.C.; Sônego, F.; Souto, F.O.; Freitas, A.; Verri, W.A.; Auxiliadora-Martins, M.; Basile-Filho, A.; McKenzie, A.N.; Xu, D.; Cunha, F.Q.; et al. Interleukin-33 Attenuates Sepsis by Enhancing Neutrophil Influx to the Site of Infection. Nat. Med. 2010, 16, 708–712. [Google Scholar] [CrossRef]
- Packiriswamy, N.; Lee, T.; Raghavendra, P.B.; Durairaj, H.; Wang, H.; Parameswaran, N. G-Protein-Coupled Receptor Kinase-5 Mediates Inflammation but Does Not Regulate Cellular Infiltration or Bacterial Load in a Polymicrobial Sepsis Model in Mice. J. Innate Immun. 2013, 5, 401–413. [Google Scholar] [CrossRef] [PubMed]
- Parvataneni, S.; Gonipeta, B.; Packiriswamy, N.; Lee, T.; Durairaj, H.; Parameswaran, N. Role of Myeloid-Specific G-Protein Coupled Receptor Kinase-2 in Sepsis. Int. J. Clin. Exp. Med. 2011, 4, 320–330. [Google Scholar] [PubMed]
- Patial, S.; Saini, Y.; Parvataneni, S.; Appledorn, D.M.; Dorn, G.W., 2nd; Lapres, J.J.; Amalfitano, A.; Senagore, P.; Parameswaran, N. Myeloid-specific GPCR Kinase-2 Negatively Regulates NF-κB1p105-ERK Pathway and Limits Endotoxemic Shock in Mice. J. Cell. Physiol. 2011, 226, 627–637. [Google Scholar] [CrossRef] [Green Version]
- Patial, S.; Shahi, S.; Saini, Y.; Lee, T.; Packiriswamy, N.; Appledorn, D.M.; LaPres, J.J.; Amalfitano, A.; Parameswaran, N. G-Protein Coupled Receptor Kinase 5 Mediates LipopolysacchariDe-induced NFκB Activation in Primary Macrophages and Mod-Ulates Inflammation in Vivo in Mice. J. Cell. Physiol. 2011, 226, 1323–1333. [Google Scholar] [CrossRef] [Green Version]
- Parameswaran, N.; Pao, C.S.; Leonhard, K.S.; Kang, D.S.; Kratz, M.; Ley, S.C.; Benovic, J.L. Arrestin-2 and G Protein-Coupled Receptor Kinase 5 Interact with NFκb1 p105 and Negatively Regulate Lipopolysaccharide-stimulated ERK1/2 Activation in Macrophages. J. Biol. Chem. 2006, 281, 34159–34170. [Google Scholar] [CrossRef] [Green Version]
- Patial, S.; Luo, J.; Porter, K.J.; Benovic, J.L.; Parameswaran, N. G-Protein Coupled Receptor Kinases Mediate TNF Alpha-Induced NF KappaB Signaling via Direct Interaction with and Phosphor-Ylation of I Kappa B Alpha (38.15). Am. Assoc. Immnol. 2009, 425, 169–178. [Google Scholar]
- Peregrin, S.; Jurado-Pueyo, M.; Campos, P.M.; Sanz-Moreno, V.; Ruiz-Gomez, A.; Crespo, P.; Mayor, F., Jr.; Murga, C. Phos-Phorylation of p38 by GRK2 at the Docking Groove Unveils a Novel Mechanism for Inactivating p38MAPK. Curr. Biol. 2006, 16, 2042–2047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willemen, H.L.; Eijkelkamp, N.; Wang, H.; Dantzer, R.; Dorn II, G.W.; Kelley, K.W.; Heijnen, C.J.; Kavelaars, A. Microglial/Macrophage GRK2 Determines Duration of Peripheral IL-1β-Induced Hyperalgesia: Contribution of Spinal Cord CX3CR1, p38 and IL-1 signaling. Pain 2010, 150, 550–560. [Google Scholar] [CrossRef] [Green Version]
- Jurado-Pueyo, M.; Campos, P.M.; Mayor, F.; Murga, C. GRK2-Dependent Desensitization Downstream of G Proteins. J. Recept. Signal Transduct. 2008, 28, 59–70. [Google Scholar] [CrossRef]
- Liu, S.; Premont, R.T.; Kontos, C.D.; Zhu, S.; Rockey, D.C. A Crucial Role for GRK2 in Regulation of Endothelial Cell Nitric Oxide Synthase Function in Portal Hypertension. Nat. Med. 2005, 11, 952–958. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Kobayashi, T.; Takenouchi, Y.; Matsumoto, T.; Kamata, K. Angiotensin II Causes Endothelial Dysfunction via the GRK2/Akt/eNOS Pathway in Aortas from a Murine Type 2 Diabetic Model. Pharmacol. Res. 2011, 64, 535–546. [Google Scholar] [CrossRef]
- Premont, R.T.; Claing, A.; Vitale, N.; Freeman, J.L.R.; Pitcher, J.A.; Patton, W.A.; Moss, J.; Vaughan, M.; Lefkowitz, R.J. 2-Adrenergic Receptor Regulation by GIT1, a G Protein-Coupled Receptor Kinase-Associated ADP Ribosylation Factor GTPase-Activating Protein. Proc. Natl. Acad. Sci. USA 1998, 95, 14082–14087. [Google Scholar] [CrossRef] [Green Version]
- Lafarga, V.; Aymerich, I.; Tapia, O.; Mayor, F., Jr.; Penela, P. A Novel GRK2/HDAC6 Interaction Modulates Cell Spreading and Motility. EMBO J. 2012, 31, 856–869. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Benovic, J.L. G Protein-coupled Receptor Kinase Interaction with Hsp90 Mediates Kinase Maturation. J. Biol. Chem. 2003, 278, 50908–50914. [Google Scholar] [CrossRef] [Green Version]
- De Jager, S.C.A.; Bermúdez, B.; Bot, I.; Koenen, R.R.; Bot, M.; Kavelaars, A.; De Waard, V.; Heijnen, C.J.; Muriana, F.J.G.; Weber, C.; et al. Growth Differentiation Factor 15 Deficiency Protects against Atherosclerosis by Attenuating CCR2-Mediated Macrophage Chemotaxis. J. Exp. Med. 2011, 208, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Ho, J.; Cocolakis, E.; Dumas, V.M.; Posner, B.I.; Laporte, S.A.; Lebrun, J.J. The G protein-coupled Receptor Kinase-2 is a TGFbeta-Inducible Antagonist of TGFbeta Signal Transduction. EMBO J. 2005, 24, 3247–3258. [Google Scholar] [CrossRef] [Green Version]
- Luerman, G.C.; Powell, D.W.; Uriarte, S.M.; Cummins, T.D.; Merchant, M.L.; Ward, R.A.; McLeish, K.R. Identification of Phosphoproteins Associated with Human Neutrophil Granules Following Chemotactic Peptide Stimulation. Mol. Cell. Proteom. 2011, 10, M110.001552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiedemann, R.E.; Zhu, Y.X.; Schmidt, J.; Yin, H.; Shi, C.-X.; Que, Q.; Basu, G.; Azorsa, D.; Perkins, L.M.; Braggio, E.; et al. Kinome-wide RNAi studies in human multiple myeloma identify vulnerable kinase targets, including a lymphoid-restricted kinase, GRK6. Blood 2010, 115, 1594–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauthier, C.; Tavernier, G.; Charpentier, F.; Langin, D.; Le Marec, H. Functional beta3-Adrenoceptor in the Human Heart. J. Clin. Investig. 1996, 98, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Vinge, L.E.; von Lueder, T.G.; Aasum, E.; Qvigstad, E.; Gravning, J.A.; How, O.J.; Edvardsen, T.; Bjornerheim, R.; Ahmed, M.S.; Mikkelsen, B.W.; et al. Cardiac-Restricted Expression of the Carboxyl-Terminal Fragment of GRK3 Uncovers Distinct Functions of GRK3 in Regulation of Cardiac Contractility and Growth: GR3 Controls Cardiac alpha1-Adrenergic Receptor Responsiveness. J. Biol. Chem. 2008, 283, 10601–10610. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Matkovich, S.J.; Duan, X.; Gold, J.I.; Koch, W.J.; Dorn, G.W. Nuclear Effects of G-Protein Receptor Kinase 5 on Histone Deacetylase 5–Regulated Gene Transcription in Heart Failure. Circ. Heart Fail. 2011, 4, 659–668. [Google Scholar] [CrossRef] [Green Version]
- Chen, E.P.; Bittner, H.B.; Akhter, S.A.; Koch, W.J.; Davis, R. Myocardial Function in Hearts with Transgenic Overexpression of the G Protein-Coupled Receptor Kinase 5. Ann. Thorac. Surg. 2001, 71, 1320–1324. [Google Scholar] [CrossRef]
- Ungerer, M.; Böhm, M.; Elce, J.S.; Erdmann, E.; Lohse, M.J. Altered Expression of Beta-Adrenergic Receptor Kinase and Beta 1-Adrenergic Receptors in the Failing Human Heart. Circulation 1993, 87, 454–463. [Google Scholar] [CrossRef] [Green Version]
- Ungerer, M.; Kessebohm, K.; Kronsbein, K.; Lohse, M.J.; Richardt, G. Activation of Beta-Adrenergic Receptor Kinase during Myocardial Ischemia. Circ. Res. 1996, 79, 455–460. [Google Scholar] [CrossRef]
- Gros, R.; Benovic, J.L.; Tan, C.M.; Feldman, R.D. G-Protein-Coupled Receptor Kinase Activity Is Increased in Hypertension. J. Clin. Investig. 1997, 99, 2087–2093. [Google Scholar] [CrossRef]
- Choi, D.J.; Koch, W.J.; Hunter, J.J.; Rockman, H.A. Mechanism of Beta-Adrenergic Receptor Desensitization in Cardiac Hy-Pertrophy Is Increased Beta-Adrenergic Receptor Kinase. J. Biol. Chem. 1997, 272, 17223–17229. [Google Scholar] [PubMed] [Green Version]
- Raake, P.W.; Vinge, L.E.; Gao, E.; Boucher, M.; Rengo, G.; Chen, X.; DeGeorge, B.R., Jr.; Matkovich, S.; Houser, S.R.; Most, P.; et al. G Protein-Coupled Receptor Kinase 2 Ablation in Cardiac Myocytes before or after Myocardial Infarction Prevents Heart Failure. Circ. Res. 2008, 103, 413–422. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Pesant, S.; Cohn, H.I.; Soltys, S.; Koch, W.J.; Eckhart, A.D. Negative Regulation of VEGF Signaling in Human Coronary Artery Endothelial Cells by G Protein-Coupled Receptor Kinase 5. Clin. Transl. Sci. 2009, 2, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Brinks, H.; Boucher, M.; Gao, E.; Chuprun, J.K.; Pesant, S.; Raake, P.W.; Huang, Z.M.; Wang, X.; Qiu, G.; Gumpert, A.; et al. Level of G Protein-Coupled Receptor Kinase-2 Determines Myocardial Ische-Mia/Reperfusion Injury via Pro-and Anti-apoptotic Mechanisms. Circ. Res. 2010, 107, 1140–1149. [Google Scholar] [CrossRef] [Green Version]
- Suo, W.Z.; Li, L. Dysfunction of G Protein-Coupled Receptor Kinases in Alzheimer’s Disease. Sci. World J. 2010, 10, 1667–1678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vroon, A.; Kavelaars, A.; Limmroth, V.; Lombardi, M.S.; Goebel, M.U.; Van Dam, A.-M.; Caron, M.G.; Schedlowski, M.; Heijnen, C.J. G Protein-Coupled Receptor Kinase 2 in Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis. J. Immunol. 2005, 174, 4400–4406. [Google Scholar] [CrossRef] [Green Version]
- Bychkov, E.R.; Gurevich, V.V.; Joyce, J.N.; Benovic, J.L.; Gurevich, E.V. Arrestins and Two Receptor Kinases Are Upregulated in Parkinson’s Disease with Dementia. Neurobiol. Aging 2008, 29, 379–396. [Google Scholar] [CrossRef] [Green Version]
- Obrenovich, M.E.; Palacios, H.H.; Gasimov, E.; Leszek, J.; Aliev, G. The GRK2 Overexpression Is a Primary Hallmark of Mitochondrial Lesions during Early Alzheimer Disease. Cardiovasc. Psychiatry Neurol. 2009, 2009, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Sorensen, S.D.; Conn, P.J. G Protein-Coupled Receptor Kinases Regulate Metabotropic Glutamate Receptor 5 Function and Ex-pression. Neuropharmacology 2003, 44, 699–706. [Google Scholar] [CrossRef]
- Degos, V.; Peineau, S.; Nijboer, C.; Kaindl, A.M.; Sigaut, S.; Favrais, G.; Plaisant, F.; Teissier, N.; Gouadon, E.; Lombet, A.; et al. G Protein-Coupled Receptor Kinase 2 and Group I Metabotropic Glutamate Receptors Mediate Inflammation-Induced Sensitization to Excitotoxic Neurodegeneration. Ann. Neurol. 2013, 73, 667–678. [Google Scholar] [CrossRef]
- Eijkelkamp, N.; Heijnen, C.J.; Willemen, H.L.; Deumens, R.; Joosten, E.A.; Kleibeuker, W.; den Hartog, I.J.; van Velthoven, C.T.; Nijboer, C.; Nassar, M.A.; et al. GRK2: A Novel Cell-Specific Regulator of Severity and Duration of Inflammatory Pain. J. Neurosci. 2010, 30, 2138–2149. [Google Scholar] [CrossRef]
- Ferrari, L.F.; Bogen, O.; Alessandri–Haber, N.; Levine, E.; Gear, R.W.; Levine, J.D. Transient Decrease in Nociceptor grk2 Expression Produces Long-Term Enhancement in Inflammatory Pain. Neuroscience 2012, 222, 392–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Rasul, I.; Sun, Y.; Wu, G.; Li, L.; Premont, R.T.; Suo, W.Z. GRK5 Deficiency Leads to Reduced Hippocampal Acetylcholine Level via Impaired Presynaptic M2/M4 Autoreceptor Desensitization. J. Biol. Chem. 2009, 284, 19564–19571. [Google Scholar] [CrossRef] [Green Version]
- Nakaya, M.; Tajima, M.; Kosako, H.; Nakaya, T.; Hashimoto, A.; Watari, K.; Nishihara, H.; Ohba, M.; Komiya, S.; Tani, N.; et al. GRK6 Deficiency in Mice Causes Autoimmune Disease Due to Impaired Apoptotic Cell Clearance. Nat. Commun. 2013, 4, 1532. [Google Scholar] [CrossRef] [Green Version]
- Lappano, R.; Maggiolini, M. G Protein-Coupled Receptors: Novel Targets for Drug Discovery in Cancer. Nat. Rev. Drug Discov. 2010, 10, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Dorsam, R.T.; Gutkind, J.S. G-Protein-Coupled Receptors and Cancer. Nat. Rev. Cancer 2007, 7, 79–94. [Google Scholar] [CrossRef]
- Penela, P.; Murga, C.; Ribas, C.; Lafarga, V.; Mayor, F., Jr. The Complex G Protein-Coupled Receptor Kinase 2 (GRK2) Interactome Unveils New Physiopathological Targets. Br. J. Pharmacol. 2010, 160, 821–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurevich, E.V.; Tesmer, J.J.; Mushegian, A.; Gurevich, V.V. G Protein-Coupled Receptor Kinases: More Than Just Kinases and Not Only for GPCRs. Pharmacol. Ther. 2012, 133, 40–69. [Google Scholar] [CrossRef] [Green Version]
- Métayé, T.; Menet, E.; Guilhot, J.; Kraimps, J.-L. Expression and Activity of G Protein-Coupled Receptor Kinases in Differentiated Thyroid Carcinoma. J. Clin. Endocrinol. Metab. 2002, 87, 3279–3286. [Google Scholar] [CrossRef]
- Ma, Y.; Han, C.-C.; Huang, Q.; Sun, W.-Y.; Wei, W. GRK2 Overexpression Inhibits IGF1-Induced Proliferation and Migration of Human Hepatocellular Carcinoma Cells by Downregulating EGR1. Oncol. Rep. 2016, 35, 3068–3074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Z.; Hurtt, R.; Gu, T.; Bodzin, A.S.; Koch, W.J.; Doria, C. GRK2 Negatively Regulates IGF-1R Signaling Pathway and Cyclins’ Expression in HepG2 Cells. J. Cell. Physiol. 2013, 228, 1897–1901. [Google Scholar] [CrossRef]
- Zhou, L.; Wang, M.-Y.; Liang, Z.-Y.; Zhou, W.-X.; You, L.; Pan, B.-J.; Liao, Q.; Zhao, Y.-P. G-Protein-Coupled Receptor Kinase 2 in Pancreatic Cancer: Clinicopathologic and Prognostic Significance. Hum. Pathol. 2016, 56, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Bin, W.Z. Expression of GRK2 and β-Arrestin2 in the Dorsal Root Ganglion Neurons and the Regulated Effect by Nerve Growth Factor in Rats with Bone Cancer. Pain. Chin. J. Cancer Prevent. Treat. 2011, 18, 4. [Google Scholar]
- Hu, M.; Wang, C.; Li, W.; Lu, W.; Bai, Z.; Qin, D.; Yan, Q.; Zhu, J.; Krueger, B.J.; Renne, R.; et al. A KSHV microRNA Directly Targets G Protein-Coupled Receptor Kinase 2 to Promote the Migration and Invasion of Endothelial Cells by Inducing CXCR2 and Activating AKT Signaling. PLoS Pathog. 2015, 11, e1005171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogues, L.; Salcedo, A.; Mendiola, M.; Hardission, D.; Mayor, F.; Penela, P. G Protein-Coupled Receptor Kinase 2 (GRK2) Con-Tributes to Cellular Transformation and Breast Tumor Progression. Eur. J. Cancer 2012, 48, S80–S81. [Google Scholar] [CrossRef]
- Nogués, L.; Reglero, C.; Rivas, V.; Salcedo, A.; Lafarga, V.; Neves, M.; Ramos, P.; Mendiola, M.; Berjón, A.; Stamatakis, K.; et al. G Protein-coupled Receptor Kinase 2 (GRK2) Promotes Breast Tumorigenesis Through a HDAC6-Pin1 Axis. EBioMedicine 2016, 13, 132–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayáns, M.P.; Somoza-Martín, J.; Barros-Angueira, F.; Gayoso-Diz, P.; Otero-Rey, E.; Gándra-Rey, J.; Garcia-Garcia, A. Activity of β2-Adrenergic Receptor in Oral Squamous Cell Carcinoma Is Mediated by Overexpression of the ADRBK2 Gene: A Pilot Study. Biotech. Histochem. 2011, 87, 179–186. [Google Scholar] [CrossRef]
- Nakata, H.; Kinoshita, Y.; Kishi, K.; Fukuda, H.; Kawanami, C.; Matsushima, Y.; Asahara, M.; Okada, A.; Maekawa, T.; Chiba, T. Involvement of Beta-Adrenergic Receptor Kinase-1 in Homologous Desensitization of Histamine H2 Receptors in Human Gastric Carcinoma Cell Line MKN-45. Digestion 1996, 57, 406–410. [Google Scholar] [CrossRef]
- Rivas, V.; Chapuli, R.; Mayor, F.; Penela, P. 346 Role of GRK2 in Developing Vasculature and Pathological Angiogenesis. Eur. J. Cancer 2012, 48, S84. [Google Scholar] [CrossRef]
- Robinson, J.D.; Pitcher, J.A. G Protein-Coupled Receptor Kinase 2 (GRK2) Is a Rho-Activated Scaffold Protein for the ERK MAP Kinase Cascade. Cell Signal. 2013, 25, 2831–2839. [Google Scholar] [CrossRef] [Green Version]
- Prowatke, I.; Devens, F.; Benner, A.; Gröne, E.F.; Mertens, D.; Gröne, H.J.; Lichter, P.; Joos, S. Expression Analysis of Imbal-Anced Genes in Prostate Carcinoma Using Tissue Microarrays. Br. J. Cancer 2007, 96, 82–88. [Google Scholar] [CrossRef]
- Drake, J.M.; Paull, E.O.; Graham, N.A.; Lee, J.K.; Smith, B.A.; Titz, B.; Stoyanova, T.; Faltermeier, C.M.; Uzunangelov, V.; Carlin, D.E.; et al. Phosphoproteome Integration Reveals Patient-Specific Networks in Prostate Cancer. Cell 2016, 166, 1041–1054. [Google Scholar] [CrossRef] [Green Version]
- Woerner, B.M.; Luo, J.; Brown, K.R.; Jackson, E.; Dahiya, S.M.; Mischel, P.; Benovic, J.L.; Piwnica-Worms, D.; Rubin, J.B. Suppression of G-protein–Coupled Receptor Kinase 3 Expression Is a Feature of Classical GBM That Is Required for Maximal Growth. Mol. Cancer Res. 2012, 10, 156–166. [Google Scholar] [CrossRef] [Green Version]
- King, D.W.; Steinmetz, R.; Wagoner, H.A.; Hannon, T.S.; Chen, L.Y.; Eugster, E.A.; Pescovitz, O.H. Differential Expression of GRK Isoforms in Nonmalignant and Malignant Human Granulosa Cells. Endocrine 2003, 22, 135–142. [Google Scholar] [CrossRef]
- Miyagawa, Y.; Ohguro, H.; Odagiri, H.; Maruyama, I.; Maeda, T.; Maeda, A.; Sasaki, M.; Nakazawa, M. Aberrantly Expressed Recoverin Is Functionally Associated with G-Protein-Coupled Receptor Kinases in Cancer Cell Lines. Biochem. Biophys. Res. Commun. 2003, 300, 669–673. [Google Scholar] [CrossRef]
- Sánchez-Más, J.; Guillo, L.A.; Zanna, P.; Jiménez-Cervantes, C.; García-Borrón, J.C. Role of G Protein-Coupled Receptor Kinases in the Homologous Desensitization of the Human and Mouse Melanocortin 1 Receptors. Mol. Endocrinol. 2005, 19, 1035–1048. [Google Scholar] [CrossRef] [Green Version]
- Fitzhugh, D.; McGinnis, M.; Timoshchenko, R.; Lininger, R.; DeMore, N.; Serody, J.; Tarrant, T. G Protein Coupled Receptor Kinase 3 (GRK3) Negatively Regulates CXCL12/CXCR4 Signaling and Tumor Migration in Breast Cancer. J. Allergy Clin. Immunol. 2011, 127, AB230. [Google Scholar] [CrossRef]
- Li, W.; Ai, N.; Wang, S.; Bhattacharya, N.; Vrbanac, V.; Collins, M.; Signoretti, S.; Hu, Y.; Boyce, F.M.; Gravdal, K.; et al. GRK3 is Essential for Metastatic Cells and Promotes Prostate Tumor Progression. Proc. Natl. Acad. Sci. USA 2014, 111, 1521–1526. [Google Scholar] [CrossRef] [Green Version]
- Dautzenberg, F.M.; Braun, S.; Hauger, R.L. GRK3 Mediates Desensitization of CRF1 Receptors: A Potential Mechanism Regu-Lating Stress Adaptation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 280, R935–R946. [Google Scholar] [CrossRef] [Green Version]
- Matsubayashi, J.; Takanashi, M.; Oikawa, K.; Fujita, K.; Tanaka, M.; Xu, M.; De Blasi, A.; Bouvier, M.; Kinoshita, M.; Kuroda, M.; et al. Expression of G Protein-Coupled Receptor Kinase 4 Is Associated with Breast Cancer Tumourigenesis. J. Pathol. 2008, 216, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Kim, J.; Kaur, R.; Tan, I.; Bloch, O.; Sun, M.Z.; Safaee, M.; Oh, M.C.; Sughrue, M.; Phillips, J.; et al. G-Protein Coupled Receptor Kinase (GRK)-5 Regulates Proliferation of Glioblastoma-Derived Stem Cells. J. Clin. Neurosci. 2013, 20, 1014–1018. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Zhang, H.; Liu, J.; Rubin, J.B.; Cho, Y.J.; Shu, H.K.; Schniederjan, M.; MacDonald, T.J. Growth Factor Receptor-Src-Mediated Suppression of GRK6 Dysregulates CXCR4 Signaling and Promotes Medulloblastoma Migration. Mol. Cancer 2013, 12, 18. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Chakraborty, P.; Wang, Z.; Daaka, Y. G-Protein Coupled Receptor Kinase 5 Regulates Prostrate Tumor Growth. J. Urol. 2012, 187, 322–329. [Google Scholar] [CrossRef]
- Chen, X.; Zhu, H.; Yuan, M.; Fu, J.; Zhou, Y.; Ma, L. G-protein-coupled Receptor Kinase 5 Phosphorylates p53 and Inhibits DNA Damage-induced Apoptosis. J. Biol. Chem. 2010, 285, 12823–12830. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-C.; Tsai, F.-M.; Shyu, R.-Y.; Tsai, Y.-M.; Wang, C.-H.; Jiang, S.-Y. G Protein-Coupled Receptor Kinase 5 Mediates Tazarotene-Induced Gene 1-Induced Growth Suppression of Human Colon Cancer Cells. BMC Cancer 2011, 11, 175. [Google Scholar] [CrossRef] [Green Version]
- Geras-Raaka, E.; Varma, A.; Clark-Lewis, I.; Gershengorn, M.C. Kaposi’s Sarcoma-Associated Herpesvirus (KSHV) Chemokine VMIP-II and Human SDF-1alpha Inhibit Signaling by KSHV G Protein-Coupled Receptor. Biochem. Biophys. Res. Commun. 1998, 253, 725–727. [Google Scholar] [CrossRef]
- Li, Y.-P. GRK6 Expression in Patients with Hepatocellular Carcinoma. Asian Pac. J. Trop. Med. 2013, 6, 220–223. [Google Scholar] [CrossRef] [Green Version]
- Qiu, X.; Chen, J.; Zhang, Z.; You, Y.; Wang, Z. Aberrant GRK6 Promoter Methylation Is Associated with Poor Prognosis in Hypopharyngeal Squamous Cell Carcinoma. Oncol. Rep. 2016, 35, 1027–1033. [Google Scholar] [CrossRef] [Green Version]
- Raghuwanshi, S.K.; Smith, N.; Rivers, E.J.; Thomas, A.J.; Sutton, N.; Hu, Y.; Mukhopadhyay, S.; Chen, X.L.; Leung, T.; Richardson, R.M.; et al. G Protein-Coupled Receptor Kinase 6 Deficiency Promotes Angiogenesis, Tumor Progression, and Metastasis. J. Immunol. 2013, 190, 5329–5336. [Google Scholar] [CrossRef] [Green Version]
- Yao, S.; Zhong, L.; Liu, J.; Feng, J.; Bian, T.; Zhang, Q.; Chen, J.; Lv, X.; Chen, J.; Liu, Y. Prognostic Value of Decreased GRK6 Expression in Lung Adenocarcinoma. J. Cancer Res. Clin. Oncol. 2016, 142, 2541–2549. [Google Scholar] [CrossRef]
- Maeda, T.; Maeda, A.; Maruyama, I.; Ogawa, K.I.; Kuroki, Y.; Sahara, H.; Sato, N.; Ohguro, H. Mechanisms of Photoreceptor Cell Death in Cancer-Associated Retinopathy. Investig. Ophthalmol. Vis. Sci. 2001, 42, 705–712. [Google Scholar]
- Zhang, H.; Li, S.; Doan, T.; Rieke, F.; Detwiler, P.B.; Frederick, J.M.; Baehr, W. Deletion of PrBP/Delta Impedes Transport of GRK1 and PDE6 Catalytic Subunits to Photoreceptor Outer Segments. Proc. Natl. Acad. Sci. USA 2007, 104, 8857–8862. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Frederick, J.M.; Baehr, W. Unc119 Gene Deletion Partially Rescues the grk1 Transport Defect of PDE6d (-/-) Cones. Adv. Exp. Med. Biol. 2014, 801, 487–493. [Google Scholar]
- Mukherjee, D.; Zhao, J. The Role of Chemokine Receptor CXCR4 in Breast Cancer Metastasis. Am. J. Cancer Res. 2013, 3, 46–57. [Google Scholar]
- Chen, K.; Fu, C.; Chen, C.; Liu, L.; Ren, H.; Han, Y.; Yang, J.; He, D.; Zhou, L.; Yang, Z.; et al. Role of GRK4 in the Regulation of Arterial AT1Receptor in Hypertension. Hypertension 2014, 63, 289–296. [Google Scholar] [CrossRef] [Green Version]
- Villar, V.A.; Jones, J.E.; Armando, I.; Palmes-Saloma, C.; Yu, P.; Pascua, A.M.; Keever, L.; Arnaldo, F.B.; Wang, Z.; Luo, Y.; et al. G Protein-Coupled Receptor Kinase 4 (GRK4) Regulates the Phosphorylation and Function of the Dopamine D3 Receptor. J. Biol. Chem. 2009, 284, 21425–21434. [Google Scholar] [CrossRef] [Green Version]
- Harvey, J.W. The Feline Blood Film. J. Feline Med. Surg. 2017, 19, 747–757. [Google Scholar] [CrossRef]
- Senzel, L.; Gnatenko, D.V.; Bahou, W.F. The Platelet Proteome. Curr. Opin. Hematol. 2009, 16, 329–333. [Google Scholar] [CrossRef]
- White, J.G.; Key, N.S.; King, R.A.; Vercellotti, G.M. The White Platelet Syndrome: A New Autosomal Dominant Platelet Disorderi. Structural Abnormalities. Platelets 2004, 15, 173–184. [Google Scholar] [CrossRef]
- Shattil, S.J.; Kashiwagi, H.; Pampori, N. Integrin Signaling: The Platelet Paradigm. Blood 1998, 91, 2645–2657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Delaney, M.K.; O’Brien, K.A.; Du, X. Signaling During Platelet Adhesion and Activation. Arter. Thromb. Vasc. Biol. 2010, 30, 2341–2349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaffran, Y.; Meyer, S.C.; Negrescu, E.; Reddy, K.B.; Fox, J.E. Signaling across the Platelet Adhesion Receptor Glycoprotein Ib-IX Induces Alpha Iibbeta 3 Activation Both in Platelets and a Transfected Chinese Hamster Ovary Cell System. J. Biol. Chem. 2000, 275, 16779–16787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, T.; Kambayashi, J.; Okuma, M.; Tandon, N.N. Activation of the GP IIb-IIIa Complex Induced by Platelet Adhesion to Collagen Is Mediated by Both Alpha2Beta1 Integrin and GP VI. J. Biol. Chem. 1999, 274, 11897–11903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Offermanns, S. Activation of Platelet Function Through G Protein–Coupled Receptors. Circ. Res. 2006, 99, 1293–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, J.; Etherington, M.; Jamieson, S. Refractory State of Platelet Aggregation with Major Operations. Lancet 1971, 298, 741–743. [Google Scholar] [CrossRef]
- Chaudhary, P.K.; Kim, S.; Jee, Y.; Lee, S.-H.; Park, K.-M.; Kim, S. Role of GRK6 in the Regulation of Platelet Activation through Selective G Protein-Coupled Receptor (GPCR) Desensitization. Int. J. Mol. Sci. 2020, 21, 3932. [Google Scholar] [CrossRef]
- Liggett, S.B.; Freedman, N.J.; Schwinn, D.A.; Lefkowitz, R.J. Structural Basis for Receptor Subtype-Specific Regulation Revealed by a Chimeric Beta 3/Beta 2-Adrenergic Receptor. Proc. Natl. Acad. Sci. USA 1993, 90, 3665–3669. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Gupta, S.; Cooper, M.; DeHelian, D.; Zhao, X.; Naik, M.U.; Wurtzel, J.G.T.; Stalker, T.J.; Goldfinger, L.E.; Benovic, J.; et al. GRK6 Regulates the Hemostatic Response to Injury through Its Rate-Limiting Effects on Gpcr Signaling in Platelets. Blood Adv. 2020, 4, 76–86. [Google Scholar] [CrossRef]
- Hardy, A.R.; Conley, P.B.; Luo, J.; Benovic, J.L.; Poole, A.W.; Mundell, S.J. P2Y1 and P2Y12 Receptors for ADP Desensitize by Distinct Kinase-Dependent Mechanisms. Blood 2005, 105, 3552–3560. [Google Scholar] [CrossRef]
- Li, D.; D’Angelo, L.; Chavez, M.; Woulfe, D.S. Arrestin-2 Differentially Regulates PAR4 and ADP Receptor Signaling in Platelets. J. Biol. Chem. 2011, 286, 3805–3814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaff, M.; Receveur, N.; Bourdon, C.; Ohlmann, P.; Lanza, F.; Gachet, C.; Mangin, P.H. β-Arrestin-1 Participates in Thrombosis and Regulates Integrin aIIbβ3 Signalling without Affecting P2y Receptors Desensitisation and Function. Thromb. Haemost. 2012, 107, 735–748. [Google Scholar] [CrossRef] [Green Version]
- Hutchinson, J.L.; Zhao, X.; Hill, R.; Mundell, S.J. Arrestin-3 Differentially Regulates Platelet GPCR Subsets. Platelets 2019, 31, 641–645. [Google Scholar] [CrossRef]
- Amable, P.R.; Carias, R.B.V.; Teixeira, M.V.T.; da Cruz Pacheco, I.; do Amaral, R.J.F.C.; Granjeiro, J.M.; Borojevic, R. Platelet-Rich Plasma Preparation for Regenerative Medicine: Optimization and Quantification of Cytokines and Growth Factors. Stem Cell Res. Ther. 2013, 4, 67. [Google Scholar] [CrossRef] [Green Version]
- Bambace, N.M.; Holmes, C.E. The Platelet Contribution to Cancer Progression. J. Thromb. Haemost. 2011, 9, 237–249. [Google Scholar] [CrossRef]
- Wagner, D.D.; Frenette, P.S. The Vessel Wall and Its Interactions. Blood 2008, 111, 5271–5281. [Google Scholar] [CrossRef]
- Golebiewska, E.M.; Poole, A.W. Platelet Secretion: From Haemostasis to Wound Healing and Beyond. Blood Rev. 2015, 29, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Ware, J.; Corken, A.; Khetpal, R. Platelet Function beyond Hemostasis and Thrombosis. Curr. Opin. Hematol. 2013, 20, 451–456. [Google Scholar] [CrossRef] [Green Version]
- McMorran, B.J.; Wieczorski, L.; Drysdale, K.E.; Chan, J.-A.; Huang, H.M.; Smith, C.; Mitiku, C.; Beeson, J.G.; Burgio, G.; Foote, S.J. Platelet Factor 4 and Duffy Antigen Required for Platelet Killing of Plasmodium falciparum. Science 2012, 338, 1348–1351. [Google Scholar] [CrossRef]
- Yeaman, M.R. Platelets in Defense against Bacterial Pathogens. Cell. Mol. Life Sci. 2009, 67, 525–544. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, J.R.; Foster, T.J.; Cox, D. The Interaction of Bacterial Pathogens with Platelets. Nat. Rev. Genet. 2006, 4, 445–457. [Google Scholar] [CrossRef] [PubMed]
- Hamzeh-Cognasse, H.; Damien, P.; Chabert, A.; Pozzetto, B.; Cognasse, F.; Garraud, O. Platelets and Infections—Complex Interactions with Bacteria. Front. Immunol. 2015, 6, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blair, P.; Flaumenhaft, R. Platelet α-granules: Basic Biology and Clinical Correlates. Blood Rev. 2009, 23, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.-Q.; Yeaman, M.R.; Selsted, M.E. Antimicrobial Peptides from Human Platelets. Infect. Immun. 2002, 70, 6524–6533. [Google Scholar] [CrossRef] [Green Version]
- Manne, B.K.; Denorme, F.; Middleton, E.A.; Portier, I.; Rowley, J.W.; Stubben, C.J.; Petrey, A.C.; Tolley, N.D.; Guo, L.; Cody, M.J.; et al. Platelet Gene Expression and Function in Patients with COVID-19. Blood 2020, 136, 1317–1329. [Google Scholar] [CrossRef] [PubMed]
- Chavakis, T.; Langer, H.F. Platelets and Neurovascular Inflammation. Thromb. Haemost. 2013, 110, 888–893. [Google Scholar] [CrossRef] [PubMed]
- Behari, M.; Shrivastava, M. Role of Platelets in Neurodegenerative Diseases: A Universal Pathophysiology. Int. J. Neurosci. 2013, 123, 287–299. [Google Scholar] [CrossRef]
- Klouche, M.; Klinger, M.F.; Kühnel, W.; Wilhelm, D. Endocytosis, Storage, and Release of IgE by Human Platelets: Differences in Patients with Type I Allergy and Nonatopic Subjects. J. Allergy Clin. Immunol. 1997, 100, 235–241. [Google Scholar] [CrossRef]
- Pitchford, S.C.; Yano, H.; Lever, R.; Riffo-Vasquez, Y.; Ciferri, S.; Rose, M.J.; Giannini, S.; Momi, S.; Spina, D.; O’Connor, B.; et al. Platelets Are Essential for Leukocyte Recruitment in Allergic Inflammation. J. Allergy Clin. Immunol. 2003, 112, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Pitchford, S.C.; Riffo-Vasquez, Y.; Sousa, A.; Momi, S.; Gresele, P.; Spina, D.; Page, C.P. Platelets Are Necessary for Airway Wall Remodeling in a Murine Model of Chronic Allergic Inflammation. Blood 2004, 103, 639–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benton, A.S.; Kumar, N.; Lerner, J.; Wiles, A.A.; Foerster, M.; Teach, S.J.; Freishtat, R.J. Airway Platelet Activation Is Associated with Airway Eosinophilic Inflammation in Asthma. J. Investig. Med. 2010, 58, 987–990. [Google Scholar] [CrossRef]
- Montenont, E.; Echagarruga, C.; Allen, N.; Araldi, E.; Suarez, Y.; Berger, J.S. Platelet WDR1 Suppresses Platelet Activity and Is Associated with Cardiovascular Disease. Blood 2016, 128, 2033–2042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNicol, A.; Israels, S.J. Beyond Hemostasis: The Role of Platelets in Inflammation, Malignancy and Infection. Cardiovasc. Haematol. Dis. Drug Targets 2008, 8, 99–117. [Google Scholar] [CrossRef] [Green Version]
- Sharma, D.; Brummel-Ziedins, K.E.; Bouchard, B.A.; Holmes, C.E. Platelets in Tumor Progression: A Host Factor That Offers Multiple Potential Targets in the Treatment of Cancer. J. Cell. Physiol. 2014, 229, 1005–1015. [Google Scholar] [CrossRef]
- Bendas, G.; Borsig, L. Cancer Cell Adhesion and Metastasis: Selectins, Integrins, and the Inhibitory Potential of Heparins. Int. J. Cell Biol. 2012, 2012, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Dang, S.; Hong, T.; Tang, J.; Fan, J.; Bu, D.; Sun, Y.; Wang, Z.; Wisniewski, T. A Humanized Single-Chain Antibody against Beta 3 Integrin Inhibits Pulmonary Metastasis by Preferentially Fragmenting Activated Platelets in the Tumor Microen-Vironment. Blood 2012, 120, 2889–2898. [Google Scholar] [CrossRef] [Green Version]
- Ware, J. Fragmenting the Platelet to Reduce Metastasis. Blood 2012, 120, 2779–2780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothwell, P.M.; Wilson, M.; Price, J.F.; Belch, J.F.; Meade, T.W.; Mehta, Z. Effect of Daily Aspirin on Risk of Cancer Metastasis: A Study of Incident Cancers during Randomised Controlled Trials. Lancet 2012, 379, 1591–1601. [Google Scholar] [CrossRef]
- Hurlé, M.A. Changes in the Expression of G Protein-Coupled Receptor Kinases and Beta-Arrestin 2 in Rat Brain during Opioid Tolerance and Supersensitivity. J. Neurochem. 2001, 77, 486–492. [Google Scholar] [CrossRef]
- Díaz, A.; Pazos, A.; Flórez, J.; Ayesta, F.J.; Santana, V.; Hurlé, M.A. Regulation of Mu-Opioid Receptors, G-Protein-Coupled Receptor Kinases and Beta-Arrestin 2 in the Rat Brain after Chronic Opioid Receptor Antagonism. Neuroscience 2002, 112, 345–353. [Google Scholar] [CrossRef]
- Suo, Z.; Cox, A.A.; Bartelli, N.; Rasul, I.; Festoff, B.W.; Premont, R.T.; Arendash, G.W. GRK5 Deficiency Leads to Early Alz-heimer-Like Pathology and Working Memory Impairment. Neurobiol. Aging 2007, 28, 1873–1888. [Google Scholar] [CrossRef]
- Ungerer, M.; Parruti, G.; Böhm, M.; Puzicha, M.; DeBlasi, A.; Erdmann, E.; Lohse, M.J. Expression of Beta-Arrestins and Be-Ta-Adrenergic Receptor Kinases in the Failing Human Heart. Circ. Res. 1994, 74, 206–213. [Google Scholar] [CrossRef] [Green Version]
- Grange-Midroit, M.; García-Sevilla, J.A.; Ferrer-Alcón, M.; La Harpe, R.; Huguelet, P.; Guimón, J. Regulation of GRK 2 and 6, Beta-Arrestin-2 and Associated Proteins in the Prefrontal Cortex of Drug-Free and Antidepressant Drug-Treated Subjects with Major Depression. Brain Res. Mol. Brain Res. 2003, 111, 31–41. [Google Scholar] [CrossRef]
- Tesmer, V.M.; Lennarz, S.; Mayer, G.; Tesmer, J.J.G. Molecular Mechanism for Inhibition of G Protein-Coupled Receptor Kinase 2 by a Selective RNA Aptamer. Structures 2012, 20, 1300–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homan, K.T.; Tesmer, J.J.G. Molecular Basis for Small Molecule Inhibition of G Protein-Coupled Receptor Kinases. ACS Chem. Biol. 2015, 10, 246–256. [Google Scholar] [CrossRef] [PubMed]
- Waldschmidt, H.V.; Homan, K.T.; Cruz-Rodríguez, O.; Cato, M.C.; Waninger-Saroni, J.; Larimore, K.M.; Cannavo, A.; Song, J.; Cheung, J.Y.; Kirchhoff, P.D.; et al. Structure-Based Design, Synthesis, and Biological Evaluation of Highly Selective and Potent G Protein-Coupled Receptor Kinase 2 Inhibitors. J. Med. Chem. 2016, 59, 3793–3807. [Google Scholar] [CrossRef] [Green Version]
- Thal, D.M.; Yeow, R.Y.; Schoenau, C.; Huber, J.; Tesmer, J.J.G. Molecular Mechanism of Selectivity among G Protein-Coupled Receptor Kinase 2 Inhibitors. Mol. Pharmacol. 2011, 80, 294–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thal, D.M.; Homan, K.T.; Chen, J.; Wu, E.K.; Hinkle, P.M.; Huang, Z.M.; Chuprun, J.K.; Song, J.; Gao, E.; Cheung, J.Y.; et al. Paroxetine Is a Direct Inhibitor of G Protein-Coupled Receptor Kinase 2 and Increases Myocardial Contractility. ACS Chem. Biol. 2012, 7, 1830–1839. [Google Scholar] [CrossRef]
Figure 1.
The complex G protein-coupled receptor kinases (GRKs) reactome. GRKs regulate diverse signaling pathways by interaction with proteins. GRKs interact with GPCRs in a canonical pathway. In addition, evidence has suggested that GRKs modulate signaling by interacting with other protein substrates in a non-canonical manner. They have a regulatory function with Gβ subunits, other interacting proteins, MeTransferase, and other protein kinases (PKs).
Figure 1.
The complex G protein-coupled receptor kinases (GRKs) reactome. GRKs regulate diverse signaling pathways by interaction with proteins. GRKs interact with GPCRs in a canonical pathway. In addition, evidence has suggested that GRKs modulate signaling by interacting with other protein substrates in a non-canonical manner. They have a regulatory function with Gβ subunits, other interacting proteins, MeTransferase, and other protein kinases (PKs).

Figure 2.
Structural domain distribution of the GRK isoforms. All GRKs (60–80 KDa) have a short N-terminal domain consisting of an α-N-terminal domain followed by an N-terminal regulator of G-protein signaling domain (RH: RGS homology) where the G protein-coupled receptor (GPCR) binds. A central catalytic domain for GRKs catalysis and a unique variable-length C-terminal domain (~105 to 230 amino acids) are important for receptor recognition. In contrast to other isoforms, the β-adrenergic kinase (GRK2 and GRK3) has a pleckstrin homology domain (PH) necessary for terminating Gβγ complex-related downstream signaling. The numbers above the domains represent amino acid residue as reported by Lodowski et al. [21].
Figure 2.
Structural domain distribution of the GRK isoforms. All GRKs (60–80 KDa) have a short N-terminal domain consisting of an α-N-terminal domain followed by an N-terminal regulator of G-protein signaling domain (RH: RGS homology) where the G protein-coupled receptor (GPCR) binds. A central catalytic domain for GRKs catalysis and a unique variable-length C-terminal domain (~105 to 230 amino acids) are important for receptor recognition. In contrast to other isoforms, the β-adrenergic kinase (GRK2 and GRK3) has a pleckstrin homology domain (PH) necessary for terminating Gβγ complex-related downstream signaling. The numbers above the domains represent amino acid residue as reported by Lodowski et al. [21].

Figure 3.
Model depicting GRKs and arrestins in the signaling, desensitization and internalization.The activated 7-TM receptor/GPCR is phosphorylated by GRK enabling arrestin recruitment and terminating G-protein coupling, finally desensitizing GPCR downstream signaling by mediating the arrestin–receptor complex to clathrin-coated pits. This promotes receptor internalization. The internalized receptor is finally sorted either by degradation or recycling. Refer to the text for a detailed mechanism. Modified from Mohan et al. [39].
Figure 3.
Model depicting GRKs and arrestins in the signaling, desensitization and internalization.The activated 7-TM receptor/GPCR is phosphorylated by GRK enabling arrestin recruitment and terminating G-protein coupling, finally desensitizing GPCR downstream signaling by mediating the arrestin–receptor complex to clathrin-coated pits. This promotes receptor internalization. The internalized receptor is finally sorted either by degradation or recycling. Refer to the text for a detailed mechanism. Modified from Mohan et al. [39].


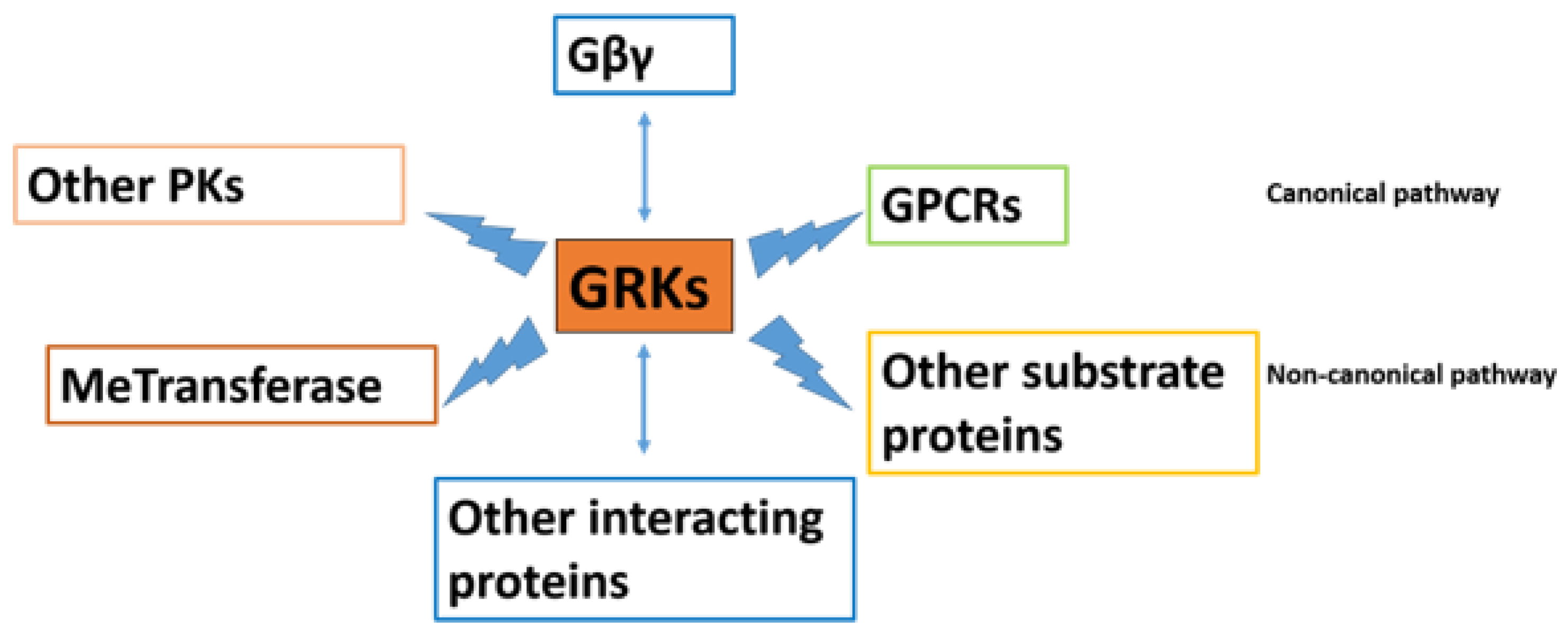{kind=link}
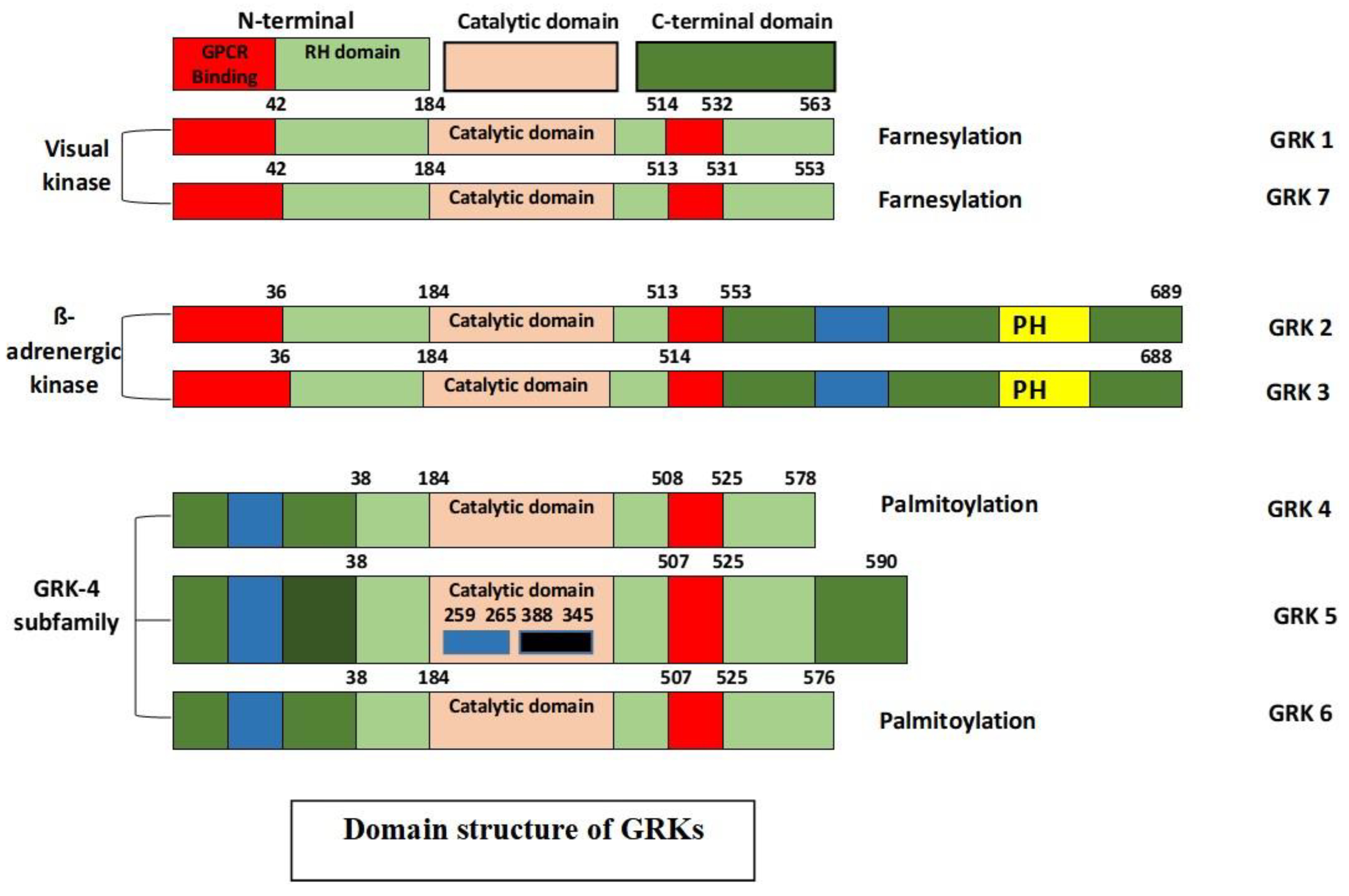{kind=link}
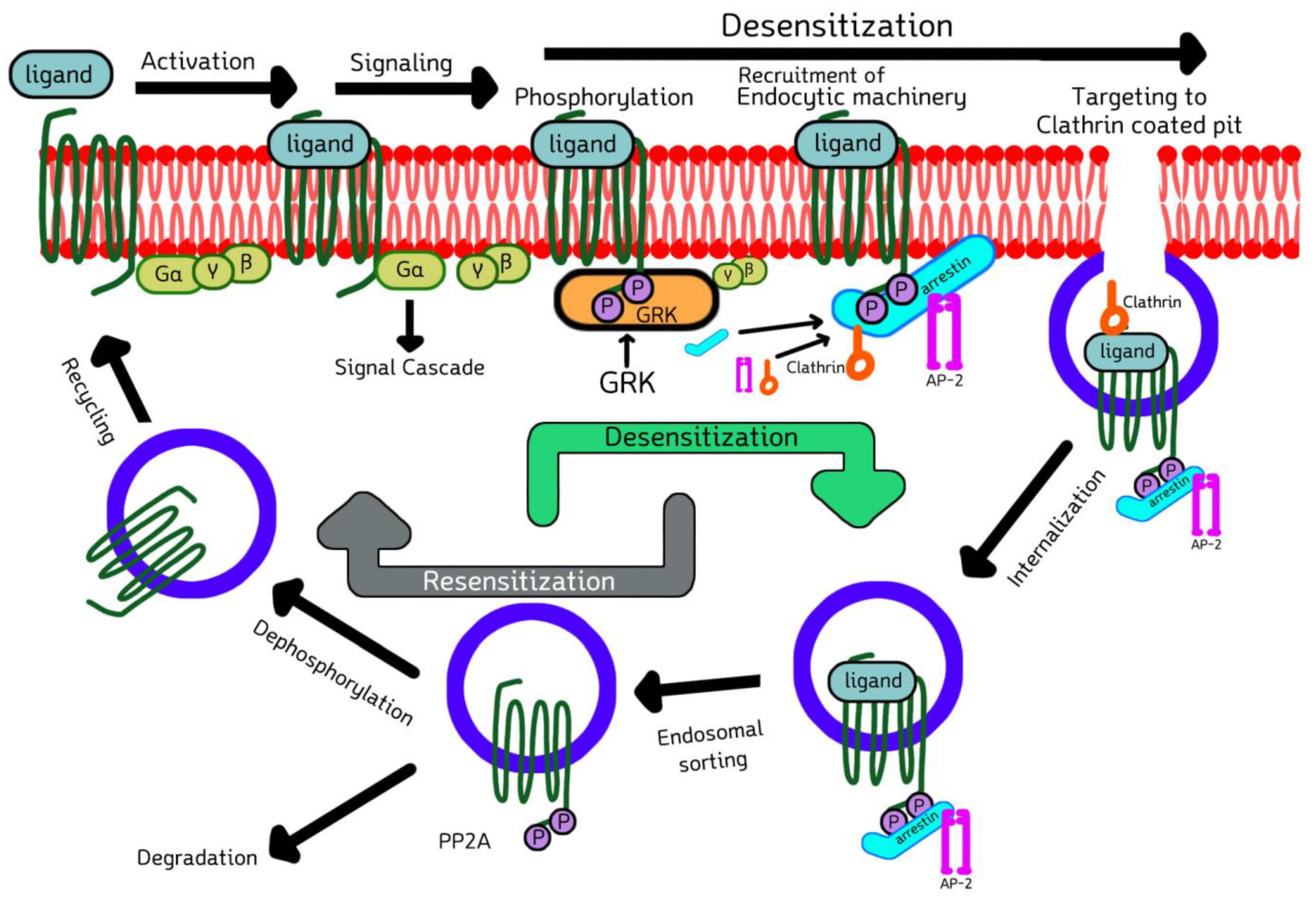{kind=link}
Table 1.
GRK isoforms and their established functions in immune cells and inflammation.
| GRK Isoform | Interacting Partner(s) | Associated Signalling Pathway/Cellular Response | References |
|---|---|---|---|
| GRK2 | NF-kB p105 subunit and inhibitor (IkB-α) phosphorylation | TLR4-induced and Tumor Necrosis Factor-α (TNF-α) pathways | [79,81,82] |
| p38 phosphorylation Raf1, MEK1, ERK2, RhoA, RKIP, GIT | P38 mitogen-activated protein kinases (MAPK) pathways Extracellular signal-regulated kinase (ERK) pathways | [83,84,85] | |
| Serine-threonine kinase Akt phosphorylation | Akt-nitric oxide (NO) pathways | [86,87] | |
| Ezrin/radixin/moesin phosphorylation | Actin cytoskeleton | [64,65] | |
| ADP ribosylation factor (ARF)-specific GTPase-activating proteins (GIT) | Focal adhesion dynamic | [63,88] | |
| Histone deacetylase 6 (HDAC6) phosphorylation | Microtubules network | [89] | |
| Heat shock protein 90 (Hsp90) | Regulation of GRK expression | [90] | |
| Receptor-regulated Smads (R-Smads) phosphorylation | Transforming growth factor β (TGF-β) pathways | [91,92] | |
| GRK3 | HSP90 | Regulation of GRK expression | [90] |
| GRK5 | ERM (moesin phosphorylation) | Actin cytoskeleton | [69] |
| GIT1 | Regulation of receptor endocytosis | [88] | |
| HSP90, HSP70 | Regulation of GRK expression and CXCR4 endocytosis | ||
| NF-kB p105 subunit and IkB-α phosphorylation | TLR4-induced and TNF-α pathways | [90,91] | |
| Src Tyrosine kinase | GRK phosphorylation and neutrophils exocytosis | [93] | |
| GRK6 | HSP90 | Regulation of GRK expression | [90,94] |
Table 2.
An overview of established roles of GRK isoforms in various types of cancer.
| GRK Subtype | Type of Cancer | Interacting Partner (s) | Molecular Mechanism | Function | Biological Model | References |
|---|---|---|---|---|---|---|
| GRK2 | Thyroid carcinoma | TSHR | ND | Decrease proliferation through rapid desensitization | Differentiated thyroid carcinoma patients and cell lines | [120] |
| Hepatocellular carcinoma cell | IGFI-R | Decrease proliferation and migration | [121] | |||
| Human hepatocellular carcinoma (HepG2) | IGFI-R | Decrease cell cycle progression | [122] | |||
| Pancreatic cancer | N/A | ND | --T-stage and poor survival rate, increased proliferation | Pancreatic carcinoma patients, ductal adenocarcinoma patients and cell lines | [123] | |
| Breast carcinosarcoma | NGFR | Decrease bone cancer pain | [124] | |||
| Kaposi’s sarcoma-associated herpesvirus infected tumor cell | CXCR2 | Desensitization and AKT signaling | Decrease migration and invasion | Patients and cell lines | [125] | |
| Basal breast cancer with Her-2 amplification/infiltrating ductal carcinoma | Her-2/ER-α | Increase the promoting of mitogenic, anti-apoptic activities- survival and progression | [126] | |||
| Luminal and basal breast cancer | HDAC6/Pin1 | AKT/ERK cascades | Increase sensitivity of breast cancer cells to traditional chemotherapeutic treatment | Invasive ductal carcinoma patients, cell lines orthotopic and xenograftmouse models | [127] | |
| Breast cancer | CXCR4 | Desensitization and signaling | Decrease metastasis | Breast cancer patients, cell lines orthotopic mouse models | [128] | |
| Human gastric carcinoma cell line (MKN-45) | H2 receptor | --poor differentiation | [129] | |||
| Human breast cancer | N/A | Increase tumor growth and decrease angiogenesis | [130,131] | |||
| Prostrate | ND | Differentiation | Adenocarcinoma patients | [132] | ||
| Prostrate | ND | ND | ND | Neuroendocrine prostrate and metastatic castrastion-resistant prostate cancer patients | [133] | |
| Glioblastoma | ND | ND | Mesenchymal glioblastoma patients. | [134] | ||
| GRK2/4 | Ovary | ND | ND | Granulosa cell cancer patients | [135] | |
| GRK2/5/6 | Gastric cancer (SSTW-2) | recoverin | --tumor progression, metastasis | [136] | ||
| GRK2/6 | melanoma | Melanocortin 1 receptor | --determinant for skin cancer | [137] | ||
| GRK3 | Breast cancers (MDA-MB-231, MDA-MB-468 | CXCR4 | Decrease metastasis increase migration | Breast cancer patients, cell lines orthotopic mouse models | [138] | |
| Prostate cancer (PC3) | N/A | Downmodulation of angiogenesis inhibitors | Increase metastasis, tumor progression, angiogenesis | Metastatic castration-resistant prostate cancer patients, cell lines and orthotopic mouse models | [139] | |
| Retinoblastoma (Y-79) | CRFI receptor | Increase stress adaptation | [140] | |||
| Oral squamous carcinoma | β2-adrenergic receptor | --tumor malignancy and invasion | [128] | |||
| Glioblastoma | CXCR4 desensitization and signaling | desensitization and signaling | Increased proliferation | Classical Glioblastoma patients | [134] | |
| GRK4 | Ovarian malignant granulosa cell tumor | FSHR | --benign and malignant transformation in tumor development | [135] | ||
| Breast cancer | Arrestin2 receptor | Mediated ERK & JNK signaling | Increase proliferation | Ductal carcinoma patients and cell lines | [141] | |
| GRK5 | glioblastoma | N/A | Proliferation rate and WHO grade | Glioblastoma multiform patients and cell lines | [142] | |
| Thyroid carcinoma | TSHR | TSHR desensitization and signaling | Decrease proliferation through slow desensitization, increase proliferation | Differentiated thyroid carcinoma patients | [120] | |
| Prostate cancer (PC3) | Cyclin D1 | G2/M progression | Decrease proliferation, cell cycle | Cell lines and xenograft mouse tumors | [143] | |
| Prostate cancer (PC3, DU145, LNCaP) | Moesin | Moesin phosphorylation | Decrease migration, invasion Increase cell adhesion | Cell lines and xenograft mouse tumors | [69] | |
| Prostate cancer | N/A | Increase tumor growth, invasion, and metastasis | [144] | |||
| Osteosarcoma (U2OS, Saos-2) | P53 | Phosphorylation and degradation | Decrease cell apoptosis and radiosensitivity | Cell lines | [145] | |
| Colon | PGE2 | Desensitization and signaling | Increased proliferation | Cell lines | [146] | |
| Kaposi’s sarcoma | KSHV-GPCR | Desensitization and signaling | Increased proliferation | Cell lines | [147] | |
| GRK6 | Heptocellular carcinoma | N/A | --proliferation maker in early diagnosis | [148] | ||
| Hypopharyngeal squamous cell carcinoma (FaDu) | Methyl transferase | Methylation of GRK6 | --cancer progression Decrease invasion | [149] | ||
| Medulloblastoma | CXCR4/ EGFR/ PDGFR-Src | Increase migration | [143] | |||
| Lung cancer | CXCR2 | Decrease cancer development | [150] | |||
| Lung | ND | ND | Decreased survival | Adenocarcinoma patients | [151] | |
| Medullo-Blastoma | CXCR4 | Desensitization and signaling | Increased migration | Medulloblastoma patients and cell lines | [143] | |
| Myeloma | STAT3 | phosphorylation | Increased survival | Primary multiple myeloma patients and cell lines | [94] | |
| GRK1/7 | recoverin | --cancer-associated retinopathy | [152] |
N/A: not provided by the research, --: has association with.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Chaudhary, P.K.; Kim, S. The GRKs Reactome: Role in Cell Biology and Pathology. Int. J. Mol. Sci. 2021, 22, 3375. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073375
AMA Style
Chaudhary PK, Kim S. The GRKs Reactome: Role in Cell Biology and Pathology. International Journal of Molecular Sciences. 2021; 22(7):3375. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073375
Chicago/Turabian StyleChaudhary, Preeti Kumari, and Soochong Kim. 2021. "The GRKs Reactome: Role in Cell Biology and Pathology" International Journal of Molecular Sciences 22, no. 7: 3375. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073375
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.