
miR-200c-3p Regulates Epitelial-to-Mesenchymal Transition in Epicardial Mesothelial Cells by Targeting Epicardial Follistatin-Related Protein 1

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
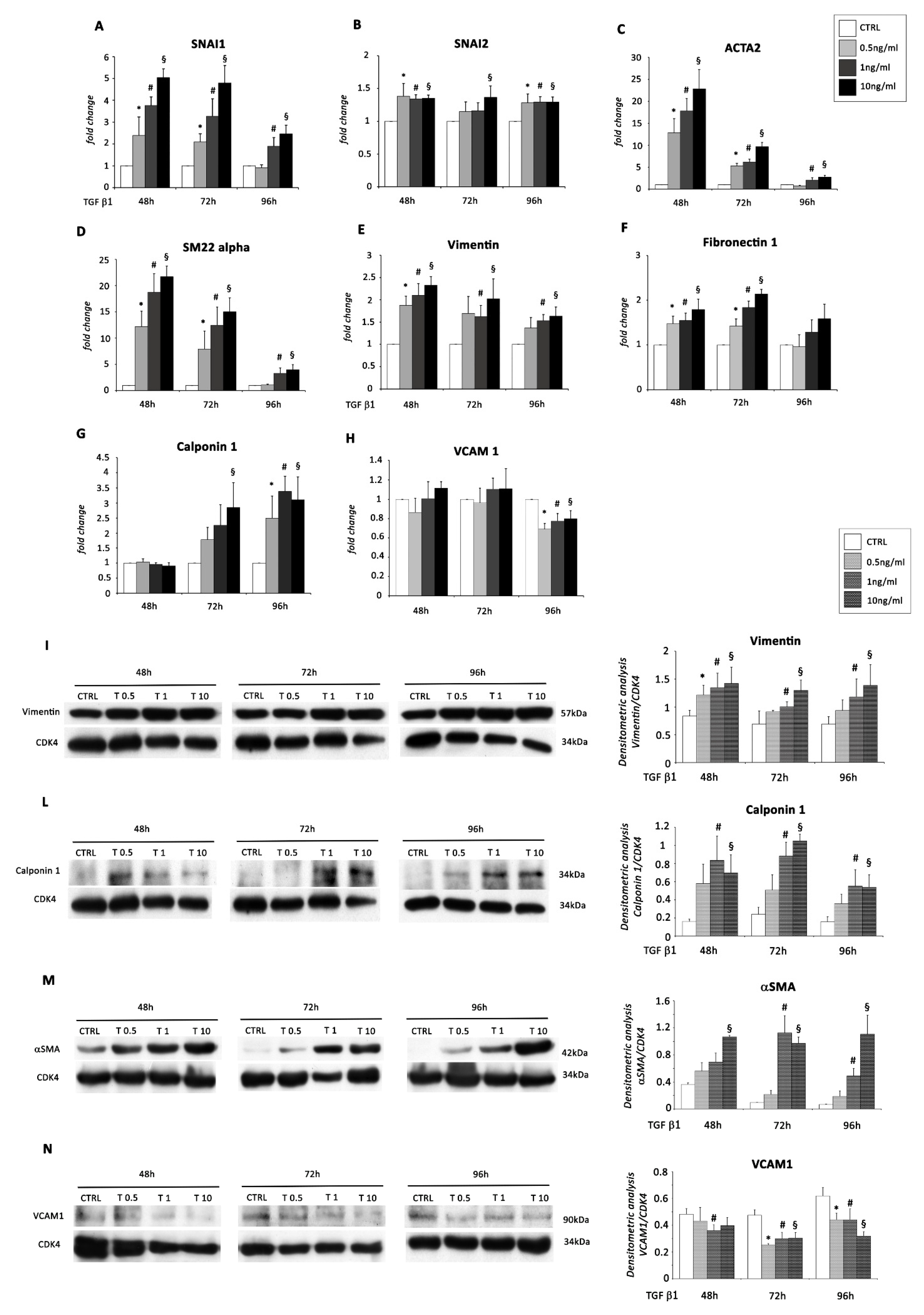
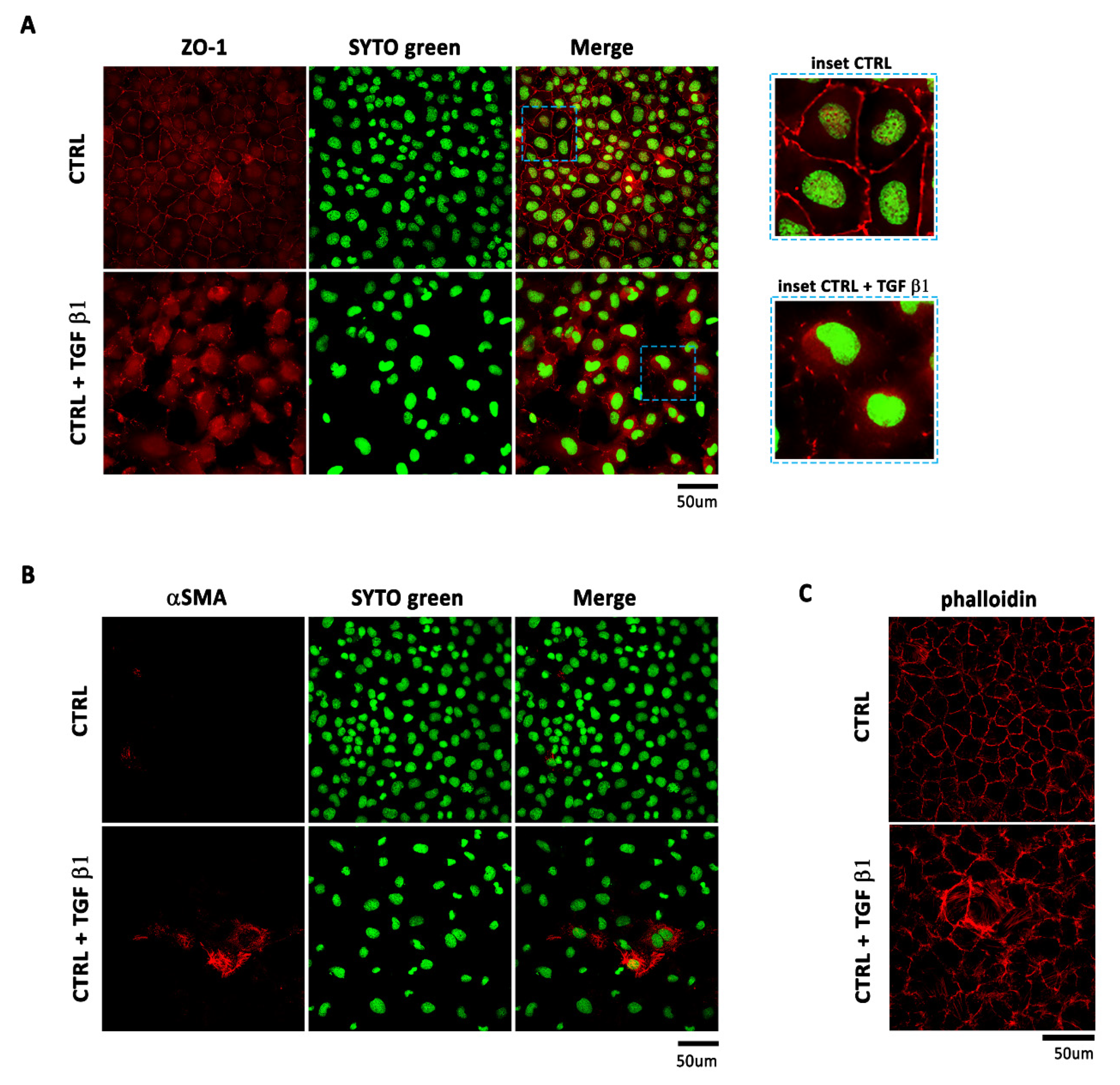
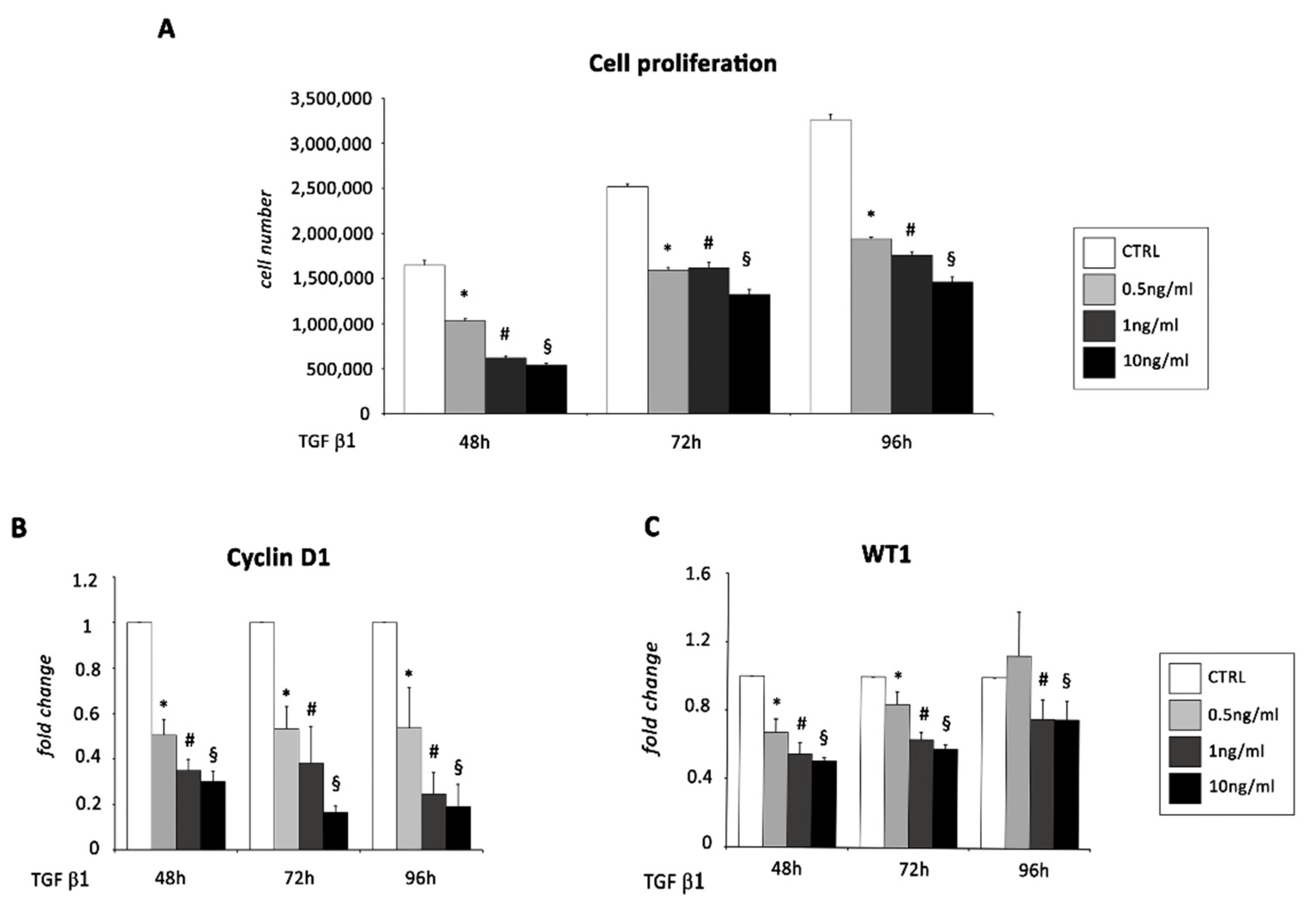
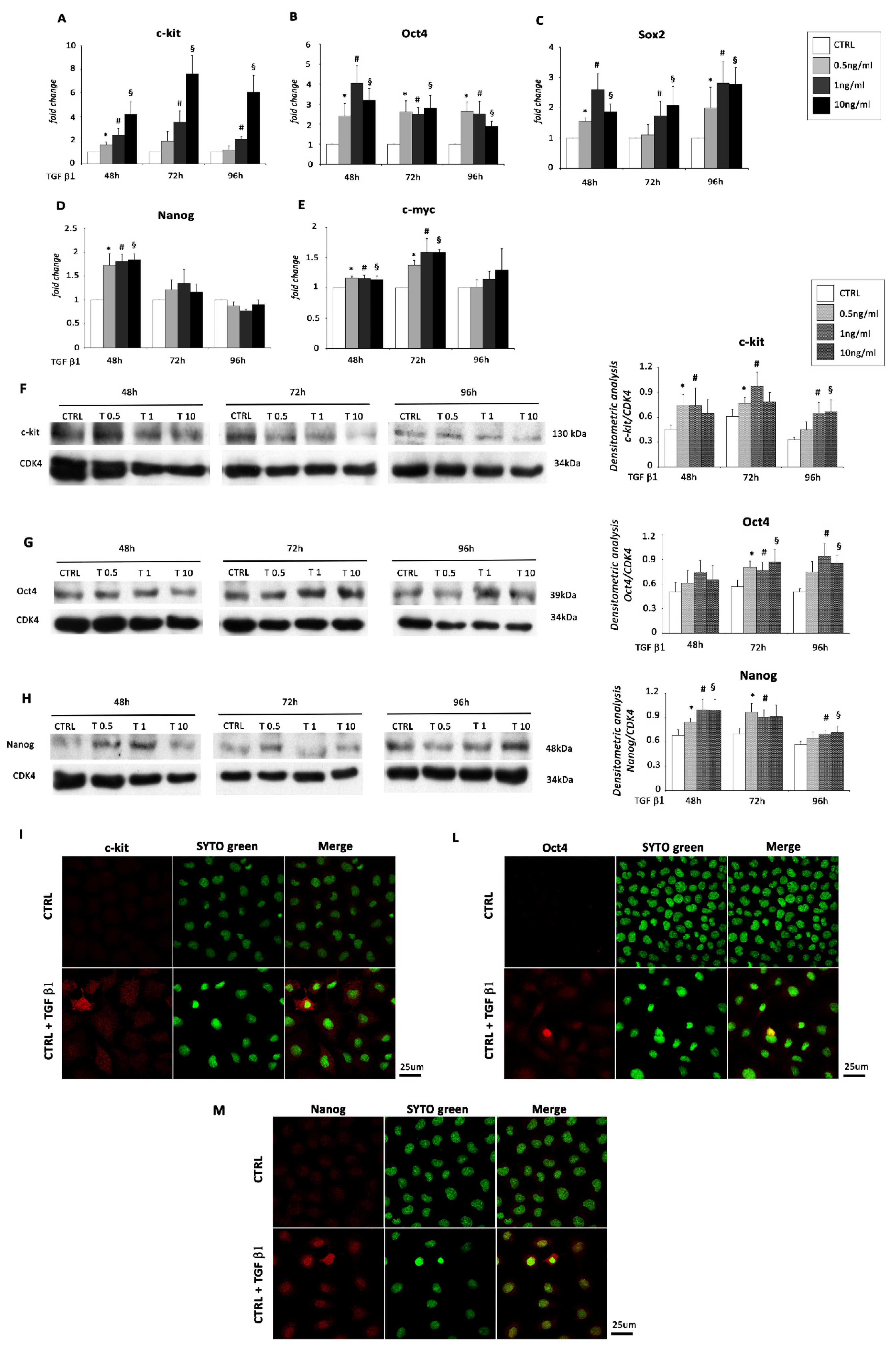
2.1. TGF β1 Treatment Induces Mesenchyme Formation and Stem Cell-Like Acquisition in EMCs
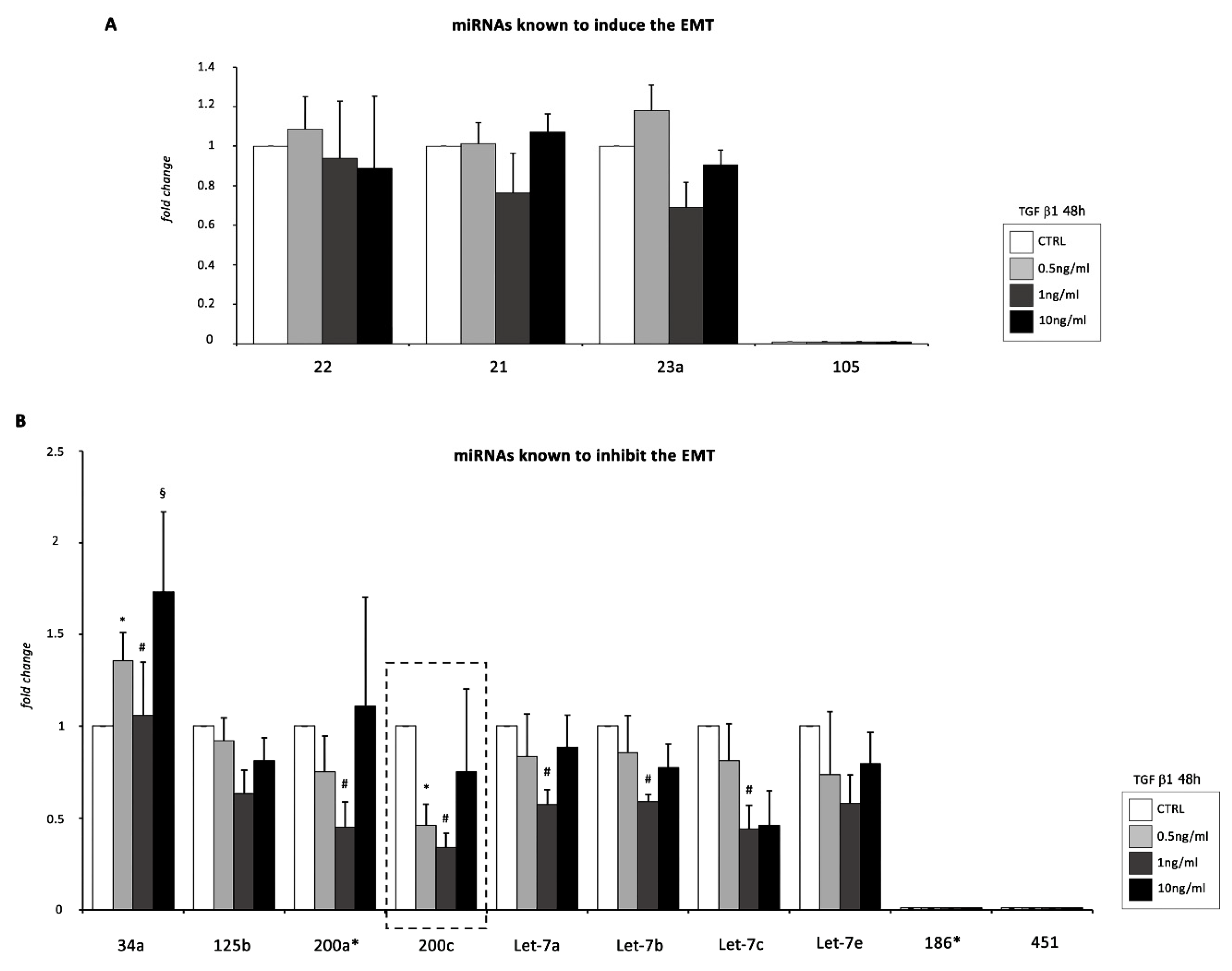
2.2. miR-200c Is the Major Modulated miRNA during TGF β1-Mediated EMT of EMCs
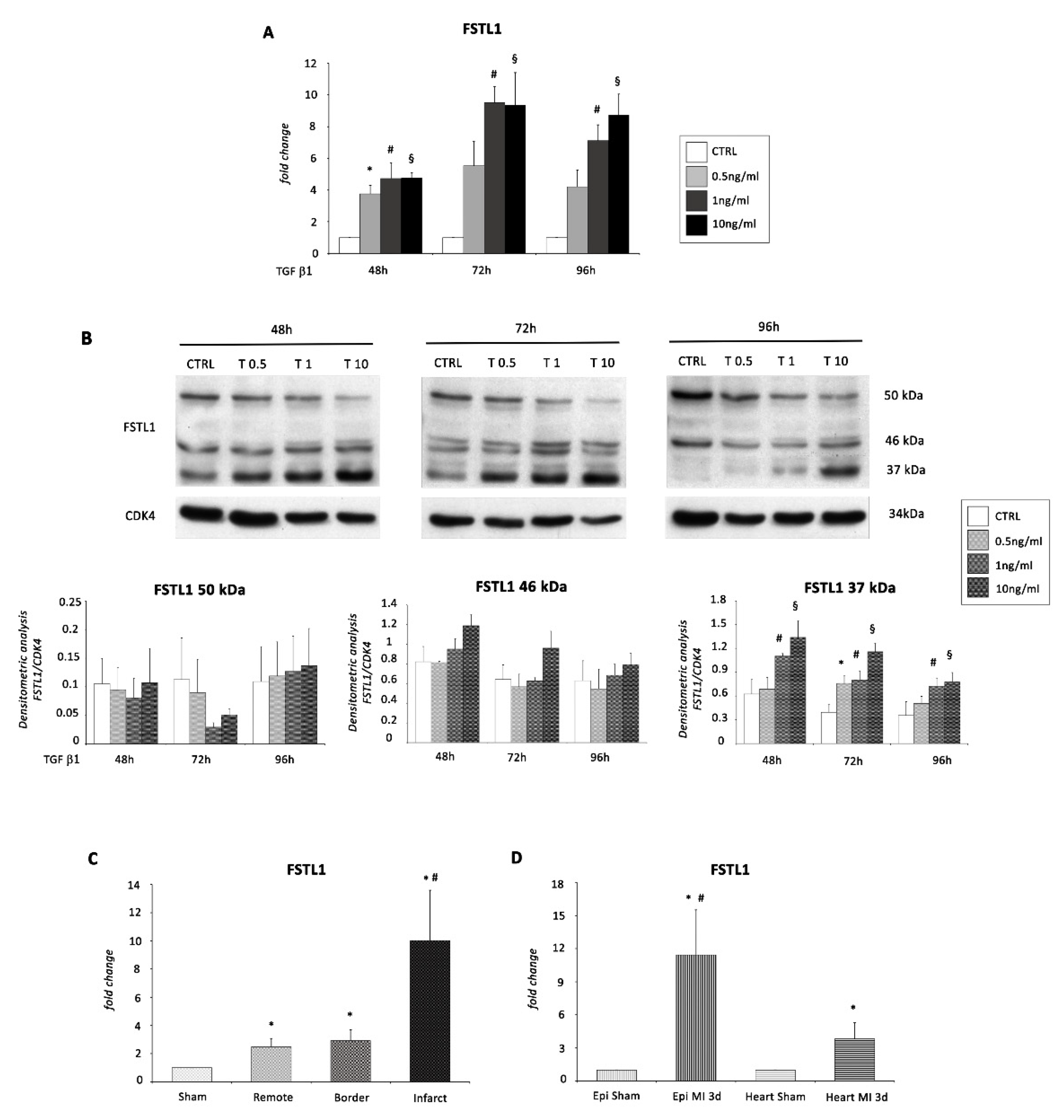
2.3. FSTL1 Is Induced in TGF β1-Mediated EMT of EMCs
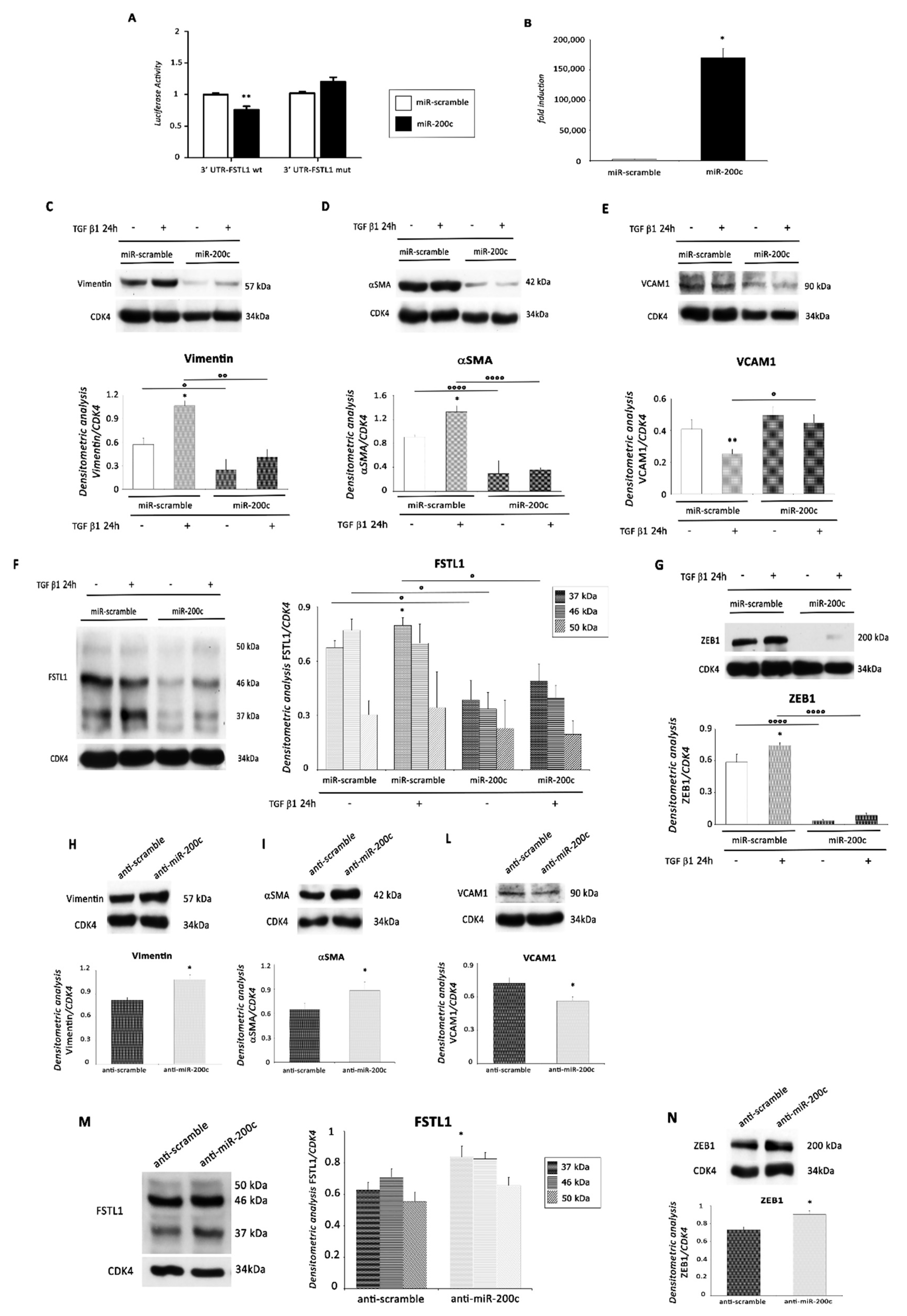
2.4. miR-200c Inhibits EMT by Targeting FSTL1
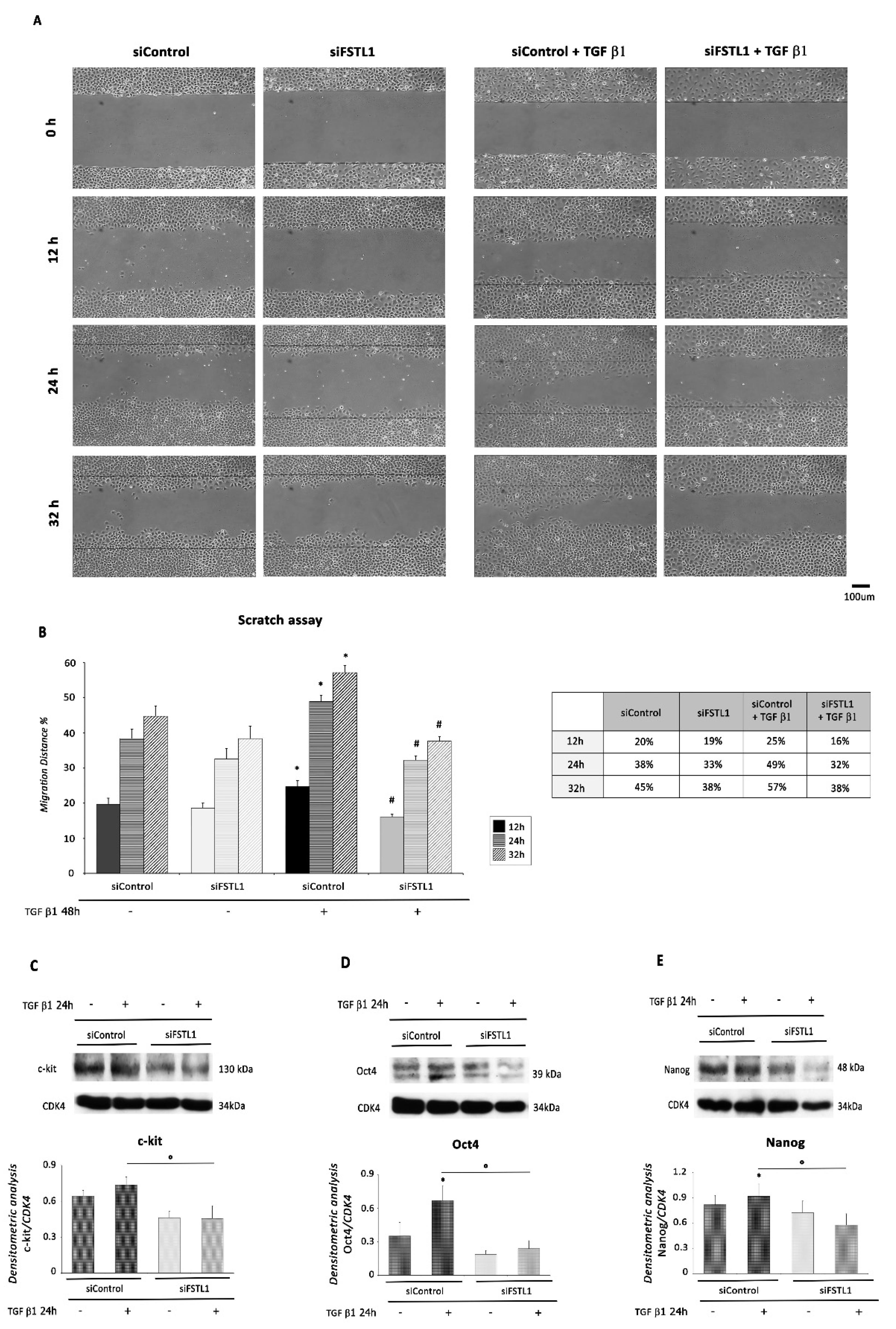
2.5. FSTL1 Is Required and Sufficient for the Acquisition of EMT-Associated and Stem-Like Traits
3. Discussion
4. Materials and Methods
4.1. Animal Model and In Vivo Study
4.2. Epicardial Cell Isolation
4.3. Cell Culture and Treatment Protocols
4.3.1. TGF β1 Treatment
4.3.2. Stable Modulation of miR200c Expression
4.3.3. Generation of FLST1-Silenced EMCs
4.4. In Vitro Cell Proliferation Assay
4.5. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
4.5.1. Total RNA Isolation and Quantification
4.5.2. Quantitative Gene Expression Analysis
4.5.3. Quantitative Mature miRNA Expression Analysis
4.6. Western Blot Assay
4.7. Immunofluorescence
4.8. Luciferase Assay
4.9. Scratch Assay
4.10. Data Collections and Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tsai, J.H.; Yang, J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013, 27, 2192–2206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forte, E.; Miraldi, F.; Chimenti, I.; Angelini, F.; Zeuner, A.; Giacomello, A.; Mercola, M.; Messina, E. TGFbeta-dependent epithelial-to-mesenchymal transition is required to generate cardiospheres from human adult heart biopsies. Stem Cells Dev. 2012, 21, 3081–3090. [Google Scholar] [CrossRef] [Green Version]
- Foglio, E.; Puddighinu, G.; Fasanaro, P.; D’Arcangelo, D.; Perrone, G.A.; Mocini, D.; Campanella, C.; Coppola, L.; Logozzi, M.; Azzarito, T. Exosomal clusterin, identified in the pericardial fluid, improves myocardial performance following MI through epicardial activation, enhanced arteriogenesis and reduced apoptosis. Int. J. Cardiol. 2015, 197, 333–347. [Google Scholar] [CrossRef]
- Castaldo, C.; Di Meglio, F.; Nurzynska, D.; Romano, G.; Maiello, C.; Bancone, C.; Müller, P.; Böhm, M.; Cotrufo, M.; Montagnani, S. CD117-Positive Cells in Adult Human Heart Are Localized in the Subepicardium, and Their Activation Is Associated with Laminin-1 and α6Integrin Expression. Stem Cells 2008, 26, 1723–1731. [Google Scholar] [CrossRef] [PubMed]
- Di Meglio, F.; Castaldo, C.; Nurzynska, D.; Romano, V.; Miraglia, R.; Montagnani, S. Epicardial cells are missing from the surface of hearts with ischemic cardiomyopathy: A useful clue about the self-renewal potential of the adult human heart? Int. J. Cardiol. 2010, 145, e44–e46. [Google Scholar] [CrossRef]
- Limana, F.; Bertolami, C.; Mangoni, A.; Di Carlo, A.; Avitabile, D.; Mocini, D.; Iannelli, P.; De Mori, R.; Marchetti, C.; Pozzoli, O.; et al. Myocardial infarction induces embryonic reprogramming of epicardial c-kit+ cells: Role of the pericardial fluid. J. Mol. Cell. Cardiol. 2010, 48, 609–618. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boon, R.A.; Dimmeler, S. MicroRNAs in myocardial infarction. Nat. Rev. Cardiol. 2015, 12, 135–142. [Google Scholar] [CrossRef]
- Sun, T.; Dong, Y.-H.; Du, W.; Shi, C.-Y.; Wang, K.; Tariq, M.-A.; Wang, J.-X.; Li, P.-F. The Role of MicroRNAs in Myocardial Infarction: From Molecular Mechanism to Clinical Application. Int. J. Mol. Sci. 2017, 18, 745. [Google Scholar] [CrossRef] [Green Version]
- Musavi Shenas, M.H.; Eghbal-Fard, S.; Mehrisofiani, V.; Yazdani, N.A.; Farzam, O.R.; Marofi, F.; Yousefi, M. MicroRNAs and signaling networks involved in epithelial-mesenchymal transition. J. Cell Physiol 2019, 234, 5775–5785. [Google Scholar] [CrossRef]
- Bronnum, H.; Andersen, D.C.; Schneider, M.; Nossent, A.Y.; Nielsen, S.B.; Sheikh, S.P. Islet-1 is a dual regulator of fibrogenic epithelial-to-mesenchymal transition in epicardial mesothelial cells. Exp. Cell Res. 2013, 319, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Brønnum, H.; Andersen, D.C.; Schneider, M.; Sandberg, M.B.; Eskildsen, T.; Nielsen, S.B.; Kalluri, R.; Sheikh, S.P. miR-21 Promotes Fibrogenic Epithelial-to-Mesenchymal Transition of Epicardial Mesothelial Cells Involving Programmed Cell Death 4 and Sprouty-1. PLoS ONE 2013, 8, e56280. [Google Scholar] [CrossRef] [Green Version]
- Seeger, T.; Xu, Q.F.; Muhly-Reinholz, M.; Fischer, A.; Kremp, E.; Zeiher, A.M.; Dimmeler, S. Inhibition of let-7 augments the recruitment of epicardial cells and improves cardiac function after myocardial infarction. J. Mol. Cell Cardiol. 2016, 94, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Gregory, A.P.; Bracken, P.C.; Bert, G.A.; Goodall, J.G. MicroRNAs as regulators of epithelial-mesenchymal transition. Cell Cycle 2008, 7, 3112–3118. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Li, Y.; Liu, G. MicroRNA-200c exacerbates the ischemia/reperfusion injury of heart through targeting the glutaminase (GLS)-mediated glutamine metabolism. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 3282–3289. [Google Scholar]
- Chen, Z.; Zhang, S.; Guo, C.; Li, J.; Sang, W. Downregulation of miR-200c protects cardiomyocytes from hypoxia-induced apoptosis by targeting GATA-4. Int. J. Mol. Med. 2017, 39, 1589–1596. [Google Scholar] [CrossRef]
- Singh, G.B.; Raut, S.K.; Khanna, S.; Kumar, A.; Sharma, S.; Prasad, R.; Khullar, M. MicroRNA-200c modulates DUSP-1 expression in diabetes-induced cardiac hypertrophy. Mol. Cell. Biochem. 2016, 424, 1–11. [Google Scholar] [CrossRef]
- Mattiotti, A.; Prakash, S.; Barnett, P.; Hoff, M.J.B.V.D. Follistatin-like 1 in development and human diseases. Cell. Mol. Life Sci. 2018, 75, 2339–2354. [Google Scholar] [CrossRef]
- Oshima, Y.; Ouchi, N.; Sato, K.; Izumiya, Y.; Pimentel, D.R.; Walsh, K. Follistatin-Like 1 Is an Akt-Regulated Cardioprotective Factor That Is Secreted by the Heart. Circulation 2008, 117, 3099–3108. [Google Scholar] [CrossRef] [Green Version]
- Wei, K.; Serpooshan, V.; Hurtado, C.; Diez-Cuñado, M.; Zhao, M.; Maruyama, S.; Zhu, W.; Fajardo, G.; Noseda, M.; Nakamura, K.; et al. Epicardial FSTL1 reconstitution regenerates the adult mammalian heart. Nature 2015, 525, 479–485. [Google Scholar] [CrossRef] [Green Version]
- Ogura, Y.; Ouchi, N.; Ohashi, K.; Shibata, R.; Kataoka, Y.; Kambara, T.; Kito, T.; Maruyama, S.; Yuasa, D.; Matsuo, K.; et al. Therapeutic impact of follistatin-like 1 on myocardial ischemic injury in preclinical models. Circulation 2012, 126, 1728–1738. [Google Scholar] [CrossRef] [PubMed]
- Shimano, M.; Ouchi, N.; Nakamura, K.; Van Wijk, B.; Ohashi, K.; Asaumi, Y.; Higuchi, A.; Pimentel, D.R.; Sam, F.; Murohara, T.; et al. Cardiac myocyte follistatin-like 1 functions to attenuate hypertrophy following pressure overload. Proc. Natl. Acad. Sci. USA 2011, 108, E899–E906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, H.; Cui, G.; Li, Y.; Ye, W.; Sun, Y.; Zhang, Z.; Li, J.; Xu, G.; Zeng, X.; Zhang, Y.; et al. Follistatin-like 1 protects mesenchymal stem cells from hypoxic damage and enhances their therapeutic efficacy in a mouse myocardial infarction model. Stem Cell Res. Ther. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Takeichi, M.; Nimura, K.; Mori, M.; Nakagami, H.; Kaneda, Y. The transcription factors Tbx18 and Wt1 control the epicardial epithelial-mesenchymal transition through bi-directional regulation of Slug in murine primary epicardial cells. PLoS ONE 2013, 8, e57829. [Google Scholar]
- Rodriguez-Aznar, E.; Wiesmüller, L.; Sainz, J.B.; Hermann, P.C. EMT and Stemness—Key Players in Pancreatic Cancer Stem Cells. Cancers 2019, 11, 1136. [Google Scholar] [CrossRef] [Green Version]
- Germani, A.; Foglio, E.; Capogrossi, M.C.; Russo, M.A.; Limana, F. Generation of cardiac progenitor cells through epicardial to mesenchymal transition. J. Mol. Med. 2015, 93, 735–748. [Google Scholar] [CrossRef]
- Liu, T.; Liu, Y.; Miller, M.; Cao, L.; Zhao, J.; Wu, J.; Wang, J.; Liu, L.; Li, S.; Zou, M.; et al. Autophagy plays a role in FSTL1-induced epithelial mesenchymal transition and airway remodeling in asthma. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 313, L27–L40. [Google Scholar] [CrossRef] [Green Version]
- Jin, T.; Zhang, Y.; Zhang, T. MiR-524-5p Suppresses Migration, Invasion, and EMT Progression in Breast Cancer Cells Through Targeting FSTL1. Cancer Biother. Radiopharm. 2020, 35, 789–801. [Google Scholar] [CrossRef]
- Shibanuma, M.; Mashimo, J.; Mita, A.; Kuroki, T.; Nose, K. Cloning from a mouse osteoblastic cell line of a set of transforming-growth-factor-beta 1-regulated genes, one of which seems to encode a follistatin-related polypeptide. Eur. J. Biochem. 1993, 217, 13–19. [Google Scholar] [CrossRef]
- Rosenberg, M.I.; Georges, S.A.; Asawachaicharn, A.; Analau, E.; Tapscott, S.J. MyoD inhibits Fstl1 and Utrn expression by inducing transcription of miR-206. J. Cell Biol. 2006, 175, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.M.; Zhang, A.R.; Xu, M.; Lou, J.; Qiu, W.Q. TLR-4/miRNA-32-5p/FSTL1 signaling regulates mycobacterial survival and inflammatory responses in Mycobacterium tuberculosis-infected macrophages. Exp. Cell Res. 2017, 352, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.-L.; Shi, G.-R.; Xie, J.; Du, X.-Z.; Yang, A.H. MicroRNA-27a Inhibits Cell Migration and Invasion of Fibroblast-Like Synoviocytes by Targeting Follistatin-Like Protein 1 in Rheumatoid Arthritis. Mol. Cells 2016, 39, 611–618. [Google Scholar] [CrossRef]
- Xiao, Y.; Zhang, Y.; Chen, Y.; Li, J.; Zhang, Z.; Sun, Y.; Shen, H.; Zhao, Z.; Huang, Z.; Zhang, W.; et al. Inhibition of MicroRNA-9-5p Protects Against Cardiac Remodeling Following Myocardial Infarction in Mice. Hum. Gene Ther. 2019, 30, 286–301. [Google Scholar] [CrossRef]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef]
- Cheng, S.; Huang, Y.; Lou, C.; He, Y.; Zhang, Y.; Zhang, Q. FSTL1 enhances chemoresistance and maintains stemness in breast cancer cells via integrin β3/Wnt signaling under miR-137 regulation. Cancer Biol. Ther. 2018, 20, 328–337. [Google Scholar] [CrossRef] [Green Version]
- Magadum, A.; Singh, N.; Kurian, A.A.; Sharkar, M.T.K.; Chepurko, E.; Zangi, L. Ablation of a Single N-Glycosylation Site in Human FSTL 1 Induces Cardiomyocyte Proliferation and Cardiac Regeneration. Mol. Ther. Nucleic Acids 2018, 13, 133–143. [Google Scholar] [CrossRef]
- Limana, F.; Germani, A.; Zacheo, A.; Kajstura, J.; Di Carlo, A.; Borsellino, G.; Leoni, O.; Palumbo, R.; Battistini, L.; Rastaldo, R.; et al. Exogenous High-Mobility Group Box 1 Protein Induces Myocardial Regeneration After Infarction via Enhanced Cardiac C-Kit + Cell Proliferation and Differentiation. Circ. Res. 2005, 97, e73–e83. [Google Scholar] [CrossRef] [Green Version]
- Eid, H.; Larson, D.M.; Springhorn, J.P.; Attawia, M.A.; Nayak, R.C.; Smith, T.W.; Kelly, R.A. Role of epicardial mesothelial cells in the modification of phenotype and function of adult rat ventricular myocytes in primary coculture. Circ. Res. 1992, 71, 40–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Follenzi, A.; Naldini, L. [26] Generation of HIV-1 derived lentiviral vectors. Methods Enzymol. 2002, 346, 454–465. [Google Scholar] [PubMed]
- Limana, F.; Zacheo, A.; Mocini, D.; Mangoni, A.; Borsellino, G.; Diamantini, A.; De Mori, R.; Battistini, L.; Vigna, E.; Santini, M.; et al. Identification of myocardial and vascular precursor cells in human and mouse epicardium. Circ. Res. 2007, 101, 1255–1265. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| WT1 | cagatgaacctaggagctaccttaaa | tgcccttctgtccatttca |
| TBX18 | caccgaggccgacgaagacc | cgtcctcacagctgcccgc |
| TCF21 | cattcacccagtcaacctga | ccacttccttcaggtcattctc |
| VCAM1 | tcttacctgtgcgctgtgac | actggatcttcagggaatgagt |
| SNAI1 | cttgtgtctgcacgacctgt | caggagaatggcttctcacc |
| SNAI2 | cattgccttgtgtctgcaag | agaaaggcttttccccagtg |
| ACTA2 | ctctcttccagccatctttcat | tataggtggtttcgtggatgc |
| SM22-alpha | ccttccagtccacaaacgac | gtaggatggacccttgttgg |
| VIMENTIN | tgcgccagcagtatgaaa | tgcgccagcagtatgaaa |
| FIBRONECTIN1 | cggagagagtgcccctacta | cgatattggtgaatcgcaga |
| CALPONIN 1 | cggcttgtctgctgaagtaa | accccctcaatccactctct |
| CYCLIN D1 | agaaggagattgtgccatcc | ctcttcgcacttctgctcct |
| CKIT | gatctgctctgcgtcctgtt | cttgcagatggctgagacg |
| NANOG | gcctccagcagatgcaag | ggttttgaaaccaggtcttaacc |
| OCT4 | gttggagaaggtggaaccaa | ctccttctgcagggctttc |
| SOX2 | acggcagctacagcatga | gacgtcgtagcggtgcat |
| FLST1 | cagccatcaacatcaccact | atgagggcgtcaacacaga |
| ZEB 1 | aggtgatccagccaaacg | ggtggcgtggagtcagag |
| ZEB 2 | ccagaggaaacaaggatttcag | aggcctgacatgtagtcttgtg |
| GAPDH | tgccaagtatgatgacatcaagaag | ggtcctcagtgtagcccaagat |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pontemezzo, E.; Foglio, E.; Vernucci, E.; Magenta, A.; D’Agostino, M.; Sileno, S.; Astanina, E.; Bussolino, F.; Pellegrini, L.; Germani, A.; et al. miR-200c-3p Regulates Epitelial-to-Mesenchymal Transition in Epicardial Mesothelial Cells by Targeting Epicardial Follistatin-Related Protein 1. Int. J. Mol. Sci. 2021, 22, 4971. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094971
Pontemezzo E, Foglio E, Vernucci E, Magenta A, D’Agostino M, Sileno S, Astanina E, Bussolino F, Pellegrini L, Germani A, et al. miR-200c-3p Regulates Epitelial-to-Mesenchymal Transition in Epicardial Mesothelial Cells by Targeting Epicardial Follistatin-Related Protein 1. International Journal of Molecular Sciences. 2021; 22(9):4971. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094971
Chicago/Turabian StylePontemezzo, Elena, Eleonora Foglio, Enza Vernucci, Alessandra Magenta, Marco D’Agostino, Sara Sileno, Elena Astanina, Federico Bussolino, Laura Pellegrini, Antonia Germani, and et al. 2021. "miR-200c-3p Regulates Epitelial-to-Mesenchymal Transition in Epicardial Mesothelial Cells by Targeting Epicardial Follistatin-Related Protein 1" International Journal of Molecular Sciences 22, no. 9: 4971. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094971