


Novel Planar Pt(II) Cyclometallated Cytotoxic Complexes with G-Quadruplex Stabilisation and Luminescent Properties
 ,
, 
Abstract
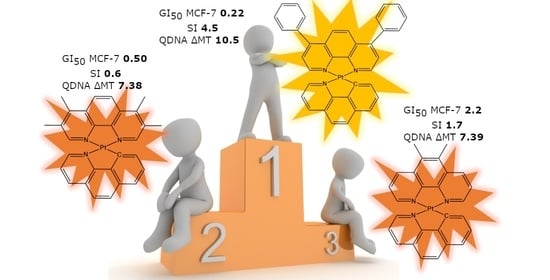
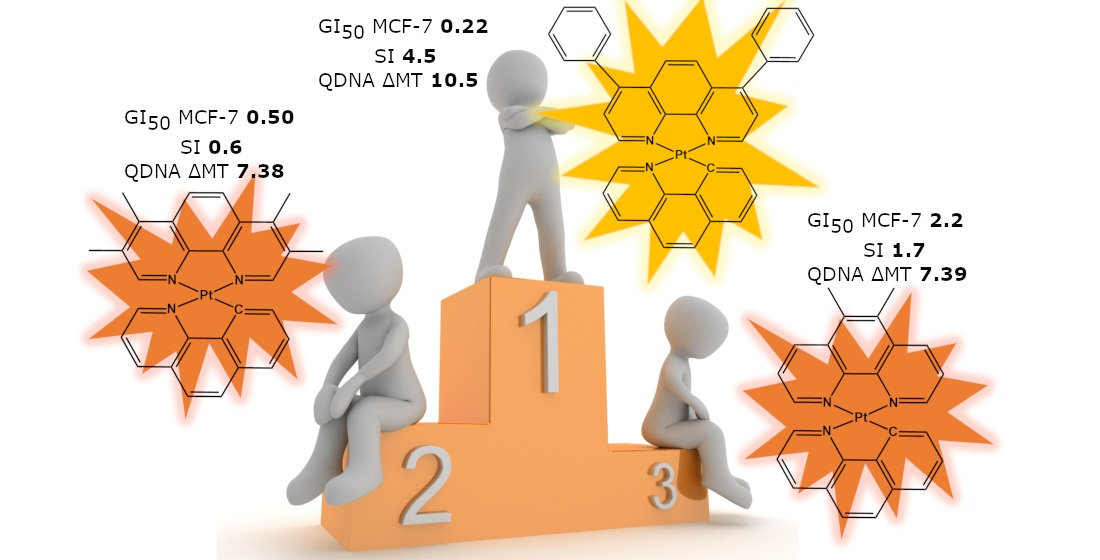
:
1. Introduction
2. Experimental Section
2.1. Materials and Methods
2.2. Synthesis
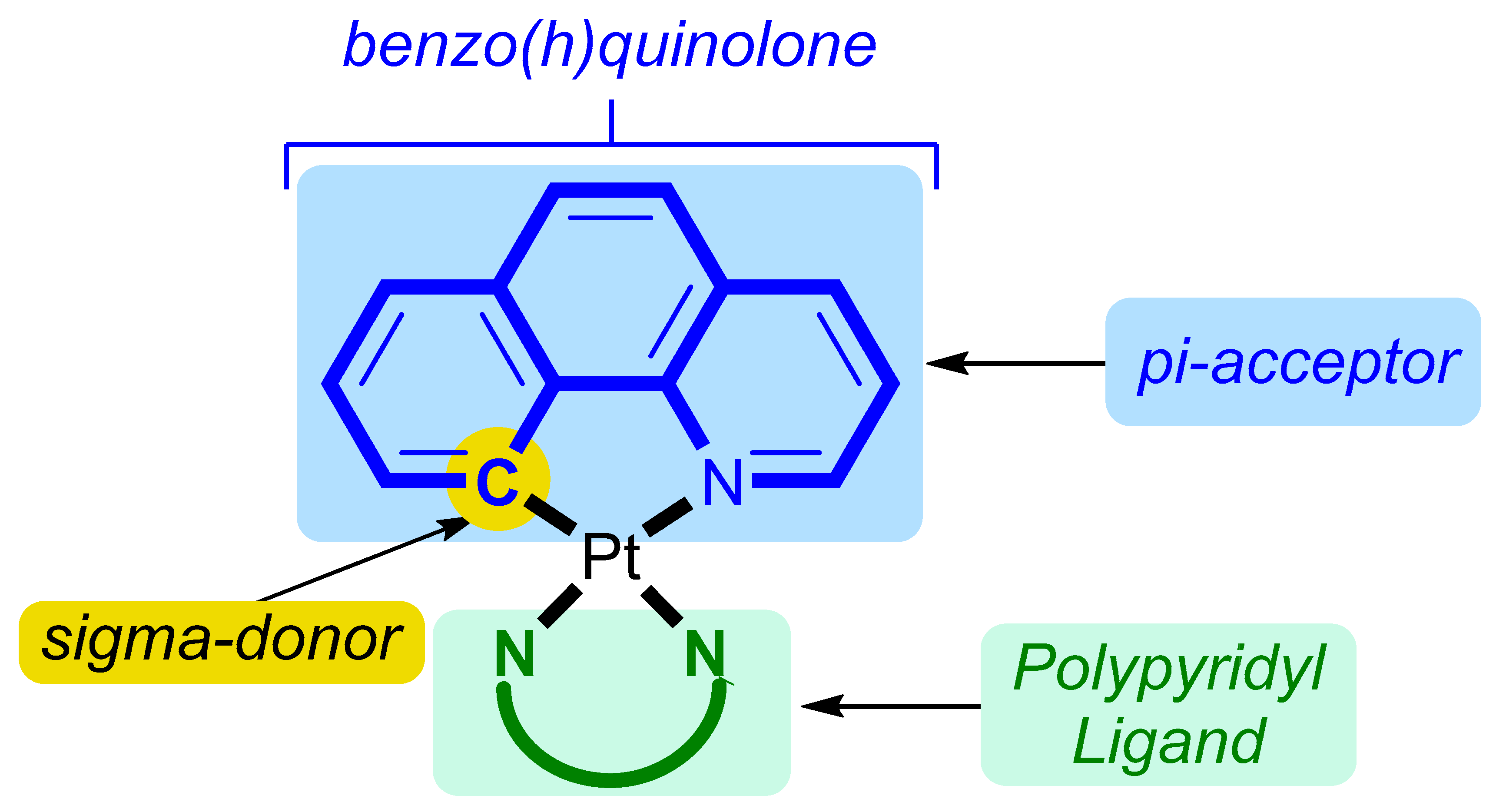
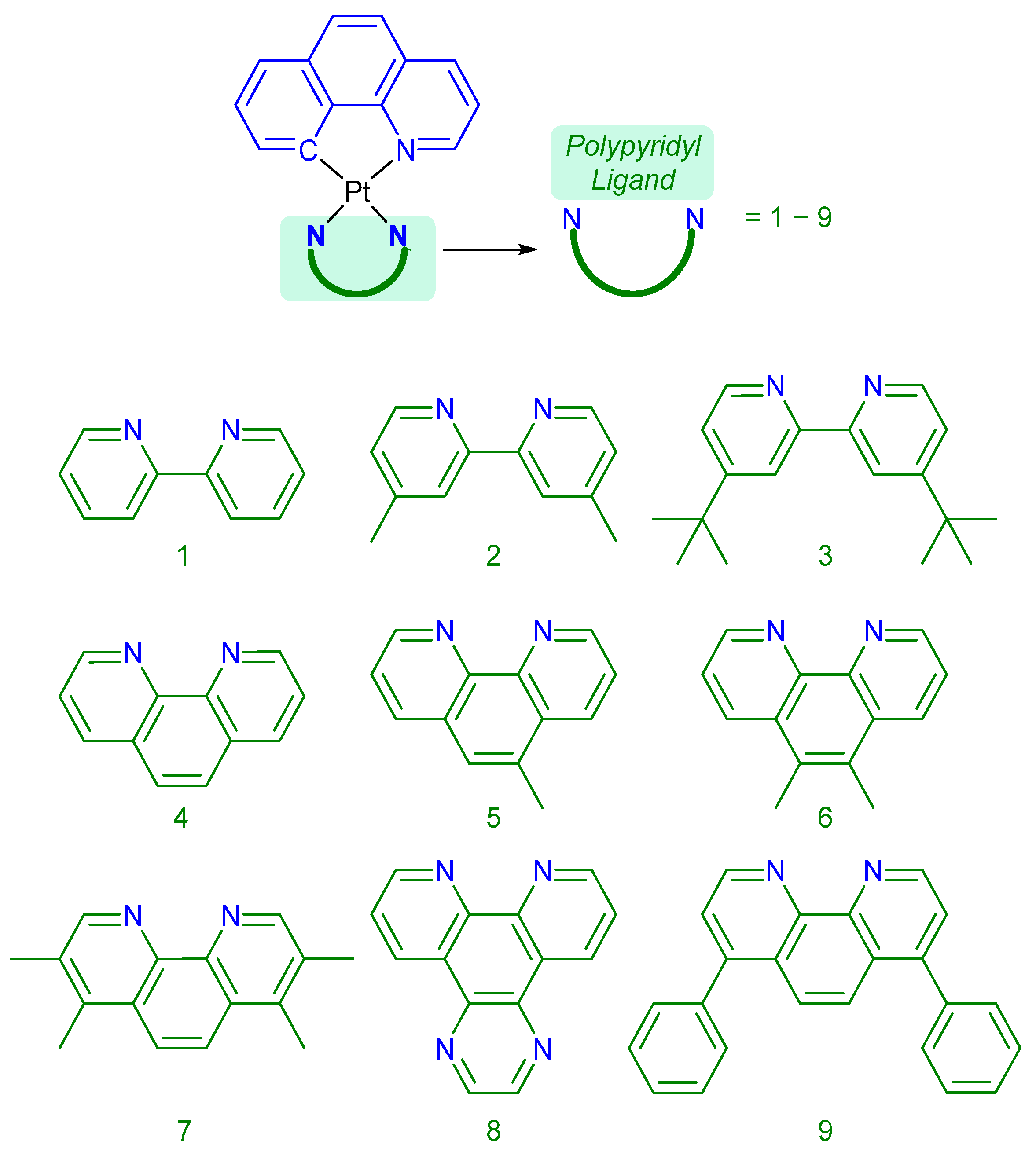
2.2.1. Synthesis of [Pt(Bequ)(Cl)2]−
2.2.2. Synthesis of [Pt(Bequ)(PL)]+ Complexes
2.3. Cytotoxicity Methodology
2.4. Biophysical Characterisation
3. Results and Discussion
3.1. Synthesis and Characterisation
3.2. Biophysical Characterisation
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Weiderpass, E.; Soerjomataram, I. The Ever-Increasing Importance of Cancer as a Leading Cause of Premature Death Worldwide. Cancer 2021, 127, 3029–3030. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Cancer (IARC); T.I.A. for R. on Global Cancer Observatory. Available online: https://gco.iarc.fr/ (accessed on 7 February 2022).
- Wheate, N.J.; Walker, S.; Craig, G.E.; Oun, R. The Status of Platinum Anticancer Drugs in the Clinic and in Clinical Trials. Dalton Trans. 2010, 39, 8113–8127. [Google Scholar] [CrossRef] [PubMed]
- Siddik, Z.H. Cisplatin: Mode of Cytotoxic Action and Molecular Basis of Resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef]
- Kartalou, M.; Essigmann, J.M. Mechanisms of Resistance to Cisplatin. Mutat. Res. Mol. Mech. Mutagen. 2001, 478, 23–43. [Google Scholar] [CrossRef]
- Balasubramanian, S.; Hurley, L.H.; Neidle, S. Targeting G-Quadruplexes in Gene Promoters: A Novel Anticancer Strategy? Nat. Rev. Drug Discov. 2011, 10, 261–275. [Google Scholar] [CrossRef]
- Ou, T.; Lu, Y.; Tan, J.; Huang, Z.; Wong, K.-Y.; Gu, L. G-Quadruplexes: Targets in Anticancer Drug Design. ChemMedChem 2008, 3, 690–713. [Google Scholar] [CrossRef]
- Düchler, M. G-Quadruplexes: Targets and Tools in Anticancer Drug Design. J. Drug Target. 2012, 20, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Monchaud, D.; Teulade-Fichou, M.-P. A Hitchhiker’s Guide to G-Quadruplex Ligands. Org. Biomol. Chem. 2008, 6, 627–636. [Google Scholar] [CrossRef]
- De Cian, A.; Cristofari, G.; Reichenbach, P.; De Lemos, E.; Monchaud, D.; Teulade-Fichou, M.-P.; Shin-ya, K.; Lacroix, L.; Lingner, J.; Mergny, J.-L. Reevaluation of Telomerase Inhibition by Quadruplex Ligands and Their Mechanisms of Action. Proc. Natl. Acad. Sci. USA 2007, 104, 17347–17352. [Google Scholar] [CrossRef] [Green Version]
- Deo, K.M.; Ang, D.L.; McGhie, B.; Rajamanickam, A.; Dhiman, A.; Khoury, A.; Holland, J.; Bjelosevic, A.; Pages, B.; Gordon, C.; et al. Platinum Coordination Compounds with Potent Anticancer Activity. Coord. Chem. Rev. 2018, 375, 148–163. [Google Scholar] [CrossRef]
- McGhie, B.S.; Sakoff, J.; Gilbert, J.; Aldrich-Wright, J.R. Synthesis and Characterisation of Platinum(IV) Polypyridyl Complexes with Halide Axial Ligands. Inorg. Chim. Acta 2019, 495, 118964. [Google Scholar] [CrossRef]
- Khoury, A.; Deo, K.M.; Aldrich-Wright, J.R. Recent Advances in Platinum-Based Chemotherapeutics That Exhibit Inhibitory and Targeted Mechanisms of Action. J. Inorg. Biochem. 2020, 207, 111070. [Google Scholar] [CrossRef]
- Deo, K.M.; Sakoff, J.; Gilbert, J.; Zhang, Y.; Wright, J.R.A. Synthesis, Characterisation and Influence of Lipophilicity on Cellular Accumulation and Cytotoxicity of Unconventional Platinum(IV) Prodrugs as Potent Anticancer Agents. Dalton Trans. 2019, 48, 17228–17240. [Google Scholar] [CrossRef]
- Ang, D.L.; Harper, B.W.J.; Cubo, L.; Mendoza, O.; Vilar, R.; Aldrich-Wright, J. Quadruplex DNA-Stabilising Dinuclear Platinum(II) Terpyridine Complexes with Flexible Linkers. Chem.-Eur. J. 2016, 22, 2317–2325. [Google Scholar] [CrossRef]
- Wilson, T.; Costa, P.J.; Félix, V.; Williamson, M.P.; Thomas, J.A. Structural Studies on Dinuclear Ruthenium(II) Complexes That Bind Diastereoselectively to an Antiparallel Folded Human Telomere Sequence. Available online: https://0-pubs-acs-org.brum.beds.ac.uk/doi/full/10.1021/jm401119b (accessed on 18 August 2022).
- Holland, J. Synthesis of Platinum(II) Based G-Quadruplex Stabilisers. Master’s Thesis, Western Sydney University, Sydney, Australia, 2017. [Google Scholar]
- Tan, C.-P.; Zhong, Y.-M.; Ji, L.-N.; Mao, Z.-W. Phosphorescent Metal Complexes as Theranostic Anticancer Agents: Combining Imaging and Therapy in a Single Molecule. Chem. Sci. 2021, 12, 2357–2367. [Google Scholar] [CrossRef]
- Qiu, K.; Chen, Y.; Rees, T.W.; Ji, L.; Chao, H. Organelle-Targeting Metal Complexes: From Molecular Design to Bio-Applications. Coord. Chem. Rev. 2019, 378, 66–86. [Google Scholar] [CrossRef]
- Ding, S.; Qiao, X.; Suryadi, J.; Marrs, G.S.; Kucera, G.L.; Bierbach, U. Using Fluorescent Post-Labeling To Probe the Subcellular Localization of DNA-Targeted Platinum Anticancer Agents. Angew. Chem. Int. Ed. 2013, 52, 3350–3354. [Google Scholar] [CrossRef]
- Xu, G.-X.; Mak, E.C.-L.; Lo, K.K.-W. Photofunctional Transition Metal Complexes as Cellular Probes, Bioimaging Reagents and Phototherapeutics. Inorg. Chem. Front. 2021, 8, 4553–4579. [Google Scholar] [CrossRef]
- Tarleton, M.; Gilbert, J.; Robertson, M.J.; McCluskey, A.; Sakoff, J.A. Library Synthesis and Cytotoxicity of a Family of 2-Phenylacrylonitriles and Discovery of an Estrogen Dependent Breast Cancer Lead Compound. MedChemComm 2011, 2, 31–37. [Google Scholar] [CrossRef]
- Brouwer, A.M. Standards for Photoluminescence Quantum Yield Measurements in Solution (IUPAC Technical Report). Pure Appl. Chem. 2011, 83, 2213–2228. [Google Scholar] [CrossRef]
- González, V.; Hurley, L.H. The C-MYC NHE III(1): Function and Regulation. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 111–129. [Google Scholar] [CrossRef]
- Ambrus, A.; Chen, D.; Dai, J.; Jones, R.A.; Yang, D. Solution Structure of the Biologically Relevant G-Quadruplex Element in the Human c-MYC Promoter. Implications for G-Quadruplex Stabilization. Biochemistry 2005, 44, 2048–2058. [Google Scholar] [CrossRef]
- Wang, Y.; Patel, D.J. Solution Structure of the Human Telomeric Repeat d [AG3 (T2AG3) 3] G-Tetraplex. Structure 1993, 1, 263–282. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
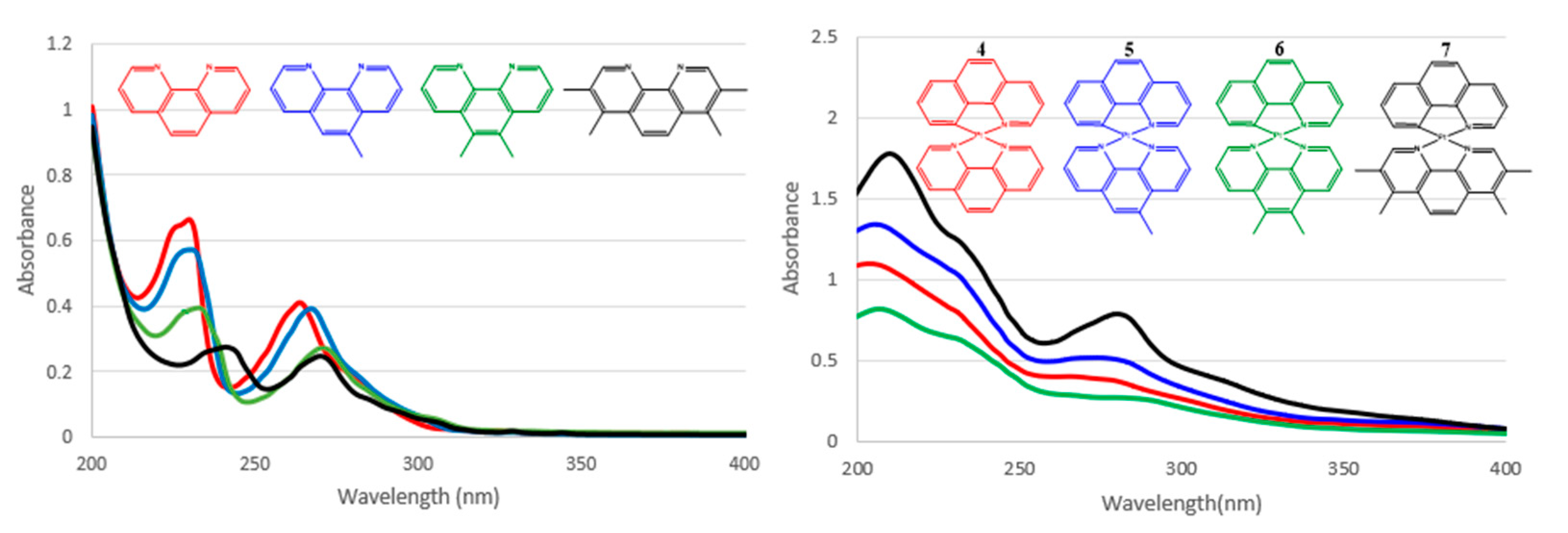{kind=link}
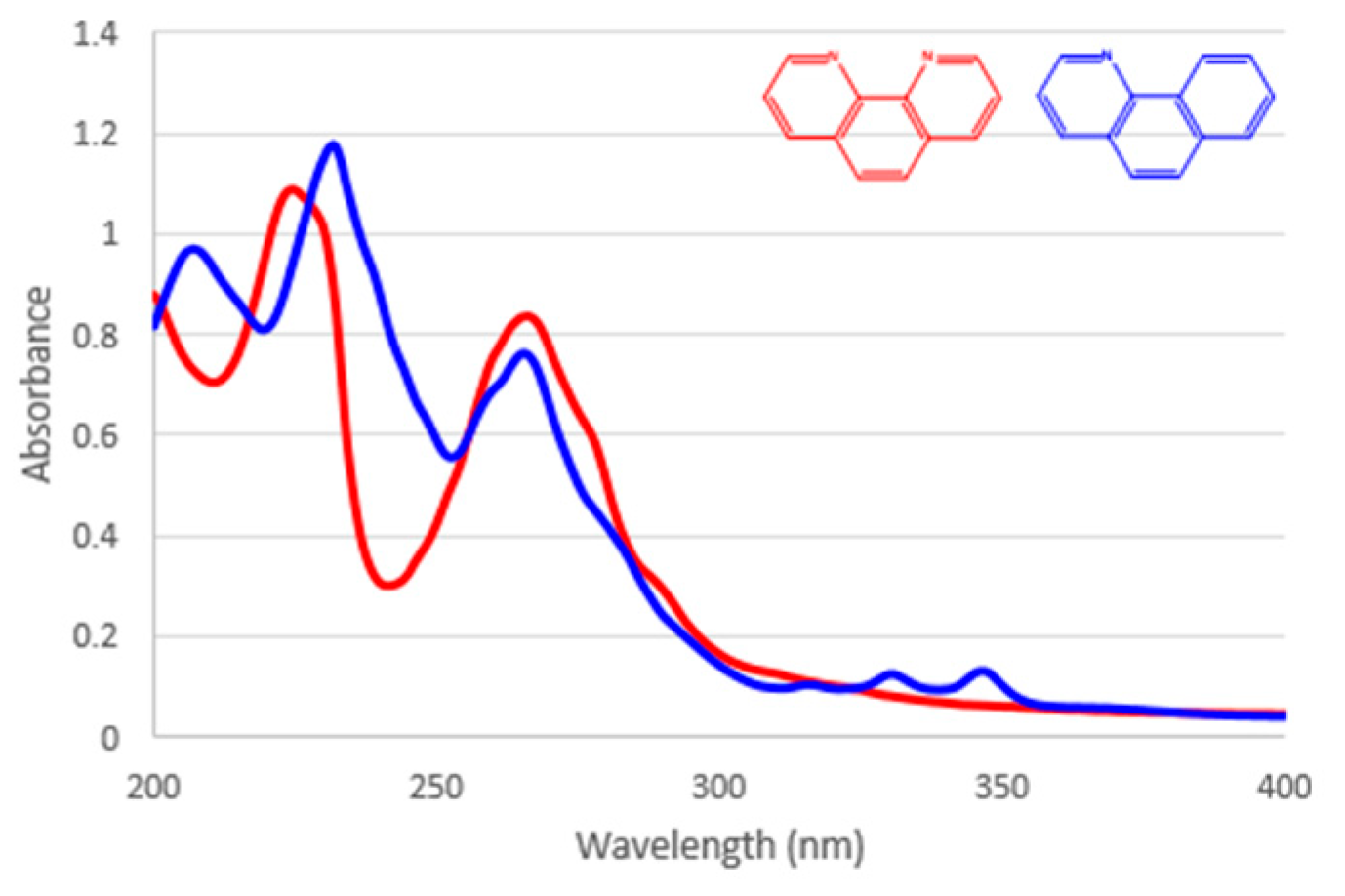{kind=link}
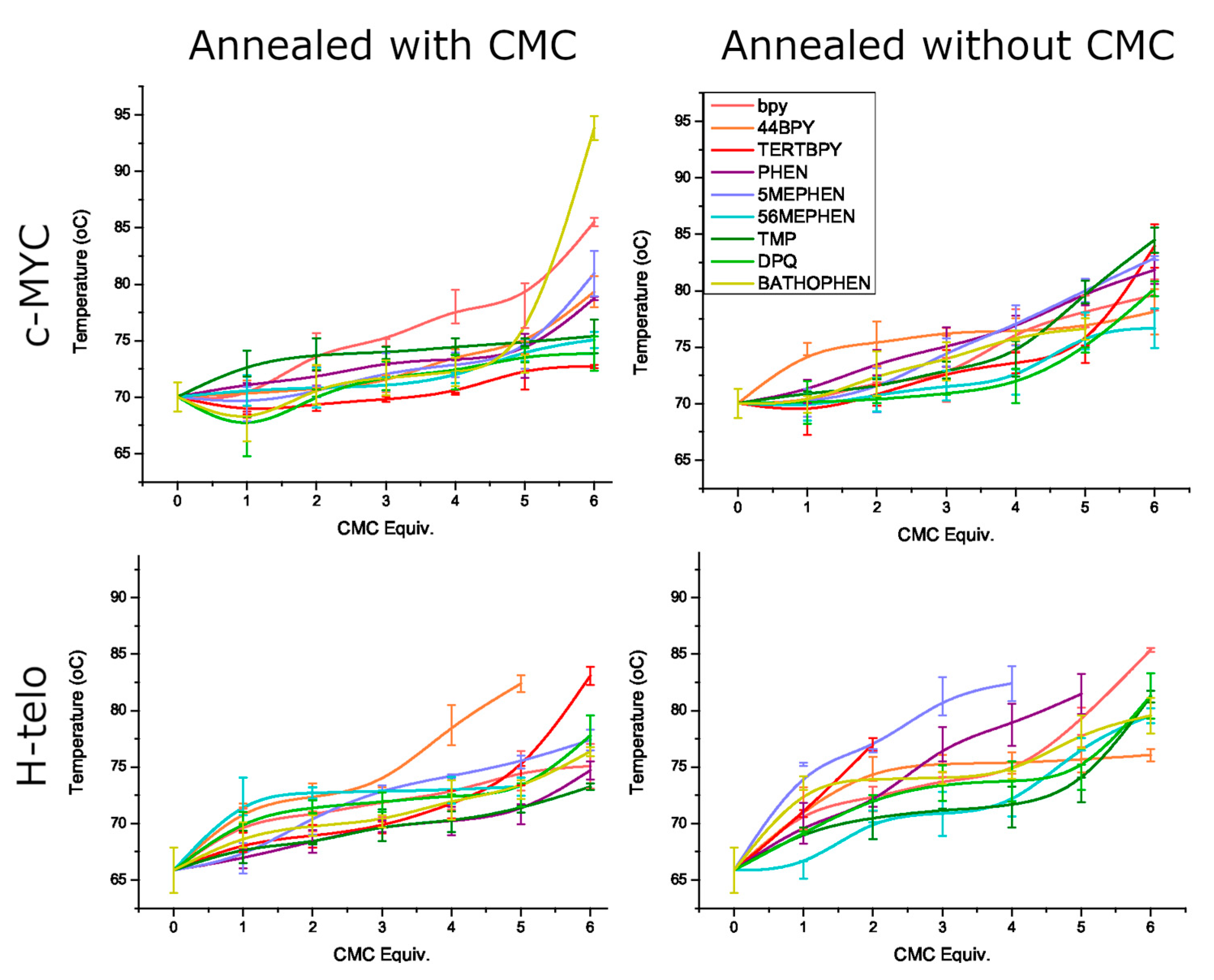{kind=link}
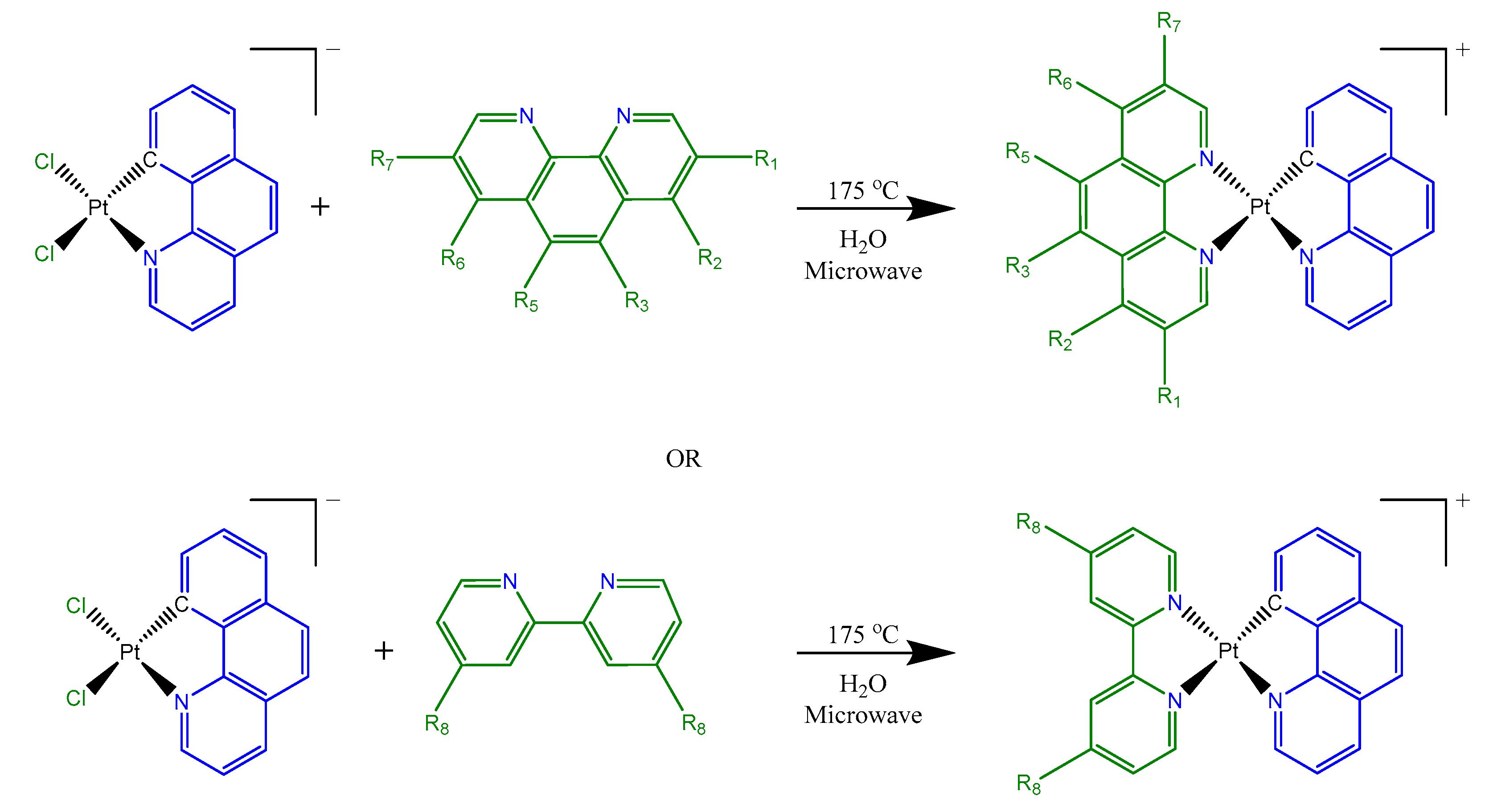{kind=link}
| CMC | Molecular Formula | Yield(%) | ESI-MS (m/z) | Lipophilicity (log kw) | Surface Area VdW/SAS (Ų) | UV/λmax (nm) (ε/mol−1·dm3·cm−1) × 102 | Fluorescence | ||
|---|---|---|---|---|---|---|---|---|---|
| [M]+ Calc. (Found) | Exmax | Emmax | ΦF | ||||||
| 1 | C23H16N3Pt | 84.05 | 529.0992 (529.0981) | 1.04 ± 0.15 | 773.9 | 206 (330 ± 0.18), 233 (341 ± 0.19) | 324.02 | 652.02 | 0.015 ± 0.121 |
| 2 | C25H20N3Pt | 86.02 | 557.1305 (557.1295) | 1.63 ± 0.20 | 842.9 | 208 (401 ± 0.32), 232 (282 ± 0.33) | 336.00 | 634.92 | 0.024 ± 0.175 |
| 3 | C32H31N3Pt | 83.59 | 657.2315 (657.2319) | 2.35 ± 0.16 | 989.4 | 208 (315 ± 0.14), 232 (252 ± 0.16) | 395.07 | 670.00 | 0.016 ± 0.187 |
| 4 | C25H16N3Pt | 82.49 | 553.0992 (553.0989) | 1.12 ± 0.11 | 803.9 | 204 (442 ± 0.16), 265 (163 ± 0.15) | 325.05 | 654.05 | 0.021 ± 0.236 |
| 5 | C26H18N3Pt | 86.23 | 567.1149 (567.1147) | 1.30 ± 0.16 | 830.8 | 206 (402 ± 0.11), 274 (153 ± 0.19) | 394.24 | 645.05 | 0.014 ± 0.222 |
| 6 | C27H20N3Pt | 85.56 | 581.1305 (581.1308) | 1.04 ± 0.25 | 856.2 | 207 (398 ± 0.15), 284 (130 ± 0.12) | 325.97 | 651.01 | 0.026 ± 0.155 |
| 7 | C29H24N3Pt | 82.89 | 609.1618 (609.1611) | 2.62 ± 0.30 | 909.8 | 210 (816 ± 0.12), 280 (359 ± 0.18) | 391.96 | 641.01 | 0.023 ± 0.136 |
| 8 | C27H16N5Pt | 78.65 | 605.1054 (605.1057) | 1.13 ± 0.14 | 864.1 | 206 (263 ± 0.26), 254 (121 ± 0.21) | 332.00 | 635.07 | 0.060 ± 0.172 |
| 9 | C37H24N3Pt | 79.89 | 705.1618 (705.1616) | 2.73 ± 0.34 | 1117.4 | 229 (104 ± 0.19), 254 (495 ± 0.24) | 258.00 | 699.00 | 0.064 ± 0.249 |
| Cell Line | Complex | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | Cisplatin | |
| HT29 | 10 ± 2.3 | 13 ± 0.3 | 3.8 ± 0.2 | 11 ± 0.7 | 2.1 ± 0.1 | 2.1 ± 0.1 | 1.1 ± 0.3 | 4.2 ± 0.3 | 0.29 ± 0.01 | nd |
| U87 | 12 ± 0.3 | 20 ± 0.9 | 3.9 ± 0.0 | 14 ± 0.3 | 2.5 ± 0.2 | 2.0 ± 0.1 | 0.38 ± 0.02 | 7.5 ± 0.8 | 0.33 ± 0.03 | 3.8 ± 1.1 |
| H460 | 3.5 ± 0.3 | 12 ± 0.3 | 3.9 ± 0.4 | 3.8 ± 0.2 | 1.3 ± 0.03 | 0.67 ± 0.14 | 0.29 ± 0.03 | 3.5 ± 0.4 | 0.36 ± 0.03 | 0.9 ± 0.2 |
| A431 | 3.8 ± 0.2 | 14 ± 0.3 | 5.7 ± 0.5 | 4.5 ± 0.5 | 2.2 ± 0.2 | 1.7 ± 0.2 | 0.42 ± 0.01 | 5.7 ± 0.6 | 0.36 ± 0.02 | 2.4 ± 0.3 |
| Du145 | 3.2 ± 0.4 | 13 ± 0.9 | 4.4 ± 0.2 | 4.6 ± 0.7 | 1.8 ± 0.1 | 1.4 ± 0.4 | 0.32 ± 0.04 | 8.1 ± 1.2 | 0.57 ± 0.04 | 1.2 ± 0.1 |
| BE2-C | 2.3 ± 0.4 | 5.1 ± 1.2 | 2.0 ± 0.3 | 2.7 ± 0.3 | 1.1 ± 0.3 | 0.59 ± 0.28 | 0.17 ± 0.05 | 2.5 ± 0.4 | 0.22 ± 0.03 | 1.9 ± 0.2 |
| SJ-G2 | 2.8 ± 0.1 | 12 ± 0.7 | 3.1 ± 0.03 | 3.1 ± 0.09 | 2.1 ± 0.1 | 1.4 ± 0.3 | 0.27 ± 0.01 | 3.1 ± 0.1 | 0.26 ± 0.00 | 0.4 ± 0.1 |
| MIA | 13.7 ± 0 | 19 ± 2 | 5.1 ± 0.97 | 14 ± 0 | 2.3 ± 0.3 | 2.3 ± 0.2 | 0.49 ± 0.10 | 12 ± 1 | 0.30 ± 0.01 | 7.5 ± 1.3 |
| ADDP | 4.3 ± 1.3 | 14 ± 2 | 3.5 ± 1.0 | 4.9 ± 1.6 | 1.7 ± 0.6 | 1.4 ± 0.6 | 0.49 ± 0.21 | 3.3 ± 0.9 | 0.30 ± 0.04 | 1.2 ± 0.1 |
| MCF-7 | 2.1 ± 0.09 | 12 ± 0.3 | 2.4 ± 0.3 | 2.9 ± 0.32 | 1.9 ± 0.29 | 1.3 ± 0.12 | 0.82 ± 0.39 | 2.2 ± 0.2 | 0.22 ± 0.009 | 6.5 ± 0.8 |
| MCF10A | 3.6 ± 0.4 | 13 ± 0 | 7.5 ± 2.2 | 4.1 ± 0.7 | 2.6 ± 0.15 | 2.2 ± 0.38 | 0.50 ± 0.01 | 5.0 ± 0.5 | 1.00 ± 0.23 | nd |
| SI | 1.7 | 1.1 | 3.1 | 1.4 | 1.4 | 1.7 | 0.6 | 2.3 | 4.5 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McGhie, B.S.; Sakoff, J.; Gilbert, J.; Gordon, C.P.; Aldrich-Wright, J.R. Novel Planar Pt(II) Cyclometallated Cytotoxic Complexes with G-Quadruplex Stabilisation and Luminescent Properties. Int. J. Mol. Sci. 2022, 23, 10469. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231810469
McGhie BS, Sakoff J, Gilbert J, Gordon CP, Aldrich-Wright JR. Novel Planar Pt(II) Cyclometallated Cytotoxic Complexes with G-Quadruplex Stabilisation and Luminescent Properties. International Journal of Molecular Sciences. 2022; 23(18):10469. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231810469
Chicago/Turabian StyleMcGhie, Brondwyn S., Jennette Sakoff, Jayne Gilbert, Christopher P. Gordon, and Janice R. Aldrich-Wright. 2022. "Novel Planar Pt(II) Cyclometallated Cytotoxic Complexes with G-Quadruplex Stabilisation and Luminescent Properties" International Journal of Molecular Sciences 23, no. 18: 10469. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231810469