
Unraveling the Transcriptional Dynamics of NASH Pathogenesis Affecting Atherosclerosis

 , ,
, ,
Abstract
:1. Introduction
2. Results
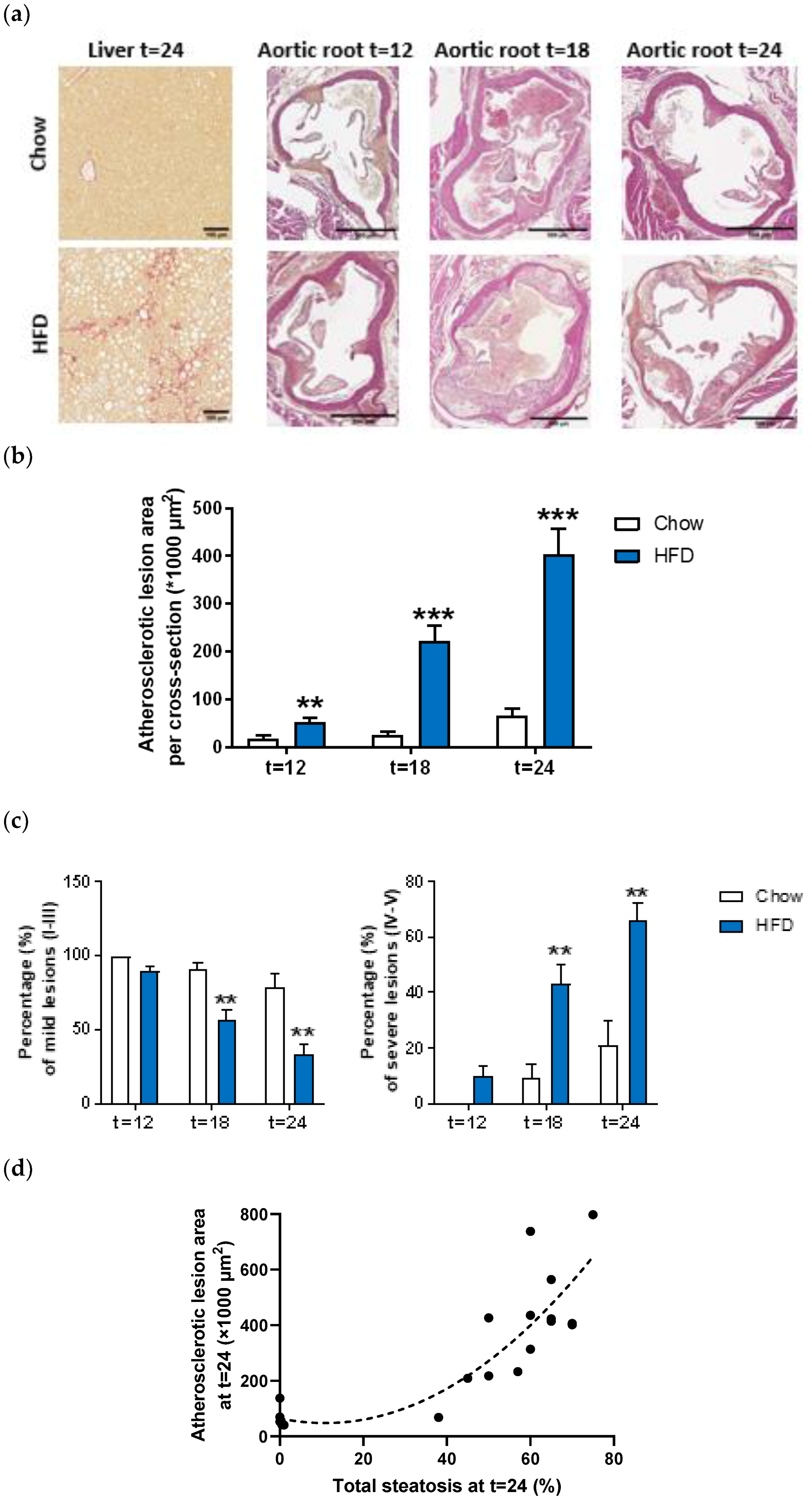
2.1. HFD Feeding Induces Features of the Metabolic Syndrome, NASH and Atherosclerosis
2.2. Transcriptome Analysis Showed Dynamics of Key Processes Involved in NASH and Atherosclerosis
2.3. Gene Regulatory Network Analysis Identified Key Hepatic Regulators Driving Atherogenic Pathways in the Aorta
3. Discussion
4. Materials and Methods
4.1. Animals and Experimental Design
4.2. Plasma Biochemical Analysis
4.3. Histology
4.4. Transcriptome Analysis
4.5. DynGENIE3
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Angulo, P.; Kleiner, D.E.; Dam-Larsen, S.; Adams, L.A.; Bjornsson, E.S.; Charatcharoenwitthaya, P.; Mills, P.R.; Keach, J.C.; Lafferty, H.D.; Stahler, A.; et al. Liver Fibrosis, but No Other Histologic Features, Is Associated with Long-term Outcomes of Patients with Nonalcoholic Fatty Liver Disease. Gastroenterology 2015, 149, 389–397.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekstedt, M.; Hagstrom, H.; Nasr, P.; Fredrikson, M.; Stal, P.; Kechagias, S.; Hultcrantz, R. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology 2015, 61, 1547–1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Csermely, A.; Petracca, G.; Beatrice, G.; Corey, K.E.; Simon, T.G.; Byrne, C.D.; Targher, G. Non-alcoholic fatty liver disease and risk of fatal and non-fatal cardiovascular events: An updated systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2021, 6, 903–913. [Google Scholar] [CrossRef]
- Mellinger, J.L.; Pencina, K.M.; Massaro, J.M.; Hoffmann, U.; Seshadri, S.; Fox, C.S.; O’Donnell, C.J.; Speliotes, E.K. Hepatic steatosis and cardiovascular disease outcomes: An analysis of the Framingham Heart Study. J. Hepatol. 2015, 63, 470–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motamed, N.; Rabiee, B.; Poustchi, H.; Dehestani, B.; Hemasi, G.R.; Khonsari, M.R.; Maadi, M.; Saeedian, F.S.; Zamani, F. Non-alcoholic fatty liver disease (NAFLD) and 10-year risk of cardiovascular diseases. Clin. Res. Hepatol. Gastroenterol. 2017, 41, 31–38. [Google Scholar] [CrossRef]
- El Hadi, H.; Di Vincenzo, A.; Vettor, R.; Rossato, M. Relationship between Heart Disease and Liver Disease: A Two-Way Street. Cells 2020, 9, 567. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.; Taskinen, M.R.; Boren, J. Crosstalk between nonalcoholic fatty liver disease and cardiometabolic syndrome. Obes. Rev. 2019, 20, 599–611. [Google Scholar] [CrossRef] [Green Version]
- D’Agostino, R.B., Sr.; Vasan, R.S.; Pencina, M.J.; Wolf, P.A.; Cobain, M.; Massaro, J.M.; Kannel, W.B. General cardiovascular risk profile for use in primary care: The Framingham Heart Study. Circulation 2008, 117, 743–753. [Google Scholar] [CrossRef] [Green Version]
- Heidemann, B.E.; Koopal, C.; Bots, M.L.; Asselbergs, F.W.; Westerink, J.; Visseren, F.L.J. The relation between VLDL-cholesterol and risk of cardiovascular events in patients with manifest cardiovascular disease. Int. J. Cardiol. 2021, 322, 251–257. [Google Scholar] [CrossRef]
- Harchaoui, K.E.; Visser, M.E.; Kastelein, J.J.; Stroes, E.S.; Dallinga-Thie, G.M. Triglycerides and cardiovascular risk. Curr. Cardiol. Rev. 2009, 5, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, S.M.; Defina, L.F.; Leonard, D.; Barlow, C.E.; Radford, N.B.; Willis, B.L.; Rohatgi, A.; McGuire, D.K.; de Lemos, J.A.; Grundy, S.M.; et al. Long-Term Association of Low-Density Lipoprotein Cholesterol with Cardiovascular Mortality in Individuals at Low 10-Year Risk of Atherosclerotic Cardiovascular Disease. Circulation 2018, 138, 2315–2325. [Google Scholar] [CrossRef] [PubMed]
- Tariq, Z.; Green, C.J.; Hodson, L. Are oxidative stress mechanisms the common denominator in the progression from hepatic steatosis towards non-alcoholic steatohepatitis (NASH)? Liver Int. 2014, 34, e180–e190. [Google Scholar] [CrossRef] [PubMed]
- Polimeni, L.; Del Ben, M.; Baratta, F.; Perri, L.; Albanese, F.; Pastori, D.; Violi, F.; Angelico, F. Oxidative stress: New insights on the association of non-alcoholic fatty liver disease and atherosclerosis. World J. Hepatol. 2015, 7, 1325–1336. [Google Scholar] [CrossRef] [PubMed]
- Arab, J.P.; Arrese, M.; Trauner, M. Recent Insights into the Pathogenesis of Nonalcoholic Fatty Liver Disease. Annu. Rev. Pathol. 2018, 13, 321–350. [Google Scholar] [CrossRef] [PubMed]
- Pasterkamp, G. Methods of accelerated atherosclerosis in diabetic patients. Heart 2013, 99, 743–749. [Google Scholar] [CrossRef]
- Bornfeldt, K.E.; Tabas, I. Insulin resistance, hyperglycemia, and atherosclerosis. Cell Metab. 2011, 14, 575–585. [Google Scholar] [CrossRef] [Green Version]
- Francque, S.M.; van der Graaff, D.; Kwanten, W.J. Non-alcoholic fatty liver disease and cardiovascular risk: Pathophysiological mechanisms and implications. J. Hepatol. 2016, 65, 425–443. [Google Scholar] [CrossRef] [Green Version]
- Gart, E.; Souto Lima, E.; Schuren, F.; de Ruiter, C.G.F.; Attema, J.; Verschuren, L.; Keijer, J.; Salic, K.; Morrison, M.C.; Kleemann, R. Diet-Independent Correlations between Bacteria and Dysfunction of Gut, Adipose Tissue, and Liver: A Comprehensive Microbiota Analysis in Feces and Mucosa of the Ileum and Colon in Obese Mice with NAFLD. Int. J. Mol. Sci. 2018, 20, 1. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, S.A.H.; Gart, E.; Vreeken, D.; Franx, B.A.A.; Wekking, L.; Verweij, V.G.M.; Worms, N.; Schoemaker, M.H.; Gross, G.; Morrison, M.C.; et al. Sex-Specific Differences in Fat Storage, Development of Non-Alcoholic Fatty Liver Disease and Brain Structure in Juvenile HFD-Induced Obese Ldlr−/−.Leiden Mice. Nutrients 2019, 11, 1861. [Google Scholar] [CrossRef] [Green Version]
- Luque-Sierra, A.; Alvarez-Amor, L.; Kleemann, R.; Martin, F.; Varela, L.M. Extra-Virgin Olive Oil with Natural Phenolic Content Exerts an Anti-Inflammatory Effect in Adipose Tissue and Attenuates the Severity of Atherosclerotic Lesions in Ldlr−/−.Leiden Mice. Mol. Nutr. Food Res. 2018, 62, e1800295. [Google Scholar] [CrossRef]
- Morrison, M.C.; Kleemann, R.; van Koppen, A.; Hanemaaijer, R.; Verschuren, L. Key Inflammatory Processes in Human NASH Are Reflected in Ldlr (-/-).Leiden Mice: A Translational Gene Profiling Study. Front. Physiol. 2018, 9, 132. [Google Scholar] [CrossRef] [PubMed]
- Morrison, M.C.; Mulder, P.; Salic, K.; Verheij, J.; Liang, W.; van Duyvenvoorde, W.; Menke, A.; Kooistra, T.; Kleemann, R.; Wielinga, P.Y. Intervention with a caspase-1 inhibitor reduces obesity-associated hyperinsulinemia, non-alcoholic steatohepatitis and hepatic fibrosis in Ldlr−/−.Leiden mice. Int. J. Obes. 2016, 40, 1416–1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, M.C.; Verschuren, L.; Salic, K.; Verheij, J.; Menke, A.; Wielinga, P.Y.; Iruarrizaga-Lejarreta, M.; Gole, L.; Yu, W.M.; Turner, S.; et al. Obeticholic Acid Modulates Serum Metabolites and Gene Signatures Characteristic of Human NASH and Attenuates Inflammation and Fibrosis Progression in Ldlr−/−.Leiden Mice. Hepatol. Commun. 2018, 2, 1513–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelgrim, C.E.; Franx, B.A.A.; Snabel, J.; Kleemann, R.; Arnoldussen, I.A.C.; Kiliaan, A.J. Butyrate Reduces HFD-Induced Adipocyte Hypertrophy and Metabolic Risk Factors in Obese Ldlr−/−.Leiden Mice. Nutrients 2017, 9, 714. [Google Scholar] [CrossRef] [Green Version]
- Salic, K.; Kleemann, R.; Wilkins-Port, C.; McNulty, J.; Verschuren, L.; Palmer, M. Apical sodium-dependent bile acid transporter inhibition with volixibat improves metabolic aspects and components of non-alcoholic steatohepatitis in Ldlr−/−.Leiden mice. PLoS ONE 2019, 14, e0218459. [Google Scholar] [CrossRef]
- Schoemaker, M.H.; Kleemann, R.; Morrison, M.C.; Verheij, J.; Salic, K.; van Tol, E.A.F.; Kooistra, T.; Wielinga, P.Y. A casein hydrolysate based formulation attenuates obesity and associated non-alcoholic fatty liver disease and atherosclerosis in Ldlr−/−.Leiden mice. PLoS ONE 2017, 12, e0180648. [Google Scholar] [CrossRef] [Green Version]
- van den Hoek, A.M.; Verschuren, L.; Worms, N.; van Nieuwkoop, A.; de Ruiter, C.; Attema, J.; Menke, A.L.; Caspers, M.P.M.; Radhakrishnan, S.; Salic, K.; et al. A Translational Mouse Model for NASH with Advanced Fibrosis and Atherosclerosis Expressing Key Pathways of Human Pathology. Cells 2020, 9, 2014. [Google Scholar] [CrossRef]
- van Koppen, A.; Verschuren, L.; van den Hoek, A.M.; Verheij, J.; Morrison, M.C.; Li, K.; Nagabukuro, H.; Costessi, A.; Caspers, M.P.M.; van den Broek, T.J.; et al. Uncovering a Predictive Molecular Signature for the Onset of NASH-Related Fibrosis in a Translational NASH Mouse Model. Cell. Mol. Gastroenterol. Hepatol. 2018, 5, 83–98.e10. [Google Scholar] [CrossRef] [Green Version]
- Collins, T.; Cybulsky, M.I. NF-kappaB: Pivotal mediator or innocent bystander in atherogenesis? J Clin Invest 2001, 107, 255–264. [Google Scholar] [CrossRef]
- Ying, H.Z.; Chen, Q.; Zhang, W.Y.; Zhang, H.H.; Ma, Y.; Zhang, S.Z.; Fang, J.; Yu, C.H. PDGF signaling pathway in hepatic fibrosis pathogenesis and therapeutics (Review). Mol. Med. Rep. 2017, 16, 7879–7889. [Google Scholar] [CrossRef] [Green Version]
- van den Hoek, A.M.; de Jong, J.; Worms, N.; van Nieuwkoop, A.; Voskuilen, M.; Menke, A.L.; Lek, S.; Caspers, M.P.M.; Verschuren, L.; Kleemann, R. Diet and exercise reduce pre-existing NASH and fibrosis and have additional beneficial effects on the vasculature, adipose tissue and skeletal muscle via organ-crosstalk. Metabolism 2021, 124, 154873. [Google Scholar] [CrossRef] [PubMed]
- Teufel, A.; Itzel, T.; Erhart, W.; Brosch, M.; Wang, X.Y.; Kim, Y.O.; von Schonfels, W.; Herrmann, A.; Bruckner, S.; Stickel, F.; et al. Comparison of Gene Expression Patterns Between Mouse Models of Nonalcoholic Fatty Liver Disease and Liver Tissues From Patients. Gastroenterology 2016, 151, 513–525.e0. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Arranz, I.; Bruzzone, C.; Noureddin, M.; Gil-Redondo, R.; Minchole, I.; Bizkarguenaga, M.; Arretxe, E.; Iruarrizaga-Lejarreta, M.; Fernandez-Ramos, D.; Lopitz-Otsoa, F.; et al. Metabolic subtypes of patients with NAFLD exhibit distinctive cardiovascular risk profiles. Hepatology 2022. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Shi, H.; Zhao, M.; Zhang, X.; Huang, W.; Wang, Y.; Zheng, L.; Xian, X.; Liu, G. Loss of LCAT activity in the golden Syrian hamster elicits pro-atherogenic dyslipidemia and enhanced atherosclerosis. Metabolism 2018, 83, 245–255. [Google Scholar] [CrossRef]
- Guo, M.; Liu, Z.; Xu, Y.; Ma, P.; Huang, W.; Gao, M.; Wang, Y.; Liu, G.; Xian, X. Spontaneous Atherosclerosis in Aged LCAT-Deficient Hamsters With Enhanced Oxidative Stress-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2829–2836. [Google Scholar] [CrossRef]
- Bourdillon, M.C.; Randon, J.; Barek, L.; Zibara, K.; Covacho, C.; Poston, R.N.; Chignier, E.; McGregor, J.L. Reduced atherosclerotic lesion size in P-selectin deficient apolipoprotein E-knockout mice fed a chow but not a fat diet. J. Biomed. Biotechnol. 2006, 2006, 49193. [Google Scholar] [CrossRef] [Green Version]
- Koike, T.; Koike, Y.; Yang, D.; Guo, Y.; Rom, O.; Song, J.; Xu, J.; Chen, Y.; Wang, Y.; Zhu, T.; et al. Human apolipoprotein A-II reduces atherosclerosis in knock-in rabbits. Atherosclerosis 2021, 316, 32–40. [Google Scholar] [CrossRef]
- Wang, Y.; Niimi, M.; Nishijima, K.; Waqar, A.B.; Yu, Y.; Koike, T.; Kitajima, S.; Liu, E.; Inoue, T.; Kohashi, M.; et al. Human apolipoprotein A-II protects against diet-induced atherosclerosis in transgenic rabbits. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 224–231. [Google Scholar] [CrossRef] [Green Version]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Nonalcoholic Steatohepatitis Clinical Research, N. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Liang, W.; Menke, A.L.; Driessen, A.; Koek, G.H.; Lindeman, J.H.; Stoop, R.; Havekes, L.M.; Kleemann, R.; van den Hoek, A.M. Establishment of a general NAFLD scoring system for rodent models and comparison to human liver pathology. PLoS ONE 2014, 9, e115922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delsing, D.J.; Offerman, E.H.; van Duyvenvoorde, W.; van Der Boom, H.; de Wit, E.C.; Gijbels, M.J.; van Der Laarse, A.; Jukema, J.W.; Havekes, L.M.; Princen, H.M. Acyl-CoA:cholesterol acyltransferase inhibitor avasimibe reduces atherosclerosis in addition to its cholesterol-lowering effect in ApoE*3-Leiden mice. Circulation 2001, 103, 1778–1786. [Google Scholar] [CrossRef] [PubMed]
- Kuhnast, S.; van der Tuin, S.J.; van der Hoorn, J.W.; van Klinken, J.B.; Simic, B.; Pieterman, E.; Havekes, L.M.; Landmesser, U.; Luscher, T.F.; Willems van Dijk, K.; et al. Anacetrapib reduces progression of atherosclerosis, mainly by reducing non-HDL-cholesterol, improves lesion stability and adds to the beneficial effects of atorvastatin. Eur. Heart J. 2015, 36, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Stary, H.C.; Chandler, A.B.; Dinsmore, R.E.; Fuster, V.; Glagov, S.; Insull, W., Jr.; Rosenfeld, M.E.; Schwartz, C.J.; Wagner, W.D.; Wissler, R.W. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 1355–1374. [Google Scholar] [CrossRef] [PubMed]
- Huynh-Thu, V.A.; Geurts, P. dynGENIE3: Dynamical GENIE3 for the inference of gene networks from time series expression data. Sci. Rep. 2018, 8, 3384. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chow | HFD | ||
|---|---|---|---|
| Body weight (g) | t = 24 | 36.7 ± 1.6 | 51.8 ± 1.0 *** |
| Blood glucose (mM) | t = 24 | 6.7 ± 0.3 | 6.9 ± 0.3 |
| Plasma insulin (ng/mL) | t = 24 | 2.0 ± 0.3 | 20.7 ± 4.3 *** |
| HOMA-IR | t = 24 | 0.6 ± 0.1 | 6.3 ± 1.4 *** |
| Plasma cholesterol (mM) | t = 24 | 7.6 ± 0.6 | 33.2 ± 2.7 *** |
| Plasma triglycerides (mM) | t = 24 | 1.3 ± 0.1 | 4.8 ± 0.8 *** |
| Total steatosis (%) | t = 6 | 0.0 ± 0.0 | 33.6 ± 3.0 *** |
| t = 12 | 0.2 ± 0.1 | 53.4 ± 2.6 *** | |
| t = 18 | 1.8 ± 0.6 | 69.3 ± 2.7 *** | |
| t = 24 | 0.3 ± 0.2 | 59.3 ± 2.8 *** | |
| Hepatic inflammation | t = 6 | 1.0 ± 0.4 | 6.1 ± 0.8 *** |
| (number of aggregates/mm2) | t = 12 | 2.0 ± 0.6 | 5.4 ± 1.2 |
| t = 18 | 2.2 ± 0.7 | 10.6 ± 3.0 ** | |
| t = 24 | 0.7 ± 0.2 | 12.1 ± 3.8 *** | |
| Hepatic fibrosis (%) | t = 6 | 0.0 ± 0.0 | 0.0 ± 0.0 |
| t = 12 | 0.0 ± 0.0 | 0.0 ± 0.0 | |
| t = 18 | 0.0 ± 0.0 | 2.5 ± 2.2 | |
| t = 24 | 0.0 ± 0.0 | 5.0 ± 2.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van den Hoek, A.M.; Özsezen, S.; Caspers, M.P.M.; van Koppen, A.; Hanemaaijer, R.; Verschuren, L. Unraveling the Transcriptional Dynamics of NASH Pathogenesis Affecting Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 8229. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23158229
van den Hoek AM, Özsezen S, Caspers MPM, van Koppen A, Hanemaaijer R, Verschuren L. Unraveling the Transcriptional Dynamics of NASH Pathogenesis Affecting Atherosclerosis. International Journal of Molecular Sciences. 2022; 23(15):8229. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23158229
Chicago/Turabian Stylevan den Hoek, Anita M., Serdar Özsezen, Martien P. M. Caspers, Arianne van Koppen, Roeland Hanemaaijer, and Lars Verschuren. 2022. "Unraveling the Transcriptional Dynamics of NASH Pathogenesis Affecting Atherosclerosis" International Journal of Molecular Sciences 23, no. 15: 8229. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23158229