
Distinct Changes in Calpain and Calpastatin during PNS Myelination and Demyelination in Rodent Models

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Calpain-Calpastatin Expression Is Developmentally Regulated in the PNS
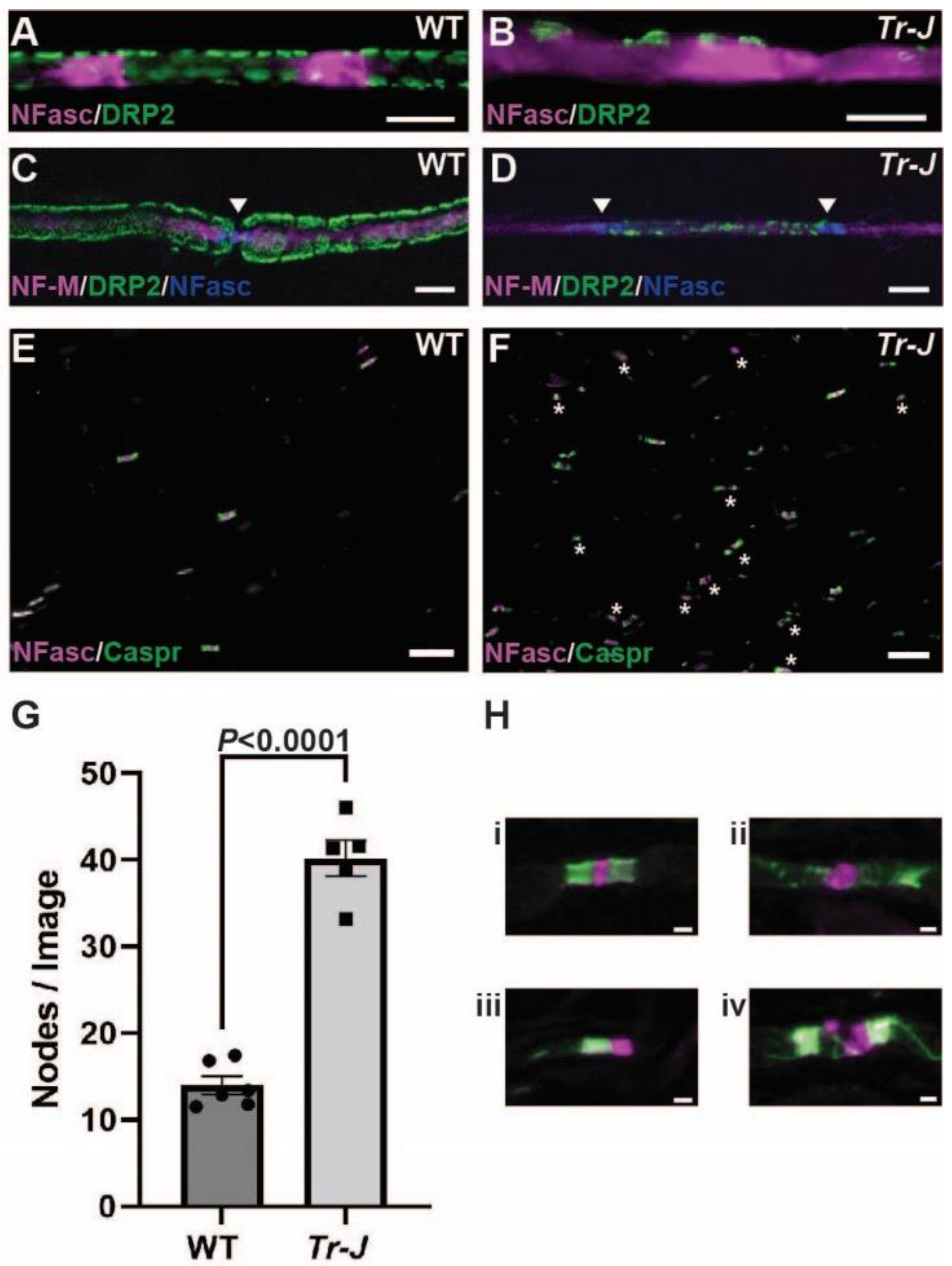
2.2. CAPN1 and Calpastatin Expression Increases in Trembler-J Mouse Model of CMT1E
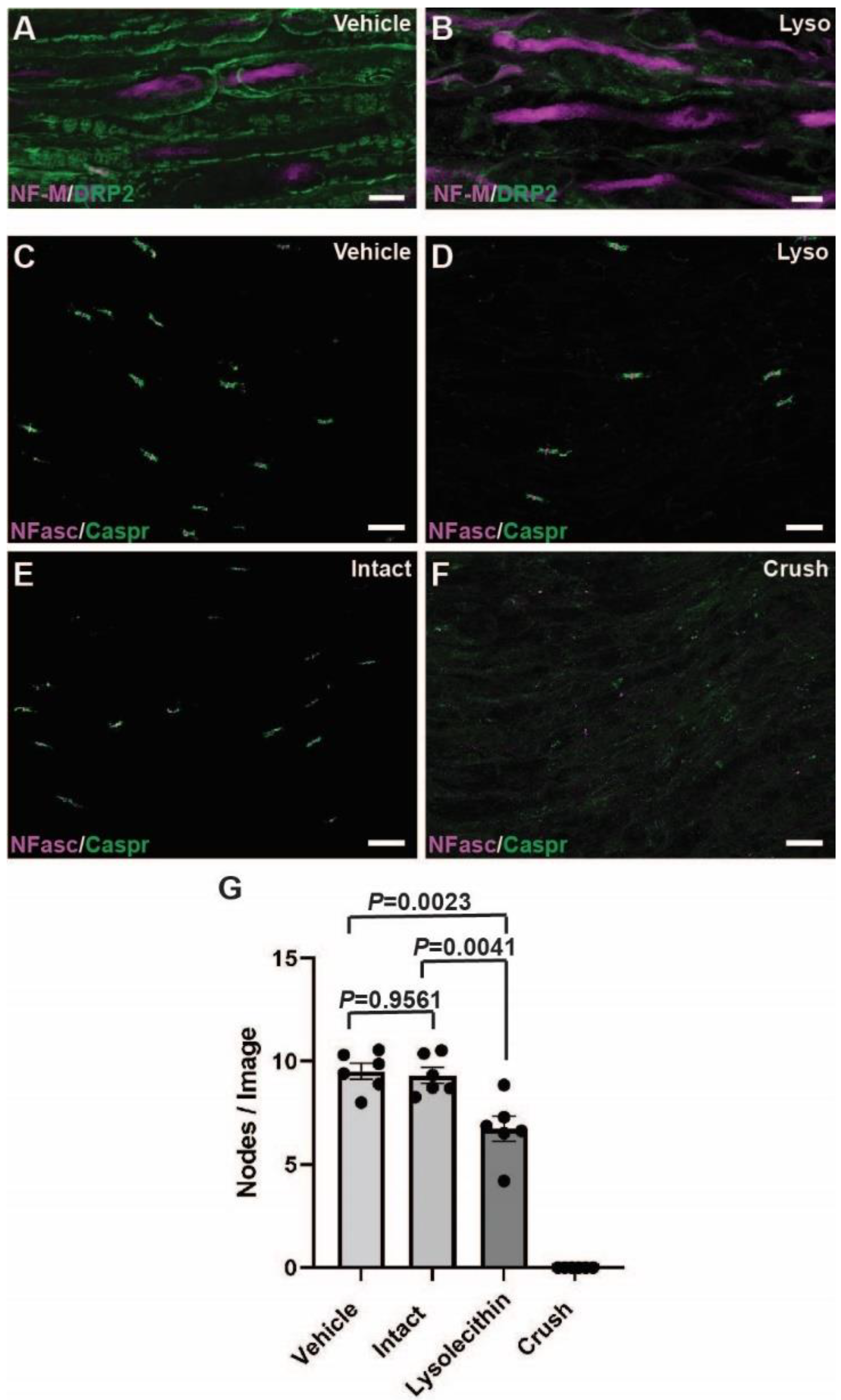
2.3. Calpain and Calpastatin Expression during Acute Focal Demyelination in PNS
2.4. Calpain and Calpastatin Expression during Acute Axonal Degeneration in PNS
3. Discussion
3.1. Calpains and Calpastatin during Development in the PNS vs. CNS
3.2. Calpains and Calpastatin during Chronic Dysmyelination in the PNS
3.3. Calpains during Acute Demyelination and Acute Axonal Degeneration in the PNS
3.4. Limitations of This Study
4. Materials and Methods
4.1. Animal Usage
4.2. Surgical Procedures
4.3. Sciatic Nerve Homogenization
4.4. Western Blotting
4.5. Immunofluorescent Imaging
4.6. Quantification of Immunohistochemical Sections
4.7. Calpain Activity Assay
4.8. Experimental Design and Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Salzer, J.L. Schwann Cell Myelination. Cold Spring Harb. Perspect. Biol. 2015, 7, a020529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasband, M.N.; Peles, E. Mechanisms of Node of Ranvier Assembly. Nat. Rev. Neurosci. 2020, 22, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Moss, K.R.; Bopp, T.S.; Johnson, A.E.; Höke, A. New Evidence for Secondary Axonal Degeneration in Demyelinating Neuropathies. Neurosci. Lett. 2021, 744, 135595. [Google Scholar] [CrossRef] [PubMed]
- Goll, D.E.; Thompson, V.F.; Li, H.; Wei, W.; Cong, J. The Calpain System. Physiol. Rev. 2003, 83, 731–801. [Google Scholar] [CrossRef]
- Ono, Y.; Sorimachi, H. Calpains—An Elaborate Proteolytic System. Biochim. Biophys. Acta 2012, 1824, 224–236. [Google Scholar] [CrossRef] [Green Version]
- Blomgren, K.; Karlsson, J.O. Developmental Changes of Calpain and Calpastatin in Rabbit Brain. Neurochem. Res. 1989, 14, 1149–1152. [Google Scholar] [CrossRef]
- Li, Y.; Bondada, V.; Joshi, A.; Geddes, J.W. Calpain 1 and Calpastatin Expression Is Developmentally Regulated in Rat Brain. Exp. Neurol. 2009, 220, 316–319. [Google Scholar] [CrossRef] [Green Version]
- Baraban, M.; Koudelka, S.; Lyons, D.A. Ca2+ Activity Signatures of Myelin Sheath Formation and Growth in Vivo. Nat. Neurosci. 2018, 21, 19–25. [Google Scholar] [CrossRef]
- Huff, T.B.; Shi, Y.; Sun, W.; Wu, W.; Shi, R.; Cheng, J.X. Real-Time CARS Imaging Reveals a Calpain-Dependent Pathway for Paranodal Myelin Retraction during High-Frequency Stimulation. PLoS ONE 2011, 6, e17176. [Google Scholar] [CrossRef] [Green Version]
- Shields, D.C.; Schaecher, K.E.; Saido, T.C.; Banik, N.L. A Putative Mechanism of Demyelination in Multiple Sclerosis by a Proteolytic Enzyme, Calpain. Proc. Natl. Acad. Sci. USA 1999, 96, 11486–11491. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Ferguson, T.A.; Schoch, K.M.; Li, J.; Qian, Y.; Shofer, F.S.; Saatman, K.E.; Neumar, R.W. Calpains Mediate Axonal Cytoskeleton Disintegration during Wallerian Degeneration. Neurobiol. Dis. 2013, 56, 34–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glass, J.D.; Culver, D.G.; Levey, A.I.; Nash, N.R. Very Early Activation of M-Calpain in Peripheral Nerve during Wallerian Degeneration. J. Neurol. Sci. 2002, 196, 9–20. [Google Scholar] [CrossRef]
- Girouard, M.P.; Simas, T.; Hua, L.; Morquette, B.; Khazaei, M.R.; Unsain, N.; Johnstone, A.D.; Rambaldi, I.; Sanz, R.L.; Di Raddo, M.-E.; et al. Collapsin Response Mediator Protein 4 (CRMP4) Facilitates Wallerian Degeneration and Axon Regeneration Following Sciatic Nerve Injury. eNeuro 2020, 7, ENEURO.0479-19.2020. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Gao, J.; Liu, J.; Siedlak, S.L.; Torres, S.; Fujioka, H.; Huntley, M.L.; Jiang, Y.; Ji, H.; Yan, T.; et al. Mitofusin 2 Regulates Axonal Transport of Calpastatin to Prevent Neuromuscular Synaptic Elimination in Skeletal Muscles. Cell. Metab. 2018, 28, 400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGonigal, R.; Rowan, E.G.; Greenshields, K.N.; Halstead, S.K.; Humphreys, P.D.; Rother, R.P.; Furukawa, K.; Willison, H.J. Anti-GD1a Antibodies Activate Complement and Calpain to Injure Distal Motor Nodes of Ranvier in Mice. Brain 2010, 133, 1944–1960. [Google Scholar] [CrossRef] [Green Version]
- Xie, W.; Uchida, H.; Nagai, J.; Ueda, M.; Chun, J.; Ueda, H. Calpain-Mediated Down-Regulation of Myelin-Associated Glycoprotein in Lysophosphatidic Acid-Induced Neuropathic Pain. J. Neurochem. 2010, 113, 1002–1011. [Google Scholar] [CrossRef]
- Yuan, X.C.; Wu, C.H.; Gao, F.; Li, H.P.; Xiang, H.C.; Zhu, H.; Pan, X.L.; Lin, L.X.; Liu, Y.S.; Yu, W.; et al. Activation and Expression of μ-Calpain in Dorsal Root Contributes to RTX-Induced Mechanical Allodynia. Mol. Pain. 2017, 13, 1744806917719169. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Larner, S.F.; Wang, K.K. Multiple AlphaII-Spectrin Breakdown Products Distinguish Calpain and Caspase Dominated Necrotic and Apoptotic Cell Death Pathways. Apoptosis 2009, 14, 1289–1298. [Google Scholar] [CrossRef]
- Castejon, M.S.; Culver, D.G.; Glass, J.D. Generation of Spectrin Breakdown Products in Peripheral Nerves by Addition of M-Calpain. Muscle Nerve 1999, 22, 905–909. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, Y.; Bi, X.; Baudry, M. Calpain-1 and Calpain-2 in the Brain: New Evidence for a Critical Role of Calpain-2 in Neuronal Death. Cells 2020, 9, 2698. [Google Scholar] [CrossRef]
- Webster, H.D. The geometry of peripheral myelin sheaths during their formation and growth in rat sciatic nerves. J. Cell Biol. 1971, 48, 348–367. [Google Scholar] [CrossRef] [PubMed]
- Siems, S.B.; Jahn, O.; Eichel, M.A.; Kannaiyan, N.; Wu, L.M.N.; Sherman, D.L.; Kusch, K.; Hesse, D.; Jung, R.B.; Fledrich, R.; et al. Proteome Profile of Peripheral Myelin in Healthy Mice and in a Neuropathy Model. elife 2020, 9, e51406. [Google Scholar] [CrossRef]
- Sherman, D.; Fabrizi, C.; Gillespie, C.; Brophy, P. Specific Disruption of a Schwann Cell Dystrophin-Related Protein Complex in a Demyelinating Neuropathy. Neuron 2001, 30, 677–687. [Google Scholar] [CrossRef] [Green Version]
- Gerber, D.; Pereira, J.A.; Gerber, J.; Tan, G.; Dimitrieva, S.; Yángüez, E.; Suter, U. Transcriptional Profiling of Mouse Peripheral Nerves to the Single-Cell Level to Build a Sciatic Nerve Atlas (Snat). elife 2021, 10, e58591. [Google Scholar] [CrossRef] [PubMed]
- Baki, A.; Tompa, P.; Alexa, A.; Molnar, O.; Friedrich, P. Autolysis Parallels Activation of µ-Calpain. Biochem. J. 1996, 318 Pt 3, 897–901. [Google Scholar] [CrossRef] [PubMed]
- Kitagaki, H.; Tomioka, S.; Yoshizawa, T.; Sorimachi, H.; Saido, T.C.; Ishiura, S.; Suzuki, K. Autolysis of Calpain Large Subunit Inducing Irreversible Dissociation of Stoichiometric Heterodimer of Calpain. Biosci. Biotechnol. Biochem. 2000, 64, 689–695. [Google Scholar] [CrossRef] [Green Version]
- Savikj, M.; Kostovski, E.; Lundell, L.S.; Iversen, P.O.; Massart, J.; Widegren, U. Altered Oxidative Stress and Antioxidant Defence in Skeletal Muscle during the First Year Following Spinal Cord Injury. Physiol. Rep. 2019, 7, e14218. [Google Scholar] [CrossRef] [Green Version]
- Meekins, G.D.; Emery, M.J.; Weiss, M.D. Nerve Conduction Abnormalities in the Trembler-j Mouse: A Model for Charcot-Marie-Tooth Disease Type 1A? J. Peripher. Nerv. Syst. 2004, 9, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Bosco, L.; Falzone, Y.M.; Previtali, S.C. Animal Models as a Tool to Design Therapeutical Strategies for CMT-like Hereditary Neuropathies. Brain Sci. 2021, 11, 1237. [Google Scholar] [CrossRef] [PubMed]
- Robertson, A.M.; Huxley, C.; King, R.H.M.; Thomas, P.K. Development of Early Postnatal Peripheral Nerve Abnormalities in Trembler-J and PMP22 Transgenic Mice. J. Anat. 1999, 195, 331–339. [Google Scholar] [CrossRef]
- Notterpek, L.; Shooter, E.M.; Snipes, G.J. Upregulation of the Endosomal-Lysosomal Pathway in the Trembler-J Neuropathy. J. Neurosci. 1997, 17, 4190–4200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devaux, J.J.; Scherer, S.S. Cellular/Molecular Altered Ion Channels in an Animal Model of Charcot-Marie-Tooth Disease Type IA. J. Neurosci. 2005, 25, 1470–1480. [Google Scholar] [CrossRef] [Green Version]
- Ewaleifoh, O.; Trinh, M.; Griffin, J.W.; Nguyen, T. A Novel System To Accelerate The Progression of Nerve Degeneration In Transgenic Mouse Models of Neuropathies. Exp. Neurol. 2012, 237, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaecher, K.E.; Shields, D.C.; Banik, N.L. Mechanism of Myelin Breakdown in Experimental Demyelination: A Putative Role for Calpain. Neurochem. Res. 2001, 26, 731–737. [Google Scholar] [CrossRef]
- Mitchell, J.; Caren, C.A. Degeneration of Non-Myelinated Axons in the Rat Sciatic Nerve Following Lysolecithin Injection. Acta Neuropathol. 1982, 56, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Hall, S.M.; Gregson, N.A. The in Vivo and Ultrastructural Effects of Injection of Lysophosphatidyl Choline into Myelinated Peripheral Nerve Fibres of the Adult Mouse. J. Cell Sci. 1971, 9, 769–789. [Google Scholar] [CrossRef] [PubMed]
- Nakata, M.; Baba, H.; Kanai, K.; Hoshi, T.; Sawai, S.; Hattori, T.; Kuwabara, S. Changes in Na+ Channel Expression and Nodal Persistent Na+ Currents Associated with Peripheral Nerve Regeneration in Mice. Muscle Nerve 2008, 37, 721–730. [Google Scholar] [CrossRef]
- Menorca, R.M.G.; Fussell, T.S.; Elfar, J.C. Nerve Physiology: Mechanisms of Injury and Recovery. Hand Clin. 2013, 29, 317–330. [Google Scholar] [CrossRef] [Green Version]
- Brosius Lutz, A.; Barres, B.A. Developmental Cell Review Contrasting the Glial Response to Axon Injury in the Central and Peripheral Nervous Systems. Dev. Cell 2014, 28, 7–17. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Wang, X.; Xu, F.; Bahr, B.A.; Shibata, M.; Uchiyama, Y.; Hagberg, H.; Blomgren, K. The Influence of Age on Apoptotic and Other Mechanisms of Cell Death after Cerebral Hypoxia–Ischemia. Cell. Death Differ. 2005, 12, 162–176. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, A.K.; Banik, N.L.; Lobo, D.C.; Terry, E.C.; Hogan, E.L. Calcium-Activated Neutral Proteinase (Calpain) in Rat Brain during Development: Compartmentation and Role in Myelination. Brain Res. Dev. Brain Res. 1993, 71, 107–113. [Google Scholar] [CrossRef]
- Simonson, L.; Baudry, M.; Siman, R.; Lynch, G. Regional Distribution of Soluble Calcium Activated Proteinase Activity in Neonatal and Adult Rat Brain. Brain Res. 1985, 327, 153–159. [Google Scholar] [CrossRef]
- Li, Z.; Banik, N.L. The Localization of Mcalpain in Myelin: Immunocytochemical Evidence in Different Areas of Rat Brain and Nerves. Brain Res. 1995, 697, 112–121. [Google Scholar] [CrossRef]
- Mata, M.; Kupina, N.; Fink, D.J. Calpain II in Rat Peripheral Nerve. Brain Res. 1991, 564, 328–331. [Google Scholar] [CrossRef] [Green Version]
- Court, F.A.; Sherman, D.L.; Pratt, T.; Garry, E.M.; Ribchester, R.R.; Cottrell, D.F.; Fleetwood-Walker, S.M.; Brophy, P.J. Restricted Growth of Schwann Cells Lacking Cajal Bands Slows Conduction in Myelinated Nerves. Nature 2004, 431, 191–195. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Hersheson, J.; Lopez, D.; Hammer, M.; Liu, Y.; Lee, K.H.; Pinto, V.; Seinfeld, J.; Wiethoff, S.; Sun, J.; et al. Defects in the CAPN1 Gene Result in Alterations in Cerebellar Development and Cerebellar Ataxia in Mice and Humans. Cell. Rep. 2016, 16, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Schröder, J.M.; Bohl, J.; von Bardeleben, U. Changes of the Ratio between Myelin Thickness and Axon Diameter in Human Developing Sural, Femoral, Ulnar, Facial, and Trochlear Nerves. Acta Neuropathol. 1988, 76, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Misko, A.; Ferguson, T.; Notterpek, L. Matrix Metalloproteinase Mediated Degradation of Basement Membrane Proteins in Trembler J Neuropathy Nerves. J. Neurochem. 2002, 83, 885–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vosler, P.S.; Brennan, C.S.; Chen, J. Calpain-Mediated Signaling Mechanisms in Neuronal Injury and Neurodegeneration. Mol. Neurobiol. 2008, 38, 78–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yehuda, B.; Gradus Pery, T.; Ophir, E.; Blumenfeld-Katzir, T.; Sheinin, A.; Alon, Y.; Danino, N.; Perlson, E.; Nevo, U. Neuronal Activity in the Sciatic Nerve Is Accompanied by Immediate Cytoskeletal Changes. Front. Mol. Neurosci. 2021, 14, 249. [Google Scholar] [CrossRef]
- Isabella Pörn-Ares, M.; Samali, A.; Orrenius, S. Cleavage of the Calpain Inhibitor, Calpastatin, during Apoptosis. Cell. Death Differ. 1998, 5, 1028–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, F.; Zhang, C.; Wang, Q.; Zeng, T.; Xie, K. Alterations in Neurofilaments Content and Calpains Activity of Sciatic Nerve of Carbon Disulfide-Treated Rats. Arch. Toxicol. 2009, 83, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Hantke, J.; Carty, L.; Wagstaff, L.; Turmaine, M.; Wilton, D.; Quintes, S.; Koltzenburg, M.; Baas, F.; Mirsky, R.; Jessen, K. c-Jun Activation in Schwann Cells Protects against Loss of Sensory Axons in Inherited Neuropathy. Brain 2014, 137, 2922–2937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banik, N.L.; Chou, C.-H.; Deibler, G.E.; Krutzch, H.C.; Hogan, E.L. Peptide Bond Specificity of Calpain: Proteolysis of Human Myelin Basic Protein. J. Neurosci. Res. 1994, 37, 489–496. [Google Scholar] [CrossRef]
- Sato, S.; Yanagisawa, K.; Miyatake, T. Conversion of Myelin-Associated Glycoprotein (MAG) to a Smaller Derivative by Calcium Activated Neutral Protease (CANP)-like Enzyme in Myelin and Inhibition by E-64 Analogue. Neurochem. Res. 1984, 9, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Czogalla, A.; Sikorski, A. Spectrin and Calpain: A “target” and a “Sniper” in the Pathology of Neuronal Cells. Cell. Mol. Life Sci. 2005, 62, 1913–1924. [Google Scholar] [CrossRef]
- Mårtensson, L.B.; Blom, C.L.; Dahlin, L.B. Ca2+ Involvement in Activation of Extracellular-Signal-Regulated-Kinase 1/2 and m-Calpain after Axotomy of the Sciatic Nerve. Neural Regen. Res. 2017, 12, 623. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Guyton, M.K.; Smith, A.; Wallace, G.; McDowell, M.L.; Matzelle, D.D.; Ray, S.K.; Banik, N.L. Calpain Inhibitor Attenuated Optic Nerve Damage in Acute Optic Neuritis in Rats. J. Neurochem. 2013, 124, 133–146. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.W.; Ray, S.K.; Das, A.; Nozaki, K.; Rohrer, B.; Banik, N.L. Calpain Inhibition as a Possible New Therapeutic Target in Multiple Sclerosis. AIMS Mol. Sci. 2017, 4, 446–462. [Google Scholar] [CrossRef]
- Yamazaki, R.; Osanai, Y.; Kouki, T.; Shinohara, Y.; Huang, J.K.; Ohno, N. Macroscopic Detection of Demyelinated Lesions in Mouse PNS with Neutral Red Dye. Sci. Rep. 2021, 11, 16906. [Google Scholar] [CrossRef]
- Gargareta, V.-I.; Reuschenbach, J.; Siems, S.B.; Sun, T.; Piepkorn, L.; Mangana, C.; Späte, E.; Goebbels, S.; Huitinga, I.; Möbius, W.; et al. Conservation and Divergence of Myelin Proteome and Oligodendrocyte Transcriptome Profiles between Humans and Mice. elife 2022, 11, 77019. [Google Scholar] [CrossRef]
- Monje, P.V. The Properties of Human Schwann Cells: Lessons from in Vitro Culture and Transplantation Studies. Glia 2020, 68, 797–810. [Google Scholar] [CrossRef]
- Rasband, M.N.; Trimmer, J.S.; Schwarz, T.L.; Levinson, S.R.; Ellisman, M.H.; Schachner, M.; Shrager, P. Potassium Channel Distribution, Clustering, and Function in Remyelinating Rat Axons. J. Neurosci. 1998, 18, 36–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yermakov, L.M.; Drouet, D.E.; Griggs, R.B.; Elased, K.M.; Susuki, K. Type 2 Diabetes Leads to Axon Initial Segment Shortening in Db/db Mice. Front. Cell. Neurosci. 2018, 12, 146. [Google Scholar] [CrossRef]
- Butler, T.A.J.; Paul, J.W.; Chan, E.C.; Smith, R.; Tolosa, J.M. Misleading Westerns: Common Quantification Mistakes in Western Blot Densitometry and Proposed Corrective Measures. Biomed. Res. Int. 2019, 2019, 5214821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otani, Y.; Yermakov, L.M.; Dupree, J.L.; Susuki, K. Chronic Peripheral Nerve Compression Disrupts Paranodal Axoglial Junctions. Muscle Nerve 2017, 55, 544–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griggs, R.B.; Yermakov, L.M.; Drouet, D.E.; Nguyen, D.V.M.; Susuki, K. Methylglyoxal Disrupts Paranodal Axoglial Junctions via Calpain Activation. ASN Neuro. 2018, 10, 1759091418766175. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen | Dilution | Species | Manufacturer Information & RRID Citation |
|---|---|---|---|
| CAPN1 | 1:1000 (WB) | Rabbit | Abcam #ab28258 RRID:AB_72581 |
| CAPN2 | 1:1000 (WB) | Rabbit | Cell Signaling Technology #2539 RRID:AB_2069843 |
| Calpastatin | 1:1000 (WB) | Rabbit | Cell Signaling Technology #4146 RRID:AB_2244162 |
| DRP2 | 1:1000 (WB) 1:400 (IF) | Rabbit | Gift from Peter Brophy |
| GAPDH | 1:1000 (WB) | Mouse | Enzo Life Sciences #ADI-CSA-335-E RRID:AB_2039148 |
| Neurofascin | 1:400 (IF) | Chicken | R&D Systems #AF3235 RRID:AB_10890736 |
| Neurofilament-M | 1:1000 (WB) 1:400(IF) | Mouse | Cell Signaling Technology #2838 RRID:AB_561191 |
| AnkyrinG | 1:400 (IF) | Mouse | UC Davis/NIH NeuroMab Cat. #75-146 RRID:AB_1067303 |
| Caspr | 1:400 (IF) | Rabbit | Abcam #ab3415 RRID:AB_869934 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miller, J.A.; Drouet, D.E.; Yermakov, L.M.; Elbasiouny, M.S.; Bensabeur, F.Z.; Bottomley, M.; Susuki, K. Distinct Changes in Calpain and Calpastatin during PNS Myelination and Demyelination in Rodent Models. Int. J. Mol. Sci. 2022, 23, 15443. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232315443
Miller JA, Drouet DE, Yermakov LM, Elbasiouny MS, Bensabeur FZ, Bottomley M, Susuki K. Distinct Changes in Calpain and Calpastatin during PNS Myelination and Demyelination in Rodent Models. International Journal of Molecular Sciences. 2022; 23(23):15443. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232315443
Chicago/Turabian StyleMiller, John A., Domenica E. Drouet, Leonid M. Yermakov, Mahmoud S. Elbasiouny, Fatima Z. Bensabeur, Michael Bottomley, and Keiichiro Susuki. 2022. "Distinct Changes in Calpain and Calpastatin during PNS Myelination and Demyelination in Rodent Models" International Journal of Molecular Sciences 23, no. 23: 15443. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232315443