
Cancer Cells Haploinsufficient for ATM Are Sensitized to PARP Inhibitors by MET Inhibition

 , , and
, , and 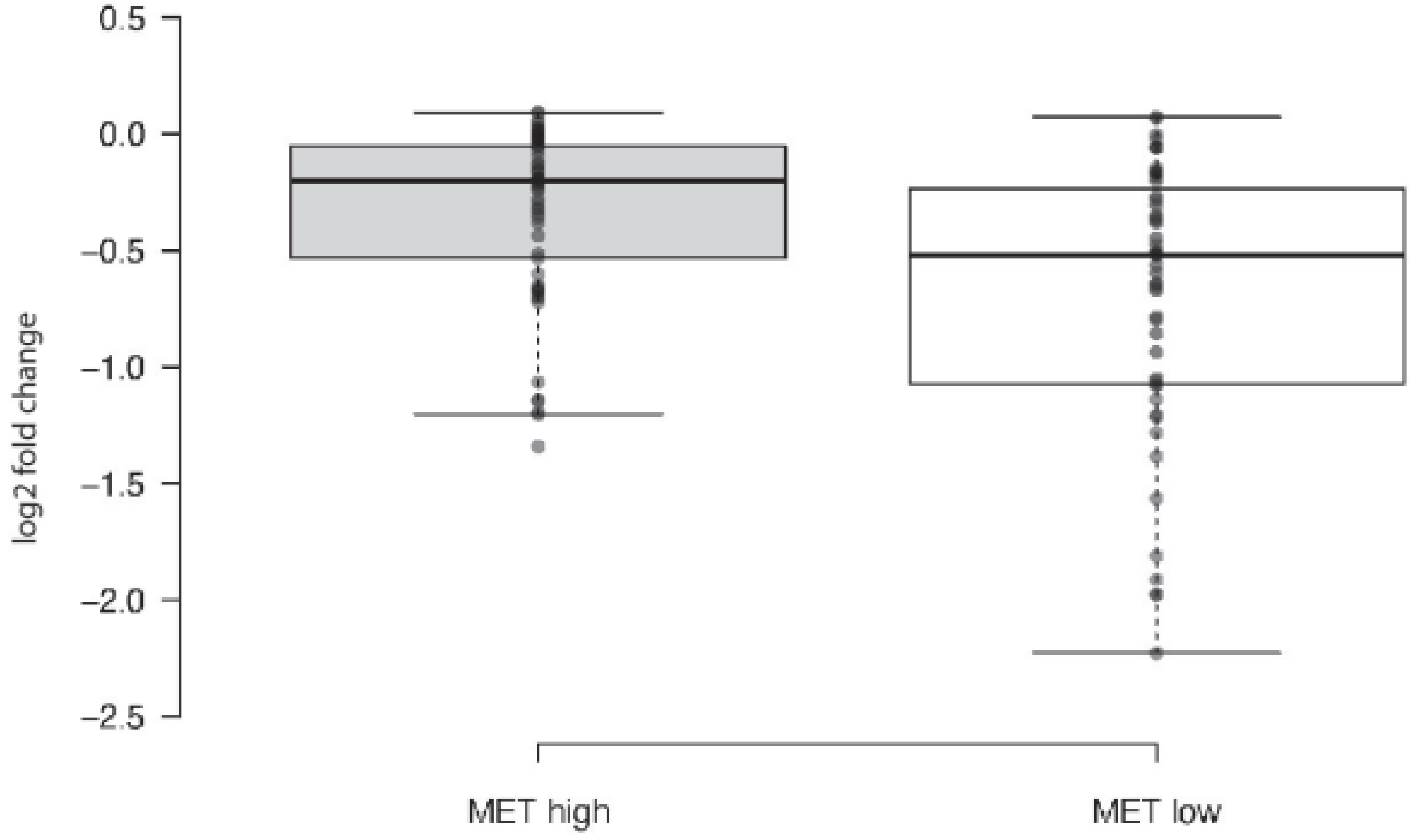{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
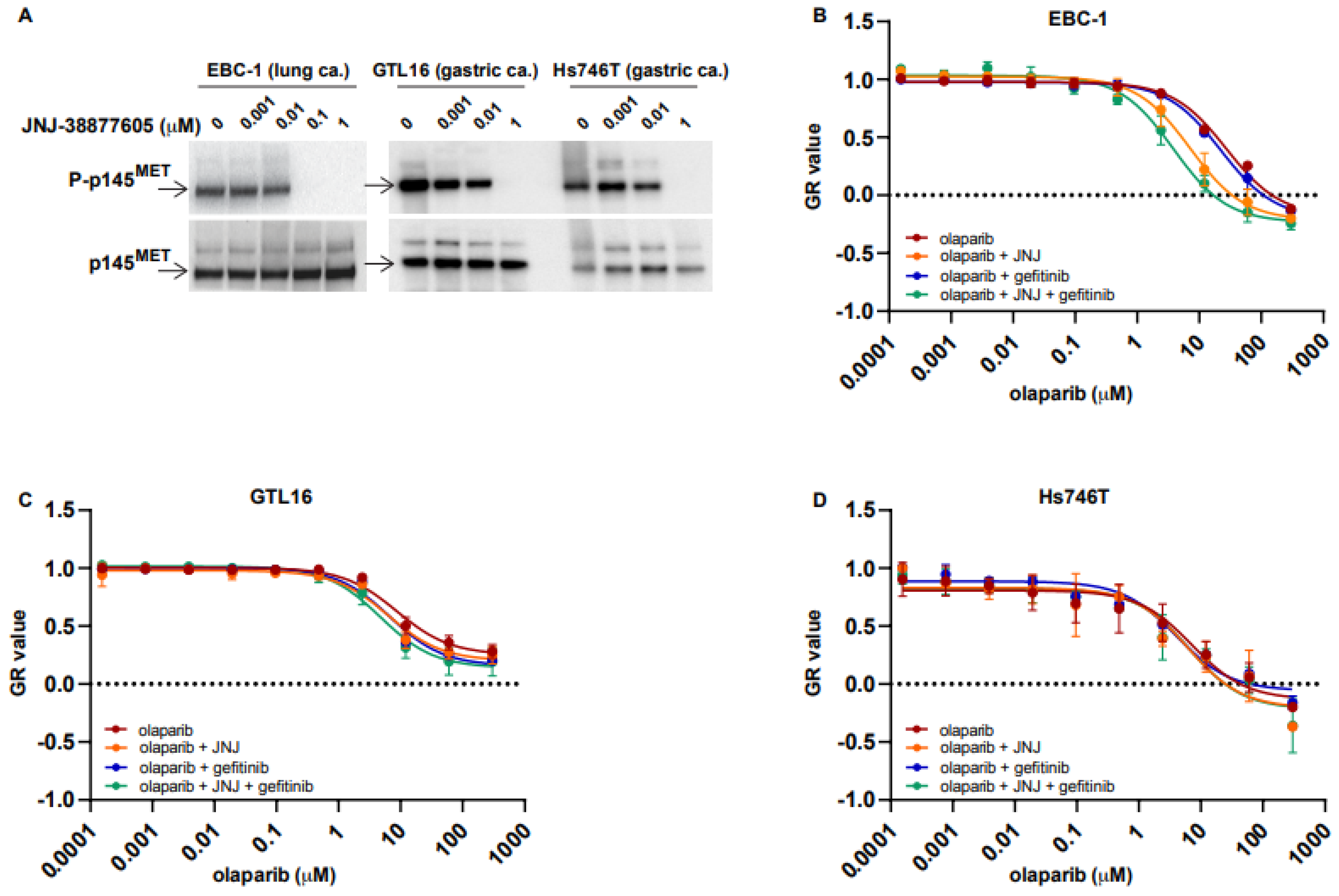
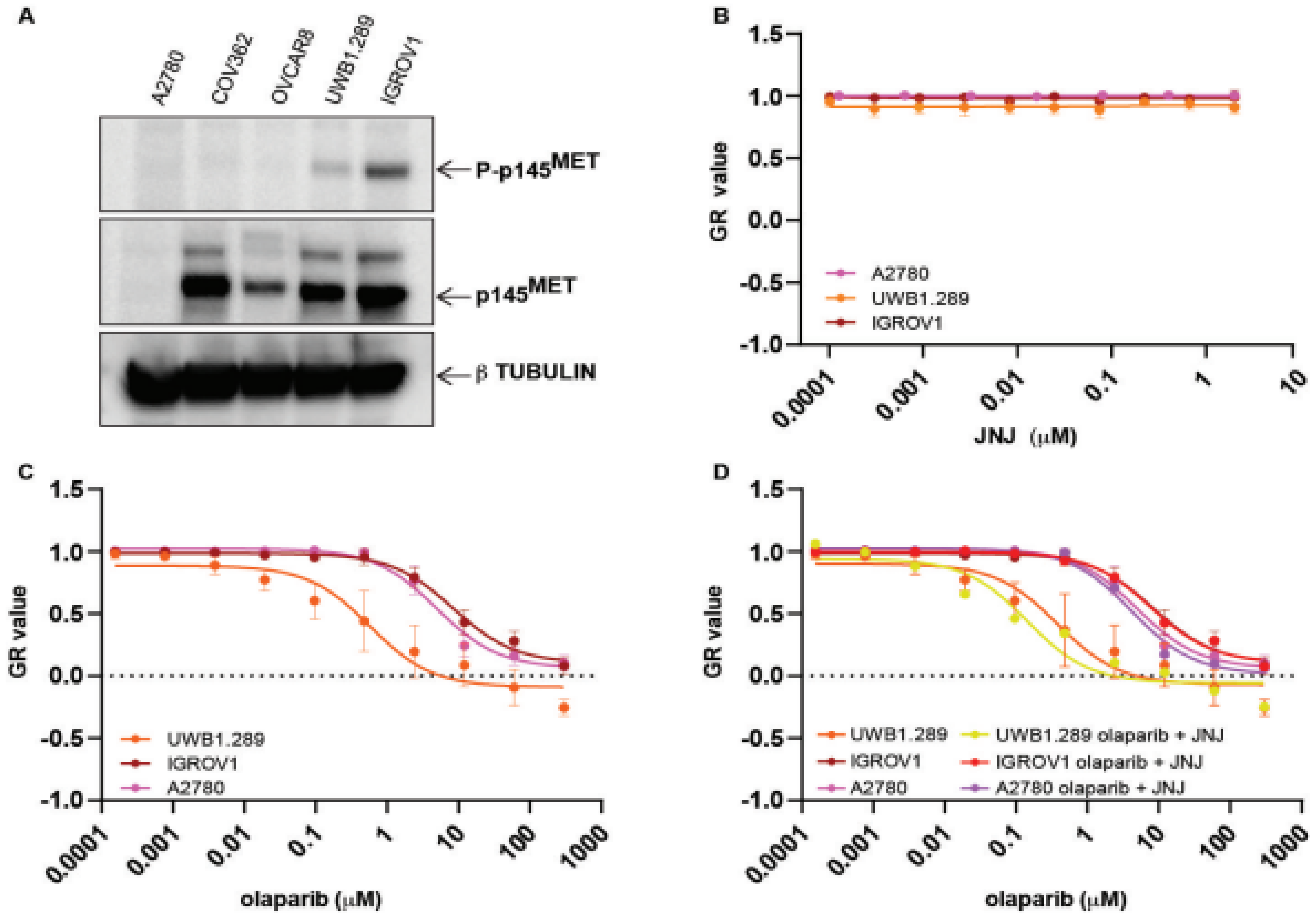
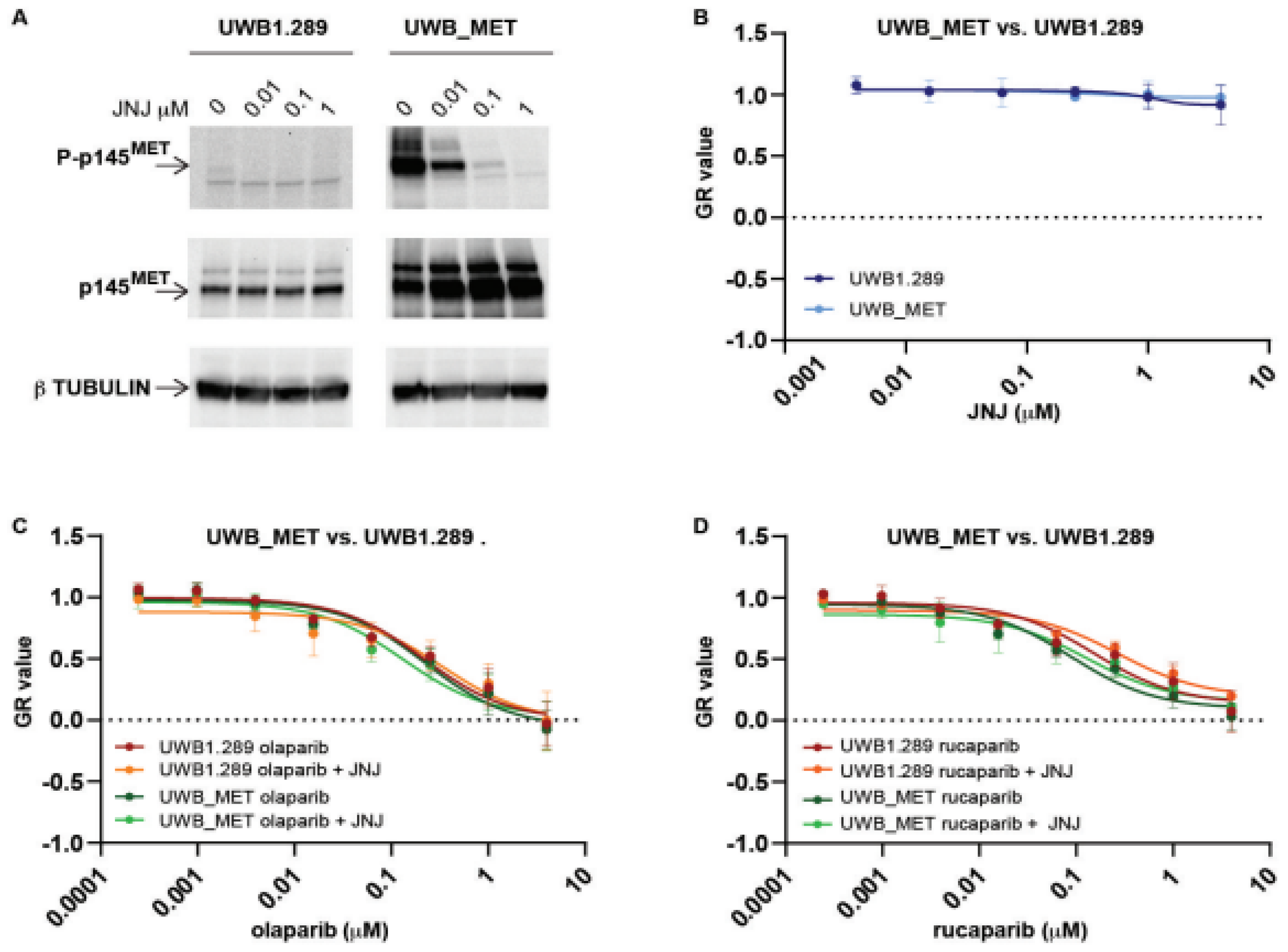
2.1. MET Overexpressing Cells Are Made Susceptible to PARPi by MET Inhibition
2.2. Exogenous MET Activation Is Not Sufficient to Convert PARPi Susceptible Cells to Be Resistant
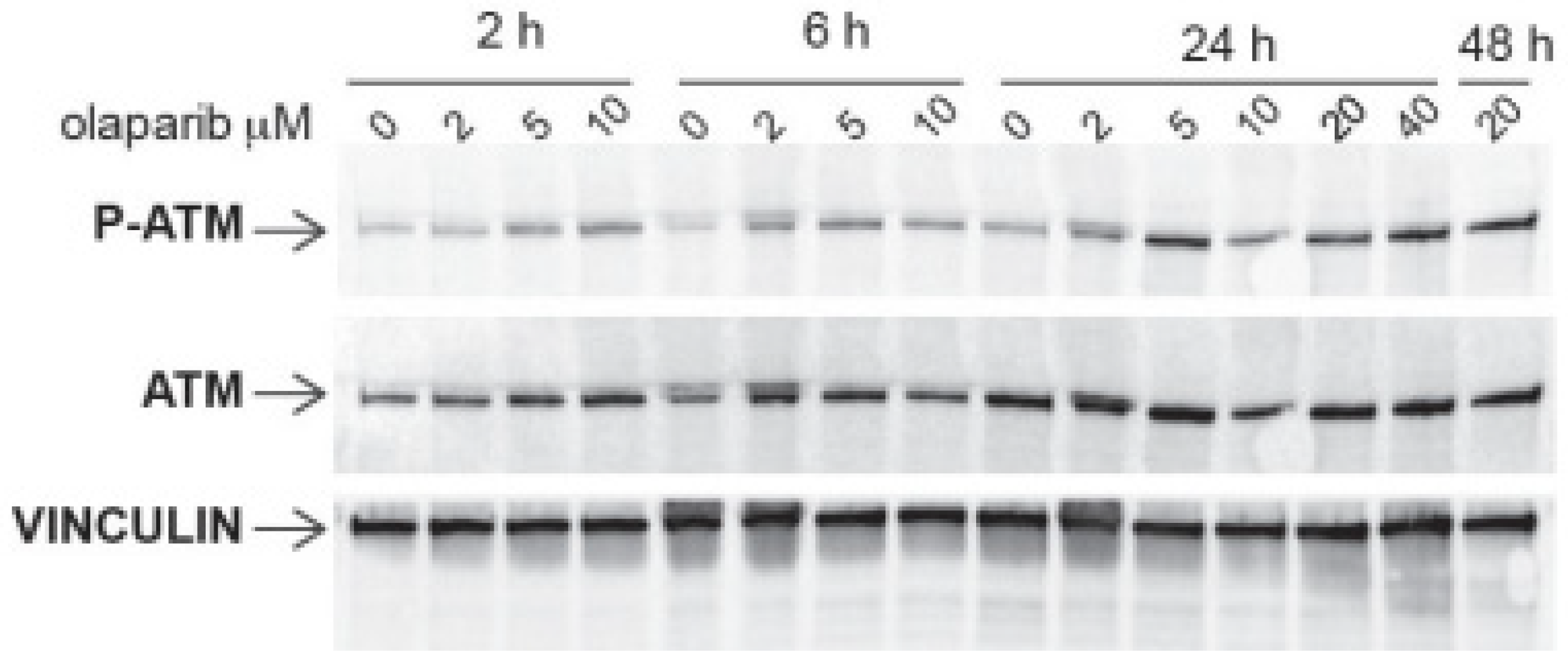
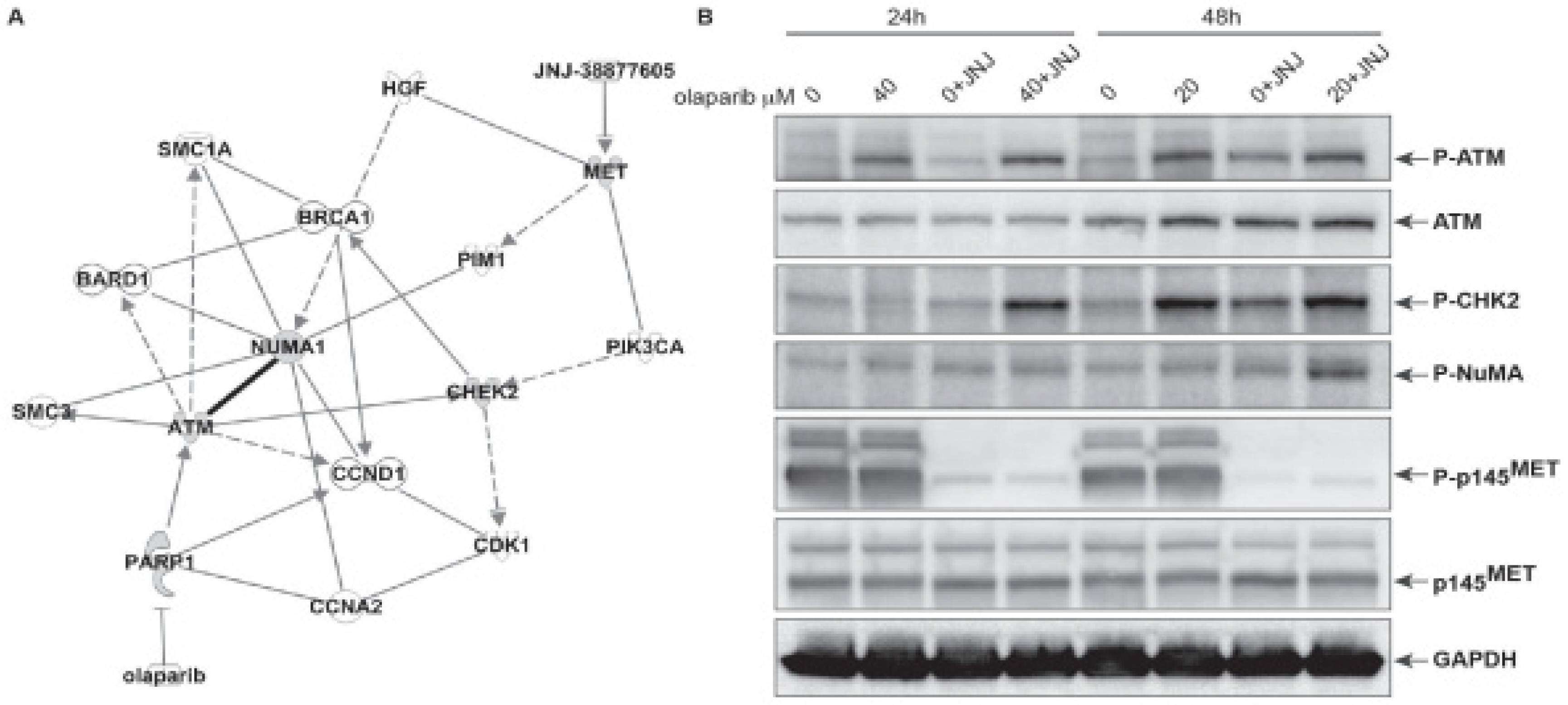
2.3. In MET Overexpressing Cells with ATM Mutation, Combined PARP and MET Inhibition Affects a Downstream Cascade Leading to the Phosphorylation of the Nuclear Mitotic Apparatus (NuMA) Protein
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Reagents
4.2. Immunofluorescence
4.3. Western Blot Analysis
4.4. CellTiter-Glo® Viability Assays
4.5. Crystal Violet Cytotoxicity Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Faraoni, I.; Graziani, G. Role of BRCA Mutations in Cancer Treatment with Poly(ADP-ribose) Polymerase (PARP) Inhibitors. Cancers 2018, 10, 487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Cleary, J.M.; Aguirre, A.J.; Shapiro, G.I.; D’Andrea, A.D. Biomarker-Guided Development of DNA Repair Inhibitors. Mol. Cell 2020, 78, 1070–1085. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Ceccaldi, R.; Shapiro, G.I.; D’Andrea, A.D. Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer. Cancer Discov. 2015, 5, 1137–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Andrea, A.D. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair 2018, 71, 172–176. [Google Scholar] [CrossRef]
- Morrison, C.; Sonoda, E.; Takao, N.; Shinohara, A.; Yamamoto, K.; Takeda, S. The controlling role of ATM in homologous recombinational repair of DNA damage. EMBO J. 2000, 19, 463–471. [Google Scholar] [CrossRef]
- Balmus, G.; Pilger, D.; Coates, J.; Demir, M.; Sczaniecka-Clift, M.; Barros, A.C.; Woods, M.; Fu, B.; Yang, F.; Chen, E.; et al. ATM orchestrates the DNA-damage response to counter toxic non-homologous end-joining at broken replication forks. Nat. Commun. 2019, 10, 87. [Google Scholar] [CrossRef] [Green Version]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [Green Version]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Chaudhary, M.W.; Al-Baradie, R.S. Ataxia-telangiectasia: Future prospects. Appl. Clin. Genet. 2014, 7, 159–167. [Google Scholar]
- Watson, I.R.; Takahashi, K.; Futreal, P.A.; Chin, L. Emerging patterns of somatic mutations in cancer. Nat. Rev. Genet. 2013, 14, 703–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, M.; Kipps, T.; Kurzrock, R. ATM Mutations in Cancer: Therapeutic Implications. Mol. Cancer Ther. 2016, 15, 1781–1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hills, S.A.; Diffley, J.F. DNA replication and oncogene-induced replicative stress. Curr. Biol. 2014, 24, R435–R444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toulany, M.; Kasten-Pisula, U.; Brammer, I.; Wang, S.; Chen, J.; Dittmann, K.; Baumann, M.; Dikomey, E.; Rodemann, H.P. Blockage of epidermal growth factor receptor-phosphatidylinositol 3-kinase-AKT signaling increases radiosensitivity of K-RAS mutated human tumor cells in vitro by affecting DNA repair. Clin. Cancer Res. 2006, 12, 4119–4126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bacco, F.; Luraghi, P.; Medico, E.; Reato, G.; Girolami, F.; Perera, T.; Gabriele, P.; Comoglio, P.M.; Boccaccio, C. Induction of MET by ionizing radiation and its role in radioresistance and invasive growth of cancer. J. Natl. Cancer Inst. 2011, 103, 645–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torti, D.; Trusolino, L. Oncogene addiction as a foundational rationale for targeted anti-cancer therapy: Promises and perils. EMBO Mol. Med. 2011, 3, 623–636. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.S.; Park, M.; Blair, D.G.; Tainsky, M.A.; Huebner, K.; Croce, C.M.; Vande Woude, G.F. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature 1984, 311, 29–33. [Google Scholar] [CrossRef]
- Comoglio, P.M.; Trusolino, L.; Boccaccio, C. Known and novel roles of the MET oncogene in cancer: A coherent approach to targeted therapy. Nat. Rev. Cancer 2018, 18, 341–358. [Google Scholar] [CrossRef]
- Bensimon, A.; Koch, J.P.; Francica, P.; Roth, S.M.; Riedo, R.; Glück, A.A.; Orlando, E.; Blaukat, A.; Aebersold, D.M.; Zimmer, Y.; et al. Deciphering MET-dependent modulation of global cellular responses to DNA damage by quantitative phosphoproteomics. Mol. Oncol. 2020, 14, 1185–1206. [Google Scholar] [CrossRef]
- De Bacco, F.; D’Ambrosio, A.; Casanova, E.; Orzan, F.; Neggia, R.; Albano, R.; Verginelli, F.; Cominelli, M.; Poliani, P.L.; Luraghi, P.; et al. MET inhibition overcomes radiation resistance of glioblastoma stem-like cells. EMBO Mol. Med. 2016, 8, 550–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Y.; Yamaguchi, H.; Wei, Y.; Hsu, J.L.; Wang, H.L.; Hsu, Y.H.; Lin, W.C.; Yu, W.H.; Leonard, P.G.; Lee, G.R.; et al. Blocking c-Met-mediated PARP1 phosphorylation enhances anti-tumor effects of PARP inhibitors. Nat. Med. 2016, 22, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.K.; Du, Y.; Sun, L.; Hsu, J.L.; Wang, Y.H.; Gao, Y.; Huang, J.; Hung, M.C. H2O2 induces nuclear transport of the receptor tyrosine kinase c-MET in breast cancer cells via a membrane-bound retrograde trafficking mechanism. J. Biol. Chem. 2019, 294, 8516–8528. [Google Scholar] [CrossRef] [PubMed]
- Ghandi, M.; Huang, F.W.; Jané-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R.; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef]
- Bertotti, A.; Burbridge, M.F.; Gastaldi, S.; Galimi, F.; Torti, D.; Medico, E.; Giordano, S.; Corso, S.; Rolland-Valognes, G.; Lockhart, B.P.; et al. Only a subset of Met-activated pathways are required to sustain oncogene addiction. Sci. Signal. 2009, 2, ra80. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013, 41, D955–D961. [Google Scholar] [CrossRef] [Green Version]
- Hafner, M.; Niepel, M.; Chung, M.; Sorger, P.K. Growth rate inhibition metrics correct for confounders in measuring sensitivity to cancer drugs. Nat. Methods 2016, 13, 521–527. [Google Scholar] [CrossRef] [Green Version]
- Stordal, B.; Timms, K.; Farrelly, A.; Gallagher, D.; Busschots, S.; Renaud, M.; Thery, J.; Williams, D.; Potter, J.; Tran, T.; et al. BRCA1/2BRCA1/2 mutation analysis in 41 ovarian cell lines reveals only one functionally deleterious BRCA1 mutation. Mol. Oncol. 2013, 7, 567–579. [Google Scholar] [CrossRef] [Green Version]
- Burgess, B.T.; Anderson, A.M.; McCorkle, J.R.; Wu, J.; Ueland, F.R.; Kolesar, J.M. Olaparib Combined with an ATR or Chk1 Inhibitor as a Treatment Strategy for Acquired Olaparib-Resistant. Diagnostics 2020, 10, 121. [Google Scholar] [CrossRef] [Green Version]
- Ledermann, J.A. PARP inhibitors in ovarian cancer. Ann. Oncol. 2016, 27 (Suppl. S1), i40–i44. [Google Scholar] [CrossRef]
- Choi, Y.E.; Meghani, K.; Brault, M.E.; Leclerc, L.; He, Y.J.; Day, T.A.; Elias, K.M.; Drapkin, R.; Weinstock, D.M.; Dao, F.; et al. Platinum and PARP Inhibitor Resistance Due to Overexpression of MicroRNA-622 in BRCA1-Mutant Ovarian Cancer. Cell Rep. 2016, 14, 429–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riches, L.C.; Trinidad, A.G.; Hughes, G.; Jones, G.N.; Hughes, A.M.; Thomason, A.G.; Gavine, P.; Cui, A.; Ling, S.; Stott, J.; et al. Pharmacology of the ATM Inhibitor AZD0156: Potentiation of Irradiation and Olaparib Responses Preclinically. Mol. Cancer Ther. 2020, 19, 13–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd, R.L.; Wijnhoven, P.W.G.; Ramos-Montoya, A.; Wilson, Z.; Illuzzi, G.; Falenta, K.; Jones, G.N.; James, N.; Chabbert, C.D.; Stott, J.; et al. Combined PARP and ATR inhibition potentiates genome instability and cell death in ATM-deficient cancer cells. Oncogene 2020, 39, 4869–4883. [Google Scholar] [CrossRef] [PubMed]
- Al-Ahmadie, H.; Iyer, G.; Hohl, M.; Asthana, S.; Inagaki, A.; Schultz, N.; Hanrahan, A.J.; Scott, S.N.; Brannon, A.R.; McDermott, G.C.; et al. Synthetic lethality in ATM-deficient RAD50-mutant tumors underlies outlier response to cancer therapy. Cancer Discov. 2014, 4, 1014–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smida, M.; Fece de la Cruz, F.; Kerzendorfer, C.; Uras, I.Z.; Mair, B.; Mazouzi, A.; Suchankova, T.; Konopka, T.; Katz, A.M.; Paz, K.; et al. MEK inhibitors block growth of lung tumours with mutations in ataxia-telangiectasia mutated. Nat. Commun. 2016, 7, 13701. [Google Scholar] [CrossRef]
- Ji, X.; Mukherjee, S.; Landi, M.T.; Bosse, Y.; Joubert, P.; Zhu, D.; Gorlov, I.; Xiao, X.; Han, Y.; Gorlova, O.; et al. Protein-altering germline mutations implicate novel genes related to lung cancer development. Nat. Commun. 2020, 11, 2220. [Google Scholar] [CrossRef]
- Mao, J.H.; Wu, D.; DelRosario, R.; Castellanos, A.; Balmain, A.; Perez-Losada, J. Atm heterozygosity does not increase tumor susceptibility to ionizing radiation alone or in a p53 heterozygous background. Oncogene 2008, 27, 6596–6600. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Wang, Y.; Jiang, W.; Liu, X.; Dubois, R.L.; Lin, C.S.; Ludwig, T.; Bakkenist, C.J.; Zha, S. Kinase-dead ATM protein causes genomic instability and early embryonic lethality in mice. J. Cell Biol. 2012, 198, 305–313. [Google Scholar] [CrossRef] [Green Version]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef]
- Bååth, M.; Jönsson, J.M.; Westbom Fremer, S.; Martín de la Fuente, L.; Tran, L.; Malander, S.; Kannisto, P.; Måsbäck, A.; Honeth, G.; Hedenfalk, I. MET Expression and Cancer Stem Cell Networks Impact Outcome in High-Grade Serous Ovarian Cancer. Genes 2021, 12, 742. [Google Scholar] [CrossRef]
- Sheta, R.; Bachvarova, M.; Plante, M.; Renaud, M.C.; Sebastianelli, A.; Gregoire, J.; Navarro, J.M.; Perez, R.B.; Masson, J.Y.; Bachvarov, D. Development of a 3D functional assay and identification of biomarkers, predictive for response of high-grade serous ovarian cancer (HGSOC) patients to poly-ADP ribose polymerase inhibitors (PARPis): Targeted therapy. J. Transl. Med. 2020, 18, 439. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Dai, Z.; Wang, L.; Gao, X.; Yang, L.; Wang, Z.; Wang, Q.; Liu, Z. MET inhibition enhances PARP inhibitor efficacy in castration-resistant prostate cancer by suppressing the ATM/ATR and PI3K/AKT pathways. J. Cell. Mol. Med. 2021, 25, 11157–11169. [Google Scholar] [CrossRef] [PubMed]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8, 564601. [Google Scholar] [CrossRef] [PubMed]
- Van Leen, E.V.; Di Pietro, F.; Bellaïche, Y. Oriented cell divisions in epithelia: From force generation to force anisotropy by tension, shape and vertices. Curr. Opin. Cell Biol. 2020, 62, 9–16. [Google Scholar] [CrossRef]
- Gehmlich, K.; Haren, L.; Merdes, A. Cyclin B degradation leads to NuMA release from dynein/dynactin and from spindle poles. EMBO Rep. 2004, 5, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Kotak, S.; Busso, C.; Gönczy, P. NuMA interacts with phosphoinositides and links the mitotic spindle with the plasma membrane. EMBO J. 2014, 33, 1815–1830. [Google Scholar] [CrossRef] [Green Version]
- Kotak, S.; Gönczy, P. NuMA phosphorylation dictates dynein-dependent spindle positioning. Cell Cycle 2014, 13, 177–178. [Google Scholar] [CrossRef] [Green Version]
- Salvador Moreno, N.; Liu, J.; Haas, K.M.; Parker, L.L.; Chakraborty, C.; Kron, S.J.; Hodges, K.; Miller, L.D.; Langefeld, C.; Robinson, P.J.; et al. The nuclear structural protein NuMA is a negative regulator of 53BP1 in DNA double-strand break repair. Nucleic Acids Res. 2019, 47, 2703–2715. [Google Scholar] [CrossRef] [Green Version]
- Palazzo, L.; Della Monica, R.; Visconti, R.; Costanzo, V.; Grieco, D. ATM controls proper mitotic spindle structure. Cell Cycle 2014, 13, 1091–1100. [Google Scholar] [CrossRef] [Green Version]
- Sharif-Askari, B.; Amrein, L.; Aloyz, R.; Panasci, L. PARP3 inhibitors ME0328 and olaparib potentiate vinorelbine sensitization in breast cancer cell lines. Breast Cancer Res. Treat. 2018, 172, 23–32. [Google Scholar] [CrossRef]
- Boehler, C.; Dantzer, F. PARP-3, a DNA-dependent PARP with emerging roles in double-strand break repair and mitotic progression. Cell Cycle 2011, 10, 1023–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giordano, S.; Di Renzo, M.F.; Ferracini, R.; Chiado-Piat, L.; Comoglio, P.M. p145, a protein with associated tyrosine kinase activity in a human gastric carcinoma cell line. Mol. Cell. Biol. 1988, 8, 3510–3517. [Google Scholar] [PubMed] [Green Version]
- Prat, M.; Crepaldi, T.; Pennacchietti, S.; Bussolino, F.; Comoglio, P.M. Agonistic monoclonal antibodies against the Met receptor dissect the biological responses to HGF. J. Cell Sci. 1998, 111 Pt 2, 237–247. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Ambrosio, C.; Erriquez, J.; Capellero, S.; Cignetto, S.; Alvaro, M.; Ciamporcero, E.; Di Renzo, M.F.; Perera, T.; Valabrega, G.; Olivero, M. Cancer Cells Haploinsufficient for ATM Are Sensitized to PARP Inhibitors by MET Inhibition. Int. J. Mol. Sci. 2022, 23, 5770. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105770
D’Ambrosio C, Erriquez J, Capellero S, Cignetto S, Alvaro M, Ciamporcero E, Di Renzo MF, Perera T, Valabrega G, Olivero M. Cancer Cells Haploinsufficient for ATM Are Sensitized to PARP Inhibitors by MET Inhibition. International Journal of Molecular Sciences. 2022; 23(10):5770. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105770
Chicago/Turabian StyleD’Ambrosio, Concetta, Jessica Erriquez, Sonia Capellero, Simona Cignetto, Maria Alvaro, Eric Ciamporcero, Maria Flavia Di Renzo, Timothy Perera, Giorgio Valabrega, and Martina Olivero. 2022. "Cancer Cells Haploinsufficient for ATM Are Sensitized to PARP Inhibitors by MET Inhibition" International Journal of Molecular Sciences 23, no. 10: 5770. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105770