
Diphenyl-Methane Based Thyromimetic Inhibitors for Transthyretin Amyloidosis


 , ,
, ,  and
and 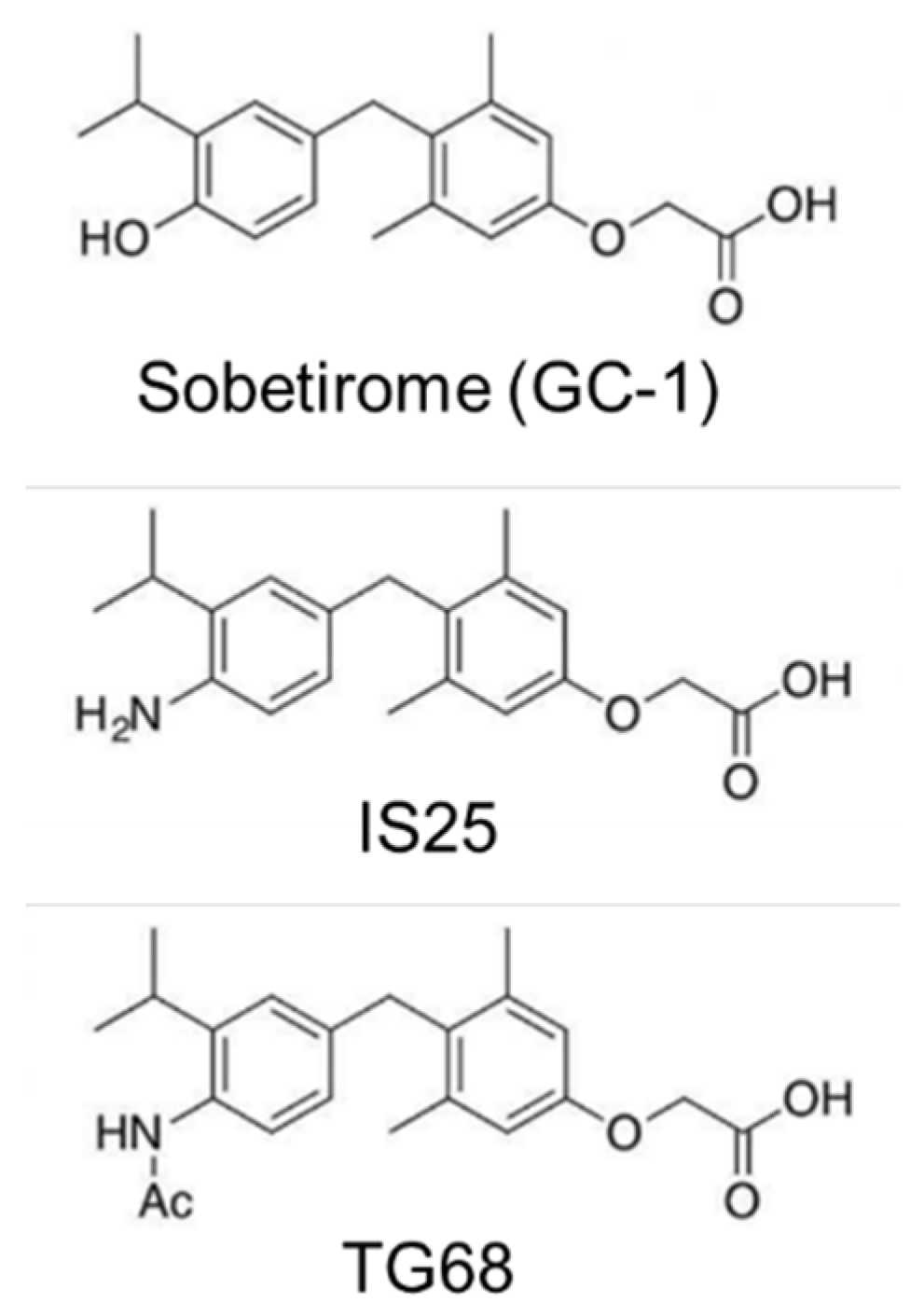{kind=link}
{kind=link}
{kind=link}
{kind=link}
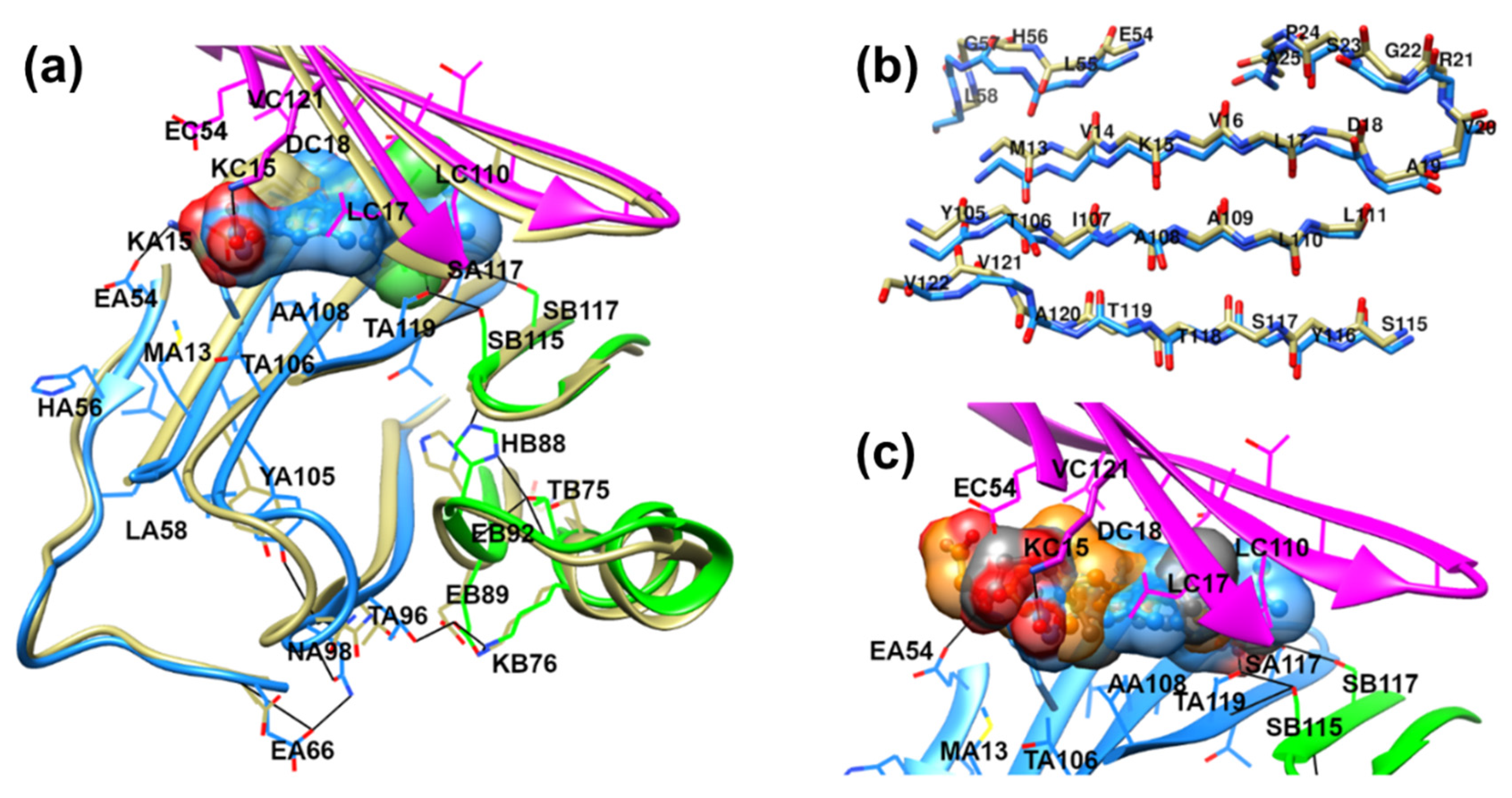{kind=link}
Abstract
:1. Introduction
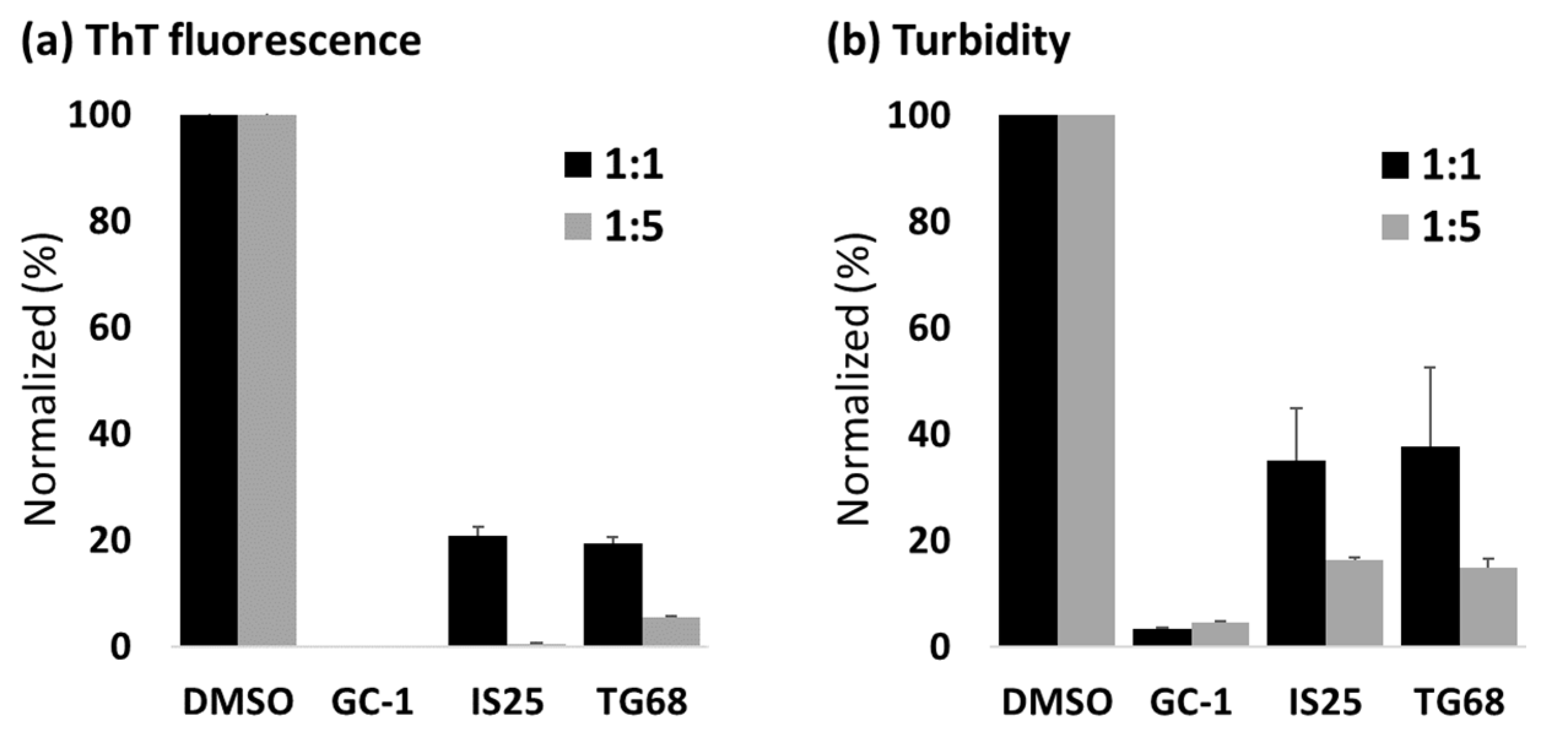
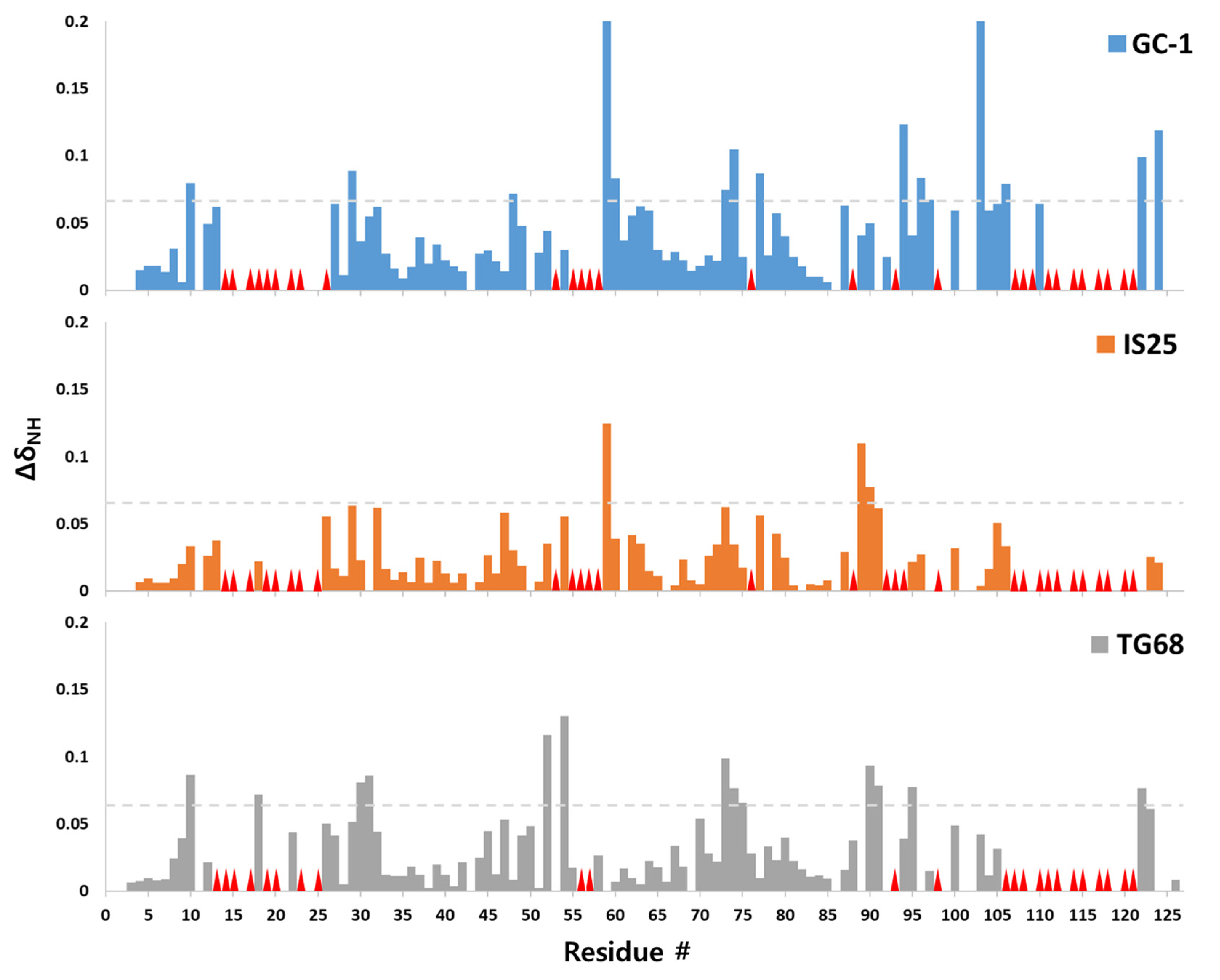
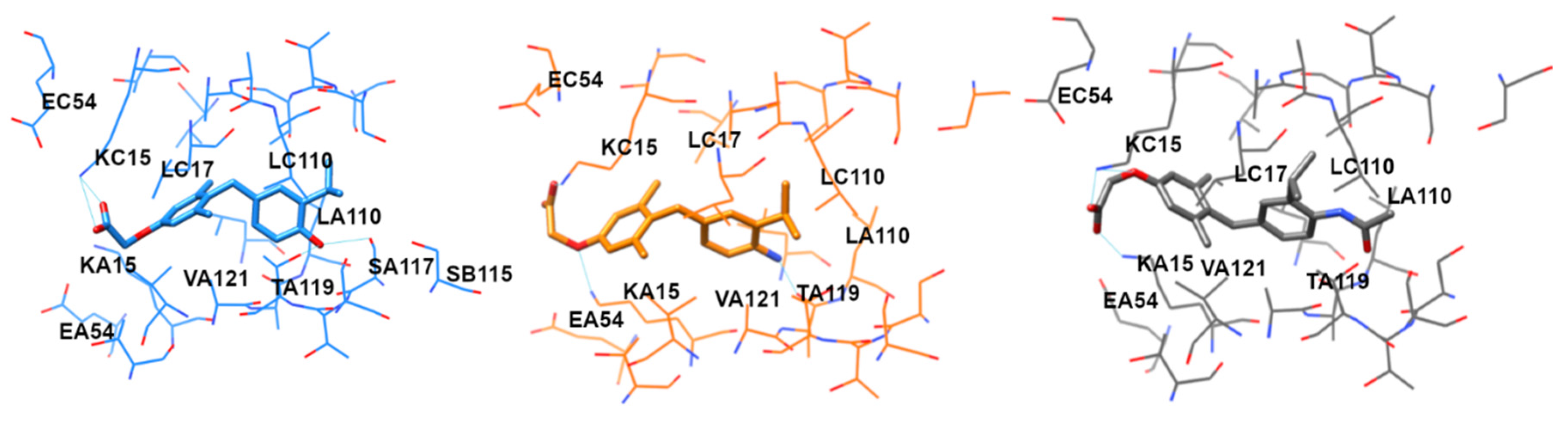
2. Results
3. Discussion
4. Materials and Methods
4.1. Sample Preparation
4.2. TTR Aggregation Assay
4.3. NMR Spectroscopy
4.4. Docking Simulation
4.5. Molecular Dynamics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| [U-13N, U-15N] | Uniformly 13C- and 15N-labeled |
| BBB | Blood brain barrier |
| BMRB | Biological Magnetic Resonance Data Bank |
| DMSO | Dimethyl sulfoxide |
| EDTA | Ethylenediaminetetraacetic acid |
| GC-1 HSQC MD | Sobiterome Heteronuclear single quantum coherence Molecular dynamics |
| MES | 2-(N-morpholino)-ethanesulfonic acid |
| NMR | Nuclear magnetic resonance |
| PDB | Protein data bank |
| RMSD TH | Root-mean-square deviation Thyroid hormone |
| ThT | Thioflavin T |
| TROSY | Transverse relaxation-optimized spectroscopy |
| TRβ | Thyroid hormone receptor β |
| TTR | Transthyretin |
| WT | Wild-type |
References
- Blake, C.C.F.; Geisow, M.J.; Oatley, S.J.; Rérat, B.; Rérat, C. Structure of prealbumin: Secondary, tertiary and quaternary interactions determined by Fourier refinement at 1.8 Å. J. Mol. Biol. 1978, 121, 339–356. [Google Scholar] [CrossRef]
- Wojtczak, A.; Cody, V.; Luft, J.R.; Pangborn, W. Structures of human transthyretin complexed with thyroxine at 2.0 Å resolution and 3′,5′-dinitro-N-acetyl-L-thyronine at 2.2 Å resolution. Acta Crystallogr. Sect. D Biol. Crystallogr. 1996, 52, 758–765. [Google Scholar] [CrossRef]
- Johnson, S.M.; Connelly, S.; Fearns, C.; Powers, E.T.; Kelly, J.W. The transthyretin amyloidoses: From delineating the molecular mechanism of aggregation linked to pathology to a regulatory-agency-approved drug. J. Mol. Biol. 2012, 421, 185–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, Z.; Colón, W.; Kelly, J.W. The acid-mediated denaturation pathway of transthyretin yields a conformational intermediate that can self-assemble into amyloid. Biochemistry 1996, 35, 6470–6482. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Koike, H.; Slama, M.; Coelho, T. Hereditary transthyretin amyloidosis: A model of medical progress for a fatal disease. Nat. Rev. Neurol. 2019, 15, 387–404. [Google Scholar] [CrossRef] [PubMed]
- Connors, L.H.; Lim, A.; Prokaeva, T.; Roskens, V.A.; Costello, C.E. Tabulation of human transthyretin (TTR) variants. Amyloid 2003, 10, 160–184. [Google Scholar] [CrossRef]
- Buxbaum, J.N.; Ruberg, F.L. Transthyretin V122I (pV142I) cardiac amyloidosis: An age-dependent autosomal dominant cardiomyopathy too common to be overlooked as a cause of significant heart disease in elderly African Americans. Genet. Med. 2017, 19, 733–742. [Google Scholar] [CrossRef] [Green Version]
- Akinboboye, O.; Shah, K.; Warner, A.L.; Damy, T.; Taylor, H.A.; Gollob, J.; Powell, C.; Karsten, V.; Vest, J.; Maurer, M.S. DISCOVERY: Prevalence of transthyretin (TTR) mutations in a US-centric patient population suspected of having cardiac amyloidosis. Amyloid 2020, 27, 223–230. [Google Scholar] [CrossRef]
- Carvalho, A.; Rocha, A.; Lobato, L. Liver transplantation in transthyretin amyloidosis: Issues and challenges. Liver Transplant. 2015, 21, 282–292. [Google Scholar] [CrossRef] [Green Version]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.-C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef]
- Miroy, G.J.; Lai, Z.; Lashuel, H.A.; Peterson, S.A.; Strang, C.; Kelly, J.W. Inhibiting transthyretin amyloid fibril formation via protein stabilization. Proc. Natl. Acad. Sci. USA 1996, 93, 15051–15056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razavi, H.; Palaninathan, S.K.; Powers, E.T.; Wiseman, R.L.; Purkey, H.E.; Mohamedmohaideen, N.N.; Deechongkit, S.; Chiang, K.P.; Dendle, M.T.A.; Sacchettini, J.C.; et al. Benzoxazoles as transthyretin amyloid fibril inhibitors: Synthesis, evaluation, and mechanism of action. Angew. Chemie Int. Ed. 2003, 42, 2758–2761. [Google Scholar] [CrossRef]
- Bulawa, C.E.; Connelly, S.; DeVit, M.; Wang, L.; Weigel, C.; Fleming, J.A.; Packman, J.; Powers, E.T.; Wiseman, R.L.; Foss, T.R.; et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc. Natl. Acad. Sci. USA 2012, 109, 9629–9634. [Google Scholar] [CrossRef] [Green Version]
- Casal, I.; Monteiro, S.; Beirão, J.M. Tafamidis in hereditary ATTR amyloidosis—our experience on monitoring the ocular manifestations. Amyloid 2016, 23, 262–263. [Google Scholar] [CrossRef]
- Salvi, F.; Volpe, R.; Pastorelli, F.; Bianchi, A.; Vella, A.; Rapezzi, C.; Mascalchi, M. Failure of tafamidis to halt progression of Ala36Pro TTR oculomeningovascular amyloidosis. J. Stroke Cerebrovasc. Dis. 2018, 27, e212–e214. [Google Scholar] [CrossRef]
- Sant’Anna, R.; Gallego, P.; Robinson, L.Z.; Pereira-Henriques, A.; Ferreira, N.; Pinheiro, F.; Esperante, S.; Pallares, I.; Huertas, O.; Almeida, M.R.; et al. Repositioning tolcapone as a potent inhibitor of transthyretin amyloidogenesis and associated cellular toxicity. Nat. Commun. 2016, 7, 10787. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, F.; Varejão, N.; Esperante, S.; Santos, J.; Velázquez-Campoy, A.; Reverter, D.; Pallarès, I.; Ventura, S. Tolcapone, a potent aggregation inhibitor for the treatment of familial leptomeningeal amyloidosis. FEBS J. 2021, 288, 310–324. [Google Scholar] [CrossRef] [PubMed]
- Corazza, A.; Verona, G.; Waudby, C.A.; Mangione, P.P.; Bingham, R.P.; Uings, I.; Canetti, D.; Nocerino, P.; Taylor, G.W.; Pepys, M.B.; et al. Binding of Monovalent and Bivalent Ligands by Transthyretin Causes Different Short- And Long-Distance Conformational Changes. J. Med. Chem. 2019, 62, 8274–8283. [Google Scholar] [CrossRef] [Green Version]
- Mangione, P.P.; Porcari, R.; Gillmore, J.D.; Pucci, P.; Monti, M.; Porcari, M.; Giorgetti, S.; Marchese, L.; Raimondi, S.; Serpell, L.C.; et al. Proteolytic cleavage of Ser52Pro variant transthyretin triggers its amyloid fibrillogenesis. Proc. Natl. Acad. Sci. USA 2014, 111, 1539–1544. [Google Scholar] [CrossRef] [Green Version]
- Marcoux, J.; Mangione, P.P.; Porcari, R.; Degiacomi, M.T.; Verona, G.; Taylor, G.W.; Giorgetti, S.; Raimondi, S.; Sanglier-Cianférani, S.; Benesch, J.L.; et al. A novel mechano-enzymatic cleavage mechanism underlies transthyretin amyloidogenesis. EMBO Mol. Med. 2015, 7, 1337–1349. [Google Scholar] [CrossRef]
- Schwarzman, A.L.; Gregori, L.; Vitek, M.P.; Lyubski, S.; Strittmatter, W.J.; Enghilde, J.J.; Bhasin, R.; Silverman, J.; Weisgraber, K.H.; Coyle, P.K. Transthyretin sequesters amyloid beta protein and prevents amyloid formation. Proc. Natl. Acad. Sci. USA 1994, 91, 8368–8372. [Google Scholar] [CrossRef] [Green Version]
- Buxbaum, J.N.; Ye, Z.; Reixach, N.; Friske, L.; Levy, C.; Das, P.; Golde, T.; Masliah, E.; Roberts, A.R.; Bartfai, T. Transthyretin protects Alzheimer’s mice from the behavioral and biochemical effects of Aβ toxicity. Proc. Natl. Acad. Sci. USA 2008, 105, 2681–2686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Buxbaum, J.N. Transthyretin and the brain re-visited: Is neuronal synthesis of transthyretin protective in Alzheimer’s disease? Mol. Neurodegener. 2011, 6, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro, C.A.; Oliveira, S.M.; Guido, L.F.; Magalhães, A.; Valencia, G.; Arsequell, G.; Saraiva, M.J.; Cardoso, I. Transthyretin stabilization by iododiflunisal promotes amyloid-β peptide clearance, decreases its deposition, and ameliorates cognitive deficits in an Alzheimer’s disease mouse model. J. Alzheimer’s Dis. 2014, 39, 357–370. [Google Scholar] [CrossRef] [PubMed]
- Santos, L.M.; Rodrigues, D.; Alemi, M.; Silva, S.C.; Ribeiro, C.A.; Cardoso, I. Resveratrol administration increases transthyretin protein levels, ameliorating AD features: The importance of transthyretin tetrameric stability. Mol. Med. 2016, 22, 597–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alemi, M.; Silva, S.C.; Santana, I.; Cardoso, I. Transthyretin stability is critical in assisting beta amyloid clearance– Relevance of transthyretin stabilization in Alzheimer’s disease. CNS Neurosci. Ther. 2017, 23, 605–619. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Song, Y.; Sanders, C.R.; Buxbaum, J.N. Transthyretin suppresses amyloid-β secretion by interfering with processing of the amyloid-β protein precursor. J. Alzheimer’s Dis. 2016, 52, 1263–1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, A.; Chang, J.C.; Xu, P.; Gindinova, K.; Cho, Y.; Sun, W.; Wu, X.; Li, Y.M.; Greengard, P.; Kelly, J.W.; et al. Brain permeable tafamidis amide analogs for stabilizing TTR and reducing APP cleavage. ACS Med. Chem. Lett. 2020, 11, 1973–1979. [Google Scholar] [CrossRef]
- Brenta, G.; Danzi, S.; Klein, I. Potential therapeutic applications of thyroid hormone analogs. Nat. Clin. Pract. Endocrinol. Metab. 2007, 3, 632–640. [Google Scholar] [CrossRef]
- Baxter, J.D.; Webb, P. Thyroid hormone mimetics: Potential applications in atherosclerosis, obesity and type 2 diabetes. Nat. Rev. Drug Discov. 2009, 8, 308–320. [Google Scholar] [CrossRef]
- Gauthier, B.R.; Sola-García, A.; Cáliz-Molina, M.Á.; Lorenzo, P.I.; Cobo-Vuilleumier, N.; Capilla-González, V.; Martin-Montalvo, A. Thyroid hormones in diabetes, cancer, and aging. Aging Cell 2020, 19, 1–25. [Google Scholar] [CrossRef]
- Saponaro, F.; Sestito, S.; Runfola, M.; Rapposelli, S.; Chiellini, G. Selective thyroid hormone receptor-beta (TRβ) agonists: New perspectives for the treatment of metabolic and neurodegenerative disorders. Front. Med. 2020, 7, 331. [Google Scholar] [CrossRef] [PubMed]
- Zucchi, R. Thyroid Hormone Analogues: An Update. Thyroid 2020, 30, 1099–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiellini, G.; Apriletti, J.W.; Yoshihara, H.A.; Baxter, J.D.; Ribeiro, R.C.J.; Scanlan, T.S. A high-affinity subtype-selective agonist ligand for the thyroid hormone receptor. Chem. Biol. 1998, 5, 299–306. [Google Scholar] [CrossRef] [Green Version]
- Lammel Lindemann, J.; Webb, P. Sobetirome: The past, present and questions about the future. Expert Opin. Ther. Targets 2016, 20, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Trost, S.U.; Swanson, E.; Gloss, B.; Wang-Iverson, D.B.; Zhang, H.; Volodarsky, T.; Grover, G.J.; Baxter, J.D.; Chiellini, G.; Scanlan, T.S.; et al. The thyroid hormone receptor-β-selective agonist GC-1 differentially affects plasma lipids and cardiac activity. Endocrinology 2000, 141, 3057–3064. [Google Scholar] [CrossRef]
- Takahashi, N.; Asano, Y.; Maeda, K.; Watanabe, N. In Vivo evaluation of 1-benzyl-4-aminoindole-based thyroid hormone receptor β agonists: Importance of liver selectivity in drug discovery. Biol. Pharm. Bull. 2014, 37, 1103–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellusci, L.; Laurino, A.; Sabatini, M.; Sestito, S.; Lenzi, P.; Raimondi, L.; Rapposelli, S.; Biagioni, F.; Fornai, F.; Salvetti, A.; et al. New insights into the potential roles of 3-iodothyronamine (T1AM) and newly developed thyronamine-like TAAR1 agonists in neuroprotection. Front. Pharmacol. 2017, 8, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Meinig, J.M.; Ferrara, S.J.; Banerji, T.; Banerji, T.; Sanford-Crane, H.S.; Bourdette, D.; Scanlan, T.S. Targeting fatty-acid amide hydrolase with prodrugs for CNS-selective therapy. ACS Chem. Neurosci. 2017, 8, 2468–2476. [Google Scholar] [CrossRef]
- Runfola, M.; Sestito, S.; Bellusci, L.; La Pietra, V.; D’Amore, V.M.; Kowalik, M.A.; Chiellini, G.; Gul, S.; Perra, A.; Columbano, A.; et al. Design, synthesis and biological evaluation of novel TRβ selective agonists sustained by ADME-toxicity analysis. Eur. J. Med. Chem. 2020, 188, 112006. [Google Scholar] [CrossRef]
- Runfola, M.; Sestito, S.; Gul, S.; Chiellini, G.; Rapposelli, S. Collecting data through high throughput in vitro early toxicity and off-target liability assays to rapidly identify limitations of novel thyromimetics. Data Br. 2020, 29, 105206. [Google Scholar] [CrossRef]
- Perra, A.; Kowalik, M.A.; Cabras, L.; Runfola, M.; Sestito, S.; Migliore, C.; Giordano, S.; Chiellini, G.; Rapposelli, S.; Columbano, A. Potential role of two novel agonists of thyroid hormone receptor-β on liver regeneration. Cell Prolif. 2020, 53, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Trivella, D.B.B.; Sairre, M.I.; Foguel, D.; Lima, L.M.T.R.; Polikarpov, I. The binding of synthetic triiodo l-thyronine analogs to human transthyretin: Molecular basis of cooperative and non-cooperative ligand recognition. J. Struct. Biol. 2011, 173, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Colon, W.; Kelly, J.W. Partial denaturation of transthyretin is sufficient for amyloid fibril formation in vitro. Biochemistry 1992, 31, 8654–8660. [Google Scholar] [CrossRef] [PubMed]
- Gade Malmos, K.; Blancas-Mejia, L.M.; Weber, B.; Buchner, J.; Ramirez-Alvarado, M.; Naiki, H.; Otzen, D. ThT 101: A primer on the use of thioflavin T to investigate amyloid formation. Amyloid 2017, 24, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Saelices, L.; Johnson, L.M.; Liang, W.Y.; Sawaya, M.R.; Cascio, D.; Ruchala, P.; Whitelegge, J.; Jiang, L.; Riek, R.; Eisenberg, D.S. Uncovering the mechanism of aggregation of human transthyretin. J. Biol. Chem. 2015, 290, 28932–28943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.T.; Yen, Y.J.; Ricardo, F.; Chang, Y.; Wu, P.H.; Huang, S.J.; Lin, K.P.; Yu, T.Y. Biophysical characterization and modulation of Transthyretin Ala97Ser. Ann. Clin. Transl. Neurol. 2019, 6, 1961–1970. [Google Scholar] [CrossRef] [Green Version]
- Trivella, D.B.B.; dos Reis, C.V.; Lima, L.M.T.R.; Foguel, D.; Polikarpov, I. Flavonoid interactions with human transthyretin: Combined structural and thermodynamic analysis. J. Struct. Biol. 2012, 180, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Gales, L.; Macedo-Ribeiro, S.; Arsequell, G.; Valencia, G.; Saraiva, M.J.; Damas, A.M. Human transthyretin in complex with iododiflunisal: Structural features associated with a potent amyloid inhibitor. Biochem. J. 2005, 388, 615–621. [Google Scholar] [CrossRef] [Green Version]
- Saldaño, T.E.; Zanotti, G.; Parisi, G.; Fernandez-Alberti, S. Evaluating the effect of mutations and ligand binding on transthyretin homotetramer dynamics. PLoS ONE 2017, 12, e0181019. [Google Scholar] [CrossRef] [Green Version]
- Childers, M.C.; Daggett, V. Edge strand dissociation and conformational changes in Transthyretin under amyloidogenic conditions. Biophys. J. 2020, 119, 1995–2009. [Google Scholar] [CrossRef]
- Ferreira, N.; Cardoso, I.; Domingues, M.R.; Vitorino, R.; Bastos, M.; Bai, G.; Saraiva, M.J.; Almeida, M.R. Binding of epigallocatechin-3-gallate to transthyretin modulates its amyloidogenicity. FEBS Lett. 2009, 583, 3569–3576. [Google Scholar] [CrossRef] [Green Version]
- Miyata, M.; Sato, T.; Kugimiya, M.; Sho, M.; Nakamura, T.; Ikemizu, S.; Chirifu, M.; Mizuguchi, M.; Nabeshima, Y.; Suwa, Y.; et al. The Crystal Structure of the Green Tea Polyphenol (−)-Epigallocatechin Gallate−Transthyretin Complex Reveals a Novel Binding Site Distinct from the Thyroxine Binding Site. Biochemistry 2010, 49, 6104–6114. [Google Scholar] [CrossRef]
- Ferreira, N.; Saraiva, M.J.; Almeida, M.R. Natural polyphenols inhibit different steps of the process of transthyretin (TTR) amyloid fibril formation. FEBS Lett. 2011, 585, 2424–2430. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, N.; Pereira-Henriques, A.; Almeida, M.R. Transthyretin chemical chaperoning by flavonoids: Structure–activity insights towards the design of potent amyloidosis inhibitors. Biochem. Biophys. Rep. 2015, 3, 123–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, N.; Saraiva, M.J.; Almeida, M.R. Epigallocatechin-3-Gallate as a Potential Therapeutic Drug for TTR-Related Amyloidosis: “In Vivo” Evidence from FAP Mice Models. PLoS ONE 2012, 7, e29933. [Google Scholar] [CrossRef]
- Kristen, A.V.; Lehrke, S.; Buss, S.; Mereles, D.; Steen, H.; Ehlermann, P.; Hardt, S.; Giannitsis, E.; Schreiner, R.; Haberkorn, U.; et al. Green tea halts progression of cardiac transthyretin amyloidosis: An observational report. Clin. Res. Cardiol. 2012, 101, 805–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- aus dem Siepen, F.; Bauer, R.; Aurich, M.; Buss, S.; Steen, H.; Altland, K.; Kristen, A.; Katus, H.A. Green tea extract as a treatment for patients with wild-type transthyretin amyloidosis: An observational study. Drug Des. Devel. Ther. 2015, 9, 6319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamski-Werner, S.L.; Palaninathan, S.K.; Sacchettini, J.C.; Kelly, J.W. Diflunisal analogues stabilize the native state of Transthyretin. Potent inhibition of Amyloidogenesis. J. Med. Chem. 2004, 47, 355–374. [Google Scholar] [CrossRef] [PubMed]
- Das, J.K.; Mall, S.S.; Bej, A.; Mukherjee, S. Conformational flexibility tunes the propensity of transthyretin to form fibrils through non-native intermediate states. Angew. Chemie Int. Ed. 2014, 53, 12781–12784. [Google Scholar] [CrossRef]
- Zanotti, G.; Vallese, F.; Ferrari, A.; Menozzi, I.; Saldaño, T.E.; Berto, P.; Fernandez-Alberti, S.; Berni, R. Structural and dynamics evidence for scaffold asymmetric flexibility of the human transthyretin tetramer. PLoS ONE 2017, 12, e0187716. [Google Scholar] [CrossRef] [Green Version]
- Leach, B.I.; Zhang, X.; Kelly, J.W.; Dyson, H.J.; Wright, P.E. NMR Measurements Reveal the Structural Basis of Transthyretin Destabilization by Pathogenic Mutations. Biochemistry 2018, 57, 4421–4430. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Oroz, J.; Zweckstetter, M. Structure of monomeric Transthyretin carrying the clinically important T119M mutation. Angew. Chemie Int. Ed. 2016, 55, 16168–16171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, L.Z.; Reixach, N. Quantification of quaternary structure stability in aggregation-prone proteins under physiological conditions: The transthyretin case. Biochemistry 2014, 53, 6496–6510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Kelly, J.W.; Wemmer, D.E. Native state hydrogen exchange study of suppressor and pathogenic variants of transthyretin. J. Mol. Biol. 2002, 320, 821–832. [Google Scholar] [CrossRef]
- Lee, W.; Tonelli, M.; Markley, J.L. NMRFAM-SPARKY: Enhanced software for biomolecular NMR spectroscopy. Bioinformatics 2015, 31, 1325–1327. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.; Goddard, T.; Huang, C.; Couch, G.; Greenblatt, D.; Meng, E.; Ferrin, T. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger Inc Macromodel; Schrödinger, LLC: New York, NY, USA, 2009.
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins 2003, 52, 609–623. [Google Scholar] [CrossRef]
- Ortore, G.; Martinelli, A. Identification of Transthyretin fibril formation inhibitors using structure-based virtual screening. ChemMedChem 2017, 12, 1327–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Case, D.A.; Berryman, J.T.; Betz, R.M.; Cerutti, D.S.; Cheatham, T.E.I.; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; et al. AMBER, Version 14; University of California: San Francisco, CA, USA, 2015. [Google Scholar]
- Roe, D.R.; Cheatham, T.E., 3rd. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, B.; Ko, Y.H.; Runfola, M.; Rapposelli, S.; Ortore, G.; Chiellini, G.; Kim, J.H. Diphenyl-Methane Based Thyromimetic Inhibitors for Transthyretin Amyloidosis. Int. J. Mol. Sci. 2021, 22, 3488. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073488
Kim B, Ko YH, Runfola M, Rapposelli S, Ortore G, Chiellini G, Kim JH. Diphenyl-Methane Based Thyromimetic Inhibitors for Transthyretin Amyloidosis. International Journal of Molecular Sciences. 2021; 22(7):3488. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073488
Chicago/Turabian StyleKim, Bokyung, Young Ho Ko, Massimiliano Runfola, Simona Rapposelli, Gabriella Ortore, Grazia Chiellini, and Jin Hae Kim. 2021. "Diphenyl-Methane Based Thyromimetic Inhibitors for Transthyretin Amyloidosis" International Journal of Molecular Sciences 22, no. 7: 3488. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073488