
The Diagnostic Potential of Amyloidogenic Proteins


Abstract
:1. The Need for Novel Diagnostic Approaches for Neurodegeneration
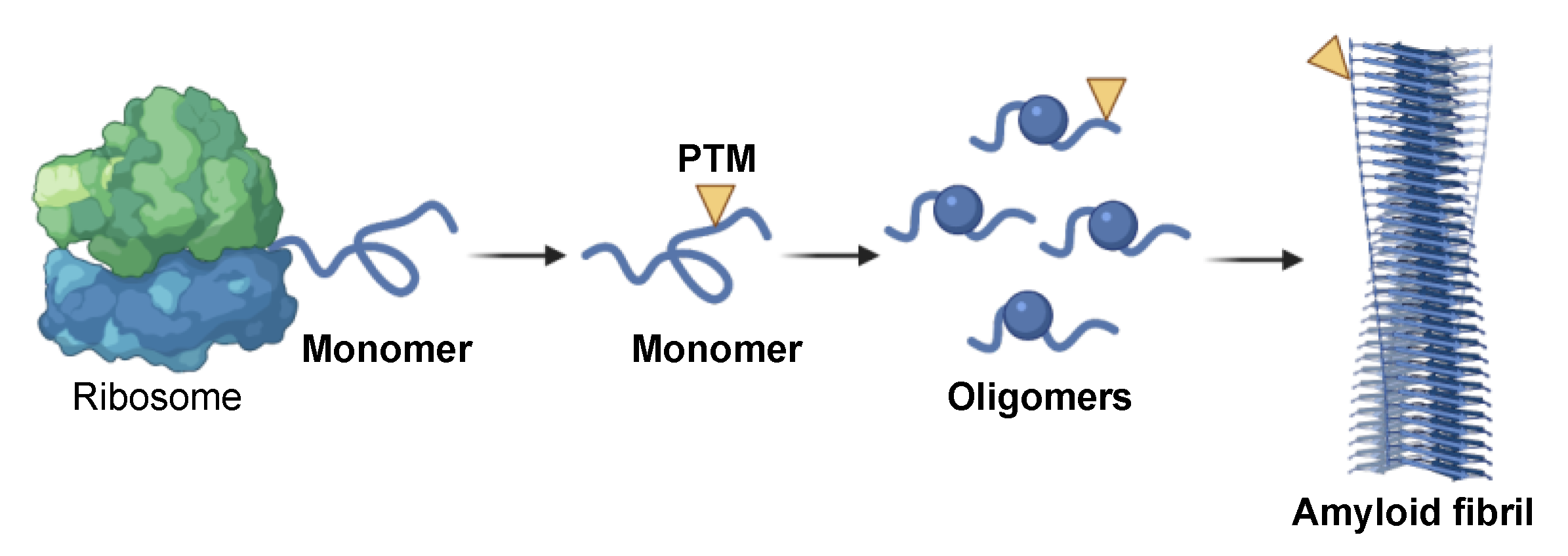
2. Amyloid Aggregation as Potential Source of Biomarkers
3. Amyloidogenic Proteins Involved in Neurodegeneration
3.1. Aβ and Tau in AD
3.2. α-Syn in PD
3.3. TDP-43 and FUS in ALS and FTD
4. Diagnostic Potential of Genetic, Structural and Chemical Features of Amyloidogenic Proteins
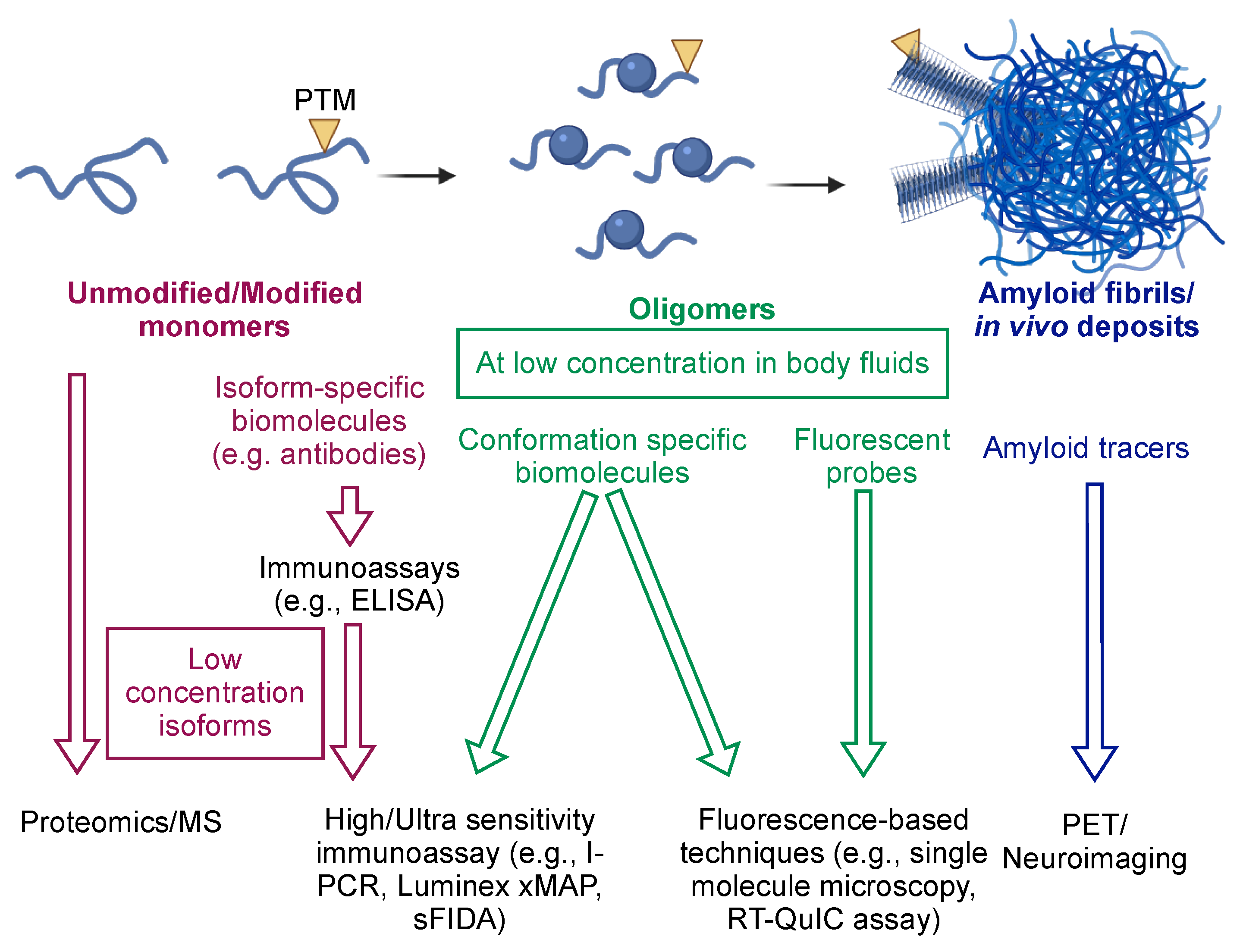
4.1. Levels of Amyloidogenic Proteins in Neurodegeneration

{kind=link}
{kind=link}
{kind=link}
| Protein | PTMs | Major Modification Sites | Key Remarks | Refs. |
|---|---|---|---|---|
| Aβ | Cleavage | 1-37, 1-38, 1-39, 1-40, 1-42, 1-43 | Aβ 40 is the most abundant; Aβ 42 aggregates more rapidly. | [30,78,79] |
| Phosphorylation | Ser8, Ser26 | Increased abundance and stability of toxic aggregates. | [30,80,81] | |
| Acetylation | Lys16, Lys28 | Altered aggregation behavior. | [38,39] | |
| Oxidation | Met35 | Regulation of oxidative stress. Slower aggregation. | [30,82] | |
| Nitration | Tyr10 | Enhanced aggregation and plaque formation. | [30,37] | |
| Isomerization | Asp1, Asp7 | Higher aggregation propensity and resistance to degradation. | [30] | |
| Racemization | Asp1, Asp23, Ser26 | Higher aggregation propensity. | [30] | |
| O-glycosylation | Tyr10 | Found in short Aβ fragments in AD patients’ CSF. Aβ1-15 and Aβ1-17 are the most abundant fragments. | [30,83] | |
| Pyroglutamate formation | Glu3, Glu11 | Increased oligomerization. It correlates with the extent of Aβ deposition. | [84,85] | |
| Tau | Phosphorylation | Thr181, Thr199, Thr217, Thr231 | Increased aggregation. It is a key event in the formation of NFT. | [27,86] |
| Acetylation | Lys174, Lys274, Lys280 | Regulation of tau function; promotion of p-tau aggregation. | [87] | |
| Oxidation | Cys322 | Enhanced PHF assembly | [88] | |
| Nitration | Tyr29 | Accumulation of oligomeric species. | [89] | |
| N-glycosylation | Under investigation. Putative sites: Asn167, Ans359, Asn410 | Higher levels in AD patients’ brains. Promotion of tau hyperphosphorylation and PHF accumulation. | [83,90] | |
| O-glycosylation | Under investigation | Lower levels in AD patients’ brains. It prevents tau hyperphosphorylation and PHF formation and PHF accumulation. | [91] | |
| Ubiquitination | Lys48, Lys63 | It has been proposed to contribute to the formation of the tangles. | [92,93,94] | |
| α-Syn | Fragmentation | C-terminally truncated α-syn of around 10–15 kDa | Accelerated aggregation. | [28] |
| Phosphorylation | Ser129 | Enhanced aggregation and toxicity. | [95,96] | |
| Acetylation | N-terminal | Increased helical propensity, altered fibril polymorphism and decreased aggregation. | [97] | |
| Oxidation | Met1, Met5, Met116, Met127 | Inhibition of fibrillation by stabilization of soluble oligomeric species. | [98] | |
| Nitration | Tyr39 | Enhanced aggregation. | [99] | |
| O-glycosylation | Under investigation. | Inhibition of aggregation. | [100] | |
| Ubiquitination | Lys6, Lys12, Lys21, Lys43 | Inhibition of aggregation. | [28] | |
| Lys10, Lys23 | Faster fibril formation. | [28] | ||
| TDP-43 | Fragmentation | C-terminally truncated TDP-43 at 25 kDa and 35 kDa fragments | Enhanced aggregation, altered RNA processing, and cellular redistribution. | [101,102] |
| Phosphorylation | Ser409, Ser410 | Enhanced aggregation and cellular mislocalization. | [29,75] | |
| Acetylation | Lys145, Lys192 | Impaired RNA binding and mitochondrial function; enhanced aggregation of phosphorylated TDP-43. | [29] | |
| Oxidation | Cys3, Cys50, Cys173, Cys175 | Enhanced oligomerization and self-association. | [29] | |
| Ubiquitination | Lys 48, Lys 63 | Enhanced cytoplasmic accumulation to higher molecular weight aggregates. | [29] |
4.2. PTMs Associated with Neurodegeneration
4.2.1. Fragmentation and Cleavage
4.2.2. Phosphorylation
4.2.3. Acetylation
4.2.4. Oxidation and Nitration
4.2.5. Glycosylation
4.2.6. Ubiquitination
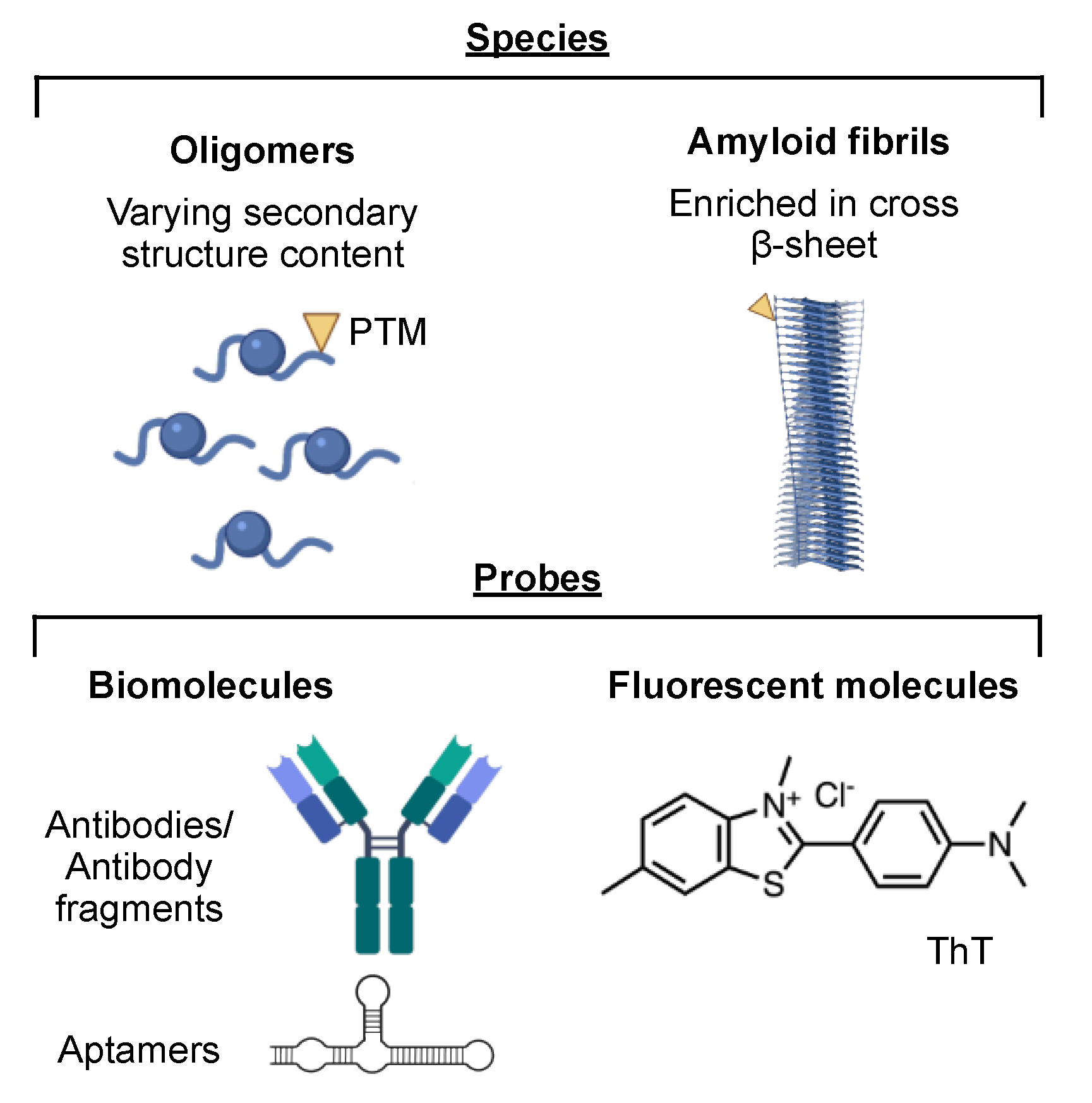
4.3. Amyloid Aggregates and Amyloid-Sensitive Probes
4.4. PET Neuroimaging of Amyloids in the Brain
4.5. Combined Detection
| Type of Diagnostic Markers | Key Remarks | Refs. | ||
|---|---|---|---|---|
| Genetic | ||||
| Protein | Gene | Mutations | Pathological implications | |
| APP | APP | E693Q, A692G | Early onset familial AD | [35,36] |
| α-Syn | SNCA | A53T, G51D, H50Q, E46K, A30P, locus amplification | Early onset familial PD | [46,47,48,156] |
| TDP-43 | TARDBP | G294A, Q331K, M337V, K181E | Sporadic and familial ALS | [58,59] |
| FUS | FUS | R521C, R521H | Early onset ALS | [63] |
| Neuroimaging | ||||
| Protein species | Analytical technique | |||
| Aβ and tau aggregates | PET with 18F-FDDNP. Lacks protein specific and is unable to distinguish different forms of neurodegeneration. | [147] | ||
| Tau aggregates | PET with 18F-AV1451 or 18F-GTP1. High affinity for tau aggregates. Not able to distinguish different forms of tauopathies. | [147] | ||
| Aβ aggregates | PET with 11C-PiB and Florbetapir (18F). High affinity for Aβ plaques. Not able to distinguish different forms of neurodegeneration. | [147,148] | ||
| Biomarkers | ||||
| Protein variants | Analytical techniques | |||
| CSF Aβ42 | Immunoassays. In the majority of cases, it is present at lower concentration in patients affected by AD and other forms of dementia. It is not accurate in distinguishing different forms of neurodegeneration. | [78] | ||
| CSF Aβ42/Aβ40 ratio | Immunoassays. Slightly better diagnostic performance for AD than Aβ42 alone. Usually, it is used in combination with other potential biomarkers (e.g., tau). | [78,103] | ||
| CSF Aβ42/Aβ38 ratio | Immunoassays. Better diagnostic performance for AD than Aβ42 alone. Also used in combination with other potential biomarkers (e.g., p-tau). | [78,103,107] | ||
| CSF total tau | Immunoassays. Generally present at higher concentrations in the CSF of AD patients. It is not disease-specific when used alone and usually measured in association with other amyloidogenic proteins (e.g., Aβ42). | [67] | ||
| CSF p-tau217, p-tau181 | Immunoassays and MS. Their concentrations correlate with AD. P-tau181 is generally used in combination with other potential biomarkers, including total tau and Aβ42, or with the Aβ42/Aβ38 ratio. | [107] | ||
| Plasma p-tau217 | Immunoassays and MS. Able to discriminate AD from other forms of neurodegeneration. | [86] | ||
| CSF and plasma p-α-syn129 | Immunoassays. Good diagnostic performance for PD when used in combination with other potential biomarkers, including tau and α-syn oligomers. | [109,110,154] | ||
| CSF Aβ oligomers and α-syn oligomers | Immunoassays and single-molecule approaches. Good potential diagnostic performance for AD or PD, respectively. However, their use as biomarkers is not widely implemented yet. Some works have used them in combination with other potential biomarkers. | [109,132,154] | ||
5. Recent Advances in Detection Technology
6. Conclusions and Potential Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Graham, M.E. The securitisation of dementia: Socialities of securitisation on secure dementia care units. Ageing Soc. 2021, 41, 439–455. [Google Scholar] [CrossRef]
- Agrawal, M.; Biswas, A. Molecular diagnostics of neurodegenerative disorders. Front. Mol. Biosci. 2015, 2, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, R.; Zubair, H.; Pursell, S.; Shahab, M. Neurodegenerative diseases: Regenerative mechanisms and novel therapeutic approaches. Brain Sci. 2018, 8, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reul, S.; Lohmann, H.; Wiendl, H.; Duning, T.; Johnen, A. Can cognitive assessment really discriminate early stages of Alzheimer’s and behavioural variant frontotemporal dementia at initial clinical presentation? Alzheimers Res. Ther. 2017, 9, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Steenoven, I.; Koel-Simmelink, M.J.; Vergouw, L.J.; Tijms, B.M.; Piersma, S.R.; Pham, T.V.; Bridel, C.; Ferri, G.-L.; Cocco, C.; Noli, B.; et al. Identification of novel cerebrospinal fluid biomarker candidates for dementia with Lewy bodies: A proteomic approach. Mol. Neurodegener. 2020, 15, 36. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Ge, P.; Murray, K.A.; Sheth, P.; Zhang, M.; Nair, G.; Sawaya, M.R.; Shin, W.S.; Boyer, D.R.; Ye, S.; et al. Cryo-EM of full-length α-synuclein reveals fibril polymorphs with a common structural kernel. Nat. Commun. 2018, 9, 3609. [Google Scholar] [CrossRef]
- Dobson, C.M. Protein folding and misfolding. Nature 2003, 426, 884–890. [Google Scholar] [CrossRef]
- Knowles, T.P.; Vendruscolo, M.; Dobson, C.M. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 2014, 15, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Astoricchio, E.; Alfano, C.; Rajendran, L.; Temussi, P.A.; Pastore, A. The Wide World of Coacervates: From the Sea to Neurodegeneration. Trends Biochem. Sci. 2020, 45, 706–717. [Google Scholar] [CrossRef]
- Buratti, E.; Baralle, F.E. TDP-43: Gumming up neurons through protein–protein and protein–RNA interactions. Trends Biochem. Sci. 2012, 37, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Vogler, T.O.; Wheeler, J.R.; Nguyen, E.D.; Hughes, M.P.; Britson, K.A.; Lester, E.; Rao, B.; Dalla Betta, N.; Whitney, O.N.; Ewachiw, T.E.; et al. TDP-43 and RNA form amyloid-like myo-granules in regenerating muscle. Nature 2018, 563, 508–513. [Google Scholar] [CrossRef]
- Bhardwaj, A.; Myers, M.P.; Buratti, E.; Baralle, F.E. Characterizing TDP-43 interaction with its RNA targets. Nucleic Acids Res. 2013, 41, 5062–5074. [Google Scholar] [CrossRef] [Green Version]
- Bucciantini, M.; Rigacci, S.; Stefani, M. Amyloid aggregation: Role of biological membranes and the aggregate–membrane system. J. Phys. Chem. Lett. 2014, 5, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.W.; Drakulic, S.; Deas, E.; Ouberai, M.; Aprile, F.A.; Arranz, R.; Ness, S.; Roodveldt, C.; Guilliams, T.; De-Genst, E.J.; et al. Structural characterization of toxic oligomers that are kinetically trapped during α-synuclein fibril formation. Proc. Natl. Acad. Sci. USA 2015, 112, E1994–E2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, D.; Mehra, S.; Sahay, S.; Singh, P.K.; Maji, S.K. α-synuclein aggregation and its modulation. Int. J. Biol. Macromol. 2017, 100, 37–54. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.-f.; Xu, T.-h.; Yan, Y.; Zhou, Y.-r.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef]
- De, S.; Klenerman, D. Imaging individual protein aggregates to follow aggregation and determine the role of aggregates in neurodegenerative disease. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 870–878. [Google Scholar] [CrossRef]
- Cremades, N.; Cohen, S.I.; Deas, E.; Abramov, A.Y.; Chen, A.Y.; Orte, A.; Sandal, M.; Clarke, R.W.; Dunne, P.; Aprile, F.A.; et al. Direct observation of the interconversion of normal and toxic forms of α-synuclein. Cell 2012, 149, 1048–1059. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, D.; Singh, P.K.; Sahay, S.; Jha, N.N.; Jacob, R.S.; Sen, S.; Kumar, A.; Riek, R.; Maji, S.K. Structure based aggregation studies reveal the presence of helix-rich intermediate during α-Synuclein aggregation. Sci. Rep. 2015, 5, 9228. [Google Scholar] [CrossRef] [Green Version]
- Shea, D.; Hsu, C.-C.; Bi, T.M.; Paranjapye, N.; Childers, M.C.; Cochran, J.; Tomberlin, C.P.; Wang, L.; Paris, D.; Zonderman, J.; et al. α-Sheet secondary structure in amyloid β-peptide drives aggregation and toxicity in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2019, 116, 8895–8900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fusco, G.; Chen, S.W.; Williamson, P.T.; Cascella, R.; Perni, M.; Jarvis, J.A.; Cecchi, C.; Vendruscolo, M.; Chiti, F.; Cremades, N.; et al. Structural basis of membrane disruption and cellular toxicity by α-synuclein oligomers. Science 2017, 358, 1440–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De, S.; Wirthensohn, D.C.; Flagmeier, P.; Hughes, C.; Aprile, F.A.; Ruggeri, F.S.; Whiten, D.R.; Emin, D.; Xia, Z.; Varela, J.A.; et al. Different soluble aggregates of Aβ42 can give rise to cellular toxicity through different mechanisms. Nat. Commun. 2019, 10, 1541. [Google Scholar] [CrossRef] [Green Version]
- Bucciantini, M.; Calloni, G.; Chiti, F.; Formigli, L.; Nosi, D.; Dobson, C.M.; Stefani, M. Prefibrillar amyloid protein aggregates share common features of cytotoxicity. J. Biol. Chem. 2004, 279, 31374–31382. [Google Scholar] [CrossRef] [Green Version]
- Angelova, P.R.; Abramov, A.Y. Alpha-synuclein and beta-amyloid–different targets, same players: Calcium, free radicals and mitochondria in the mechanism of neurodegeneration. Biochem. Biophys. Res. Commun. 2017, 483, 1110–1115. [Google Scholar] [CrossRef]
- Koffie, R.M.; Meyer-Luehmann, M.; Hashimoto, T.; Adams, K.W.; Mielke, M.L.; Garcia-Alloza, M.; Micheva, K.D.; Smith, S.J.; Kim, M.L.; Lee, V.M.; et al. Oligomeric amyloid β associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc. Natl. Acad. Sci. USA 2009, 106, 4012–4017. [Google Scholar] [CrossRef] [Green Version]
- Winner, B.; Jappelli, R.; Maji, S.K.; Desplats, P.A.; Boyer, L.; Aigner, S.; Hetzer, C.; Loher, T.; Vilar, M.; Campioni, S.; et al. In vivo demonstration that α-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. USA 2011, 108, 4194–4199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, C.-X.; Liu, F.; Grundke-Iqbal, I.; Iqbal, K. Post-translational modifications of tau protein in Alzheimer’s disease. J. Neural Transm. 2005, 112, 813–838. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, X.; Li, J.-D. The Roles of Post-translational Modifications on α-Synuclein in the Pathogenesis of Parkinson’s Diseases. Front. Neurosci. 2019, 13, 381. [Google Scholar] [CrossRef] [Green Version]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular mechanisms of TDP-43 misfolding and pathology in amyotrophic lateral sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef] [PubMed]
- Kummer, M.P.; Heneka, M.T. Truncated and modified amyloid-beta species. Alzheimers Res. Ther. 2014, 6, 28. [Google Scholar] [CrossRef] [Green Version]
- Buratti, E. TDP-43 post-translational modifications in health and disease. Expert Opin. Ther. Targets 2018, 22, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Mayeux, R. Biomarkers: Potential uses and limitations. NeuroRx 2004, 1, 182–188. [Google Scholar] [CrossRef]
- Blennow, K. CSF biomarkers for Alzheimer’s disease: Use in early diagnosis and evaluation of drug treatment. Expert Rev. Mol. Diagn. 2005, 5, 661–672. [Google Scholar] [CrossRef]
- Hatami, A.; Monjazeb, S.; Milton, S.; Glabe, C.G. Familial Alzheimer’s disease mutations within the amyloid precursor protein alter the aggregation and conformation of the amyloid-β peptide. J. Biol. Chem. 2017, 292, 3172–3185. [Google Scholar] [CrossRef] [Green Version]
- Hendriks, L.; van Duijn, C.M.; Cras, P.; Cruts, M.; Van Hul, W.; van Harskamp, F.; Warren, A.; McInnis, M.G.; Antonarakis, S.E.; Martin, J.-J.; et al. Presenile dementia and cerebral haemorrhage linked to a mutation at codon 692 of the β–amyloid precursor protein gene. Nat. Genet. 1992, 1, 218–221. [Google Scholar] [CrossRef]
- Nilsberth, C.; Westlind-Danielsson, A.; Eckman, C.B.; Condron, M.M.; Axelman, K.; Forsell, C.; Stenh, C.; Luthman, J.; Teplow, D.B.; Younkin, S.G.; et al. The ‘Arctic’APP mutation (E693G) causes Alzheimer’s disease by enhanced Aβ protofibril formation. Nat. Neurosci. 2001, 4, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Kummer, M.P.; Hermes, M.; Delekarte, A.; Hammerschmidt, T.; Kumar, S.; Terwel, D.; Walter, J.; Pape, H.-C.; König, S.; Roeber, S.; et al. Nitration of tyrosine 10 critically enhances amyloid β aggregation and plaque formation. Neuron 2011, 71, 833–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilkington IV, A.W.; Schupp, J.; Nyman, M.; Valentine, S.J.; Smith, D.M.; Legleiter, J. Acetylation of Aβ40 Alters Aggregation in the Presence and Absence of Lipid Membranes. ACS Chem. Neurosci. 2019, 11, 146–161. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, R.; Yang, M.; Saikia, N.; Dutta, C.; Alharbi, W.F.; Shan, Z.; Pandey, R.; Tiwari, A. Acetylation of Aβ42 at Lysine 16 Disrupts Amyloid Formation. ACS Chem. Neurosci. 2020, 11, 1178–1191. [Google Scholar] [CrossRef]
- Oakley, S.S.; Maina, M.B.; Marshall, K.E.; Al-Hilaly, Y.K.; Harrington, C.R.; Wischik, C.M.; Serpell, L.C. Tau Filament Self-Assembly and Structure: Tau as a Therapeutic Target. Front. Neurol. 2020, 11, 1207. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M. Tau filaments in neurodegenerative diseases. FEBS Lett. 2018, 592, 2383–2391. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, A.W.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.-Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Rachakonda, V.; Pan, T.H.; Le, W.D. Biomarkers of neurodegenerative disorders: How good are they? Cell Res. 2004, 14, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Auluck, P.K.; Caraveo, G.; Lindquist, S. α-Synuclein: Membrane interactions and toxicity in Parkinson’s disease. Annu. Rev. Cell Dev. Biol. 2010, 26, 211–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goedert, M. Alpha-synuclein and neurodegenerative diseases. Nat. Rev. Neurosci. 2001, 2, 492–501. [Google Scholar] [CrossRef]
- Lesage, S.; Anheim, M.; Letournel, F.; Bousset, L.; Honoré, A.; Rozas, N.; Pieri, L.; Madiona, K.; Dürr, A.; Melki, R.; et al. G51D α-synuclein mutation causes a novel Parkinsonian–pyramidal syndrome. Ann. Neurol. 2013, 73, 459–471. [Google Scholar] [CrossRef]
- Book, A.; Guella, I.; Candido, T.; Brice, A.; Hattori, N.; Jeon, B.; Farrer, M.J.; SNCA Multiplication Investigators of the GEoPD Consortium. A meta-analysis of α-synuclein multiplication in familial parkinsonism. Front. Neurol. 2018, 9, 1021. [Google Scholar] [CrossRef]
- Surgucheva, I.; Sharov, V.S.; Surguchov, A. γ-Synuclein: Seeding of α-synuclein aggregation and transmission between cells. Biochemistry 2012, 51, 4743–4754. [Google Scholar] [CrossRef]
- Brown, J.W.; Buell, A.K.; Michaels, T.C.; Meisl, G.; Carozza, J.; Flagmeier, P.; Vendruscolo, M.; Knowles, T.P.; Dobson, C.M.; Galvagnion, C. β-Synuclein suppresses both the initiation and amplification steps of α-synuclein aggregation via competitive binding to surfaces. Sci. Rep. 2016, 6, 36010. [Google Scholar] [CrossRef]
- Schmid, A.W.; Fauvet, B.; Moniatte, M.; Lashuel, H.A. Alpha-synuclein post-translational modifications as potential biomarkers for Parkinson disease and other synucleinopathies. Mol. Cell. Proteomics 2013, 12, 3543–3558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlad, C.; Lindner, K.; Karreman, C.; Schildknecht, S.; Leist, M.; Tomczyk, N.; Rontree, J.; Langridge, J.; Danzer, K.; Ciossek, T. Autoproteolytic fragments are intermediates in the oligomerization-aggregation of Parkinson’s disease protein alpha-synuclein as revealed by ion mobility mass spectrometry. ChemBioChem 2011, 12, 2740–2744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kellie, J.F.; Higgs, R.E.; Ryder, J.W.; Major, A.; Beach, T.G.; Adler, C.H.; Merchant, K.; Knierman, M.D. Quantitative measurement of intact alpha-synuclein proteoforms from post-mortem control and Parkinson’s disease brain tissue by intact protein mass spectrometry. Sci. Rep. 2014, 4, 5797. [Google Scholar] [CrossRef] [PubMed]
- Feneberg, E.; Gray, E.; Ansorge, O.; Talbot, K.; Turner, M.R. Towards a TDP-43-based biomarker for ALS and FTLD. Mol. Neurobiol. 2018, 55, 7789–7801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Prakash, A.; Tomar, A.K.; Srivastava, A.; Kundu, B.; Lynn, A.M.; Hassan, M.I. Exploring the aggregation-prone regions from structural domains of human TDP-43. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 286–296. [Google Scholar] [CrossRef]
- Zacco, E.; Martin, S.R.; Thorogate, R.; Pastore, A. The RNA-recognition motifs of TAR DNA-binding protein 43 may play a role in the aberrant self-assembly of the protein. Front. Mol. Neurosci. 2018, 11, 372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zacco, E.; Graña-Montes, R.; Martin, S.R.; de Groot, N.S.; Alfano, C.; Tartaglia, G.G.; Pastore, A. RNA as a key factor in driving or preventing self-assembly of the TAR DNA-binding protein 43. J. Mol. Biol. 2019, 431, 1671–1688. [Google Scholar] [CrossRef] [PubMed]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-J.; Topp, S.D.; Hui, H.S.; Zacco, E.; Katarya, M.; McLoughlin, C.; King, A.; Smith, B.N.; Troakes, C.; Pastore, A.; et al. RRM adjacent TARDBP mutations disrupt RNA binding and enhance TDP-43 proteinopathy. Brain 2019, 142, 3753–3770. [Google Scholar] [CrossRef]
- McAlary, L.; Plotkin, S.S.; Yerbury, J.J.; Cashman, N.R. Prion-like propagation of protein misfolding and aggregation in amyotrophic lateral sclerosis. Front. Mol. Neurosci. 2019, 12, 262. [Google Scholar] [CrossRef] [Green Version]
- Nolan, M.; Talbot, K.; Ansorge, O. Pathogenesis of FUS-associated ALS and FTD: Insights from rodent models. Acta Neuropathol. Commun. 2016, 4, 99. [Google Scholar] [CrossRef] [Green Version]
- St George-Hyslop, P.; Lin, J.Q.; Miyashita, A.; Phillips, E.C.; Qamar, S.; Randle, S.J.; Wang, G. The physiological and pathological biophysics of phase separation and gelation of RNA binding proteins in amyotrophic lateral sclerosis and fronto-temporal lobar degeneration. Brain Res. 2018, 1693, 11–23. [Google Scholar] [CrossRef]
- Vance, C.; Rogelj, B.; Hortobágyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhoads, S.N.; Monahan, Z.T.; Yee, D.S.; Shewmaker, F.P. The role of post-translational modifications on prion-like aggregation and liquid-phase separation of FUS. Int. J. Mol. Sci. 2018, 19, 886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meeter, L.H.; Kaat, L.D.; Rohrer, J.D.; Van Swieten, J.C. Imaging and fluid biomarkers in frontotemporal dementia. Nat. Rev. Neurol. 2017, 13, 406–419. [Google Scholar] [CrossRef]
- Lee, J.C.; Kim, S.J.; Hong, S.; Kim, Y. Diagnosis of Alzheimer’s disease utilizing amyloid and tau as fluid biomarkers. Exp. Mol. Med. 2019, 51, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Sunderland, T.; Linker, G.; Mirza, N.; Putnam, K.T.; Friedman, D.L.; Kimmel, L.H.; Bergeson, J.; Manetti, G.J.; Zimmermann, M.; Tang, B.; et al. Decreased β-amyloid1-42 and increased tau levels in cerebrospinal fluid of patients with Alzheimer disease. JAMA 2003, 289, 2094–2103. [Google Scholar] [CrossRef] [Green Version]
- Mollenhauer, B.; Locascio, J.J.; Schulz-Schaeffer, W.; Sixel-Döring, F.; Trenkwalder, C.; Schlossmacher, M.G. α-Synuclein and tau concentrations in cerebrospinal fluid of patients presenting with parkinsonism: A cohort study. The Lancet Neurol. 2011, 10, 230–240. [Google Scholar] [CrossRef]
- Atik, A.; Stewart, T.; Zhang, J. Alpha-synuclein as a biomarker for Parkinson’s disease. Brain Pathol. 2016, 26, 410–418. [Google Scholar] [CrossRef]
- Mollenhauer, B. Quantification of α-synuclein in cerebrospinal fluid: How ideal is this biomarker for Parkinson’s disease? Parkinsonism Relat. Disord. 2014, 20, S76–S79. [Google Scholar] [CrossRef]
- Foulds, P.; McAuley, E.; Gibbons, L.; Davidson, Y.; Pickering-Brown, S.M.; Neary, D.; Snowden, J.S.; Allsop, D.; Mann, D.M. TDP-43 protein in plasma may index TDP-43 brain pathology in Alzheimer’s disease and frontotemporal lobar degeneration. Acta Neuropathol. 2008, 116, 141–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verstraete, E.; Kuiperij, H.B.; van Blitterswijk, M.M.; Veldink, J.H.; Schelhaas, H.J.; Van Den Berg, L.H.; Verbeek, M.M. TDP-43 plasma levels are higher in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2012, 13, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Noto, Y.-I.; Shibuya, K.; Sato, Y.; Kanai, K.; Misawa, S.; Sawai, S.; Mori, M.; Uchiyama, T.; Isose, S.; Nasu, S.; et al. Elevated CSF TDP-43 levels in amyotrophic lateral sclerosis: Specificity, sensitivity, and a possible prognostic value. Amyotroph. Lateral Scler. 2011, 12, 140–143. [Google Scholar] [CrossRef]
- Majumder, V.; Gregory, J.M.; Barria, M.A.; Green, A.; Pal, S. TDP-43 as a potential biomarker for amyotrophic lateral sclerosis: A systematic review and meta-analysis. BMC Neurol. 2018, 18, 90. [Google Scholar] [CrossRef] [PubMed]
- Steinacker, P.; Barschke, P.; Otto, M. Biomarkers for diseases with TDP-43 pathology. Mol. Cell. Neurosci. 2019, 97, 43–59. [Google Scholar] [CrossRef]
- Feneberg, E.; Steinacker, P.; Lehnert, S.; Schneider, A.; Walther, P.; Thal, D.R.; Linsenmeier, M.; Ludolph, A.C.; Otto, M. Limited role of free TDP-43 as a diagnostic tool in neurodegenerative diseases. Amyotroph. Lateral Scler. Front. Degener. 2014, 15, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Goossens, J.; Vanmechelen, E.; Trojanowski, J.Q.; Lee, V.M.; Van Broeckhoven, C.; van der Zee, J.; Engelborghs, S. TDP-43 as a possible biomarker for frontotemporal lobar degeneration: A systematic review of existing antibodies. Acta Neuropathol. Commun. 2015, 3, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Struyfs, H.; Van Broeck, B.; Timmers, M.; Fransen, E.; Sleegers, K.; Van Broeckhoven, C.; De Deyn, P.P.; Streffer, J.R.; Mercken, M.; Engelborghs, S. Diagnostic accuracy of cerebrospinal fluid amyloid-β isoforms for early and differential dementia diagnosis. J. Alzheimers Dis. 2015, 45, 813–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauridsen, C.; Sando, S.B.; Møller, I.; Berge, G.; Pomary, P.K.; Grøntvedt, G.R.; Salvesen, Ø.; Bråthen, G.; White, L.R. Cerebrospinal fluid Aβ43 is reduced in early-onset compared to late-onset Alzheimer’s disease, but has similar diagnostic accuracy to Aβ42. Front. Aging Neurosci. 2017, 9, 210. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Rezaei-Ghaleh, N.; Terwel, D.; Thal, D.R.; Richard, M.; Hoch, M.; Mc Donald, J.M.; Wüllner, U.; Glebov, K.; Heneka, M.T.; et al. Extracellular phosphorylation of the amyloid β-peptide promotes formation of toxic aggregates during the pathogenesis of Alzheimer’s disease. EMBO J. 2011, 30, 2255–2265. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Wirths, O.; Stüber, K.; Wunderlich, P.; Koch, P.; Theil, S.; Rezaei-Ghaleh, N.; Zweckstetter, M.; Bayer, T.A.; Brüstle, O.; et al. Phosphorylation of the amyloid β-peptide at Ser26 stabilizes oligomeric assembly and increases neurotoxicity. Acta Neuropathol. 2016, 131, 525–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedemann, M.; Helk, E.; Tiiman, A.; Zovo, K.; Palumaa, P.; Tõugu, V. Effect of methionine-35 oxidation on the aggregation of amyloid-β peptide. Biochem. Biophys. Rep. 2015, 3, 94–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schedin-Weiss, S.; Winblad, B.; Tjernberg, L.O. The role of protein glycosylation in Alzheimer disease. FEBS J. 2014, 281, 46–62. [Google Scholar] [CrossRef]
- Harigaya, Y.; Saido, T.C.; Eckman, C.B.; Prada, C.-M.; Shoji, M.; Younkin, S.G. Amyloid β protein starting pyroglutamate at position 3 is a major component of the amyloid deposits in the Alzheimer’s disease brain. Biochem. Biophys. Res. Commun. 2000, 276, 422–427. [Google Scholar] [CrossRef]
- Perez-Garmendia, R.; Gevorkian, G. Pyroglutamate-modified amyloid beta peptides: Emerging targets for Alzheimer s disease immunotherapy. Curr. Neuropharmacol. 2013, 11, 491–498. [Google Scholar] [CrossRef] [Green Version]
- Palmqvist, S.; Janelidze, S.; Quiroz, Y.T.; Zetterberg, H.; Lopera, F.; Stomrud, E.; Su, Y.; Chen, Y.; Serrano, G.E.; Leuzy, A.; et al. Discriminative accuracy of plasma phospho-tau217 for Alzheimer disease vs other neurodegenerative disorders. JAMA 2020, 324, 772–781. [Google Scholar] [CrossRef]
- Cohen, T.J.; Guo, J.L.; Hurtado, D.E.; Kwong, L.K.; Mills, I.P.; Trojanowski, J.Q.; Lee, V.M. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat. Commun. 2011, 2, 252. [Google Scholar] [CrossRef] [Green Version]
- Landino, L.M.; Skreslet, T.E.; Alston, J.A. Cysteine oxidation of tau and microtubule-associated protein-2 by peroxynitrite modulation of microtubule assembly kinetics by the thioredoxin reductase system. J. Biol. Chem. 2004, 279, 35101–35105. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, M.R.; Reyes, J.F.; Fu, Y.; Bigio, E.H.; Guillozet-Bongaarts, A.L.; Berry, R.W.; Binder, L.I. Tau nitration occurs at tyrosine 29 in the fibrillar lesions of Alzheimer’s disease and other tauopathies. J. Neurosci. 2006, 26, 10636–10645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Losev, Y.; Frenkel-Pinter, M.; Abu-Hussien, M.; Viswanathan, G.K.; Elyashiv-Revivo, D.; Geries, R.; Khalaila, I.; Gazit, E.; Segal, D. Differential effects of putative N-glycosylation sites in human Tau on Alzheimer’s disease-related neurodegeneration. Cell. Mol. Life Sci. 2021, 78, 2231–2245. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Shi, J.; Tanimukai, H.; Gu, J.; Gu, J.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.-X. Reduced O-GlcNAcylation links lower brain glucose metabolism and tau pathology in Alzheimer’s disease. Brain 2009, 132, 1820–1832. [Google Scholar] [CrossRef] [Green Version]
- Bancher, C.; Grundke-Iqbal, I.; Iqbal, K.; Fried, V.; Smith, H.; Wisniewski, H. Abnormal phosphorylation of tau precedes ubiquitination in neurofibrillary pathology of Alzheimer disease. Brain Res. 1991, 539, 11–18. [Google Scholar] [CrossRef]
- Kontaxi, C.; Piccardo, P.; Gill, A.C. Lysine-directed post-translational modifications of tau protein in Alzheimer’s disease and related Tauopathies. Front. Mol. Biosci. 2017, 4, 56. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.M.; Wong, E.S.; Kirkpatrick, D.S.; Pletnikova, O.; Ko, H.S.; Tay, S.-P.; Ho, M.W.; Troncoso, J.; Gygi, S.P.; Lee, M.K.; et al. Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum. Mol. Genet. 2008, 17, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. α-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.W.; Margolis, R.L.; Li, X.; Troncoso, J.C.; Lee, M.K.; Dawson, V.L.; Dawson, T.M.; Iwatsubo, T.; Ross, C.A. α-Synuclein phosphorylation enhances eosinophilic cytoplasmic inclusion formation in SH-SY5Y cells. J. Neurosci. 2005, 25, 5544–5552. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Moriarty, G.M.; Woods, L.A.; Ashcroft, A.E.; Radford, S.E.; Baum, J. N-terminal acetylation of α-synuclein induces increased transient helical propensity and decreased aggregation rates in the intrinsically disordered monomer. Protein Sci. 2012, 21, 911–917. [Google Scholar] [CrossRef] [Green Version]
- Glaser, C.B.; Yamin, G.; Uversky, V.N.; Fink, A.L. Methionine oxidation, α-synuclein and Parkinson’s disease. Biochim Biophys Acta Proteins Proteom 2005, 1703, 157–169. [Google Scholar] [CrossRef]
- Prigione, A.; Piazza, F.; Brighina, L.; Begni, B.; Galbussera, A.; DiFrancesco, J.C.; Andreoni, S.; Piolti, R.; Ferrarese, C. Alpha-synuclein nitration and autophagy response are induced in peripheral blood cells from patients with Parkinson disease. Neurosci. Lett. 2010, 477, 6–10. [Google Scholar] [CrossRef]
- Levine, P.M.; Galesic, A.; Balana, A.T.; Mahul-Mellier, A.-L.; Navarro, M.X.; De Leon, C.A.; Lashuel, H.A.; Pratt, M.R. α-Synuclein O-GlcNAcylation alters aggregation and toxicity, revealing certain residues as potential inhibitors of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2019, 116, 1511–1519. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-J.; Xu, Y.-F.; Cook, C.; Gendron, T.F.; Roettges, P.; Link, C.D.; Lin, W.-L.; Tong, J.; Castanedes-Casey, M.; Ash, P.; et al. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 7607–7612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Che, M.X.; Jiang, Y.J.; Xie, Y.Y.; Jiang, L.L.; Hu, H.Y. Aggregation of the 35-kDa fragment of TDP-43 causes formation of cytoplasmic inclusions and alteration of RNA processing. FASEB J. 2011, 25, 2344–2353. [Google Scholar] [CrossRef]
- Koychev, I.; Galna, B.; Zetterberg, H.; Lawson, J.; Zamboni, G.; Ridha, B.H.; Rowe, J.B.; Thomas, A.; Howard, R.; Malhotra, P.; et al. Aβ 42/Aβ 40 and Aβ 42/Aβ 38 Ratios Are Associated with Measures of Gait Variability and Activities of Daily Living in Mild Alzheimer’s Disease: A Pilot Study. J. Alzheimers Dis. 2018, 65, 1377–1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welge, V.; Fiege, O.; Lewczuk, P.; Mollenhauer, B.; Esselmann, H.; Klafki, H.-W.; Wolf, S.; Trenkwalder, C.; Otto, M.; Kornhuber, J.; et al. Combined CSF tau, p-tau181 and amyloid-β 38/40/42 for diagnosing Alzheimer’s disease. J. Neural Transm. 2009, 116, 203–212. [Google Scholar] [CrossRef]
- Tsuji, H.; Arai, T.; Kametani, F.; Nonaka, T.; Yamashita, M.; Suzukake, M.; Hosokawa, M.; Yoshida, M.; Hatsuta, H.; Takao, M.; et al. Molecular analysis and biochemical classification of TDP-43 proteinopathy. Brain 2012, 135, 3380–3391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinacker, P.; Hendrich, C.; Sperfeld, A.D.; Jesse, S.; von Arnim, C.A.; Lehnert, S.; Pabst, A.; Uttner, I.; Tumani, H.; Lee, V.M.-Y.; et al. TDP-43 in cerebrospinal fluid of patients with frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Arch. Neurol. 2008, 65, 1481–1487. [Google Scholar] [CrossRef] [Green Version]
- Janelidze, S.; Stomrud, E.; Smith, R.; Palmqvist, S.; Mattsson, N.; Airey, D.C.; Proctor, N.K.; Chai, X.; Shcherbinin, S.; Sims, J.R.; et al. Cerebrospinal fluid p-tau217 performs better than p-tau181 as a biomarker of Alzheimer’s disease. Nat. Commun. 2020, 11, 1683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feany, M.B.; Bender, W.W. A Drosophila model of Parkinson’s disease. Nature 2000, 404, 394–398. [Google Scholar] [CrossRef]
- Foulds, P.G.; Mitchell, J.D.; Parker, A.; Turner, R.; Green, G.; Diggle, P.; Hasegawa, M.; Taylor, M.; Mann, D.; Allsop, D. Phosphorylated α-synuclein can be detected in blood plasma and is potentially a useful biomarker for Parkinson’s disease. FASEB J. 2011, 25, 4127–4137. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shi, M.; Chung, K.A.; Zabetian, C.P.; Leverenz, J.B.; Berg, D.; Srulijes, K.; Trojanowski, J.Q.; Lee, V.M.-Y.; Siderowf, A.D.; et al. Phosphorylated α-synuclein in Parkinson’s disease. Sci. Transl. Med. 2012, 4, ra120–ra121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, T.; Sossi, V.; Aasly, J.O.; Wszolek, Z.K.; Uitti, R.J.; Hasegawa, K.; Yokoyama, T.; Zabetian, C.P.; Leverenz, J.B.; Stoessl, A.J.; et al. Phosphorylated α-synuclein in Parkinson’s disease: Correlation depends on disease severity. Acta Neuropathol. Commun. 2015, 3, 7. [Google Scholar] [CrossRef] [Green Version]
- Ayyadevara, S.; Balasubramaniam, M.; Parcon, P.A.; Barger, S.W.; Griffin, W.S.T.; Alla, R.; Tackett, A.J.; Mackintosh, S.G.; Petricoin, E.; Zhou, W.; et al. Proteins that mediate protein aggregation and cytotoxicity distinguish Alzheimer’s hippocampus from normal controls. Aging Cell 2016, 15, 924–939. [Google Scholar] [CrossRef] [PubMed]
- Öhrfelt, A.; Zetterberg, H.; Andersson, K.; Persson, R.; Secic, D.; Brinkmalm, G.; Wallin, A.; Mulugeta, E.; Francis, P.T.; Vanmechelen, E.; et al. Identification of novel α-synuclein isoforms in human brain tissue by using an online nanoLC-ESI-FTICR-MS method. Neurochem. Res. 2011, 36, 2029–2042. [Google Scholar] [CrossRef] [Green Version]
- Vinueza-Gavilanes, R.; Íñigo-Marco, I.; Larrea, L.; Lasa, M.; Carte, B.; Santamaría, E.; Fernández-Irigoyen, J.; Bugallo, R.; Aragón, T.; Aldabe, R.; et al. N-terminal acetylation mutants affect alpha-synuclein stability, protein levels and neuronal toxicity. Neurobiol. Dis. 2020, 137, 104781. [Google Scholar] [CrossRef]
- Collin, F.; Cheignon, C.; Hureau, C. Oxidative stress as a biomarker for Alzheimer’s disease. Biomark. Med. 2018, 12, 201–203. [Google Scholar] [CrossRef] [PubMed]
- Näslund, J.; Schierhorn, A.; Hellman, U.; Lannfelt, L.; Roses, A.D.; Tjernberg, L.O.; Silberring, J.; Gandy, S.E.; Winblad, B.; Greengard, P. Relative abundance of Alzheimer A beta amyloid peptide variants in Alzheimer disease and normal aging. Proc. Natl. Acad. Sci. USA 1994, 91, 8378–8382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranda, H.V.; Cássio, R.; Correia-Guedes, L.; Gomes, M.A.; Chegão, A.; Miranda, E.; Soares, T.; Coelho, M.; Rosa, M.M.; Ferreira, J.J.; et al. Posttranslational modifications of blood-derived alpha-synuclein as biochemical markers for Parkinson’s disease. Sci. Rep. 2017, 7, 13713. [Google Scholar] [CrossRef] [Green Version]
- Mangialasche, F.; Polidori, M.C.; Monastero, R.; Ercolani, S.; Camarda, C.; Cecchetti, R.; Mecocci, P. Biomarkers of oxidative and nitrosative damage in Alzheimer’s disease and mild cognitive impairment. Ageing Res. Rev. 2009, 8, 285–305. [Google Scholar] [CrossRef] [Green Version]
- Halim, A.; Brinkmalm, G.; Rüetschi, U.; Westman-Brinkmalm, A.; Portelius, E.; Zetterberg, H.; Blennow, K.; Larson, G.; Nilsson, J. Site-specific characterization of threonine, serine, and tyrosine glycosylations of amyloid precursor protein/amyloid β-peptides in human cerebrospinal fluid. Proc. Natl. Acad. Sci. USA 2011, 108, 11848–11853. [Google Scholar] [CrossRef] [Green Version]
- Gizaw, S.T.; Ohashi, T.; Tanaka, M.; Hinou, H.; Nishimura, S.-I. Glycoblotting method allows for rapid and efficient glycome profiling of human Alzheimer’s disease brain, serum and cerebrospinal fluid towards potential biomarker discovery. Biochim. Biophys. Acta Gen. Subj. 2016, 1860, 1716–1727. [Google Scholar] [CrossRef] [Green Version]
- Palmigiano, A.; Barone, R.; Sturiale, L.; Sanfilippo, C.; Bua, R.O.; Romeo, D.A.; Messina, A.; Capuana, M.L.; Maci, T.; Le Pira, F.; et al. CSF N-glycoproteomics for early diagnosis in Alzheimer’s disease. J. Proteom. 2016, 131, 29–37. [Google Scholar] [CrossRef]
- Cole, G.M.; Timiras, P.S. Ubiquitin-protein conjugates in Alzheimer’s lesions. Neurosci. Lett. 1987, 79, 207–212. [Google Scholar] [CrossRef]
- Riederer, I.M.; Schiffrin, M.; Kövari, E.; Bouras, C.; Riederer, B.M. Ubiquitination and cysteine nitrosylation during aging and Alzheimer’s disease. Brain Res. Bull. 2009, 80, 233–241. [Google Scholar] [CrossRef]
- Hasegawa, M.; Fujiwara, H.; Nonaka, T.; Wakabayashi, K.; Takahashi, H.; Lee, V.M.-Y.; Trojanowski, J.Q.; Mann, D.; Iwatsubo, T. Phosphorylated α-synuclein is ubiquitinated in α-synucleinopathy lesions. J. Biol. Chem. 2002, 277, 49071–49076. [Google Scholar] [CrossRef] [Green Version]
- Sjödin, S.; Hansson, O.; Öhrfelt, A.; Brinkmalm, G.; Zetterberg, H.; Brinkmalm, A.; Blennow, K. Mass spectrometric analysis of cerebrospinal fluid ubiquitin in Alzheimer’s disease and Parkinsonian disorders. Proteom. Clin. Appl. 2017, 11, 1700100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, K.; Grundke-Iqbal, I. Elevated levels of τ and ubiquitin in brain and cerebrospinal fluid in Alzheimer’s disease. Int. Psychogeriatr. 1997, 9, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Aprile, F.A.; Sormanni, P.; Podpolny, M.; Chhangur, S.; Needham, L.-M.; Ruggeri, F.S.; Perni, M.; Limbocker, R.; Heller, G.T.; Sneideris, T.; et al. Rational design of a conformation-specific antibody for the quantification of Aβ oligomers. Proc. Natl. Acad. Sci. USA 2020, 117, 13509–13518. [Google Scholar] [CrossRef] [PubMed]
- Sormanni, P.; Aprile, F.A.; Vendruscolo, M. Rational design of antibodies targeting specific epitopes within intrinsically disordered proteins. Proc. Natl. Acad. Sci. USA 2015, 112, 9902–9907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meli, G.; Lecci, A.; Manca, A.; Krako, N.; Albertini, V.; Benussi, L.; Ghidoni, R.; Cattaneo, A. Conformational targeting of intracellular Aβ oligomers demonstrates their pathological oligomerization inside the endoplasmic reticulum. Nat. Commun. 2014, 5, 3867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perchiacca, J.M.; Ladiwala, A.R.A.; Bhattacharya, M.; Tessier, P.M. Aggregation-resistant domain antibodies engineered with charged mutations near the edges of the complementarity-determining regions. Protein Eng. Des. Sel. 2012, 25, 591–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horrocks, M.H.; Lee, S.F.; Gandhi, S.; Magdalinou, N.K.; Chen, S.W.; Devine, M.J.; Tosatto, L.; Kjaergaard, M.; Beckwith, J.S.; Zetterberg, H.; et al. Single-molecule imaging of individual amyloid protein aggregates in human biofluids. ACS Chem. Neurosci. 2016, 7, 399–406. [Google Scholar] [CrossRef] [Green Version]
- De, S.; Whiten, D.R.; Ruggeri, F.S.; Hughes, C.; Rodrigues, M.; Sideris, D.I.; Taylor, C.G.; Aprile, F.A.; Muyldermans, S.; Knowles, T.P.; et al. Soluble aggregates present in cerebrospinal fluid change in size and mechanism of toxicity during Alzheimer’s disease progression. Acta Neuropathol. Commun. 2019, 7, 120. [Google Scholar] [CrossRef] [Green Version]
- Thompson, A.J.; Herling, T.W.; Kubánková, M.t.; Vyšniauskas, A.; Knowles, T.P.; Kuimova, M.K. Molecular rotors provide insights into microscopic structural changes during protein aggregation. J. Phys. Chem. B 2015, 119, 10170–10179. [Google Scholar] [CrossRef] [Green Version]
- Kubánková, M.; López-Duarte, I.; Bull, J.A.; Vadukul, D.M.; Serpell, L.C.; de Saint Victor, M.; Stride, E.; Kuimova, M.K. Probing supramolecular protein assembly using covalently attached fluorescent molecular rotors. Biomaterials 2017, 139, 195–201. [Google Scholar] [CrossRef]
- Needham, L.-M.; Weber, J.; Varela, J.A.; Fyfe, J.W.; Do, D.T.; Xu, C.K.; Tutton, L.; Cliffe, R.; Keenlyside, B.; Klenerman, D.; et al. ThX–a next-generation probe for the early detection of amyloid aggregates. Chem. Sci. 2020, 11, 4578–4583. [Google Scholar] [CrossRef] [Green Version]
- Babu, E.; Mareeswaran, P.M.; Sathish, V.; Singaravadivel, S.; Rajagopal, S. Sensing and inhibition of amyloid-β based on the simple luminescent aptamer–ruthenium complex system. Talanta 2015, 134, 348–353. [Google Scholar] [CrossRef]
- Tsukakoshi, K.; Abe, K.; Sode, K.; Ikebukuro, K. Selection of DNA aptamers that recognize α-synuclein oligomers using a competitive screening method. Anal. Chem. 2012, 84, 5542–5547. [Google Scholar] [CrossRef]
- Whiten, D.R.; Zuo, Y.; Calo, L.; Choi, M.L.; De, S.; Flagmeier, P.; Wirthensohn, D.C.; Kundel, F.; Ranasinghe, R.T.; Sanchez, S.E.; et al. Nanoscopic characterisation of individual endogenous protein aggregates in human neuronal cells. ChemBioChem 2018, 19, 2033–2038. [Google Scholar] [CrossRef] [PubMed]
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.L.; Johnson, M.J.; Herzenberg, L.A.; Oi, V.T. Chimeric human antibody molecules: Mouse antigen-binding domains with human constant region domains. Proc. Natl. Acad. Sci. USA 1984, 81, 6851–6855. [Google Scholar] [CrossRef] [Green Version]
- Omidfar, K.; Daneshpour, M. Advances in phage display technology for drug discovery. Expert Opin. Drug Discov. 2015, 10, 651–669. [Google Scholar] [CrossRef] [PubMed]
- Sormanni, P.; Aprile, F.A.; Vendruscolo, M. Third generation antibody discovery methods: In silico rational design. Chem. Soc. Rev. 2018, 47, 9137–9157. [Google Scholar] [CrossRef] [PubMed]
- Baran, D.; Pszolla, M.G.; Lapidoth, G.D.; Norn, C.; Dym, O.; Unger, T.; Albeck, S.; Tyka, M.D.; Fleishman, S.J. Principles for computational design of binding antibodies. Proc. Natl. Acad. Sci. USA 2017, 114, 10900–10905. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Pantazes, R.J.; Maranas, C.D. OptMAVEn–a new framework for the de novo design of antibody variable region models targeting specific antigen epitopes. PLoS ONE 2014, 9, e105954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adolf-Bryfogle, J.; Kalyuzhniy, O.; Kubitz, M.; Weitzner, B.D.; Hu, X.; Adachi, Y.; Schief, W.R.; Dunbrack, R.L., Jr. RosettaAntibodyDesign (RAbD): A general framework for computational antibody design. PLoS Comput. Biol. 2018, 14, e1006112. [Google Scholar] [CrossRef] [Green Version]
- Aprile, F.A.; Sormanni, P.; Perni, M.; Arosio, P.; Linse, S.; Knowles, T.P.; Dobson, C.M.; Vendruscolo, M. Selective targeting of primary and secondary nucleation pathways in Aβ42 aggregation using a rational antibody scanning method. Sci. Adv. 2017, 3, e1700488. [Google Scholar] [CrossRef] [Green Version]
- Ehrenberg, A.J.; Khatun, A.; Coomans, E.; Betts, M.J.; Capraro, F.; Thijssen, E.H.; Senkevich, K.; Bharucha, T.; Jafarpour, M.; Young, P.N.; et al. Relevance of biomarkers across different neurodegenerative diseases. Alzheimers Res. Ther. 2020, 12, 56. [Google Scholar] [CrossRef]
- Klunk, W.E.; Engler, H.; Nordberg, A.; Wang, Y.; Blomqvist, G.; Holt, D.P.; Bergström, M.; Savitcheva, I.; Huang, G.F.; Estrada, S.; et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 2004, 55, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Meier, S.R.; Syvänen, S.; Hultqvist, G.; Fang, X.T.; Roshanbin, S.; Lannfelt, L.; Neumann, U.; Sehlin, D. Antibody-based in vivo PET imaging detects amyloid-β reduction in Alzheimer transgenic mice after BACE-1 inhibition. J. Nucl. Med. 2018, 59, 1885–1891. [Google Scholar] [CrossRef] [Green Version]
- Nabuurs, R.J.; Rutgers, K.S.; Welling, M.M.; Metaxas, A.; De Backer, M.E.; Rotman, M.; Bacskai, B.J.; Van Buchem, M.A.; Van der Maarel, S.M.; van Der Weerd, L. In vivo detection of amyloid-β deposits using heavy chain antibody fragments in a transgenic mouse model for Alzheimer’s disease. PLoS ONE 2012, 7, e38284. [Google Scholar] [CrossRef] [Green Version]
- Spires-Jones, T.L.; Attems, J.; Thal, D.R. Interactions of pathological proteins in neurodegenerative diseases. Acta Neuropathol. 2017, 134, 187–205. [Google Scholar] [CrossRef] [PubMed]
- Colom-Cadena, M.; Gelpi, E.; Charif, S.; Belbin, O.; Blesa, R.; Martí, M.J.; Clarimon, J.; Lleó, A. Confluence of α-synuclein, tau, and β-amyloid pathologies in dementia with Lewy bodies. J. Neuropathol. Exp. Neurol. 2013, 72, 1203–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Wolf, F.; Ghanbari, M.; Licher, S.; McRae-McKee, K.; Gras, L.; Weverling, G.J.; Wermeling, P.; Sedaghat, S.; Ikram, M.K.; Waziry, R.; et al. Plasma tau, neurofilament light chain and amyloid-β levels and risk of dementia; a population-based cohort study. Brain 2020, 143, 1220–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majbour, N.K.; Vaikath, N.N.; van Dijk, K.D.; Ardah, M.T.; Varghese, S.; Vesterager, L.B.; Montezinho, L.P.; Poole, S.; Safieh-Garabedian, B.; Tokuda, T.; et al. Oligomeric and phosphorylated alpha-synuclein as potential CSF biomarkers for Parkinson’s disease. Mol. Neurodegener. 2016, 11, 7. [Google Scholar] [CrossRef] [Green Version]
- Bourbouli, M.; Rentzos, M.; Bougea, A.; Zouvelou, V.; Constantinides, V.C.; Zaganas, I.; Evdokimidis, I.; Kapaki, E.; Paraskevas, G.P. Cerebrospinal fluid TAR DNA-binding protein 43 combined with tau proteins as a candidate biomarker for amyotrophic lateral sclerosis and frontotemporal dementia spectrum disorders. Dement. Geriatr. Cogn. Disord. 2017, 44, 144–152. [Google Scholar] [CrossRef]
- Appel-Cresswell, S.; Vilarino-Guell, C.; Encarnacion, M.; Sherman, H.; Yu, I.; Shah, B.; Weir, D.; Thompson, C.; Szu-Tu, C.; Trinh, J.; et al. Alpha-synuclein p. H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov. Disord. 2013, 28, 811–813. [Google Scholar] [CrossRef]
- de Lassichère, C.C.; Mai, T.D.; Taverna, M. Antibody-free detection of amyloid beta peptides biomarkers in cerebrospinal fluid using capillary isotachophoresis coupled with mass spectrometry. J. Chromatogr. A 2019, 1601, 350–356. [Google Scholar] [CrossRef] [PubMed]
- DeMarco, M.L.; Nguyen, Q.; Fok, A.; Hsiung, G.-Y.R.; van der Gugten, J.G. An automated clinical mass spectrometric method for identification and quantification of variant and wild-type amyloid-β 1-40 and 1-42 peptides in CSF. Alzheimers Dement. 2020, 12, e12036. [Google Scholar] [CrossRef]
- Weber, D.M.; Tran, D.; Goldman, S.M.; Taylor, S.W.; Ginns, E.I.; Lagier, R.J.; Rissman, R.A.; Brewer, J.B.; Clarke, N.J. High-Throughput Mass Spectrometry Assay for Quantifying β-Amyloid 40 and 42 in Cerebrospinal Fluid. Clin. Chem. 2019, 65, 1572–1580. [Google Scholar] [CrossRef]
- Freedman, K.J.; Otto, L.M.; Ivanov, A.P.; Barik, A.; Oh, S.-H.; Edel, J.B. Nanopore sensing at ultra-low concentrations using single-molecule dielectrophoretic trapping. Nat. Commun. 2016, 7, 10217. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wilkinson, M.D.; Lin, X.; Ren, R.; Willison, K.R.; Ivanov, A.P.; Baum, J.; Edel, J.B. Single-molecule nanopore sensing of actin dynamics and drug binding. Chem. Sci. 2020, 11, 970–979. [Google Scholar] [CrossRef] [Green Version]
- Japrung, D.; Dogan, J.; Freedman, K.J.; Nadzeyka, A.; Bauerdick, S.; Albrecht, T.; Kim, M.J.; Jemth, P.; Edel, J.B. Single-molecule studies of intrinsically disordered proteins using solid-state nanopores. Anal. Chem. 2013, 85, 2449–2456. [Google Scholar] [CrossRef] [PubMed]
- Rosenstein, J.K.; Wanunu, M.; Merchant, C.A.; Drndic, M.; Shepard, K.L. Integrated nanopore sensing platform with sub-microsecond temporal resolution. Nat. Methods 2012, 9, 487–492. [Google Scholar] [CrossRef]
- Shahnawaz, M.; Tokuda, T.; Waragai, M.; Mendez, N.; Ishii, R.; Trenkwalder, C.; Mollenhauer, B.; Soto, C. Development of a biochemical diagnosis of Parkinson disease by detection of α-synuclein misfolded aggregates in cerebrospinal fluid. JAMA Neurol. 2017, 74, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Saijo, E.; Groveman, B.R.; Kraus, A.; Metrick, M.; Orrù, C.D.; Hughson, A.G.; Caughey, B. Ultrasensitive RT-QuIC Seed Amplification Assays for Disease-Associated Tau, α-Synuclein, and Prion Aggregates. In Protein Misfolding Diseases: Methods and Protocols; Gomes, C.M., Ed.; Springer: New York, NY, USA, 2019; pp. 19–37. [Google Scholar]
- Abd-Elhadi, S.; Basora, M.; Vilas, D.; Tolosa, E.; Sharon, R. Total α-synuclein levels in human blood cells, CSF, and saliva determined by a lipid-ELISA. Anal. Bioanal. Chem. 2016, 408, 7669–7677. [Google Scholar] [CrossRef]
- Bittner, T.; Zetterberg, H.; Teunissen, C.E.; Ostlund, R.E., Jr.; Militello, M.; Andreasson, U.; Hubeek, I.; Gibson, D.; Chu, D.C.; Eichenlaub, U.; et al. Technical performance of a novel, fully automated electrochemiluminescence immunoassay for the quantitation of β-amyloid (1–42) in human cerebrospinal fluid. Alzheimers Dement. 2016, 12, 517–526. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Lachno, D.R.; Hanlon, D.; Shepro, A.; Jeromin, A.; Gemani, D.; Talbot, J.A.; Racke, M.M.; Dage, J.L.; Dean, R.A. A digital enzyme-linked immunosorbent assay for ultrasensitive measurement of amyloid-β 1–42 peptide in human plasma with utility for studies of Alzheimer’s disease therapeutics. Alzheimers Res. Ther. 2016, 8, 58. [Google Scholar] [CrossRef] [Green Version]
- Kühbach, K.; Hülsemann, M.; Herrmann, Y.; Kravchenko, K.; Kulawik, A.; Linnartz, C.; Peters, L.; Wang, K.; Willbold, J.; Willbold, D.; et al. Application of an amyloid beta oligomer standard in the sFIDA assay. Front. Neurosci. 2016, 10, 8. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.-S.; Leung, Y.Y.; Chang, S.-K.; Leight, S.; Knapik-Czajka, M.; Baek, Y.; Shaw, L.M.; Lee, V.M.-Y.; Trojanowski, J.Q.; Clark, C.M. Comparison of xMAP and ELISA assays for detecting cerebrospinal fluid biomarkers of Alzheimer’s disease. J. Alzheimers Dis. 2012, 31, 439–445. [Google Scholar] [CrossRef] [Green Version]
- Singer, D.; Soininen, H.; Alafuzoff, I.; Hoffmann, R. Immuno-PCR-based quantification of multiple phosphorylated tau-epitopes linked to Alzheimer’s disease. Anal. Bioanal. Chem. 2009, 395, 2263–2267. [Google Scholar] [CrossRef] [PubMed]
- Stegurová, L.; Dráberová, E.; Bartos, A.; Dráber, P.; Řípová, D.; Dráber, P. Gold nanoparticle-based immuno-PCR for detection of tau protein in cerebrospinal fluid. J. Immunol. Methods 2014, 406, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Aoki, M.; Winblad, B.; Tjernberg, L.O. A novel approach for Aβ1–40 quantification using immuno-PCR. J. Neurosci. Methods 2012, 205, 364–367. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.-Y.; Fu, Y.; Chang, K.-H.; Lin, K.-J.; Lu, Y.-J.; Cheng, C.-M. Point-of-care devices using disease biomarkers to diagnose neurodegenerative disorders. Trends Biotechnol. 2018, 36, 290–303. [Google Scholar] [CrossRef]
- Shao, Y.; Le, W. Recent advances and perspectives of metabolomics-based investigations in Parkinson’s disease. Mol. Neurodegener. 2019, 14, 3. [Google Scholar] [CrossRef] [Green Version]
- Phelan, M.; Caamaño-Gutiérrez, E.; Gant, M.; Grosman, R.; Madine, J. Using an NMR metabolomics approach to investigate the pathogenicity of amyloid-beta and alpha-synuclein. Metabolomics 2017, 13, 151. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Liu, Z.; Ji, B.; Liu, L.; Wu, S.; Liu, X.; Wang, S.; Wang, L. Metabolite Profile of Alzheimer’s Disease in the Frontal Cortex as Analyzed by HRMAS 1H NMR. Front. Aging Neurosci. 2019, 10, 424. [Google Scholar] [CrossRef]
- Zhu, S.; Wuolikainen, A.; Wu, J.; Öhman, A.; Wingsle, G.; Moritz, T.; Andersen, P.M.; Forsgren, L.; Trupp, M. Targeted multiple reaction monitoring analysis of CSF identifies UCHL1 and GPNMB as candidate biomarkers for ALS. J. Mol. Neurosci. 2019, 69, 643–657. [Google Scholar] [CrossRef] [Green Version]
- Hilton, D.; Stephens, M.; Kirk, L.; Edwards, P.; Potter, R.; Zajicek, J.; Broughton, E.; Hagan, H.; Carroll, C. Accumulation of α-synuclein in the bowel of patients in the pre-clinical phase of Parkinson’s disease. Acta Neuropathol. 2014, 127, 235–241. [Google Scholar] [CrossRef]
- Gries, M.; Christmann, A.; Schulte, S.; Weyland, M.; Rommel, S.; Martin, M.; Baller, M.; Röth, R.; Schmitteckert, S.; Unger, M.; et al. Functional and molecular early enteric biomarkers for Parkinson’s disease in mice and men. bioRxiv 2020. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, Y.; Vadukul, D.M.; Gialama, D.; Ge, Y.; Thrush, R.; White, J.T.; Aprile, F.A. The Diagnostic Potential of Amyloidogenic Proteins. Int. J. Mol. Sci. 2021, 22, 4128. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084128
Jin Y, Vadukul DM, Gialama D, Ge Y, Thrush R, White JT, Aprile FA. The Diagnostic Potential of Amyloidogenic Proteins. International Journal of Molecular Sciences. 2021; 22(8):4128. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084128
Chicago/Turabian StyleJin, Yiyun, Devkee Mahesh Vadukul, Dimitra Gialama, Ying Ge, Rebecca Thrush, Joe Thomas White, and Francesco Antonio Aprile. 2021. "The Diagnostic Potential of Amyloidogenic Proteins" International Journal of Molecular Sciences 22, no. 8: 4128. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084128