
Pretreatment with Zonisamide Mitigates Oxaliplatin-Induced Toxicity in Rat DRG Neurons and DRG Neuron–Schwann Cell Co-Cultures


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
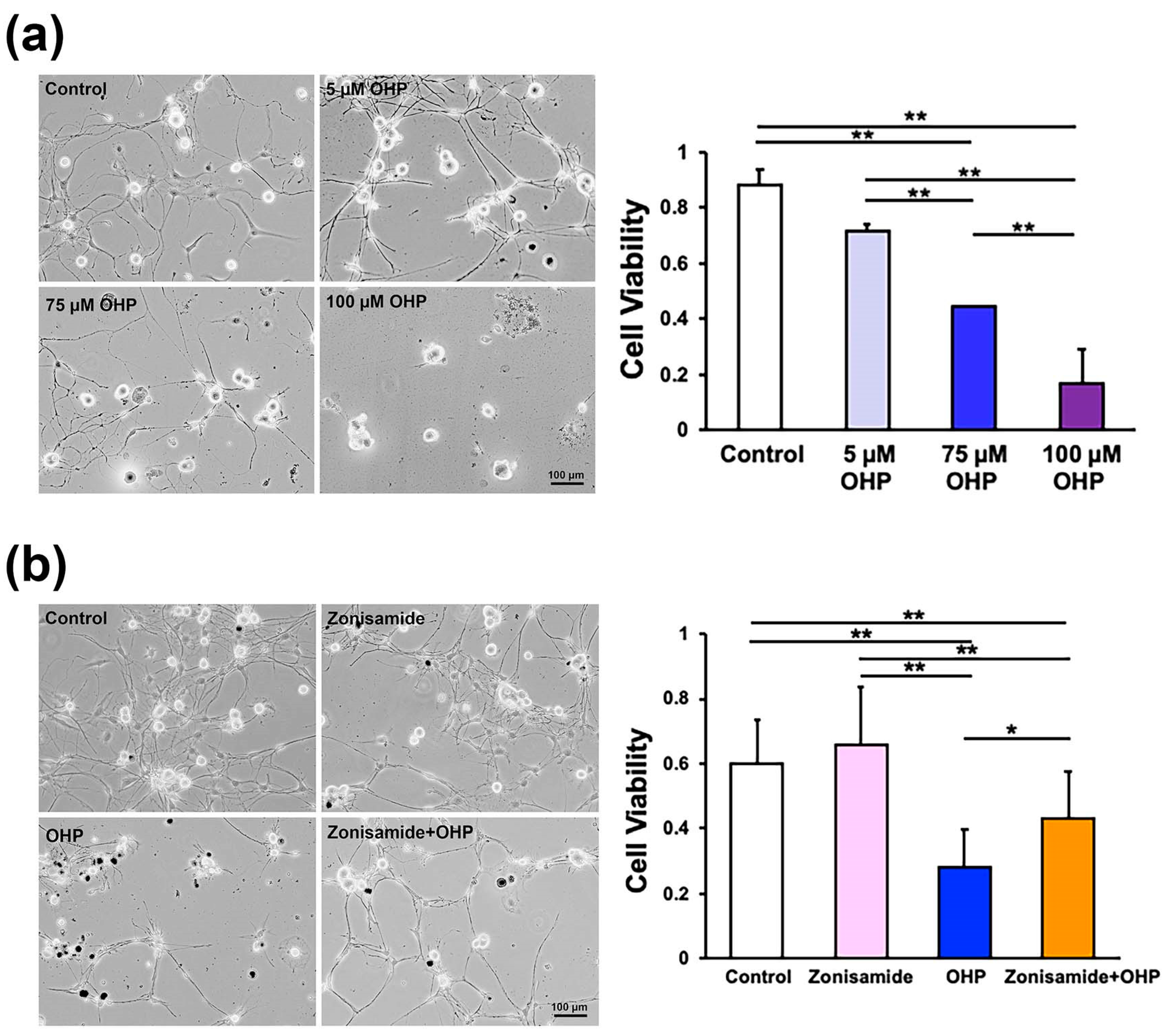
2.1. Zonisamide Pretreatment Alleviates OHP-Induced Toxicity against Primary Cultured Adult Rat DRG Neurons
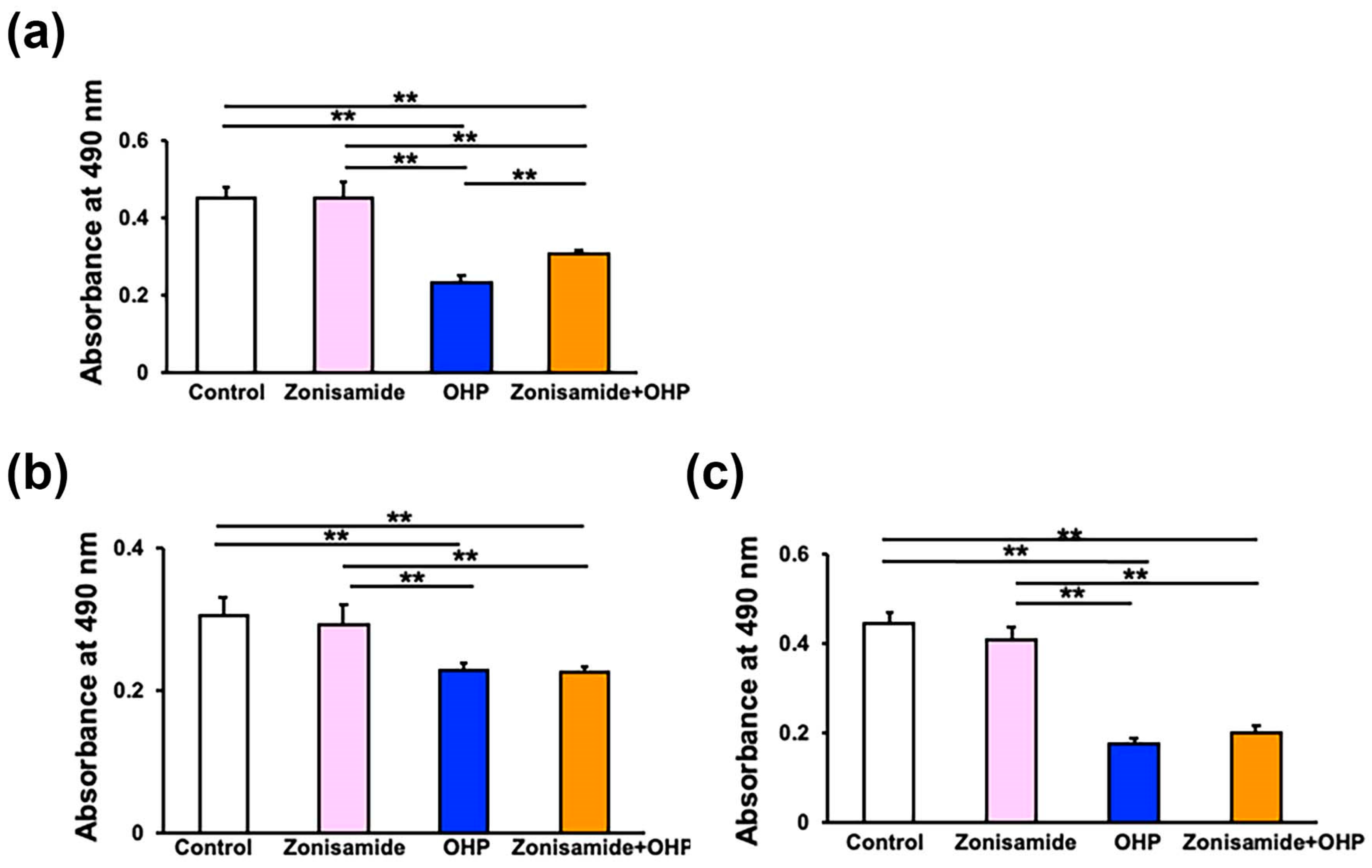
2.2. Zonisamide Pretreatment Alleviates the OHP-Induced Toxicity against Immortalized Rat DRG Neurons, ND7/23, but Not against Immortalized Rat Schwann Cells, IFRS1
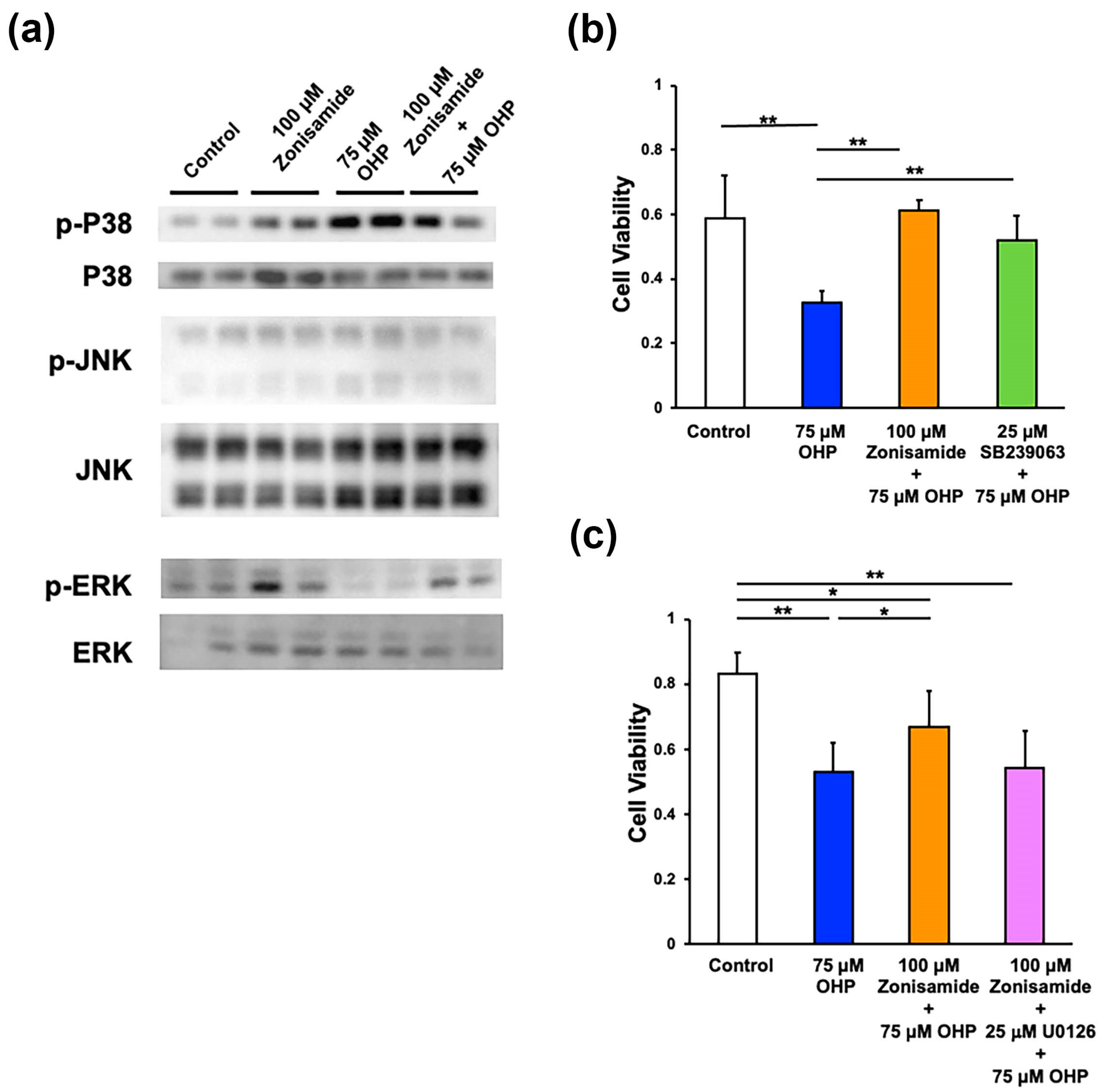
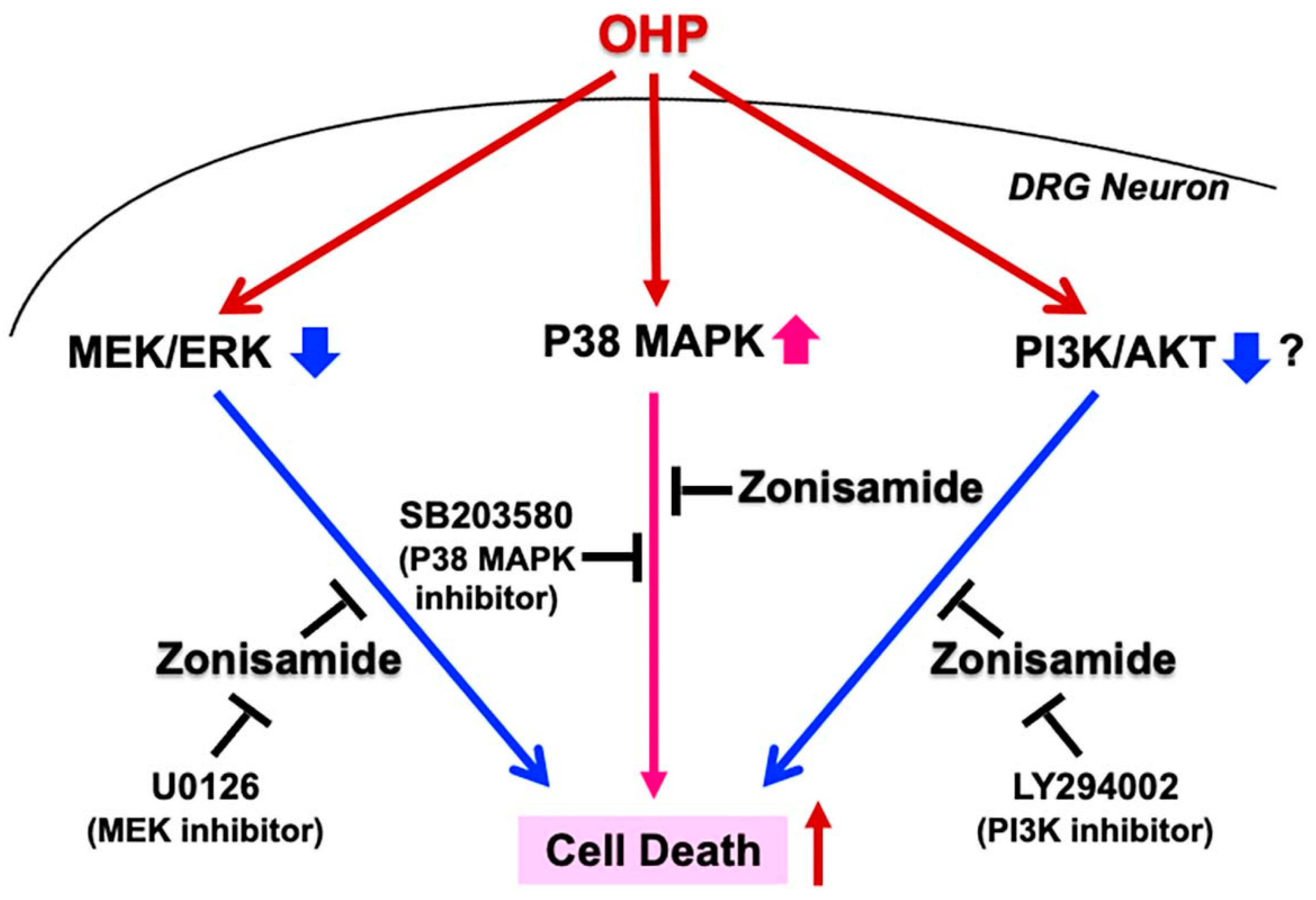
2.3. Involvement of p38 MAPK and MEK/ERK Signaling Pathways in Zonisamide’s Alleviating Effects on the OHP-Induced DRG Neuronal Death
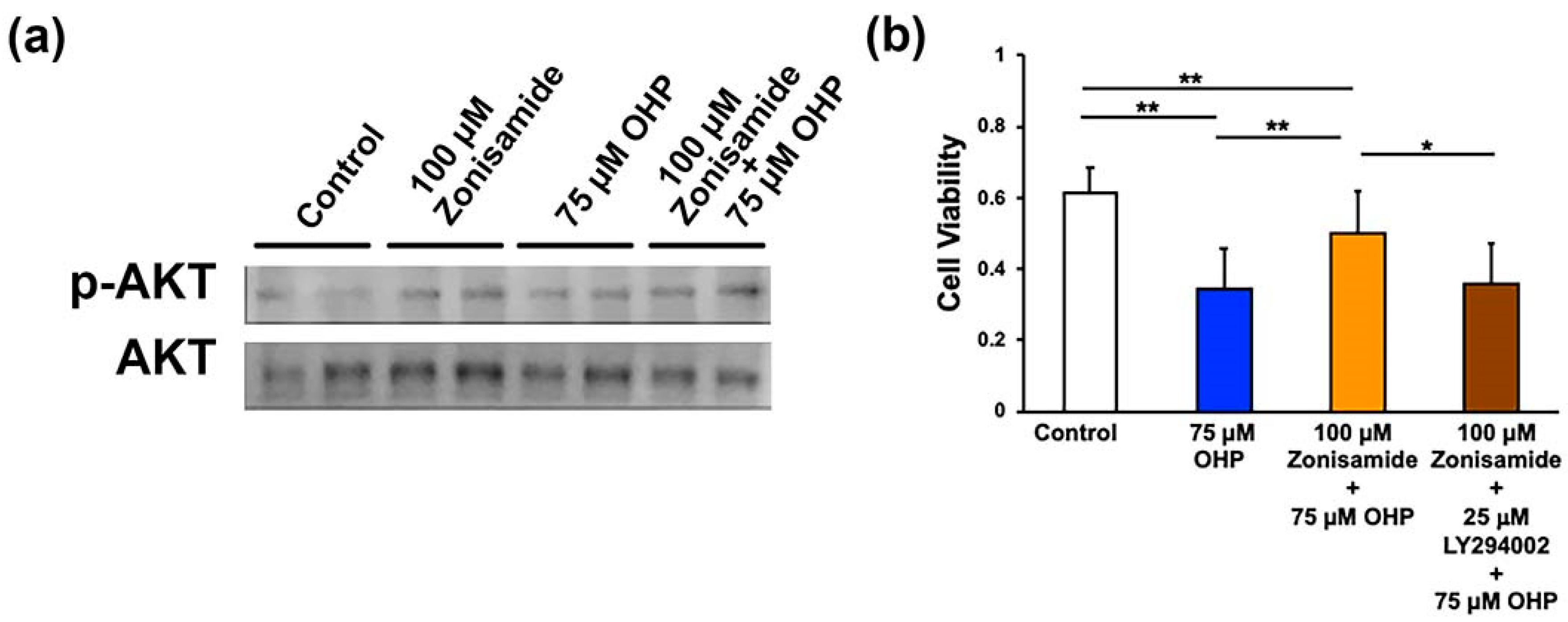
2.4. Involvement of the PI3K/AKT Signaling Pathway in Zonisamide’s Alleviating Effects on the OHP-Induced DRG Neuron Death
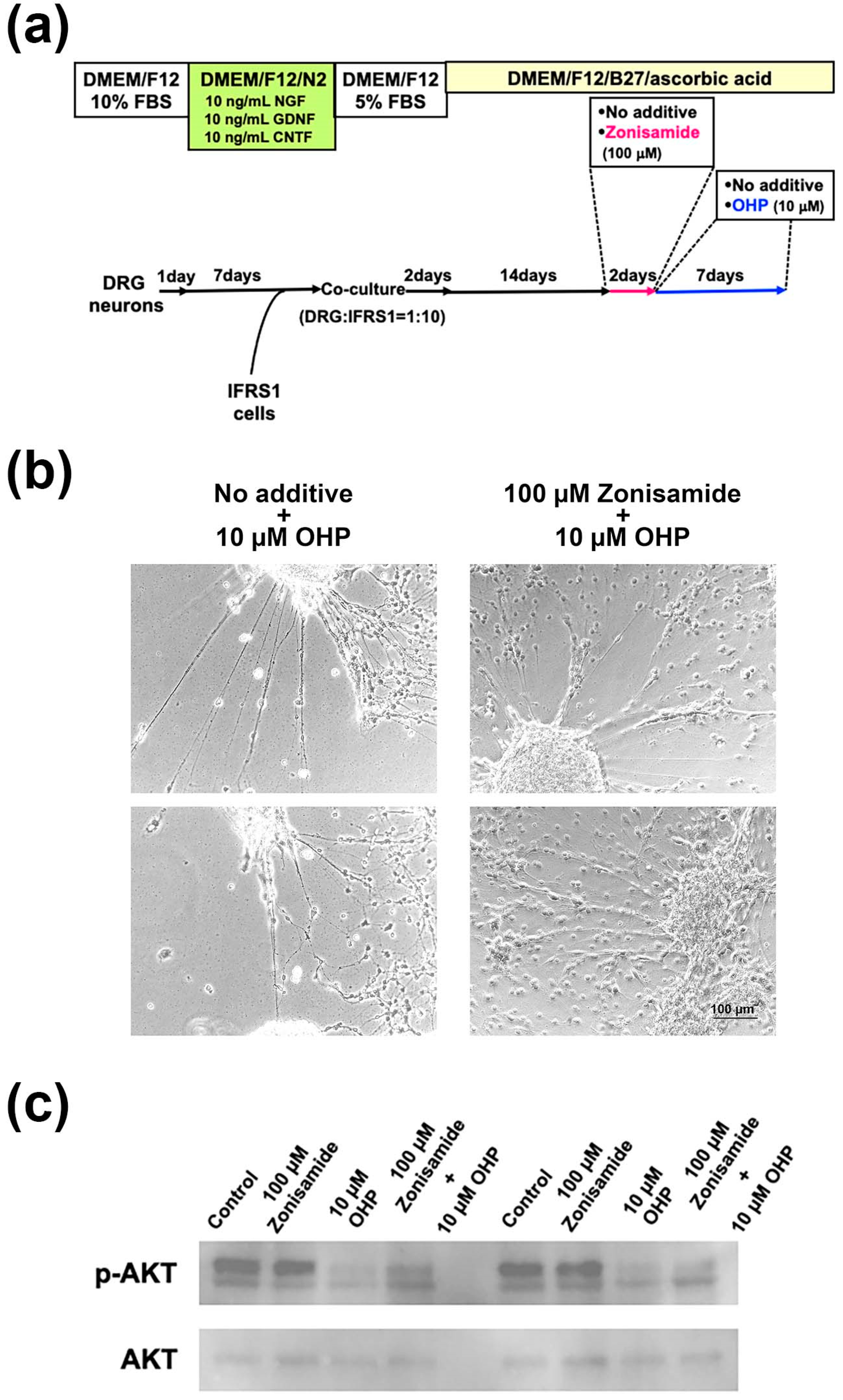
2.5. Pretreatment with Zonisamide Alleviates the OHP-Induced Neurite Degeneration and Demyelination-like Changes in DRG Neuron–IFRS1 Co-Cultures
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Primary Culture of DRG Neurons and Cell Viability Assay
4.3. Culture of ND7/23 Sensory Neuron-Like Cells and IFRS1 Schwann Cells and Cell Viability Assay
4.4. Co-Culture of DRG Neurons and IFRS1 Schwann Cells
4.5. Western Blotting Analysis
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Padhy, B.M.; Gupta, Y.K. Drug repositioning: Re-investigating existing drugs for new therapeutic indications. J. Postgrad. Med. 2011, 57, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Jain, K.K. An assessment of zonisamide as an anti-epileptic drug. Expert Opin. Pharmacother. 2000, 1, 1245–1260. [Google Scholar] [CrossRef] [PubMed]
- Murata, M.; Hasegawa, K.; Kanazawa, I.; Fukasaka, J.; Kochi, K.; Shimazu, R.; Japan Zonisamide on PD Study Group. Zonisamide improves wearing-off in Parkinson’s disease: A randomized, double-blind study. Mov. Disord. 2015, 30, 1343–1350. [Google Scholar] [CrossRef]
- Li, C.; Xue, L.; Liu, Y.; Yang, Z.; Chi, S.; Xie, A. Zonisamide for the treatment of Parkinson disease: A current update. Front. Neurosci. 2020, 14, 574652. [Google Scholar] [CrossRef]
- Yagi, H.; Ohkawara, B.; Nakashima, H.; Ito, K.; Tsushima, M.; Ishii, H.; Noto, K.; Ohta, K.; Masuda, A.; Imagama, S.; et al. Zonisamide enhances neurite elongation of primary motor neurons and facilitates peripheral nerve regeneration in vitro and in a mouse model. PLoS ONE 2015, 10, e0142786. [Google Scholar] [CrossRef] [PubMed]
- Takaku, S.; Sango, K. Zonisamide enhances neurite outgrowth from adult rat dorsal root ganglion neurons, but not proliferation or migration of Schwann cells. Histochem. Cell Biol. 2020, 153, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Bektas, N.; Arslan, R.; Ozturk, Y. Zonisamide: Antihyperalgesic efficacy, the role of serotonergic receptors on efficacy in a rat model for painful diabetic neuropathy. Life Sci. 2014, 95, 9–13. [Google Scholar] [CrossRef]
- Koshimizu, H.; Ohkawara, B.; Nakashima, H.; Ota, K.; Kanbara, S.; Inoue, T.; Tomita, H.; Sayo, A.; Kiryu-Seo, S.; Konishi, H.; et al. Zonisamide ameliorates neuropathic pain partly by suppressing microglial activation in the spinal cord in a mouse model. Life Sci. 2020, 263, 118577. [Google Scholar] [CrossRef]
- Argyriou, A.A. Updates on oxaliplatin-induced peripheral neurotoxicity (OXAIPN). Toxics 2015, 3, 187–197. [Google Scholar] [CrossRef]
- Ta, L.E.; Espeset, L.; Podratz, J.; Windebank, A.J. Neurotoxicity of oxaliplatin and cisplatin for dorsal root ganglion neurons correlates with platinum-DNA binding. Neurotoxicology 2006, 27, 992–1002. [Google Scholar] [CrossRef]
- Joseph, E.K.; Chen, X.; Bogen, O.; Levine, J.D. Oxaliplatin acts on IB4-positive nociceptors to induce an oxidative stress-dependent acute painful peripheral neuropathy. J. Pain 2008, 9, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Scuteri, A.; Galimberti, A.; Ravasi, M.; Pasini, S.; Donzelli, E.; Cavaletti, G.; Tredici, G. NGF protects dorsal root ganglion neurons from oxaliplatin by modulating JNK/Sapk and ERK1/2. Neurosci. Lett. 2010, 486, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Tian, Y.; Xu, S.; Chen, H. Oxaliplatin-induced peripheral neuropathy: Clinical features, mechanisms, prevention and treatment. J. Neurol. 2021, 268, 3269–3282. [Google Scholar] [CrossRef] [PubMed]
- Branca, J.; Carrino, D.; Gulisano, M.; Ghelardini, C.; Di Cesare Mannelli, L.; Pacini, A. Oxaliplatin-induced neuropathy: Genetic and epigenetic profile to better understand how to ameliorate this side effect. Front. Mol. Biosci. 2021, 8, 643824. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; Li, L.; Song, F.; Li, P.; Chen, M.; Ni, S.; Zhang, H.; Zhou, H.; Zeng, S.; Jiang, H. L-tetrahydropalmatine reduces oxaliplatin accumulation in the dorsal root ganglion and mitochondria through selectively inhibiting the transporter-mediated uptake thereby attenuates peripheral neurotoxicity. Toxicology 2021, 459, 152853. [Google Scholar] [CrossRef]
- Mimaki, T. Clinical pharmacology and therapeutic drug monitoring of zonisamide. Ther. Drug Monit. 1998, 20, 593–597. [Google Scholar] [CrossRef]
- Wood, J.N.; Bevan, S.J.; Coote, P.R.; Dunn, P.M.; Harmar, A.; Hogan, P.; Latchman, D.S.; Morrison, C.; Rougon, G.; Theveniau, M. Novel cell lines display properties of nociceptive sensory neurons. Proc. Biol. Sci. 1990, 241, 187–194. [Google Scholar]
- Haberberger, R.V.; Barry, C.; Matusica, D. Immortalized dorsal root ganglion neuron cell lines. Front. Cell. Neurosci. 2020, 14, 184. [Google Scholar] [CrossRef]
- Sango, K.; Yanagisawa, H.; Kawakami, E.; Takaku, S.; Ajiki, K.; Watabe, K. Spontaneously immortalized Schwann cells from adult Fischer rat as a valuable tool for exploring neuron-Schwann cell interactions. J. Neurosci. Res. 2011, 89, 898–908. [Google Scholar] [CrossRef]
- Niimi, N.; Yako, H.; Takaku, S.; Kato, H.; Matsumoto, T.; Nishito, Y.; Watabe, K.; Ogasawara, S.; Mizukami, H.; Yagihashi, S.; et al. A spontaneously immortalized Schwann cell line from aldose reductase-deficient mice as a useful tool for studying polyol pathway and aldehyde metabolism. J. Neurochem. 2018, 144, 710–722. [Google Scholar] [CrossRef]
- Maruta, T.; Nemoto, T.; Hidaka, K.; Koshida, T.; Shirasaka, T.; Yanagita, T.; Takeya, R.; Tsuneyoshi, I. Upregulation of ERK phosphorylation in rat dorsal root ganglion neurons contributes to oxaliplatin-induced chronic neuropathic pain. PLoS ONE 2019, 14, e0225586. [Google Scholar] [CrossRef]
- Takaku, S.; Tsukamoto, M.; Niimi, N.; Yako, H.; Sango, K. Exendin-4 promotes Schwann cell survival/migration and myelination in vitro. Int. J. Mol. Sci. 2021, 22, 2971. [Google Scholar] [CrossRef] [PubMed]
- Goyal, S.; Singla, S.; Kumar, D.; Menaria, G. Comparison of the Effects of Zonisamide, ethosuximide and pregabalin in the chronic constriction injury induced neuropathic pain in rats. Ann. Med. Health Sci. Res. 2015, 5, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Ohkawara, B.; Bushra, S.; Kanbara, S.; Nakashima, H.; Koshimizu, H.; Tomita, H.; Ito, M.; Masuda, A.; Ishiguro, N.; et al. Zonisamide upregulates neuregulin-1 expression and enhances acetylcholine receptor clustering at the in vitro neuromuscular junction. Neuropharmacology 2021, 195, 108637. [Google Scholar] [CrossRef]
- Grolleau, F.; Gamelin, L.; Boisdron-Celle, M.; Lapied, B.; Pelhate, M.; Gamelin, E. A possible explanation for a neurotoxic effect of the anticancer agent oxaliplatin on neuronal voltage-gated sodium channels. J. Neurophysiol. 2001, 85, 2293–2297. [Google Scholar] [CrossRef] [PubMed]
- Akamine, T.; Takaku, S.; Suzuki, M.; Niimi, N.; Yako, H.; Matoba, K.; Kawanami, D.; Utsunomiya, K.; Nishimura, R.; Sango, K. Glycolaldehyde induces sensory neuron death through activation of the c-Jun N-terminal kinase and p-38 MAP kinase pathways. Histochem. Cell Biol. 2020, 153, 111–119. [Google Scholar] [CrossRef]
- Johnson, G.L.; Lapadat, R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef] [PubMed]
- Tsubaki, M.; Takeda, T.; Tani, T.; Shimaoka, H.; Suzuyama, N.; Sakamoto, K.; Fujita, A.; Ogawa, N.; Itoh, T.; Imano, M.; et al. PKC/MEK inhibitors suppress oxaliplatin-induced neuropathy and potentiate the antitumor effects. Int. J. Cancer 2015, 137, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Yeo, J.H.; Yoon, S.Y.; Kim, S.J.; Oh, S.B.; Lee, J.H.; Beitz, A.J.; Roh, D.H. Clonidine, an alpha-2 adrenoceptor agonist relieves mechanical allodynia in oxaliplatin-induced neuropathic mice; potentiation by spinal p38 MAPK inhibition without motor dysfunction and hypotension. Int. J. Cancer 2016, 138, 2466–2476. [Google Scholar] [CrossRef]
- Fontanet, P.; Irala, D.; Alsina, F.C.; Paratcha, G.; Ledda, F. Pea3 transcription factor family members Etv4 and Etv5 mediate retrograde signaling and axonal growth of DRG sensory neurons in response to NGF. J. Neurosci. 2013, 33, 15940–15951. [Google Scholar] [CrossRef]
- Yamamoto, K.; Chiba, N.; Chiba, T.; Kambe, T.; Abe, K.; Kawakami, K.; Utsunomiya, I.; Taguchi, K. Transient receptor potential ankyrin 1 that is induced in dorsal root ganglion neurons contributes to acute cold hypersensitivity after oxaliplatin administration. Mol. Pain 2015, 11, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, J.; Zhang, Q.; Yang, C.; Xiao, L.; Xue, Z.; Zhu, J. Duloxetine, a balanced serotonin-norepinephrine reuptake inhibitor, improves painful chemotherapy-induced peripheral neuropathy by inhibiting activation of p38 MAPK and NF-κB. Front. Pharmacol. 2019, 10, 365. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Hu, L.; Wang, C.; Yang, X.; Song, L.; Jiang, C.; Li, Y.; Li, T.; Liu, W.T.; Feng, J. p38/TF/HIF-α Signaling pathway participates in the progression of CIPN in mice. BioMed Res. Int. 2019, 2019, 5347804. [Google Scholar] [CrossRef]
- Celik, H.; Kucukler, S.; Ozdemir, S.; Comakli, S.; Gur, C.; Kandemir, F.M.; Yardim, A. Lycopene protects against central and peripheral neuropathy by inhibiting oxaliplatin-induced ATF-6 pathway, apoptosis, inflammation and oxidative stress in brains and sciatic tissues of rats. Neurotoxicology 2020, 80, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Lu, Y.; Yang, C.; Zhang, Q.; Qian, Y.; Suo, J.; Cheng, P.; Zhu, J. Based on systematic pharmacology: Molecular mechanism of Siwei Jianbu decoction in preventing oxaliplatin-induced peripheral neuropathy. Neural Plast. 2020, 2020, 8880543. [Google Scholar] [CrossRef] [PubMed]
- He, Y.X.; Shen, Q.Y.; Tian, J.H.; Wu, Q.; Xue, Q.; Zhang, G.P.; Wei, W.; Liu, Y.H. Zonisamide ameliorates cognitive impairment by inhibiting ER stress in a mouse model of type 2 Diabetes Mellitus. Front. Aging Neurosci. 2020, 12, 192. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.H.; Wu, Q.; He, Y.X.; Shen, Q.Y.; Rekep, M.; Zhang, G.P.; Luo, J.D.; Xue, Q.; Liu, Y.H. Zonisamide, an antiepileptic drug, alleviates diabetic cardiomyopathy by inhibiting endoplasmic reticulum stress. Acta Pharmacol. Sin. 2021, 42, 393–403. [Google Scholar] [CrossRef]
- Jiang, S.P.; Zhang, Z.D.; Kang, L.M.; Wang, Q.H.; Zhang, L.; Chen, H.P. Celecoxib reverts oxaliplatin-induced neuropathic pain through inhibiting PI3K/Akt2 pathway in the mouse dorsal root ganglion. Exp. Neurol. 2016, 275, 11–16. [Google Scholar] [CrossRef]
- Duan, Z.; Su, Z.; Wang, H.; Pang, X. Involvement of pro-inflammation signal pathway in inhibitory effects of rapamycin on oxaliplatin-induced neuropathic pain. Mol. Pain 2018, 14, 1744806918769426. [Google Scholar] [CrossRef]
- Cao, F.L.; Liu, M.G.; Hao, J.; Li, Z.; Lu, Z.M.; Chen, J. Different roles of spinal p38 and c-Jun N-terminal kinase pathways in bee venom-induced multiple pain-related behaviors. Neurosci. Lett. 2007, 427, 50–54. [Google Scholar] [CrossRef]
- Bison, S.; Razzoli, M.; Arban, R.; Michielin, F.; Bertani, S.; Carboni, L. Effect of the p38 MAPK inhibitor SB-239063 on lipopolysaccharide-induced psychomotor retardation and peripheral biomarker alterations in rats. Eur. J. Pharmacol. 2011, 661, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Canta, A.; Chiorazzi, A.; Pozzi, E.; Fumagalli, G.; Monza, L.; Meregalli, C.; Carozzi, V.A.; Rodriguez-Menendez, V.; Oggioni, N.; Näsström, J.; et al. Calmangafodipir reduces sensory alterations and prevents intraepidermal nerve fibers loss in a mouse model of oxaliplatin induced peripheral neurotoxicity. Antioxidants 2020, 9, 594. [Google Scholar] [CrossRef]
- Maj, M.A.; Ma, J.; Krukowski, K.N.; Kavelaars, A.; Heijnen, C.J. Inhibition of mitochondrial p53 accumulation by PFT-μ prevents cisplatin-induced peripheral neuropathy. Front. Mol. Neurosci. 2017, 10, 108. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.H.; Zheng, H.; Bennett, G.J. Characterization of oxaliplatin-induced chronic painful peripheral neuropathy in the rat and comparison with the neuropathy induced by paclitaxel. Neuroscience 2012, 203, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Nishida, K.; Takeuchi, K.; Hosoda, A.; Sugano, S.; Morisaki, E.; Ohishi, A.; Nagasawa, K. Ergothioneine ameliorates oxaliplatin-induced peripheral neuropathy in rats. Life Sci. 2018, 207, 516–524. [Google Scholar] [CrossRef]
- Niimi, N.; Yako, H.; Tsukamoto, M.; Takaku, S.; Yamauchi, J.; Kawakami, E.; Yanagisawa, H.; Watabe, K.; Utsunomiya, K.; Sango, K. Involvement of oxidative stress and impaired lysosomal degradation in amiodarone-induced schwannopathy. Eur. J. Neurosci. 2016, 44, 1723–1733. [Google Scholar] [CrossRef]
- Manji, H. Drug-induced neuropathies. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2013; Volume 115, pp. 729–742. [Google Scholar]
- Imai, S.; Koyanagi, M.; Azimi, Z.; Nakazato, Y.; Matsumoto, M.; Ogihara, T.; Yonezawa, A.; Omura, T.; Nakagawa, S.; Wakatsuki, S.; et al. Taxanes and platinum derivatives impair Schwann cells via distinct mechanisms. Sci. Rep. 2017, 7, 5947. [Google Scholar] [CrossRef]
- Tsutsumi, K.; Yamashita, Y.; Ushio, S.; Kawashiri, T.; Kaname, T.; Fujita, S.; Oishi, R.; Egashira, N. Oxaliplatin induces hypomyelination and reduced neuregulin 1 expression in the rat sciatic nerve. Neurosci. Res. 2014, 80, 86–90. [Google Scholar] [CrossRef]
- Basak, S.; Desai, D.J.; Rho, E.H.; Ramos, R.; Maurel, P.; Kim, H.A. E-cadherin enhances neuregulin signaling and promotes Schwann cell myelination. Glia 2015, 63, 1522–1536. [Google Scholar] [CrossRef]
- Hackett, A.R.; Strickland, A.; Milbrandt, J. Disrupting insulin signaling in Schwann cells impairs myelination and induces a sensory neuropathy. Glia 2020, 68, 963–978. [Google Scholar] [CrossRef]
- Sango, K.; Takaku, S.; Tsukamoto, M.; Niimi, N.; Yako, H. Glucagon-like peptide-1 receptor agonists as potential myelination-inducible and anti-demyelinating remedies. Front. Cell Dev. Biol. 2022, 10, 950623. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.C.; Tai, C.H.; Pan, M.K.; Kuo, C.C. The T-type calcium channel as a new therapeutic target for Parkinson’s disease. Pflügers Arch.-Eur. J. Physiol. 2014, 466, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, L.I.; Leo, M.; Kleinschnitz, C.; Hagenacker, T. Oxaliplatin modulates the characteristics of voltage-gated calcium channels and action potentials in small dorsal root ganglion neurons of rats. Mol. Neurobiol. 2018, 55, 8842–8855. [Google Scholar] [CrossRef] [PubMed]
- Landmark, C.J. Targets for antiepileptic drugs in the synapse. Med. Sci. Monit. 2007, 13, RA1–RA7. [Google Scholar]
- Al-Massri, K.F.; Ahmed, L.A.; El-Abhar, H.S. Pregabalin and lacosamide ameliorate paclitaxel-induced peripheral neuropathy via inhibition of JAK/STAT signaling pathway and Notch-1 receptor. Neurochem. Int. 2018, 120, 164–171. [Google Scholar] [CrossRef]
- Di Cesare Mannelli, L.; Maresca, M.; Micheli, L.; Farina, C.; Scherz, M.W.; Ghelardini, C. A rat model of FOLFOX-induced neuropathy: Effects of oral dimiracetam in comparison with duloxetine and pregabalin. Cancer Chemother. Pharmacol. 2017, 80, 1091–1103. [Google Scholar] [CrossRef]
- Argyriou, A.A.; Kalofonou, F.; Litsardopoulos, P.; Anastopoulou, G.G.; Psimaras, D.; Bruna, J.; Kalofonos, H.P. Real world, open label experience with lacosamide against acute painful oxaliplatin-induced peripheral neurotoxicity. J. Peripher. Nerv. Syst. 2020, 25, 178–183. [Google Scholar] [CrossRef]
- Jugait, S.; Areti, A.; Nellaiappan, K.; Narwani, P.; Saha, P.; Velayutham, R.; Kumar, A. Neuroprotective effect of baicalein against oxaliplatin-induced peripheral neuropathy: Impact on oxidative stress, neuro-inflammation and WNT/β-catenin signaling. Mol. Neurobiol. 2022, 59, 4334–4350. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takaku, S.; Sango, K. Pretreatment with Zonisamide Mitigates Oxaliplatin-Induced Toxicity in Rat DRG Neurons and DRG Neuron–Schwann Cell Co-Cultures. Int. J. Mol. Sci. 2022, 23, 9983. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23179983
Takaku S, Sango K. Pretreatment with Zonisamide Mitigates Oxaliplatin-Induced Toxicity in Rat DRG Neurons and DRG Neuron–Schwann Cell Co-Cultures. International Journal of Molecular Sciences. 2022; 23(17):9983. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23179983
Chicago/Turabian StyleTakaku, Shizuka, and Kazunori Sango. 2022. "Pretreatment with Zonisamide Mitigates Oxaliplatin-Induced Toxicity in Rat DRG Neurons and DRG Neuron–Schwann Cell Co-Cultures" International Journal of Molecular Sciences 23, no. 17: 9983. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23179983