
Characterization of the Secretome of a Specific Cell Expressing Mutant Methionyl-tRNA Synthetase in Co-Culture Using Click Chemistry

 , and
, and
Abstract
:1. Introduction
2. Results
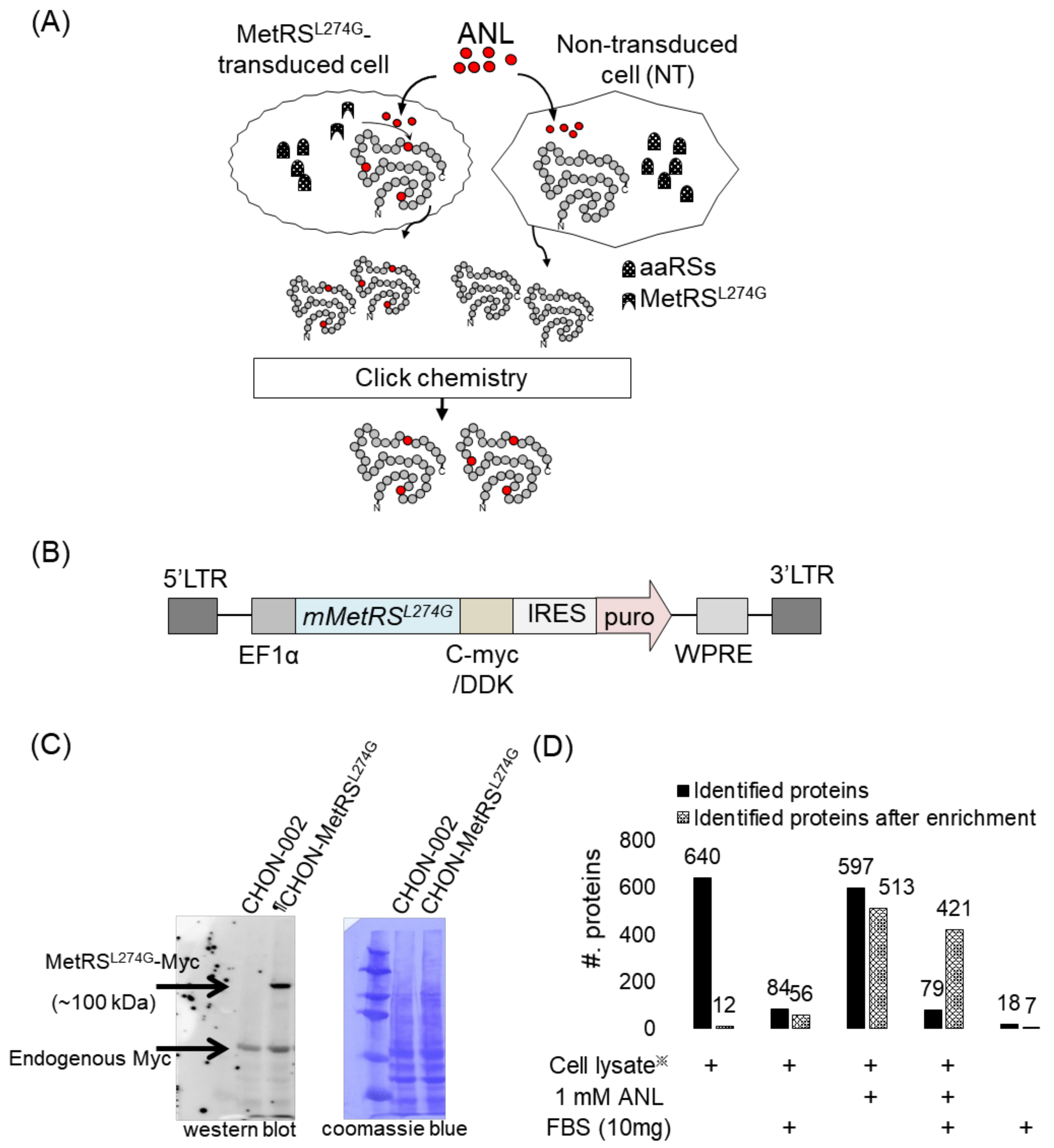
2.1. Cell-Type-Specific Labeling with Bioorthogonal and Non-Canonical Amino Acid
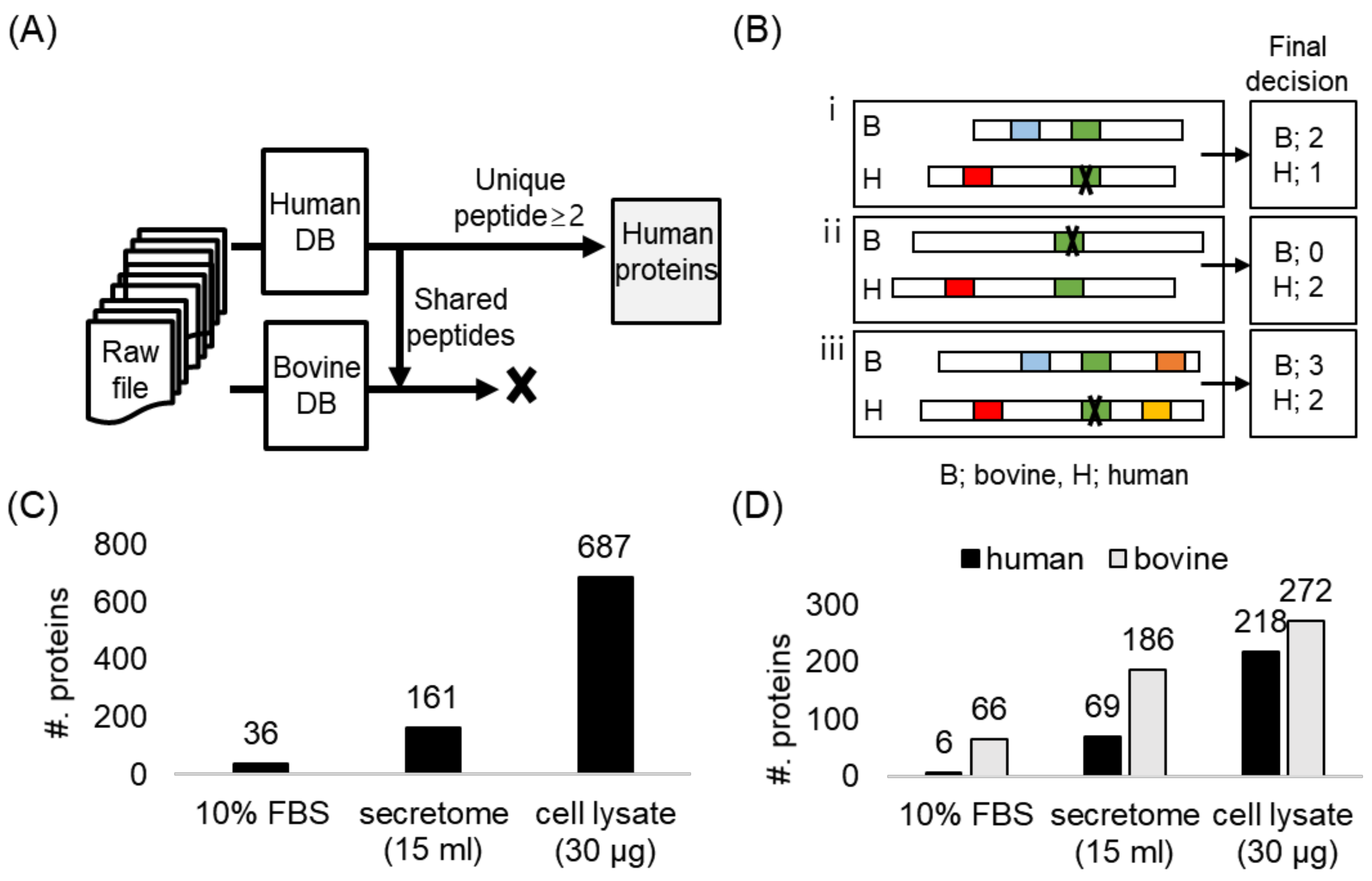
2.2. The Secretome Analysis in Serum-Supplemented Media
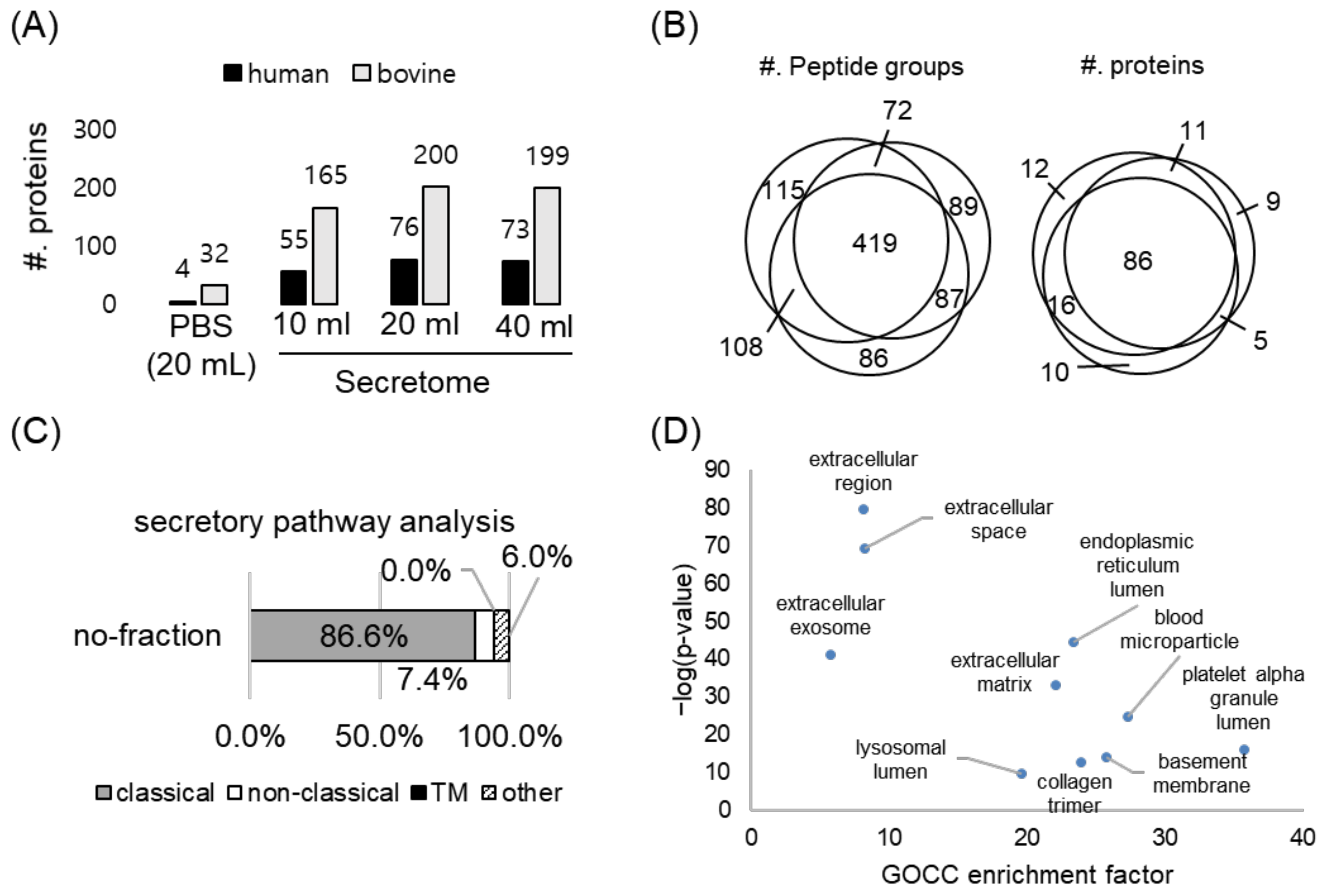
2.3. Optimization and Validation of the Co-Culture System
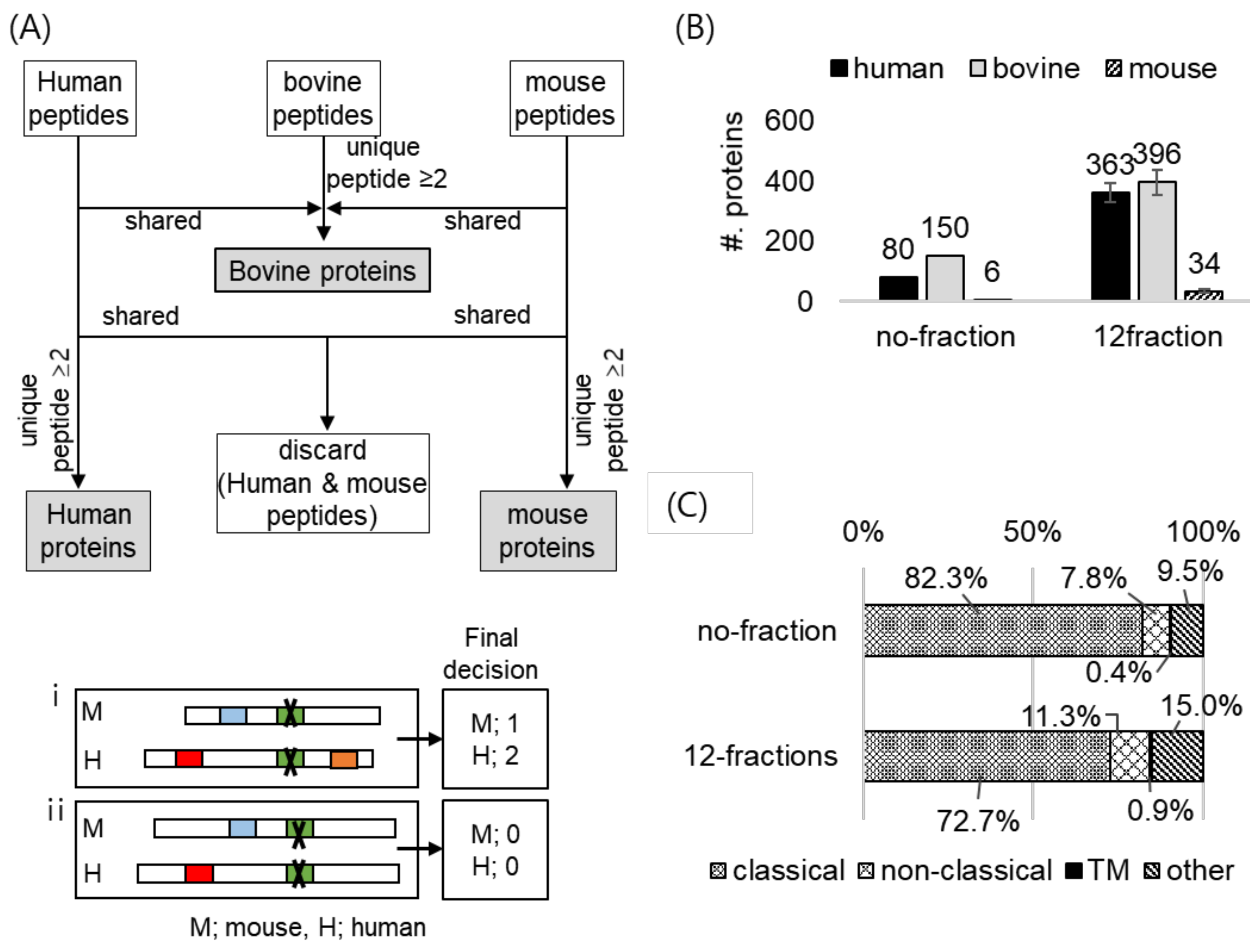
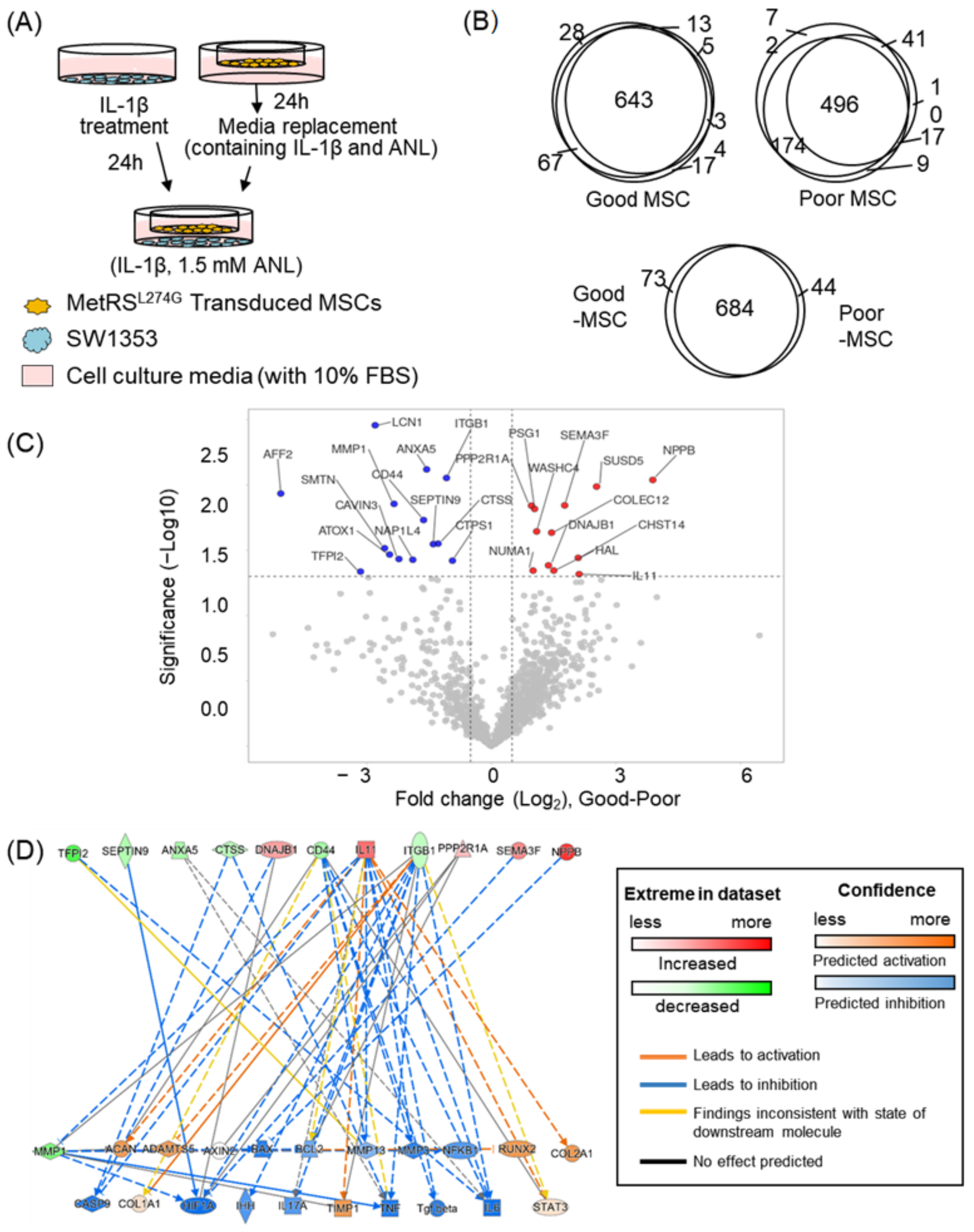
2.4. Application of the Co-Culture System; Analysis of MSC Secretome

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Normal Chondrocyte Cell (vs. OA-Induced Cell) ¶ | Predicted Alterations by Secretome vs. Literature $ | Reference |
|---|---|---|---|
| MMP3 | − | Mat. | [25,26] |
| MMP13 | − | Mat. | [20] |
| ADAMTS5 | − | Opp. | [25] |
| Caspase9 | − | Mat. | [27] |
| Bax | − | Mat. | [28,29] |
| Bcl-2 | N.C | Mat. | [30] |
| COL2a1 | + | Mat. | [23,25] |
| ACAN | + | Mat. | [23] |
| SOX9 | + | N.D | [23] |
| HIF1α | + | Opp. | [31] |
| TNFα | − | Mat. | [32] |
| IL-1β | − | N.D | [33] |
| IL-4 | + | Mat. | [34] |
| TGFβ | − | Mat. | [24] |
| COL1a1 | − | Opp. | [23] |
| BMP2 | − | N.D | [23] |
| RUNX2 | − | Opp. | [23,35] |
| COL10a1 | − | N.D | [23,36] |
| IHH | − | Mat. | [23,37] |
| DKK1 | + | N.D | [23,38,39] |
| FRZB | + | N.D | [23] |
| GREM1 | +(early)/−(late) | N.D | [23] |
| AXIN2 | − | N.D | [23] |
| TIMP1 | + | Mat. | |
| IL-6 | − | Mat. | [21,22,40] |
| LIF | − | N.D | |
| IL-17 | − | Mat. | [33] |
| MMP1 | − | Mat. | [20,25] |
| NF-kB | − | Opp. | [22,41] |
| STAT3 | − | Opp. | [21,42] |
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Cell Culture
5.2. Construction of a Cell Line Expressing Mutant Methionyl-tRNA Synthetase (MetRS)
5.3. Sample Preparation for the Click Chemistry-Based Enrichment: Cell Lysate and Secretome
5.4. Secretome Preparation in Co-Culture Condition
5.5. Enrichment of ANL-Tagged Proteins Using Click Chemistry
5.6. Basic pH Reversed-Phase Fractionation
5.7. Liquid Chromatography—Tandem Mass Spectrometry (LC-MS/MS) Analysis
5.8. MS Data Analysis
5.9. Bioinformatics Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pittenger, M.F.; Discher, D.E.; Peault, B.M.; Phinney, D.G.; Hare, J.M.; Caplan, A.I. Mesenchymal stem cell perspective: Cell biology to clinical progress. NPJ Regen. Med. 2019, 4, 22. [Google Scholar] [CrossRef] [PubMed]
- Andrzejewska, A.; Lukomska, B.; Janowski, M. Concise Review: Mesenchymal Stem Cells: From Roots to Boost. Stem Cells 2019, 37, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Giordano, A.; Galderisi, U.; Marino, I.R. From the laboratory bench to the patient’s bedside: An update on clinical trials with mesenchymal stem cells. J. Cell. Physiol. 2007, 211, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Cho, S.G. New strategies for overcoming limitations of mesenchymal stem cell-based immune modulation. Int. J. Stem Cells 2015, 8, 54–68. [Google Scholar] [CrossRef]
- Gonzalez-Gonzalez, A.; Garcia-Sanchez, D.; Dotta, M.; Rodriguez-Rey, J.C.; Perez-Campo, F.M. Mesenchymal stem cells secretome: The cornerstone of cell-free regenerative medicine. World J. Stem Cells 2020, 12, 1529–1552. [Google Scholar] [CrossRef]
- Kumar, P.; Kandoi, S.; Misra, R.; Vijayalakshmi, S.; Rajagopal, K.; Verma, R.S. The mesenchymal stem cell secretome: A new paradigm towards cell-free therapeutic mode in regenerative medicine. Cytokine Growth Factor Rev. 2019, 46, 1–9. [Google Scholar]
- Brose, T.Z.; Kubosch, E.J.; Schmal, H.; Stoddart, M.J.; Armiento, A.R. Crosstalk Between Mesenchymal Stromal Cells and Chondrocytes: The Hidden Therapeutic Potential for Cartilage Regeneration. Stem Cell Rev. Rep. 2021, 17, 1647–1665. [Google Scholar] [CrossRef]
- Kastner, N.; Mester-Tonczar, J.; Winkler, J.; Traxler, D.; Spannbauer, A.; Ruger, B.M.; Goliasch, G.; Pavo, N.; Gyongyosi, M.; Zlabinger, K. Comparative Effect of MSC Secretome to MSC Co-culture on Cardiomyocyte Gene Expression Under Hypoxic Conditions in vitro. Front. Bioeng. Biotechnol. 2020, 8, 502213. [Google Scholar]
- Munoz-Perez, E.; Gonzalez-Pujana, A.; Igartua, M.; Santos-Vizcaino, E.; Hernandez, R.M. Mesenchymal Stromal Cell Secretome for the Treatment of Immune-Mediated Inflammatory Diseases: Latest Trends in Isolation, Content Optimization and Delivery Avenues. Pharmaceutics 2021, 13, 1802. [Google Scholar] [CrossRef]
- Shin, J.; Kwon, Y.; Lee, S.; Na, S.; Hong, E.Y.; Ju, S.; Jung, H.G.; Kaushal, P.; Shin, S.; Back, J.H.; et al. Common Repository of FBS Proteins (cRFP) To Be Added to a Search Database for Mass Spectrometric Analysis of Cell Secretome. J. Proteome Res. 2019, 18, 3800–3806. [Google Scholar] [CrossRef]
- Nonnis, S.; Maffioli, E.; Zanotti, L.; Santagata, F.; Negri, A.; Viola, A.; Elliman, S.; Tedeschi, G. Effect of fetal bovine serum in culture media on MS analysis of mesenchymal stromal cells secretome. EuPA Open Proteomics 2016, 10, 28–30. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Rhim, J.; Kwon, Y.; Choi, S.Y.; Shin, S.; Ha, C.W.; Lee, C. Comparative analysis of differentially secreted proteins in serum-free and serum-containing media by using BONCAT and pulsed SILAC. Sci. Rep. 2019, 9, 3096. [Google Scholar] [CrossRef] [PubMed]
- Howden, A.J.; Geoghegan, V.; Katsch, K.; Efstathiou, G.; Bhushan, B.; Boutureira, O.; Thomas, B.; Trudgian, D.C.; Kessler, B.M.; Dieterich, D.C.; et al. QuaNCAT: Quantitating proteome dynamics in primary cells. Nat. Methods 2013, 10, 343–346. [Google Scholar] [CrossRef] [PubMed]
- Landgraf, P.; Antileo, E.R.; Schuman, E.M.; Dieterich, D.C. BONCAT: Metabolic labeling, click chemistry, and affinity purification of newly synthesized proteomes. Methods Mol. Biol. 2015, 1266, 199–215. [Google Scholar]
- Erdmann, I.; Marter, K.; Kobler, O.; Niehues, S.; Abele, J.; Muller, A.; Bussmann, J.; Storkebaum, E.; Ziv, T.; Thomas, U.; et al. Cell-selective labelling of proteomes in Drosophila melanogaster. Nat. Commun. 2015, 6, 7521. [Google Scholar] [CrossRef]
- Mahdavi, A.; Hamblin, G.D.; Jindal, G.A.; Bagert, J.D.; Dong, C.; Sweredoski, M.J.; Hess, S.; Schuman, E.M.; Tirrell, D.A. Engineered Aminoacyl-tRNA Synthetase for Cell-Selective Analysis of Mammalian Protein Synthesis. J. Am. Chem Soc. 2016, 138, 4278–4281. [Google Scholar] [CrossRef]
- Azizian, N.G.; Sullivan, D.K.; Nie, L.; Pardo, S.; Molleur, D.; Chen, J.; Weintraub, S.T.; Li, Y. Selective Labeling and Identification of the Tumor Cell Proteome of Pancreatic Cancer In Vivo. J. Proteome Res. 2021, 20, 858–866. [Google Scholar] [CrossRef]
- Liu, Y.; Conboy, M.J.; Mehdipour, M.; Liu, Y.T.; Tran, T.P.; Blotnick, A.; Rajan, P.; Santos, T.C.; Conboy, I.M. Application of bio-orthogonal proteome labeling to cell transplantation and heterochronic parabiosis. Nat. Commun. 2017, 8, 643. [Google Scholar] [CrossRef]
- Alvarez-Castelao, B.; Schanzenbacher, C.T.; Hanus, C.; Glock, C.; Tom Dieck, S.; Dorrbaum, A.R.; Bartnik, I.; Nassim-Assir, B.; Ciirdaeva, E.; Mueller, A.; et al. Cell-type-specific metabolic labeling of nascent proteomes in vivo. Nat. Biotechnol. 2017, 35, 1196–1201. [Google Scholar] [CrossRef]
- Salerno, A.; Brady, K.; Rikkers, M.; Li, C.; Caamano-Gutierrez, E.; Falciani, F.; Blom, A.W.; Whitehouse, M.R.; Hollander, A.P. MMP13 and TIMP1 are functional markers for two different potential modes of action by mesenchymal stem/stromal cells when treating osteoarthritis. Stem Cells 2020, 38, 1438–1453. [Google Scholar] [CrossRef]
- Lee, S.Y.; Lee, S.H.; Na, H.S.; Kwon, J.Y.; Kim, G.Y.; Jung, K.; Cho, K.H.; Kim, S.A.; Go, E.J.; Park, M.J.; et al. The Therapeutic Effect of STAT3 Signaling-Suppressed MSC on Pain and Articular Cartilage Damage in a Rat Model of Monosodium Iodoacetate-Induced Osteoarthritis. Front. Immunol. 2018, 9, 2881. [Google Scholar] [CrossRef]
- Gadient, R.A.; Patterson, P.H. Leukemia inhibitory factor, interleukin 6, and other cytokines using the GP130 transducing receptor: Roles in inflammation and injury. Stem Cells 1999, 17, 127–137. [Google Scholar] [CrossRef]
- Zhong, L.; Huang, X.; Karperien, M.; Post, J.N. Correlation between Gene Expression and Osteoarthritis Progression in Human. Int. J. Mol. Sci. 2016, 17, 1126. [Google Scholar] [CrossRef]
- Zhen, G.; Wen, C.; Jia, X.; Li, Y.; Crane, J.L.; Mears, S.C.; Askin, F.B.; Frassica, F.J.; Chang, W.; Yao, J.; et al. Inhibition of TGF-beta signaling in mesenchymal stem cells of subchondral bone attenuates osteoarthritis. Nat. Med. 2013, 19, 704–712. [Google Scholar] [CrossRef]
- Rousseau, J.; Garnero, P. Biological markers in osteoarthritis. Bone 2012, 51, 265–277. [Google Scholar] [CrossRef]
- Attur, M.; Krasnokutsky-Samuels, S.; Samuels, J.; Abramson, S.B. Prognostic biomarkers in osteoarthritis. Curr. Opin. Rheumatol. 2013, 25, 136–144. [Google Scholar] [CrossRef]
- D’Lima, D.; Hermida, J.; Hashimoto, S.; Colwell, C.; Lotz, M. Caspase inhibitors reduce severity of cartilage lesions in experimental osteoarthritis. Arthritis Rheum 2006, 54, 1814–1821. [Google Scholar] [CrossRef]
- Miao, G.; Zang, X.; Hou, H.; Sun, H.; Wang, L.; Zhang, T.; Tan, Y.; Liu, W.; Ye, P.; Gao, L.; et al. Bax Targeted by miR-29a Regulates Chondrocyte Apoptosis in Osteoarthritis. Biomed. Res. Int. 2019, 2019, 1434538. [Google Scholar] [CrossRef]
- Karaliotas, G.I.; Mavridis, K.; Scorilas, A.; Babis, G.C. Quantitative analysis of the mRNA expression levels of BCL2 and BAX genes in human osteoarthritis and normal articular cartilage: An investigation into their differential expression. Mol. Med. Rep. 2015, 12, 4514–4521. [Google Scholar] [CrossRef]
- Trieb, K.; Sulzbacher, I.; Kubista, B. Bcl-2 correlates with localization but not outcome in human osteosarcoma. Oncol. Lett. 2013, 6, 559–561. [Google Scholar] [CrossRef]
- Zhang, F.J.; Luo, W.; Lei, G.H. Role of HIF-1alpha and HIF-2alpha in osteoarthritis. Joint Bone Spine 2015, 82, 144–147. [Google Scholar] [CrossRef]
- Chisari, E.; Yaghmour, K.M.; Khan, W.S. The effects of TNF-alpha inhibition on cartilage: A systematic review of preclinical studies. Osteoarthr. Cartil. 2020, 28, 708–718. [Google Scholar] [CrossRef]
- Na, H.S.; Park, J.S.; Cho, K.H.; Kwon, J.Y.; Choi, J.; Jhun, J.; Kim, S.J.; Park, S.H.; Cho, M.L. Interleukin-1-Interleukin-17 Signaling Axis Induces Cartilage Destruction and Promotes Experimental Osteoarthritis. Front. Immunol. 2020, 11, 730. [Google Scholar] [CrossRef]
- Lubberts, E.; Joosten, L.A.; Chabaud, M.; van Den Bersselaar, L.; Oppers, B.; Coenen-De Roo, C.J.; Richards, C.D.; Miossec, P.; van Den Berg, W.B. IL-4 gene therapy for collagen arthritis suppresses synovial IL-17 and osteoprotegerin ligand and prevents bone erosion. J. Clin. Invest. 2000, 105, 1697–1710. [Google Scholar] [CrossRef]
- Eliseev, R.A.; Dong, Y.F.; Sampson, E.; Zuscik, M.J.; Schwarz, E.M.; O’Keefe, R.J.; Rosier, R.N.; Drissi, M.H. Runx2-mediated activation of the Bax gene increases osteosarcoma cell sensitivity to apoptosis. Oncogene 2008, 27, 3605–3614. [Google Scholar] [CrossRef]
- Sun, J.; Yan, B.; Yin, W.; Zhang, X. Identification of genes associated with osteoarthritis by microarray analysis. Mol. Med. Rep. 2015, 12, 5211–5216. [Google Scholar] [CrossRef]
- Zhou, J.; Wei, X.; Wei, L. Indian Hedgehog, a critical modulator in osteoarthritis, could be a potential therapeutic target for attenuating cartilage degeneration disease. Connect. Tissue Res. 2014, 55, 257–261. [Google Scholar] [CrossRef]
- Jeong, S.Y.; Kim, D.H.; Ha, J.; Jin, H.J.; Kwon, S.J.; Chang, J.W.; Choi, S.J.; Oh, W.; Yang, Y.S.; Kim, G.; et al. Thrombospondin-2 secreted by human umbilical cord blood-derived mesenchymal stem cells promotes chondrogenic differentiation. Stem Cells 2013, 31, 2136–2148. [Google Scholar] [CrossRef]
- Niada, S.; Giannasi, C.; Gomarasca, M.; Stanco, D.; Casati, S.; Brini, A.T. Adipose-derived stromal cell secretome reduces TNFalpha-induced hypertrophy and catabolic markers in primary human articular chondrocytes. Stem Cell Res. 2019, 38, 101463. [Google Scholar] [CrossRef]
- Henrotin, Y. Osteoarthritis in year 2021: Biochemical markers. Osteoarthr. Cartil. 2022, 30, 237–248. [Google Scholar] [CrossRef]
- Waring, P.M.; Carroll, G.J.; Kandiah, D.A.; Buirski, G.; Metcalf, D. Increased levels of leukemia inhibitory factor in synovial fluid from patients with rheumatoid arthritis and other inflammatory arthritides. Arthritis Rheum. 1993, 36, 911–915. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Guo, Z.; Yuan, Y. STAT3 speeds up progression of osteoarthritis through NF-kappaB signaling pathway. Exp. Ther. Med. 2020, 19, 722–728. [Google Scholar] [PubMed]
- Ngo, J.T.; Champion, J.A.; Mahdavi, A.; Tanrikulu, I.C.; Beatty, K.E.; Connor, R.E.; Yoo, T.H.; Dieterich, D.C.; Schuman, E.M.; Tirrell, D.A. Cell-selective metabolic labeling of proteins. Nat. Chem. Biol. 2009, 5, 715–717. [Google Scholar] [CrossRef] [PubMed]
- Ngo, J.T.; Schuman, E.M.; Tirrell, D.A. Mutant methionyl-tRNA synthetase from bacteria enables site-selective N-terminal labeling of proteins expressed in mammalian cells. Proc. Natl. Acad. Sci. USA 2013, 110, 4992–4997. [Google Scholar] [CrossRef]
- Shin, J.; Kim, G.; Kabir, M.H.; Park, S.J.; Lee, S.T.; Lee, C. Use of Composite Protein Database including Search Result Sequences for Mass Spectrometric Analysis of Cell Secretome. PLoS ONE 2015, 10, e0121692. [Google Scholar] [CrossRef]
- Shin, S.; Lee, J.; Kwon, Y.; Park, K.S.; Jeong, J.H.; Choi, S.J.; Bang, S.I.; Chang, J.W.; Lee, C. Comparative Proteomic Analysis of the Mesenchymal Stem Cells Secretome from Adipose, Bone Marrow, Placenta and Wharton’s Jelly. Int. J. Mol. Sci. 2021, 22, 845. [Google Scholar] [CrossRef]
- Lo, W.C.; Chen, W.H.; Lin, T.C.; Hwang, S.M.; Zeng, R.; Hsu, W.C.; Chiang, Y.M.; Liu, M.C.; Williams, D.F.; Deng, W.P. Preferential therapy for osteoarthritis by cord blood MSCs through regulation of chondrogenic cytokines. Biomaterials 2013, 34, 4739–4748. [Google Scholar] [CrossRef]
- Koh, Y.G.; Kwon, O.R.; Kim, Y.S.; Choi, Y.J.; Tak, D.H. Adipose-Derived Mesenchymal Stem Cells With Microfracture Versus Microfracture Alone: 2-Year Follow-up of a Prospective Randomized Trial. Arthroscopy 2016, 32, 97–109. [Google Scholar] [CrossRef]
- Lamo-Espinosa, J.M.; Mora, G.; Blanco, J.F.; Granero-Molto, F.; Nunez-Cordoba, J.M.; Sanchez-Echenique, C.; Bondia, J.M.; Aquerreta, J.D.; Andreu, E.J.; Ornilla, E.; et al. Intra-articular injection of two different doses of autologous bone marrow mesenchymal stem cells versus hyaluronic acid in the treatment of knee osteoarthritis: Multicenter randomized controlled clinical trial (phase I/II). J. Transl. Med. 2016, 14, 246. [Google Scholar] [CrossRef]
- Matas, J.; Orrego, M.; Amenabar, D.; Infante, C.; Tapia-Limonchi, R.; Cadiz, M.I.; Alcayaga-Miranda, F.; Gonzalez, P.L.; Muse, E.; Khoury, M.; et al. Umbilical Cord-Derived Mesenchymal Stromal Cells (MSCs) for Knee Osteoarthritis: Repeated MSC Dosing Is Superior to a Single MSC Dose and to Hyaluronic Acid in a Controlled Randomized Phase I/II Trial. Stem Cells Transl. Med. 2019, 8, 215–224. [Google Scholar] [CrossRef]
- Arrigoni, C.; D’Arrigo, D.; Rossella, V.; Candrian, C.; Albertini, V.; Moretti, M. Umbilical Cord MSCs and Their Secretome in the Therapy of Arthritic Diseases: A Research and Industrial Perspective. Cells 2020, 9, 1343. [Google Scholar] [CrossRef]
- Wang, H.; Yan, X.; Jiang, Y.; Wang, Z.; Li, Y.; Shao, Q. The human umbilical cord stem cells improve the viability of OA degenerated chondrocytes. Mol. Med. Rep. 2018, 17, 4474–4482. [Google Scholar] [CrossRef]
- Wang, B.; Liu, W.; Li, J.J.; Chai, S.; Xing, D.; Yu, H.; Zhang, Y.; Yan, W.; Xu, Z.; Zhao, B.; et al. A low dose cell therapy system for treating osteoarthritis: In vivo study and in vitro mechanistic investigations. Bioact. Mater. 2022, 7, 478–490. [Google Scholar] [CrossRef]
- Tsuzaki, M.; Guyton, G.; Garrett, W.; Archambault, J.M.; Herzog, W.; Almekinders, L.; Bynum, D.; Yang, X.; Banes, A.J. IL-1 beta induces COX2, MMP-1, -3 and -13, ADAMTS-4, IL-1 beta and IL-6 in human tendon cells. J. Orthop. Res. 2003, 21, 256–264. [Google Scholar] [CrossRef]
- Zhang, L.; Ma, S.; Su, H.; Cheng, J. Isoliquiritigenin Inhibits IL-1beta-Induced Production of Matrix Metalloproteinase in Articular Chondrocytes. Mol. Ther. Methods Clin. Dev. 2018, 9, 153–159. [Google Scholar] [CrossRef]
- Wu, Y.; Tang, Y.; Zhang, X.; Chu, Z.; Liu, Y.; Tang, C. MMP-1 promotes osteogenic differentiation of human bone marrow mesenchymal stem cells via the JNK and ERK pathway. Int. J. Biochem. Cell Biol. 2020, 129, 105880. [Google Scholar] [CrossRef]
- Chen, W.; Sun, Y.; Gu, X.; Hao, Y.; Liu, X.; Lin, J.; Chen, J.; Chen, S. Conditioned medium of mesenchymal stem cells delays osteoarthritis progression in a rat model by protecting subchondral bone, maintaining matrix homeostasis, and enhancing autophagy. J. Tissue Eng. Regen. Med. 2019, 13, 1618–1628. [Google Scholar] [CrossRef]
- Hassan Famian, M.; Montazer Saheb, S.; Montaseri, A. Conditioned Medium of Wharton’s Jelly Derived Stem Cells Can Enhance the Cartilage Specific Genes Expression by Chondrocytes in Monolayer and Mass Culture Systems. Adv. Pharm. Bull. 2017, 7, 123–130. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shin, S.; Lee, S.; Choi, S.; Park, N.; Kwon, Y.; Jeong, J.; Ju, S.; Chang, Y.; Park, K.; Ha, C.; et al. Characterization of the Secretome of a Specific Cell Expressing Mutant Methionyl-tRNA Synthetase in Co-Culture Using Click Chemistry. Int. J. Mol. Sci. 2022, 23, 6527. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23126527
Shin S, Lee S, Choi S, Park N, Kwon Y, Jeong J, Ju S, Chang Y, Park K, Ha C, et al. Characterization of the Secretome of a Specific Cell Expressing Mutant Methionyl-tRNA Synthetase in Co-Culture Using Click Chemistry. International Journal of Molecular Sciences. 2022; 23(12):6527. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23126527
Chicago/Turabian StyleShin, Sungho, Seonjeong Lee, Sunyoung Choi, Narae Park, Yumi Kwon, Jaehoon Jeong, Shinyeong Ju, Yunsil Chang, Kangsik Park, Chulwon Ha, and et al. 2022. "Characterization of the Secretome of a Specific Cell Expressing Mutant Methionyl-tRNA Synthetase in Co-Culture Using Click Chemistry" International Journal of Molecular Sciences 23, no. 12: 6527. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23126527