
A Metal-Free, Disulfide Oxidized Form of Superoxide Dismutase 1 as a Primary Misfolded Species with Prion-Like Properties in the Extracellular Environments Surrounding Motor Neuron-Like Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
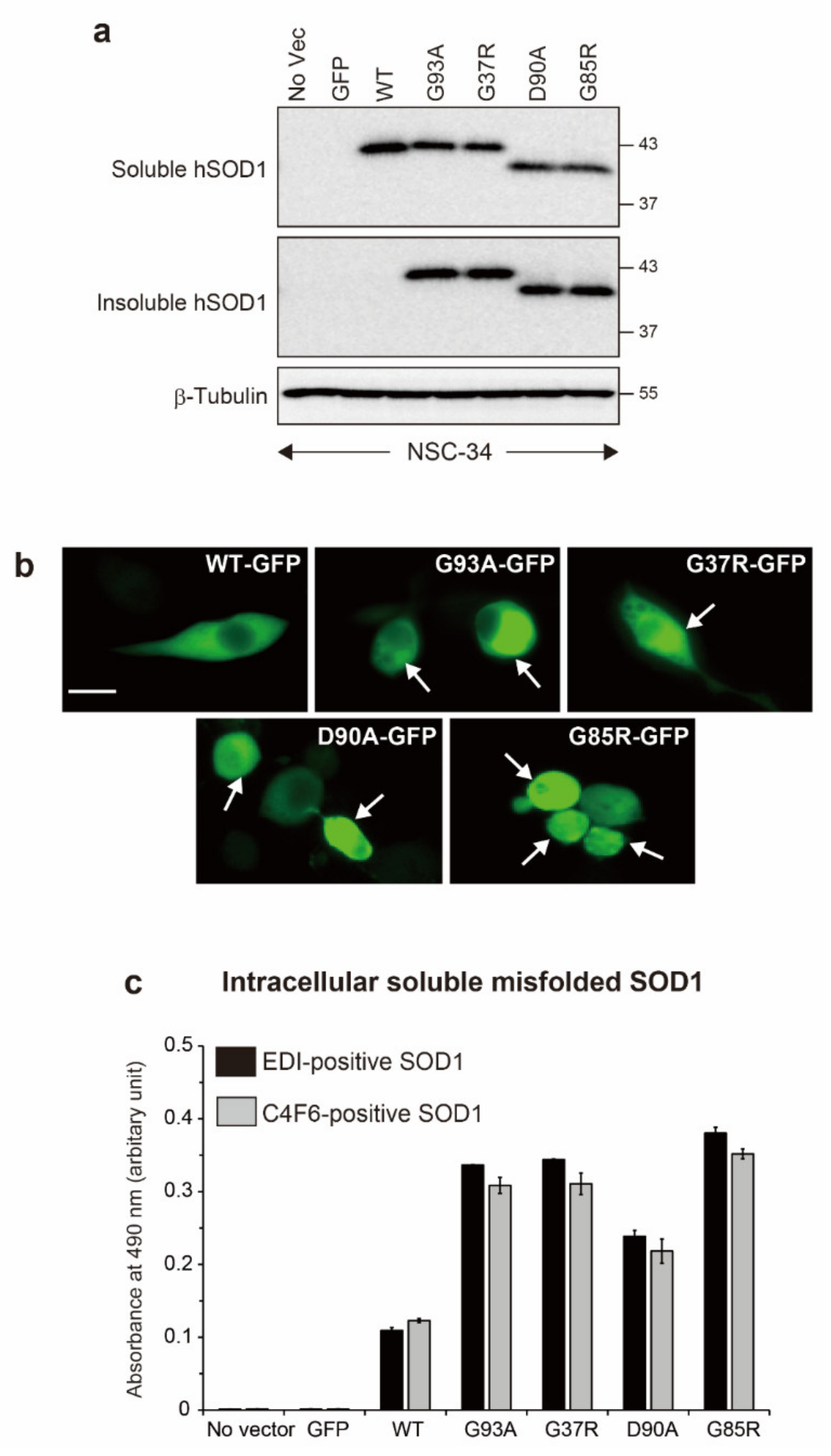
2.1. Generation of Motor Neuron Models of ALS with Intracellular Accumulation of Misfolded and Aggregated SOD1
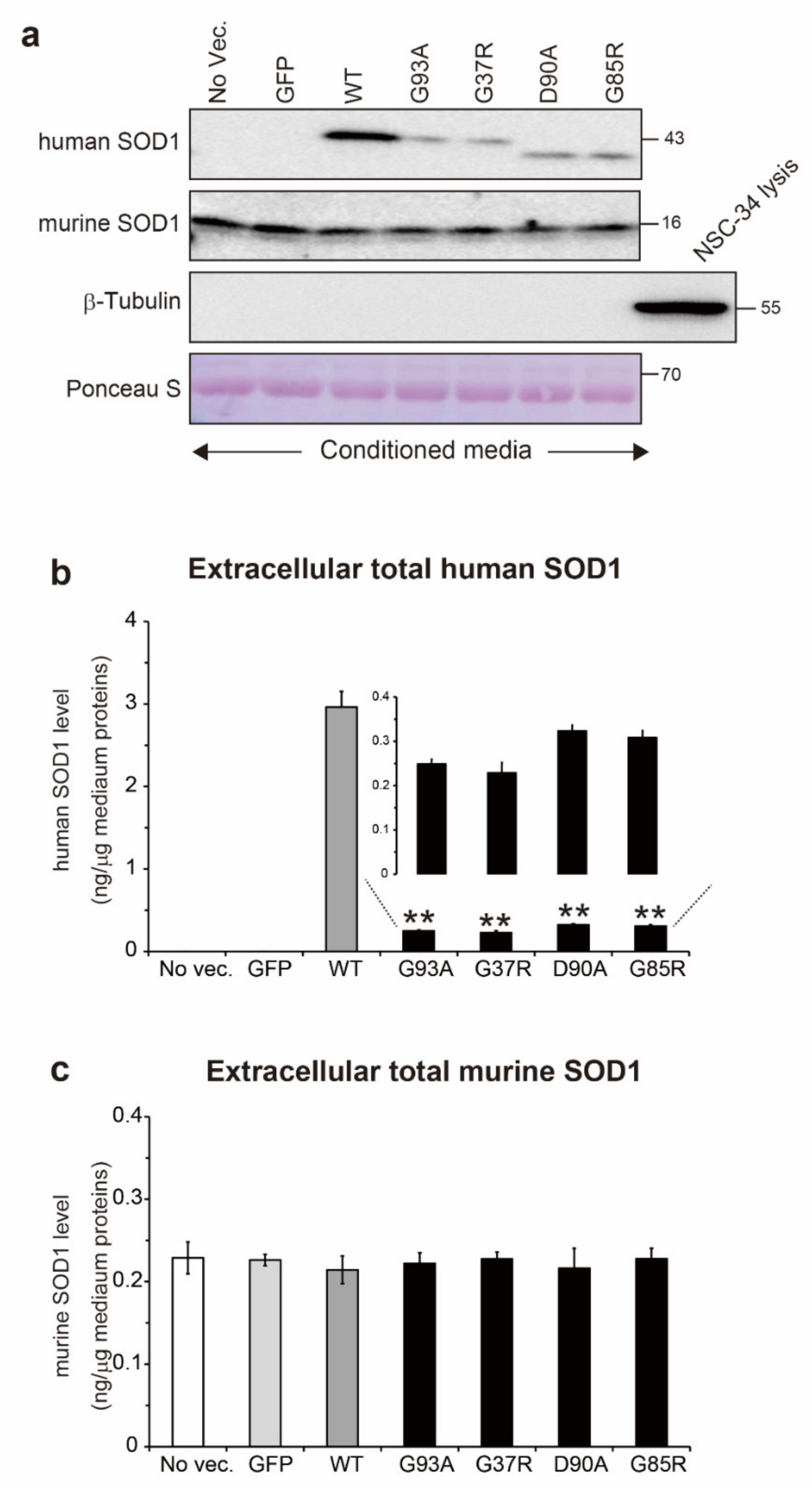
2.2. Total Protein Level of Extracellular Human and Murine SOD1 in Conditioned Medium
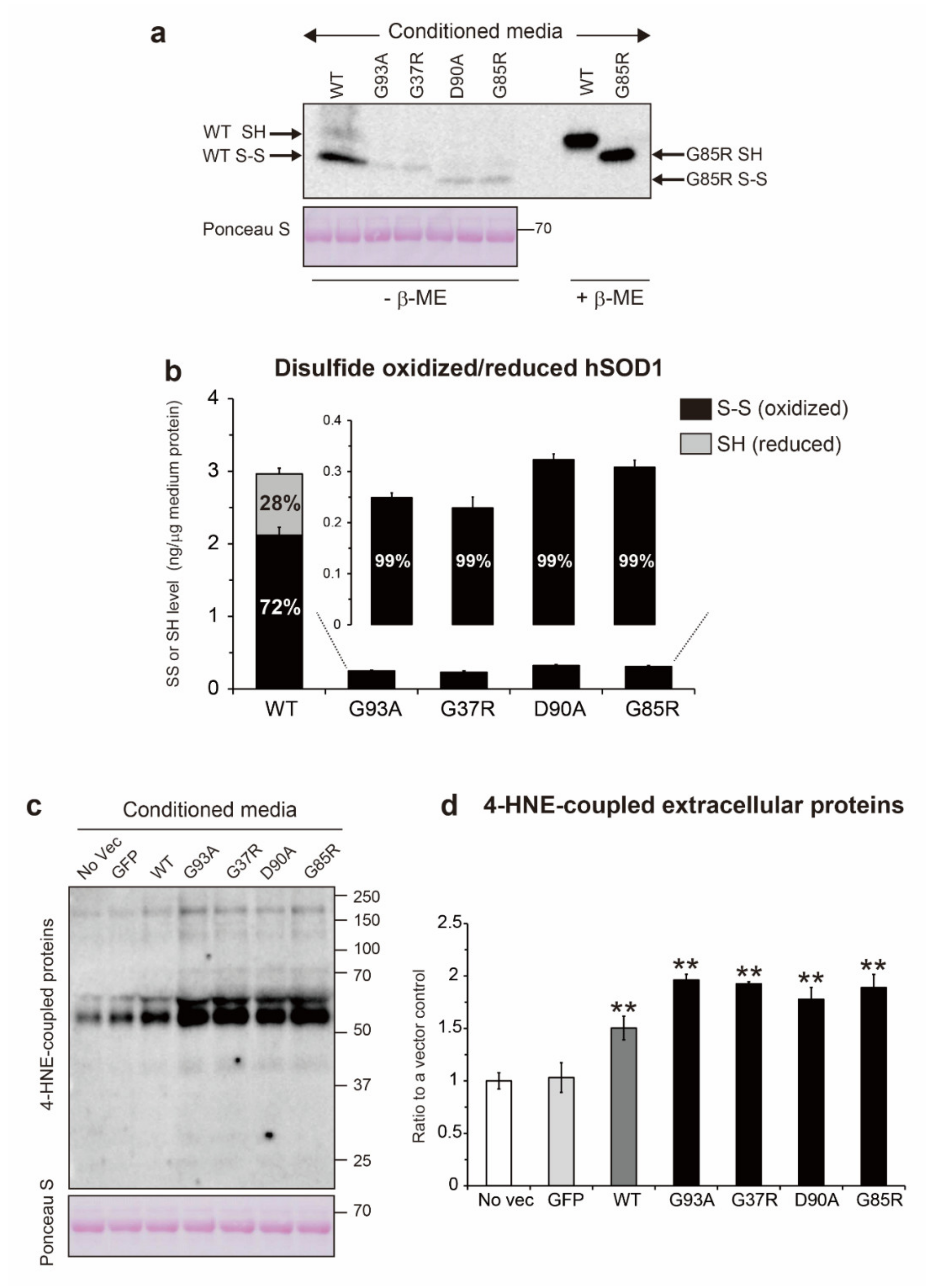
2.3. Involvement of Extracellular Oxidative Stress in the Thiol/Disulfide Redox Balance of Extracellular hSOD1
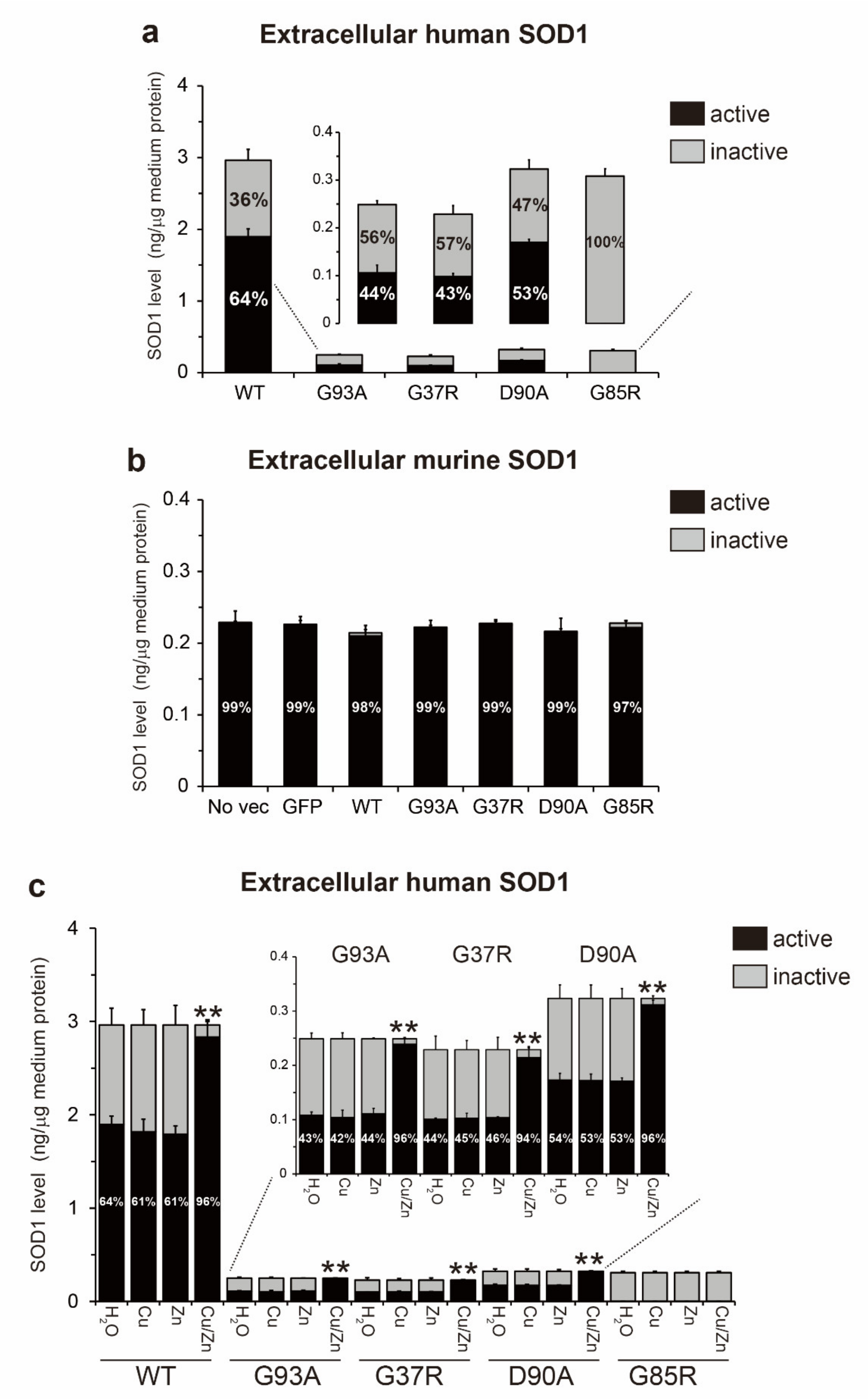
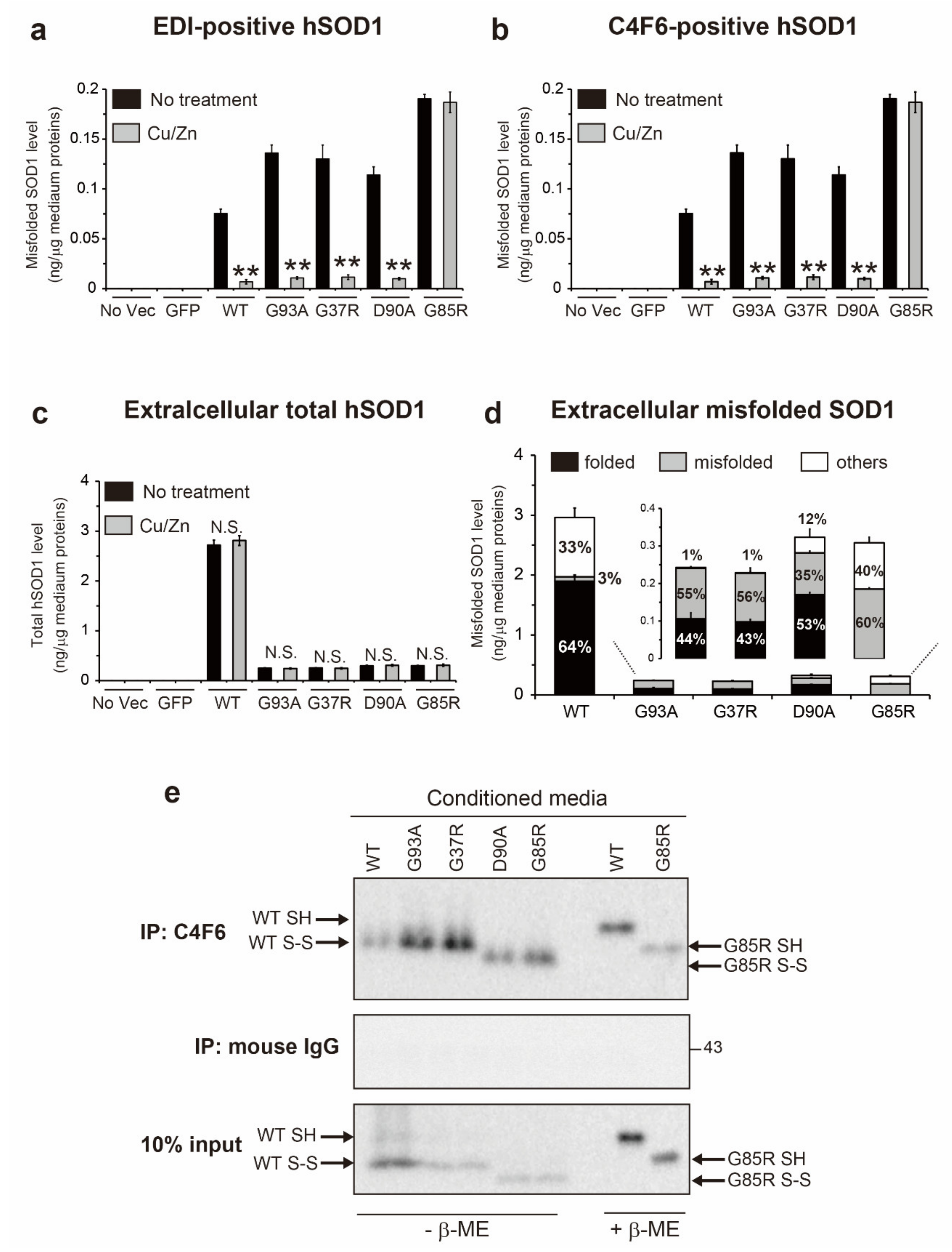
2.4. The Fraction of Non-Natively Folded Human SOD1, but not Murine SOD1, Is Increased in the Extracellular Environment
2.5. Metal-Free, Disulfide Oxidized SOD1 (apo-SOD1S-S) Is the Primary Misfolded Species in Conditioned Medium
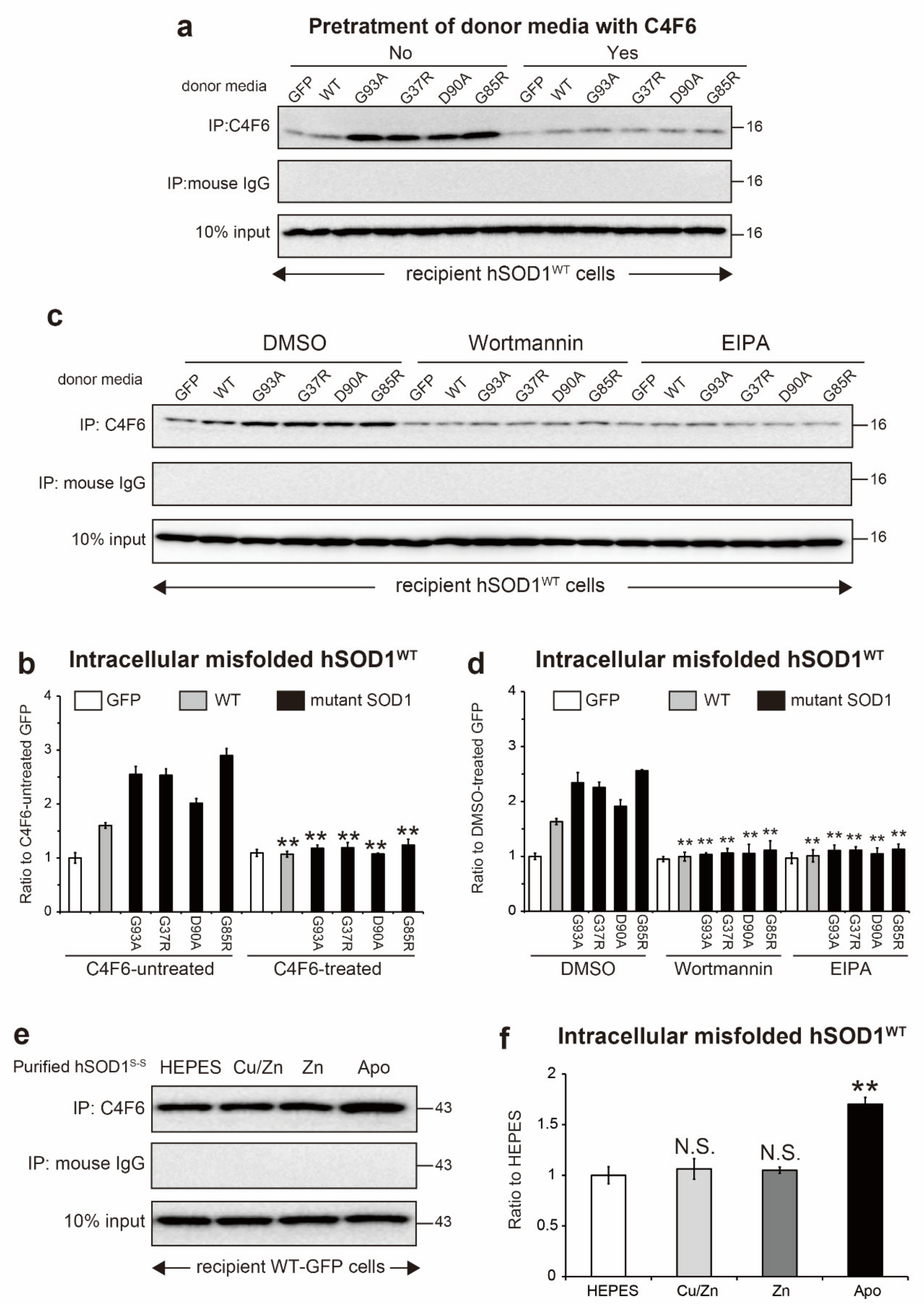
2.6. Extracellular Misfolded apo-SOD1S-S Induces Intracellular Propagation of hSOD1WT Misfolding
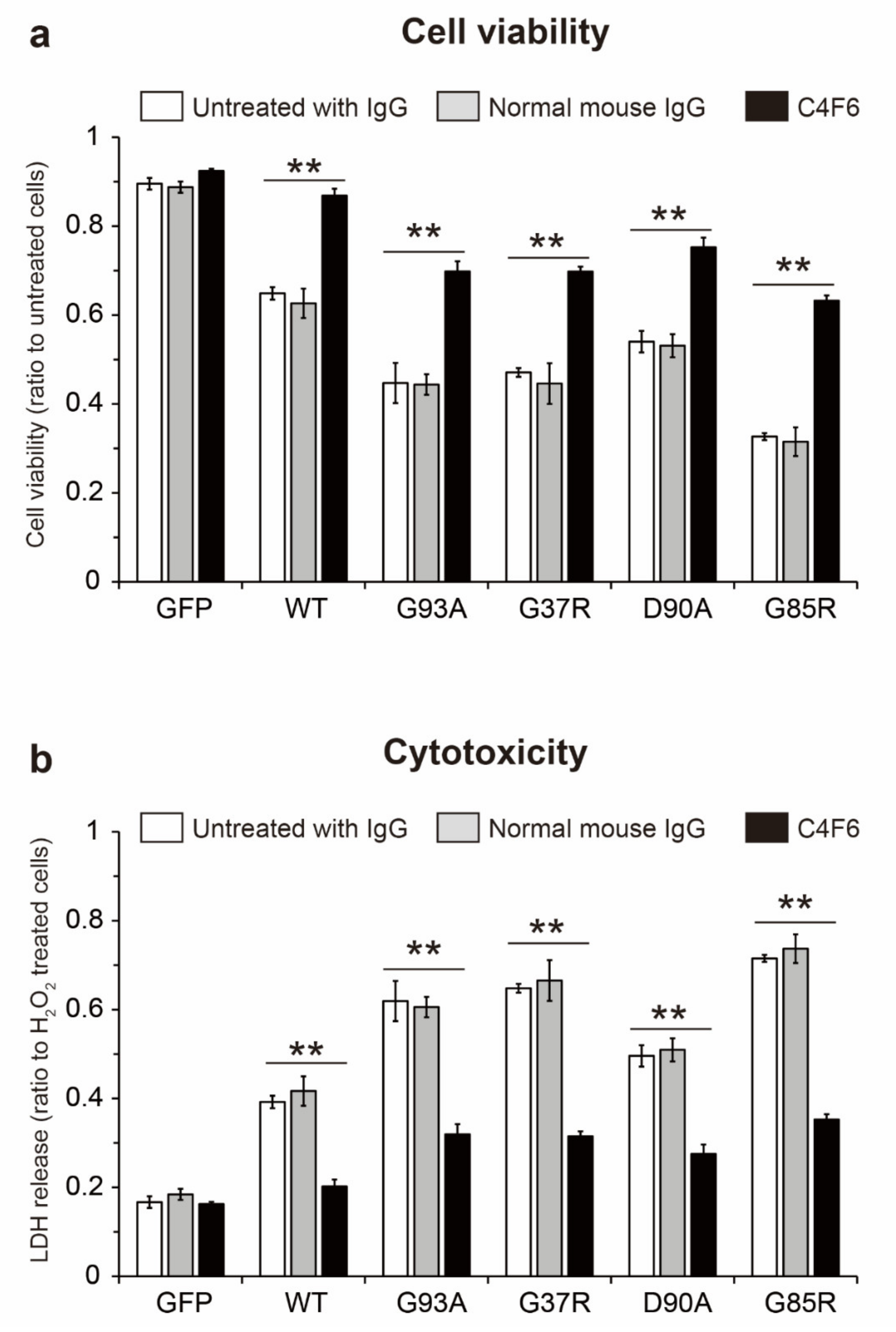
2.7. Prion-Like Propagation of Extracellular Misfolded apo-SOD1S-S Induces Cell Death in Motor Neuron-Like NSC-34 Cells
3. Discussion
4. Materials and Methods
4.1. Expression Vectors
4.2. Cell Culture and Transfection
4.3. Fluorescent Imaging
4.4. Ultrafiltration of Conditioned Medium
4.5. Protein Extraction
4.6. Western Blotting
4.7. Enzyme-Linked Immunosorbent Assay
4.8. Measurement of SOD1 Enzymatic Activity
4.9. Immunoprecipitation
4.10. Intracellular Propagation of hSOD1WT Misfolding by Conditioned Medium and Purified hSOD1S-S Proteins
4.11. Assessment of Viability and Cytotoxicity of Cells Exposed to Conditioned Medium Containing Misfolded apo-SOD1S-S
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 4-HNE | 4-Hydroxynonenal |
| ALS | Amyotrophic lateral sclerosis |
| CNS | Central nervous system |
| CSF | Cerebrospinal fluid |
| DMEM | Dulbecco’s modified Eagle’s medium |
| EIPA | 5-(N-ethyl-N-isopropyl) amiloride |
| ELISA | Enzyme-linked immunosorbent assay |
| GFP | Green fluorescent protein |
| LDH | Lactate dehydrogenase |
| HEPES | N-(2-hydroxyethyl) piperazine-N’-2-ethanesulfonic acid |
| HRP | Horseradish peroxidase |
| hSOD1 | Human superoxide dismutase 1 |
| NP-40 | Nonidet P-40 |
| PAGE | Polyacrylamide gel electrophoresis |
| PBS | Phosphate-buffered saline |
| SDS | Sodium dodecyl sulfate |
| SOD1, | Superoxide dismutase 1 |
| WT | Wild-type |
| β-ME | Beta-mercaptoethanol |
References
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef] [Green Version]
- Spires-Jones, T.L.; Attems, J.; Thal, D.R. Interactions of pathological proteins in neurodegenerative diseases. Acta Neuropathol. 2017, 134, 187–205. [Google Scholar] [CrossRef] [PubMed]
- Ravits, J.; Paul, P.; Jorg, C. Focality of upper and lower motor neuron degeneration at the clinical onset of ALS. Neurology 2007, 68, 1571–1575. [Google Scholar] [CrossRef] [PubMed]
- Vallabh, S.M.; Minikel, E.V.; Schreiber, S.L.; Lander, E.S. Towards a treatment for genetic prion disease: Trials and biomarkers. Lancet Neurol. 2020, 19, 361–368. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Münch, C.; O’Brien, J.; Bertolotti, A. Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proc. Natl. Acad. Sci. USA 2011, 108, 3548–3553. [Google Scholar] [CrossRef] [Green Version]
- Grad, L.I.; Yerbury, J.J.; Turner, B.J.; Guest, W.C.; Pokrishevsky, E.; O’Neill, M.A.; Yanai, A.; Silverman, J.M.; Zeineddine, R.; Corcoran, L.; et al. Intercellular propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-dependent and -independent mechanisms. Proc. Natl. Acad. Sci. USA 2014, 111, 3620–3625. [Google Scholar] [CrossRef] [Green Version]
- Grad, L.I.; Guest, W.C.; Yanai, A.; Pokrishevsky, E.; O’Neill, M.A.; Gibbs, E.; Semenchenko, V.; Yousefi, M.; Wishart, D.S.; Plotkin, S.S.; et al. Intermolecular transmission of superoxide dismutase 1 misfolding in living cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16398–16403. [Google Scholar] [CrossRef] [Green Version]
- Zeineddine, R.; Pundavela, J.F.; Corcoran, L.; Stewart, E.M.; Do-Ha, D.; Bax, M.; Guillemin, G.; Vine, K.L.; Hatters, D.M.; Ecroyd, H.; et al. SOD1 protein aggregates stimulate macropinocytosis in neurons to facilitate their propagation. Mol. Neurodegener. 2015, 10, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayers, J.I.; Fromholt, S.; Koch, M.; DeBosier, A.; McMahon, B.; Xu, G.; Borchelt, D.R. Experimental transmissibility of mutant SOD1 motor neuron disease. Acta Neuropathol. 2014, 128, 791–803. [Google Scholar] [CrossRef] [PubMed]
- Ayers, J.I.; Diamond, J.; Sari, A.; Fromholt, S.; Galaleldeen, A.; Ostrow, L.W.; Glass, J.D.; Hart, P.J.; Borchelt, D.R. Distinct conformers of transmissible misfolded SOD1 distinguish human SOD1-FALS from other forms of familial and sporadic ALS. Acta Neuropathol. 2016, 132, 827–840. [Google Scholar] [CrossRef] [Green Version]
- Ayers, J.I.; Fromholt, S.E.; O’Neal, V.M.; Diamond, J.H.; Borchelt, D.R. Prion-like propagation of mutant SOD1 misfolding and motor neuron disease spread along neuroanatomical pathways. Acta Neuropathol. 2016, 131, 103–114. [Google Scholar] [CrossRef]
- Bidhendi, E.E.; Bergh, J.; Zetterström, P.; Andersen, P.M.; Marklund, S.L.; Brännström, T. Two superoxide dismutase prion strains transmit amyotrophic lateral sclerosis-like disease. J. Clin. Investig. 2016, 126, 2249–2253. [Google Scholar] [CrossRef]
- Ekhtiari Bidhendi, E.; Bergh, J.; Zetterström, P.; Forsberg, K.; Pakkenberg, B.; Andersen, P.M.; Marklund, S.L.; Brännström, T. Mutant superoxide dismutase aggregates from human spinal cord transmit amyotrophic lateral sclerosis. Acta Neuropathol. 2018, 136, 939–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokuda, E.; Nomura, T.; Ohara, S.; Watanabe, S.; Yamanaka, K.; Morisaki, Y.; Misawa, H.; Furukawa, Y. A copper-deficient form of mutant Cu/Zn-superoxide dismutase as an early pathological species in amyotrophic lateral sclerosis. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2119–2130. [Google Scholar] [CrossRef]
- Tokuda, E.; Takei, Y.I.; Ohara, S.; Fujiwara, N.; Hozumi, I.; Furukawa, Y. Wild-type Cu/Zn-superoxide dismutase is misfolded in cerebrospinal fluid of sporadic amyotrophic lateral sclerosis. Mol. Neurodegener. 2019, 14, 42. [Google Scholar] [CrossRef] [Green Version]
- Ng Kee Kwong, K.C.; Mehta, A.R.; Nedergaard, M.; Chandran, S. Defining novel functions for cerebrospinal fluid in ALS pathophysiology. Acta Neuropathol. Commun. 2020, 8, 140. [Google Scholar] [CrossRef] [PubMed]
- Urushitani, M.; Sik, A.; Sakurai, T.; Nukina, N.; Takahashi, R.; Julien, J.P. Chromogranin-mediated secretion of mutant superoxide dismutase proteins linked to amyotrophic lateral sclerosis. Nat. Neurosci. 2006, 9, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, P.A.; Graffmo, K.S.; Andersen, P.M.; Brännström, T.; Lindberg, M.; Oliveberg, M.; Marklund, S.L. Disulphide-reduced superoxide dismutase-1 in CNS of transgenic amyotrophic lateral sclerosis models. Brain 2006, 129, 451–464. [Google Scholar] [CrossRef] [Green Version]
- Hayward, L.J.; Rodriguez, J.A.; Kim, J.W.; Tiwari, A.; Goto, J.J.; Cabelli, D.E.; Valentine, J.S.; Brown, R.H., Jr. Decreased metallation and activity in subsets of mutant superoxide dismutases associated with familial amyotrophic lateral sclerosis. J. Biol. Chem. 2002, 277, 15923–15931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, G.S.A.; Antonyuk, S.V.; Hasnain, S.S. The biophysics of superoxide dismutase-1 and amyotrophic lateral sclerosis. Q. Rev. Biophys. 2019, 52, e12. [Google Scholar] [CrossRef]
- Borchelt, D.R.; Lee, M.K.; Slunt, H.S.; Guarnieri, M.; Xu, Z.S.; Wong, P.C.; Brown, R.H., Jr.; Price, D.L.; Sisodia, S.S.; Cleveland, D.W. Superoxide dismutase 1 with mutations linked to familial amyotrophic lateral sclerosis possesses significant activity. Proc. Natl. Acad. Sci. USA 1994, 91, 8292–8296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marklund, S.L.; Andersen, P.M.; Forsgren, L.; Nilsson, P.; Ohlsson, P.I.; Wikander, G.; Oberg, A. Normal binding and reactivity of copper in mutant superoxide dismutase isolated from amyotrophic lateral sclerosis patients. J. Neurochem. 1997, 69, 675–681. [Google Scholar] [CrossRef] [Green Version]
- Rakhit, R.; Robertson, J.; Vande Velde, C.; Horne, P.; Ruth, D.M.; Griffin, J.; Cleveland, D.W.; Cashman, N.R.; Chakrabartty, A. An immunological epitope selective for pathological monomer-misfolded SOD1 in ALS. Nat. Med. 2007, 13, 754–759. [Google Scholar] [CrossRef]
- Ayers, J.I.; Xu, G.; Pletnikova, O.; Troncoso, J.C.; Hart, P.J.; Borchelt, D.R. Conformational specificity of the C4F6 SOD1 antibody; low frequency of reactivity in sporadic ALS cases. Acta Neuropathol. Commun. 2014, 2, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotunno, M.S.; Auclair, J.R.; Maniatis, S.; Shaffer, S.A.; Agar, J.; Bosco, D.A. Identification of a misfolded region in superoxide dismutase 1 that is exposed in amyotrophic lateral sclerosis. J. Biol. Chem. 2014, 289, 28527–28538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindberg, M.J.; Tibell, L.; Oliveberg, M. Common denominator of Cu/Zn superoxide dismutase mutants associated with amyotrophic lateral sclerosis: Decreased stability of the apo state. Proc. Natl. Acad. Sci. USA 2002, 99, 16607–16612. [Google Scholar] [CrossRef] [Green Version]
- Turner, B.J.; Atkin, J.D.; Farg, M.A.; Zang, D.W.; Rembach, A.; Lopes, E.C.; Patch, J.D.; Hill, A.F.; Cheema, S.S. Impaired extracellular secretion of mutant superoxide dismutase 1 associates with neurotoxicity in familial amyotrophic lateral sclerosis. J. Neurosci. 2005, 25, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, A.; Hayward, L.J. Familial amyotrophic lateral sclerosis mutants of copper/zinc superoxide dismutase are susceptible to disulfide reduction. J. Biol. Chem. 2003, 278, 5984–5992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furukawa, Y.; O’Halloran, T.V. Amyotrophic lateral sclerosis mutations have the greatest destabilizing effect on the apo- and reduced form of SOD1, leading to unfolding and oxidative aggregation. J. Biol. Chem. 2005, 280, 17266–17274. [Google Scholar] [CrossRef] [Green Version]
- Di Domenico, F.; Tramutola, A.; Butterfield, D.A. Role of 4-hydroxy-2-nonenal (HNE) in the pathogenesis of alzheimer disease and other selected age-related neurodegenerative disorders. Free Radic. Biol. Med. 2017, 111, 253–261. [Google Scholar] [CrossRef]
- Rae, T.D.; Torres, A.S.; Pufahl, R.A.; O’Halloran, T.V. Mechanism of Cu,Zn-superoxide dismutase activation by the human metallochaperone hCCS. J. Biol. Chem. 2001, 276, 5166–5176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.T.; Jiao, M.; Chen, J.; Liang, Y. Roles of zinc and copper in modulating the oxidative refolding of bovine copper, zinc superoxide dismutase. Acta Biochim. Biophys. Sin. 2010, 42, 183–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, W.A.; Fu, W.; Keller, J.N.; Markesbery, W.R.; Appel, S.; Smith, R.G.; Kasarskis, E.; Mattson, M.P. Protein modification by the lipid peroxidation product 4-hydroxynonenal in the spinal cords of amyotrophic lateral sclerosis patients. Ann. Neurol. 1998, 44, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Tohgi, H.; Abe, T.; Yamazaki, K.; Murata, T.; Ishizaki, E.; Isobe, C. Remarkable increase in cerebrospinal fluid 3-nitrotyrosine in patients with sporadic amyotrophic lateral sclerosis. Ann. Neurol. 1999, 46, 129–131. [Google Scholar] [CrossRef]
- Xu, W.C.; Liang, J.Z.; Li, C.; He, Z.X.; Yuan, H.Y.; Huang, B.Y.; Liu, X.L.; Tang, B.; Pang, D.W.; Du, H.N.; et al. Pathological hydrogen peroxide triggers the fibrillization of wild-type SOD1 via sulfenic acid modification of Cys-111. Cell Death Dis. 2018, 9, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anzai, I.; Tokuda, E.; Handa, S.; Misawa, H.; Akiyama, S.; Furukawa, Y. Oxidative misfolding of Cu/Zn-superoxide dismutase triggered by non-canonical intramolecular disulfide formation. Free Radic. Biol. Med. 2020, 147, 187–199. [Google Scholar] [CrossRef]
- Chattopadhyay, M.; Nwadibia, E.; Strong, C.D.; Gralla, E.B.; Valentine, J.S.; Whitelegge, J.P. The Disulfide Bond, but Not Zinc or Dimerization, Controls Initiation and Seeded Growth in Amyotrophic Lateral Sclerosis-linked Cu,Zn Superoxide Dismutase (SOD1) Fibrillation. J. Biol. Chem. 2015, 290, 30624–30636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonsson, P.A.; Ernhill, K.; Andersen, P.M.; Bergemalm, D.; Brännström, T.; Gredal, O.; Nilsson, P.; Marklund, S.L. Minute quantities of misfolded mutant superoxide dismutase-1 cause amyotrophic lateral sclerosis. Brain 2004, 127, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Sundaramoorthy, V.; Walker, A.K.; Yerbury, J.; Soo, K.Y.; Farg, M.A.; Hoang, V.; Zeineddine, R.; Spencer, D.; Atkin, J.D. Extracellular wildtype and mutant SOD1 induces ER-Golgi pathology characteristic of amyotrophic lateral sclerosis in neuronal cells. Cell Mol. Life Sci. 2013, 70, 4181–4195. [Google Scholar] [CrossRef] [Green Version]
- Benkler, C.; O’Neil, A.L.; Slepian, S.; Qian, F.; Weinreb, P.H.; Rubin, L.L. Aggregated SOD1 causes selective death of cultured human motor neurons. Sci. Rep. 2018, 8, 16393. [Google Scholar] [CrossRef] [PubMed]
- Hwang, C.; Sinskey, A.J.; Lodish, H.F. Oxidized redox state of glutathione in the endoplasmic reticulum. Science 1992, 257, 1496–1502. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, Y.; Kaneko, K.; Yamanaka, K.; O’Halloran, T.V.; Nukina, N. Complete loss of post-translational modifications triggers fibrillar aggregation of SOD1 in the familial form of amyotrophic lateral sclerosis. J. Biol. Chem. 2008, 283, 24167–24176. [Google Scholar] [CrossRef] [Green Version]
- Chattopadhyay, M.; Durazo, A.; Sohn, S.H.; Strong, C.D.; Gralla, E.B.; Whitelegge, J.P.; Valentine, J.S. Initiation and elongation in fibrillation of ALS-linked superoxide dismutase. Proc. Natl. Acad. Sci. USA 2008, 105, 18663–18668. [Google Scholar] [CrossRef] [Green Version]
- Urushitani, M.; Ezzi, S.A.; Julien, J.P. Therapeutic effects of immunization with mutant superoxide dismutase in mice models of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2007, 104, 2495–2500. [Google Scholar] [CrossRef] [Green Version]
- Johansson, A.S.; Vestling, M.; Zetterström, P.; Lang, L.; Leinartaitė, L.; Karlström, M.; Danielsson, J.; Marklund, S.L.; Oliveberg, M. Cytotoxicity of superoxide dismutase 1 in cultured cells is linked to Zn2+ chelation. PLoS ONE 2012, 7, e36104. [Google Scholar] [CrossRef] [Green Version]
- Homma, K.; Fujisawa, T.; Tsuburaya, N.; Yamaguchi, N.; Kadowaki, H.; Takeda, K.; Nishitoh, H.; Matsuzawa, A.; Naguro, I.; Ichijo, H. SOD1 as a molecular switch for initiating the homeostatic ER stress response under zinc deficiency. Mol. Cell 2013, 52, 75–86. [Google Scholar] [CrossRef] [Green Version]
- Sekhar, A.; Rumfeldt, J.A.; Broom, H.R.; Doyle, C.M.; Bouvignies, G.; Meiering, E.M.; Kay, L.E. Thermal fluctuations of immature SOD1 lead to separate folding and misfolding pathways. Elife 2015, 4, e07296. [Google Scholar] [CrossRef] [PubMed]
- Wada, K.; Fujibayashi, Y.; Tajima, N.; Yokoyama, A. Cu-ATSM, an intracellular-accessible superoxide dismutase (SOD)-like copper complex: Evaluation in an ischemia-reperfusion injury model. Biol. Pharm. Bull. 1994, 17, 701–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrawell, N.E.; Yerbury, M.R.; Plotkin, S.S.; McAlary, L.; Yerbury, J.J. CuATSM Protects Against the In Vitro Cytotoxicity of Wild-Type-Like Copper-Zinc Superoxide Dismutase Mutants but not Mutants That Disrupt Metal Binding. ACS Chem. Neurosci. 2019, 10, 1555–1564. [Google Scholar] [CrossRef] [PubMed]
- Roberts, B.R.; Lim, N.K.; McAllum, E.J.; Donnelly, P.S.; Hare, D.J.; Doble, P.A.; Turner, B.J.; Price, K.A.; Lim, S.C.; Paterson, B.M.; et al. Oral treatment with Cu(II)(atsm) increases mutant SOD1 in vivo but protects motor neurons and improves the phenotype of a transgenic mouse model of amyotrophic lateral sclerosis. J. Neurosci. 2014, 34, 8021–8031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soon, C.P.; Donnelly, P.S.; Turner, B.J.; Hung, L.W.; Crouch, P.J.; Sherratt, N.A.; Tan, J.L.; Lim, N.K.; Lam, L.; Bica, L.; et al. Diacetylbis(N(4)-methylthiosemicarbazonato) copper(II) (CuII(atsm)) protects against peroxynitrite-induced nitrosative damage and prolongs survival in amyotrophic lateral sclerosis mouse model. J. Biol. Chem. 2011, 286, 44035–44044. [Google Scholar] [CrossRef] [Green Version]
- McAllum, E.J.; Lim, N.K.; Hickey, J.L.; Paterson, B.M.; Donnelly, P.S.; Li, Q.X.; Liddell, J.R.; Barnham, K.J.; White, A.R.; Crouch, P.J. Therapeutic effects of CuII(atsm) in the SOD1-G37R mouse model of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Frontotemporal Degener. 2013, 14, 586–590. [Google Scholar] [CrossRef]
- Cashman, N.R.; Durham, H.D.; Blusztajn, J.K.; Oda, K.; Tabira, T.; Shaw, I.T.; Dahrouge, S.; Antel, J.P. Neuroblastoma x spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev. Dyn. 1992, 194, 209–221. [Google Scholar] [CrossRef]
- Furukawa, Y.; Fu, R.; Deng, H.X.; Siddique, T.; O’Halloran, T.V. Disulfide cross-linked protein represents a significant fraction of ALS-associated Cu, Zn-superoxide dismutase aggregates in spinal cords of model mice. Proc. Natl. Acad. Sci. USA 2006, 103, 7148–7153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokuda, E.; Anzai, I.; Nomura, T.; Toichi, K.; Watanabe, M.; Ohara, S.; Watanabe, S.; Yamanaka, K.; Morisaki, Y.; Misawa, H.; et al. Immunochemical characterization on pathological oligomers of mutant Cu/Zn-superoxide dismutase in amyotrophic lateral sclerosis. Mol. Neurodegener. 2017, 12, 2. [Google Scholar] [CrossRef] [Green Version]
- Rigo, A.; Viglino, P.; Rotilio, G. Kinetic study of O-2 DISMUTATION BY BOVINE SUPEROXIDE DISMUTASE. Evidence for saturation of the catalytic sites by O-2. Biochem. Biophys. Res. Commun. 1975, 63, 1013–1018. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takashima, C.; Kosuge, Y.; Inoue, M.; Ono, S.-I.; Tokuda, E. A Metal-Free, Disulfide Oxidized Form of Superoxide Dismutase 1 as a Primary Misfolded Species with Prion-Like Properties in the Extracellular Environments Surrounding Motor Neuron-Like Cells. Int. J. Mol. Sci. 2021, 22, 4155. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084155
Takashima C, Kosuge Y, Inoue M, Ono S-I, Tokuda E. A Metal-Free, Disulfide Oxidized Form of Superoxide Dismutase 1 as a Primary Misfolded Species with Prion-Like Properties in the Extracellular Environments Surrounding Motor Neuron-Like Cells. International Journal of Molecular Sciences. 2021; 22(8):4155. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084155
Chicago/Turabian StyleTakashima, Chika, Yasuhiro Kosuge, Masahisa Inoue, Shin-Ichi Ono, and Eiichi Tokuda. 2021. "A Metal-Free, Disulfide Oxidized Form of Superoxide Dismutase 1 as a Primary Misfolded Species with Prion-Like Properties in the Extracellular Environments Surrounding Motor Neuron-Like Cells" International Journal of Molecular Sciences 22, no. 8: 4155. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084155