
Use of a Molecular Switch Probe to Activate or Inhibit GIRK1 Heteromers In Silico Reveals a Novel Gating Mechanism
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
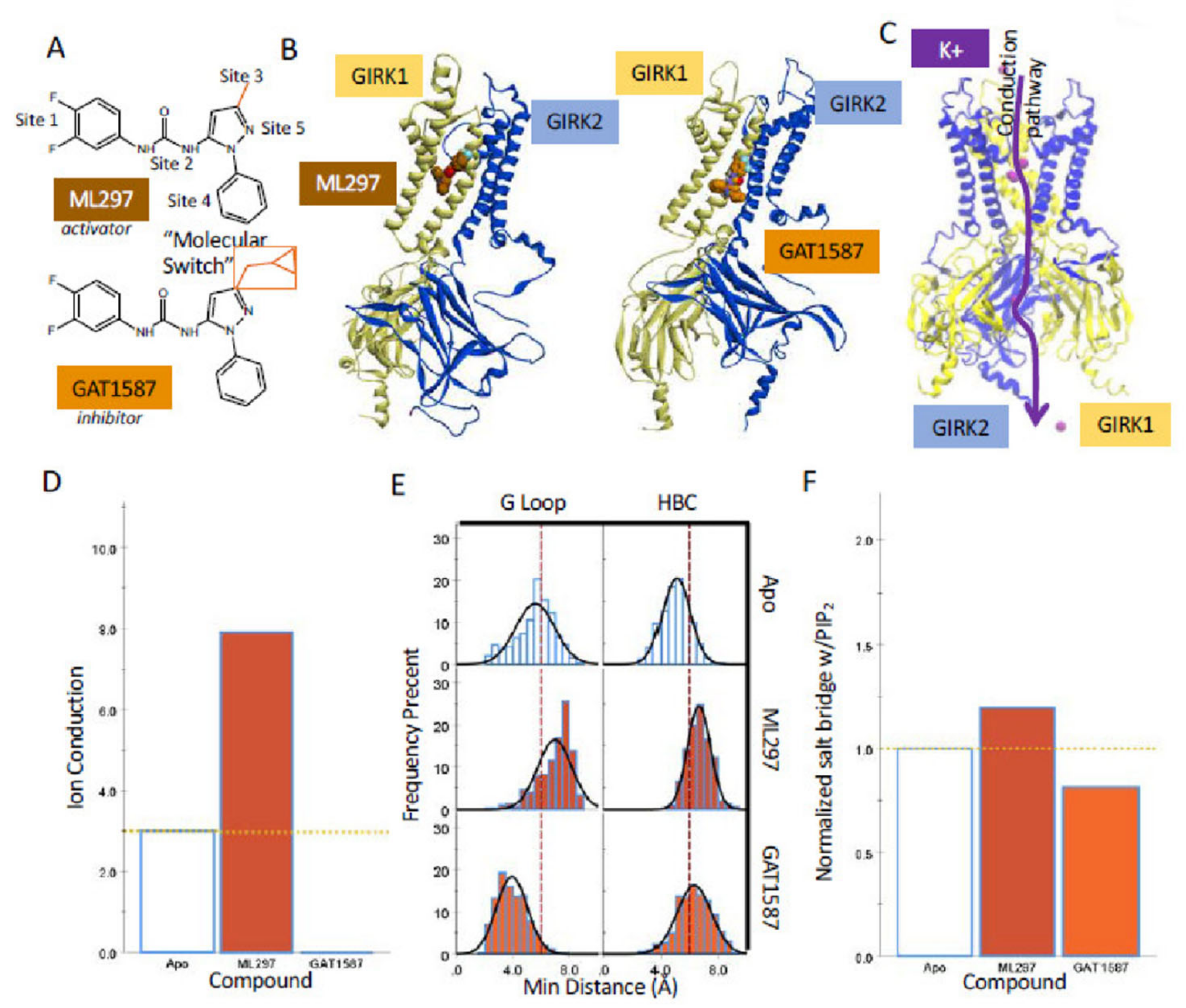
2.1. Sub-Microsecond Long Stochastic Dynamics Simulations Capture Ligand Effects, Validating the Utility of the Computational Model
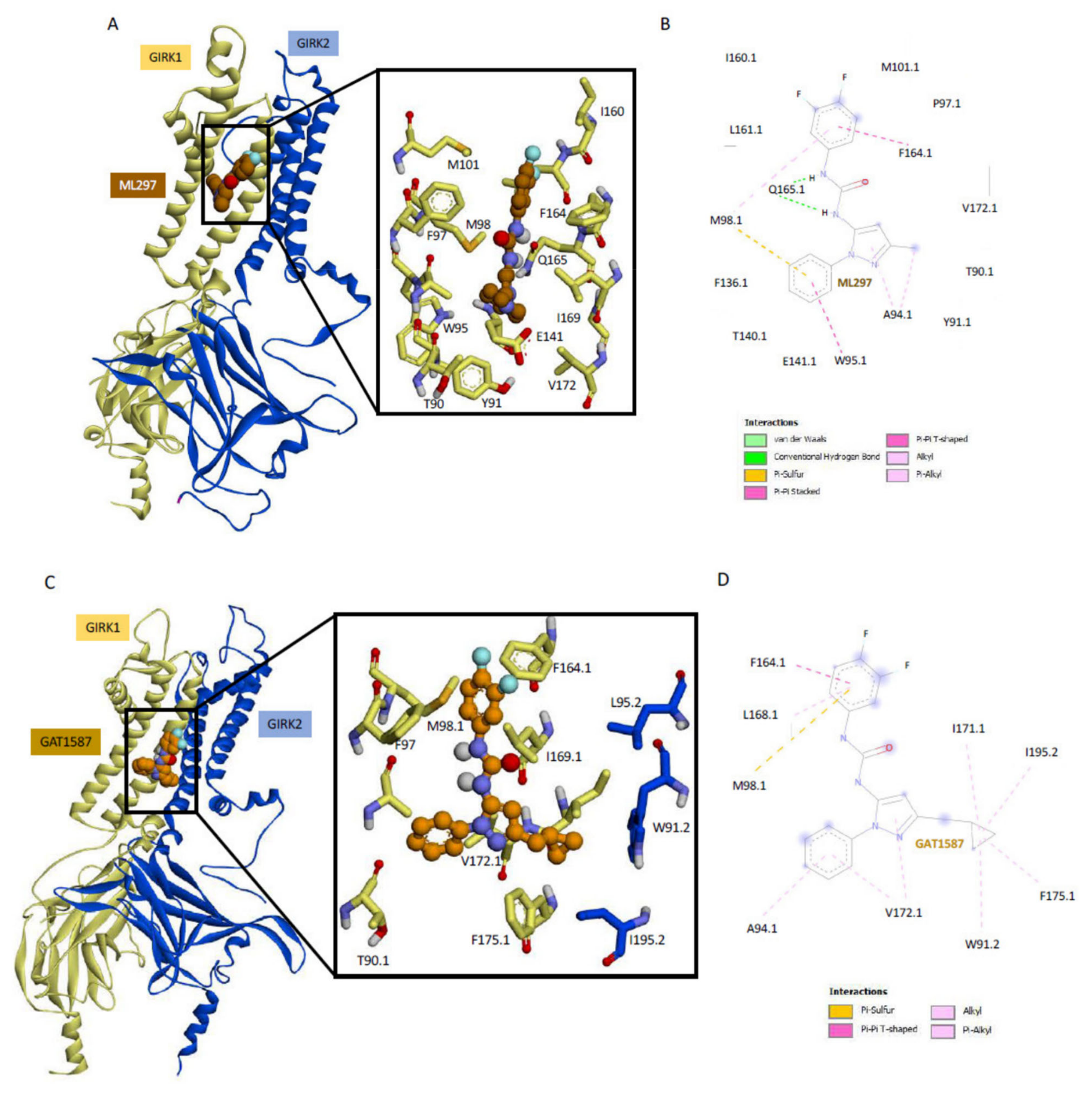
2.2. Ligand Binding to GIRK1 Subunit of Heterotetramers with GIRK2 or GIRK4 Reveals Key Differences between Residue Conformations
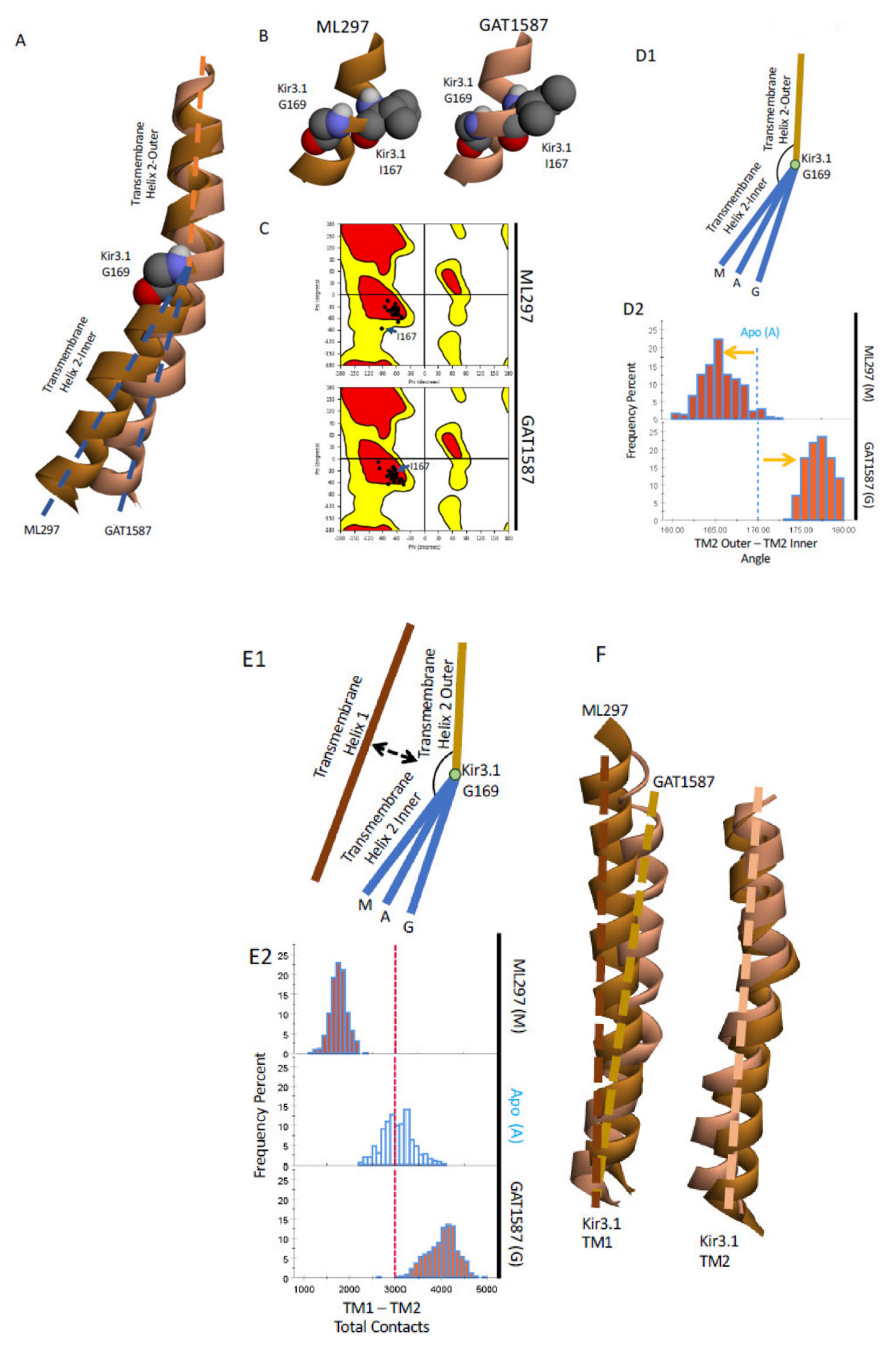
2.3. Ligand Activation Induces a Hydrophobic Chain of Residues in TM1 That Relieves Its Restrain on TM2, Allowing It to Bend and Stabilize the HBC Gate in the Open State
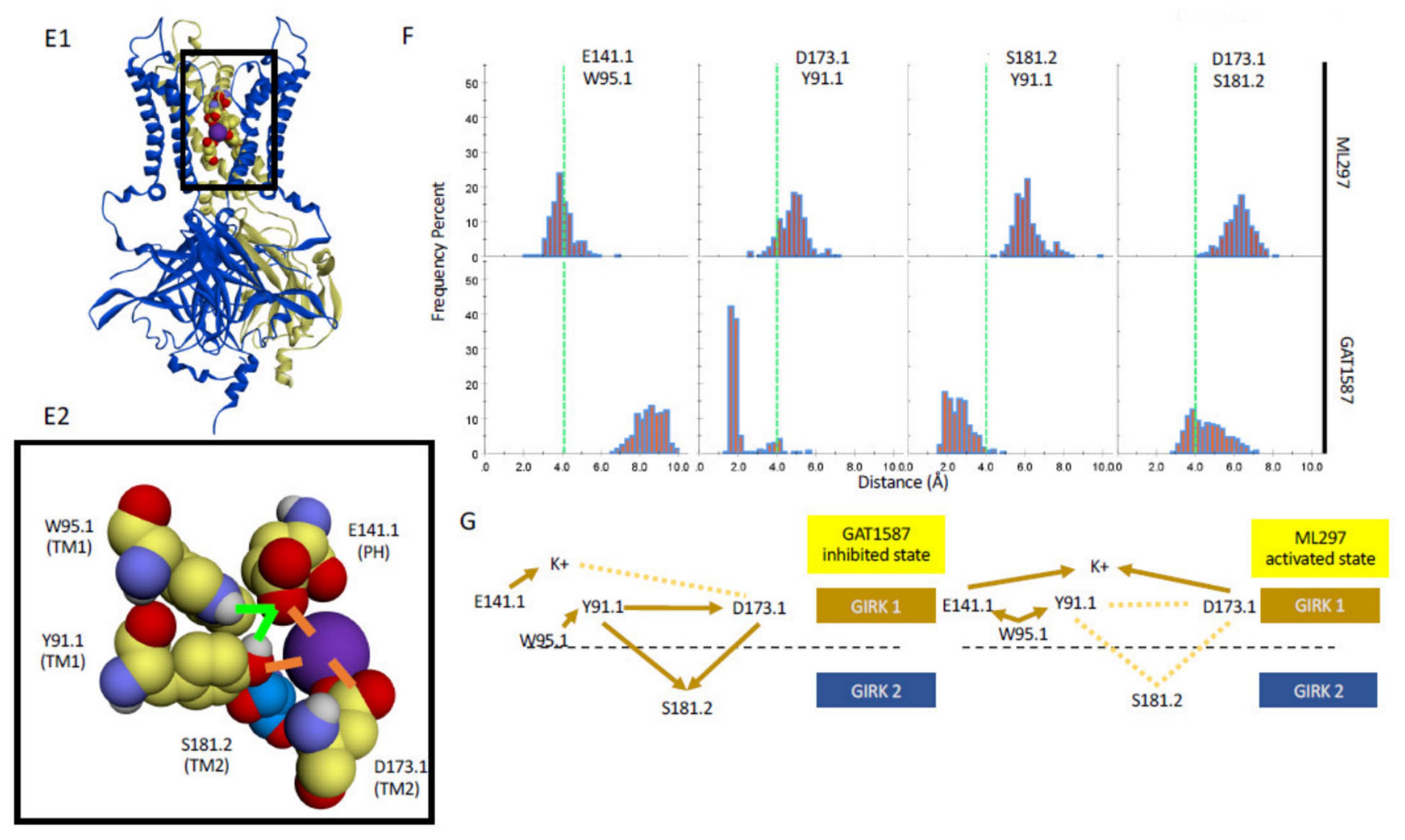
2.4. The TM1 Hydrophobic Chain Regulates K+ Ions at the E141-D173 Acidic Residue Pair of the Permeation Pathway, Depending on the Ligand Bound to the Channel
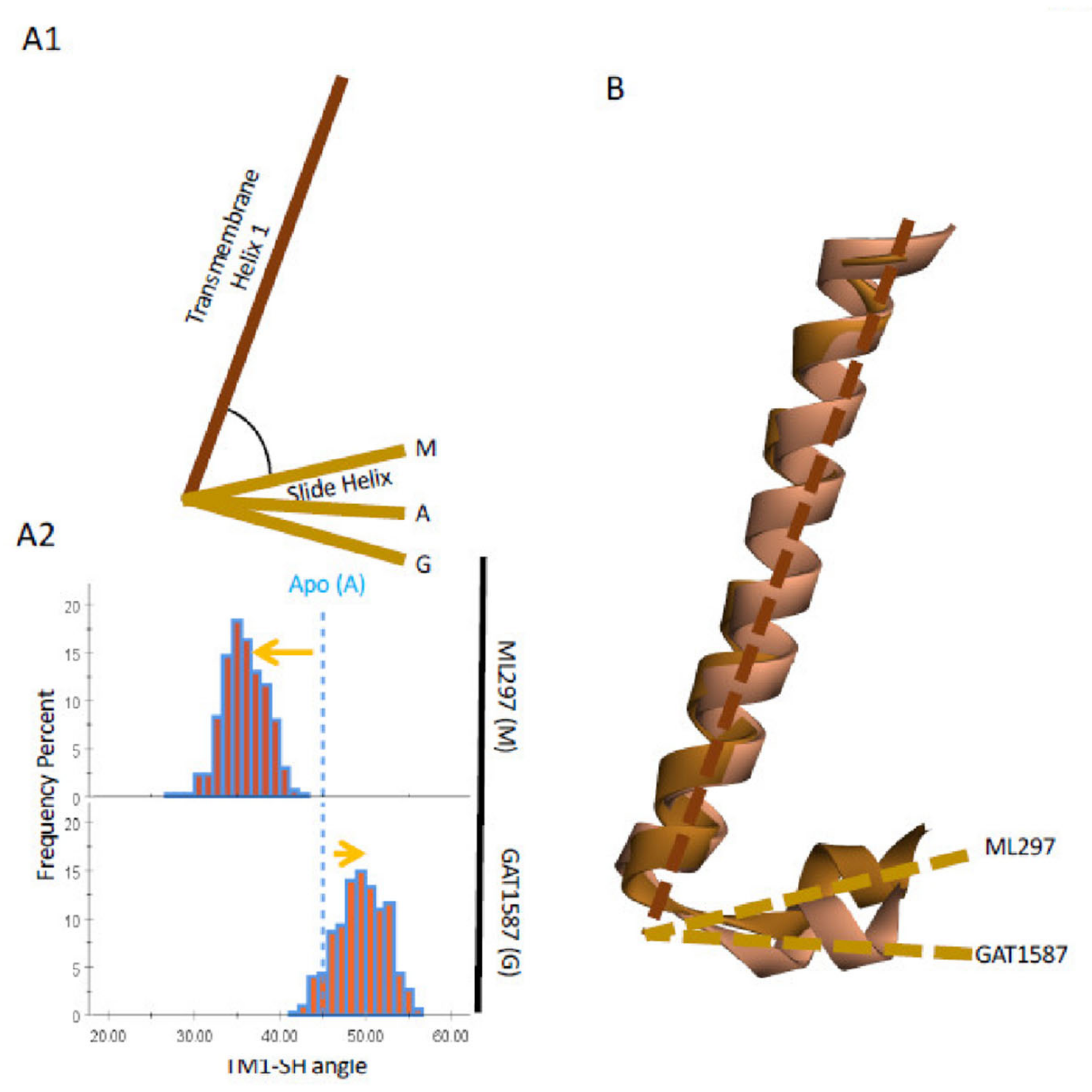
2.5. Upon ML297 Binding, TM1 Movement Is Transduced to the Slide Helix (SH) and the CD Loop, Freeing GIRK1-K188 Away the GIRK2-E315 (G-Loop) and toward PIP2 Binding
3. Discussion
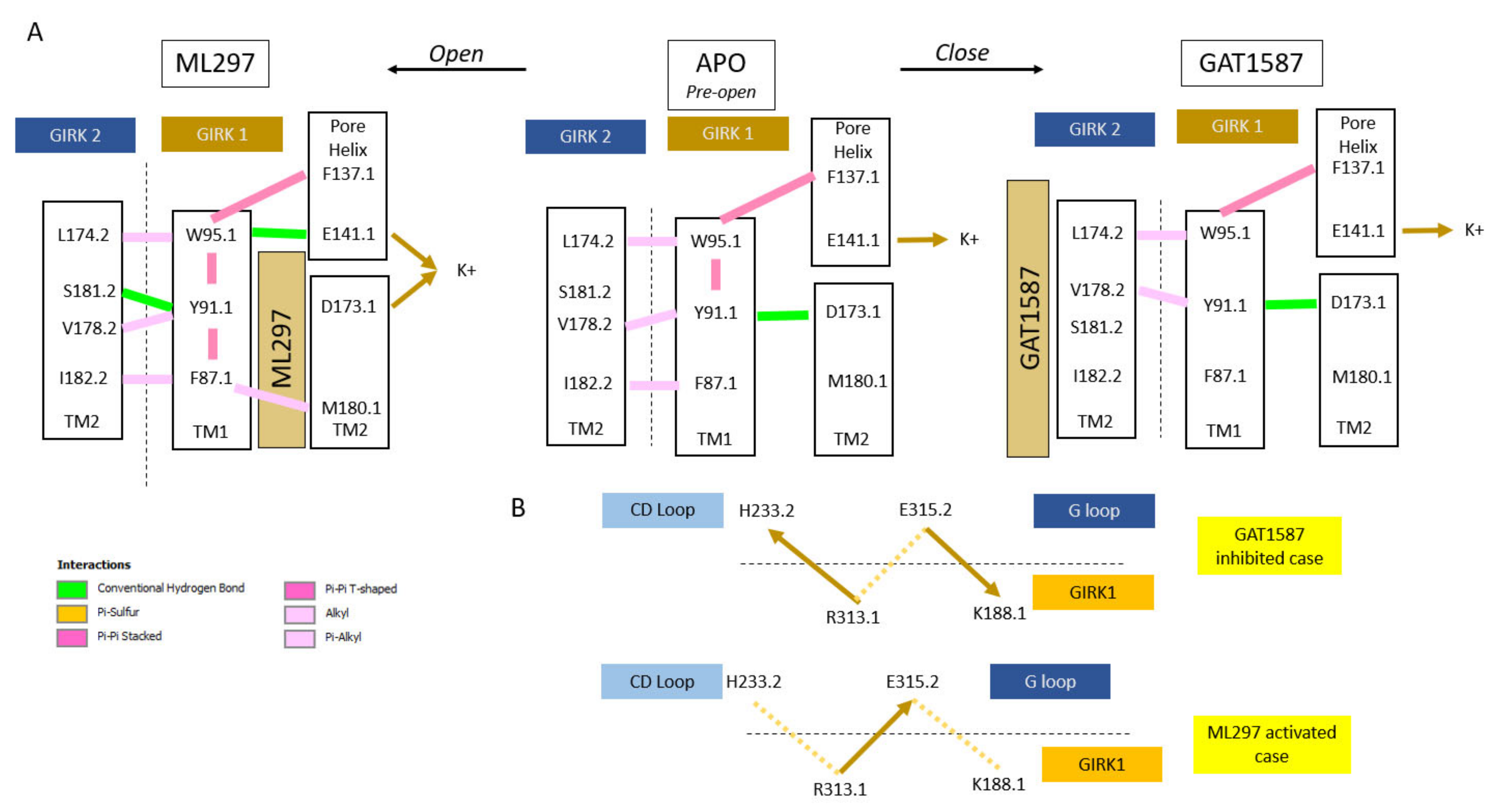
- Kir channels (characterized by two transmembrane helices -TM1 and TM2- per subunit) utilize TM1 to relieve a restrain of the pore-lining TM2 (of the same subunit type) through the formation of an open-state-dependent TM1 hydrophobic wire (for GIRK1: F87, Y91, F87). These are absolutely or highly conserved residues within Kir channels (the Y91.1 position uses either Tyr or Phe). The ML297-induced hydrophobic wire is further stabilized by TM2 hydrophobic residues of the partner subunit (the other subunit type). This conformational state is communicated to the residue preceding the residue comprising the HBC gate (for GIRK1: M180) (Figure 6A).
- The TM1 hydrophobic wire residues couple to two acidic residues along the permeation pathway. The first, E141.1 at the C-terminal end of the Pore helix (PH), is conserved among GIRK channels (it is a Gln in every other Kir channel). The second, D173 in TM2, is a GIRK1-specific residue (among Kir3 channel subtypes, and Asp, Asn or Glu in every other Kir channel). Ion conduction requires that the PH residue is stabilized by the hydrophobic wire residue while the TM2 acidic residue needs to be freed from interactions with the hydrophobic wire residue (Figure 6A).
- The TM1 movement also repositions the Slide Helix (SH), a 10-aa helical structure arranged parallel to the inner leaflet of the plasma membrane that contains two critical acidic residues (D70 and D77). A CD loop basic residue interacts with the first SH acidic residue (D70) in the presence of the inhibitory ligand but switches to the second one (D77) when the stimulatory ligand binds and the SH moves. This new salt-bridge interaction positions a critical nearby His residue in the CD loop to let go of an important basic residue that controls both gates: the stimulatory ligand frees R313.1 from the GIRK2-H233 and allows to engage the GIRK2-E315 residue away from K188 of GIRK1 stabilizing the GIRK2 G-loop open gate indirectly and allowing the GIRK1-K188 to interact with PIP2, thus stabilizing its HBC open gate directly (Figure 6B).
4. Material and Methods
4.1. Generation of the GIRK1/X Homology Models
4.2. Ligand and Co-Factor Parameterization
4.3. Ligand Docking and Model System Generation
4.4. All-Atom MD Simulations
4.5. Analysis of MD Simulations
4.6. Shortest Pathway Analysis
4.7. Chemical Synthesis
4.8. Electrophysiology
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Logothetis, D.E.; Petrou, V.I.; Zhang, M.; Mahajan, R.; Meng, X.Y.; Adney, S.K.; Cui, M.; Baki, L. Phosphoinositide control of membrane protein function: A frontier led by studies on ion channels. Annu. Rev. Physiol. 2015, 77, 81–104. [Google Scholar] [CrossRef] [PubMed]
- Logothetis, D.E.; Kurachi, Y.; Galper, J.; Neer, E.J.; Clapham, D.E. The βγ subunits of GTP-binding proteins activate the muscarinic K+ channel in heart. Nature 1987, 325, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Reuveny, E.; Slesinger, P.A.; Inglese, J.; Morales, J.M.; Iñiguez-Lluhi, J.A.; Lefkowitz, R.J.; Bourne, H.R.; Jan, Y.N.; Jan, L.Y. Activation of the cloned muscarinic potassium channel by G protein βγ subunits. Nature 1994, 370, 143–146. [Google Scholar] [CrossRef]
- Sui, J.L.; Petit-Jacques, J.; Logothetis, D.E. Activation of the atrial KACh channel by the βγ subunits of G proteins or intracellular Na+ ions depends on the presence of phosphatidylinositol phosphates. Proc. Natl. Acad. Sci. USA 1998, 95, 1307–1312. [Google Scholar] [CrossRef] [PubMed]
- Petit-Jacques, J.; Sui, J.L.; Logothetis, D.E. Synergistic activation of G protein-gated inwardly rectifying potassium channels by the βγ subunits of G proteins and Na+ and Mg2+ ions. J. Gen. Physiol. 1999, 114, 673–684. [Google Scholar] [CrossRef]
- Huang, C.L.; Feng, S.; Hilgemann, D.W. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gβγ. Nature 1998, 391, 803–806. [Google Scholar] [CrossRef] [PubMed]
- Nishida, M.; MacKinnon, R. Structural basis of inward rectification: Cytoplasmic pore of the G protein-gated inward rectifier GIRK1 at 1.8 Å resolution. Cell 2002, 111, 957–965. [Google Scholar] [CrossRef]
- Nishida, M.; Cadene, M.; Chait, B.T.; MacKinnon, R. Crystal structure of a Kir3.1prokaryotic Kir channel chimera. EMBO J. 2007, 26, 4005–4015. [Google Scholar] [CrossRef]
- Whorton, M.R.; MacKinnon, R. Crystal structure of the mammalian GIRK2 K+ channel and gating regulation by G proteins, PIP2, and sodium. Cell 2011, 147, 199–208. [Google Scholar] [CrossRef]
- Whorton, M.R.; MacKinnon, R. X-ray structure of the mammalian GIRK2-βγ G-protein complex. Nature 2013, 498, 190–197. [Google Scholar] [CrossRef] [Green Version]
- Niu, Y.; Tao, X.; Touhara, K.K.; MacKinnon, R. Cryo-EM analysis of PIP2 regulation in mammalian GIRK channels. eLife 2020, 9, e60552. [Google Scholar] [CrossRef] [PubMed]
- Jin, T.; Peng, L.; Mirshahi, T.; Rohacs, T.; Chan, K.W.; Sanchez, R.; Logothetis, D.E. The βγ subunits of G proteins gate a K+ channel by pivoted bending of a transmembrane segment. Mol. Cell. 2002, 10, 469–481. [Google Scholar] [CrossRef]
- Meng, X.Y.; Liu, S.; Cui, M.; Zhou, R.; Logothetis, D.E. The Molecular Mechanism of Opening the Helix Bundle Crossing (HBC) Gate of a Kir Channel. Sci. Rep. 2016, 6, 29399. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Jin, T.; Gazgalis, D.; Cui, M.; Logothetis, D.E. On the mechanism of GIRK2 channel gating by phosphatidylinositol bisphosphate, sodium, and the Gβγ dimer. J. Biol. Chem. 2019, 294, 18934–18948. [Google Scholar] [CrossRef] [PubMed]
- Days, E.; Kaufmann, K.; Romaine, I.; Niswender, C.; Lewis, M.; Utley, T.; Du, Y.; Sliwoski, G.; Morrison, R.; Dawson, E.S.; et al. Discovery and Characterization of a Selective Activator of the G-Protein Activated Inward-Rectifying Potassium (GIRK) Channel. In Probe Reports from the NIH Molecular Libraries Program [Internet]; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2010. [Google Scholar]
- Kaufmann, K.; Romaine, I.; Days, E.; Pascual, C.; Malik, A.; Yang, L.; Zou, B.; Du, Y.; Sliwoski, G.; Morrison, R.D.; et al. ML297 (VU0456810), the first potent and selective activator of the GIRK potassium channel, displays antiepileptic properties in mice. ACS Chem. Neurosci. 2013, 4, 1278–1286. [Google Scholar] [CrossRef]
- Wen, W.; Wu, W.; Weaver, C.D.; Lindsley, C.W. Discovery of potent and selective GIRK1/2 modulators via ‘molecular switches’ within a series of 1-(3-cyclopropyl-1phenyl-1H-pyrazol-5-yl) ureas. Bioorg. Med. Chem. Lett. 2014, 24, 5102–5106. [Google Scholar] [CrossRef]
- Wieting, J.M.; Vadukoot, A.K.; Sharma, S.; Abney, K.K.; Bridges, T.M.; Daniels, J.S.; Morrison, R.D.; Wickman, K.; Weaver, C.D.; Hopkins, C.R. Discovery and Characterization of 1H-Pyrazol-5-yl-2-phenylacetamides as Novel, Non-Urea-Containing GIRK1/2 Potassium Channel Activators. ACS Chem. Neurosci. 2017, 8, 1873–1879. [Google Scholar] [CrossRef]
- Wydeven, N.; Marron Fernandez de Velasco, E.; Du, Y.; Benneyworth, M.A.; Hearing, M.C.; Fischer, R.A.; Thomas, M.J.; Weaver, C.D.; Wickman, K. Mechanisms underlying the activation of G-protein-gated inwardly rectifying K+ (GIRK) channels by the novel anxiolytic drug, ML297. Proc. Natl. Acad. Sci. USA 2014, 111, 10755–10760. [Google Scholar] [CrossRef]
- Xu, Y.; Cantwell, L.; Molosh, A.I.; Plant, L.D.; Gazgalis, D.; Fitz, S.D.; Dustrude, E.T.; Yang, Y.; Kawano, T.; Garai, S.; et al. The small molecule GAT1508 activates brain-specific GIRK1/2 channel heteromers and facilitates conditioned fear extinction in rodents. J. Biol. Chem. 2020, 295, 3614–3634. [Google Scholar] [CrossRef]
- Li, D.L.; Hu, L.; Wang, L.; Chen, C.L. Permeation mechanisms through the selectivity filter and the open helix bundle crossing gate of GIRK2. Comput. Struct. Biotechnol. J. 2020, 18, 3950–3958. [Google Scholar] [CrossRef]
- Sadja, R.; Smadja, K.; Alagem, N.; Reuveny, E. Coupling Gβγ-dependent activation to channel opening via pore elements in inwardly rectifying potassium channels. Neuron 2001, 29, 669–680. [Google Scholar] [CrossRef]
- Chan, K.W.; Sui, J.L.; Vivaudou, M.; Logothetis, D.E. Control of channel activity through a unique amino acid residue of a G protein-gated inwardly rectifying K+ channel subunit. Proc. Natl. Acad. Sci. USA 1996, 93, 14193–14198. [Google Scholar] [CrossRef] [PubMed]
- Vivaudou, M.; Chan, K.W.; Sui, J.L.; Jan, L.Y.; Reuveny, E.; Logothetis, D.E. Probing the G-protein regulation of GIRK1 and GIRK4, the two subunits of the KACh channel, using functional homomeric mutants. J. Biol. Chem. 1997, 272, 31553–31560. [Google Scholar] [CrossRef]
- Bondi, A. Van der Waals volumes and radii. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Yi, B.A.; Lin, Y.F.; Jan, Y.N.; Jan, L.Y. Yeast screen for constitutively active mutant G protein-activated potassium channels. Neuron 2001, 29, 657–667. [Google Scholar] [CrossRef]
- Cui, M.; Cantwell, L.; Zorn, A.; Logothetis, D.E. Kir Channel Molecular Physiology, Pharmacology, and Therapeutic Implications. Handb. Exp. Pharmacol. 2021, 267, 277356. [Google Scholar]
- Lopes, C.M.; Zhang, H.; Rohacs, T.; Jin, T.; Yang, J.; Logothetis, D.E. Alterations in conserved Kir channel-PIP2 interactions underlie channelopathies. Neuron 2002, 34, 933–944. [Google Scholar] [CrossRef]
- Greenwood, J.R.; Calkins, D.; Sullivan, A.P.; Shelley, J.C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput. Aided Mol. Des. 2010, 24, 591–604. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pKa prediction and protonation state generation for drug-like molecules. J. Comp. Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247260. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general AMBER force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2019, 16, 528–552. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, K.E. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Meng, X.Y.; Zhang, H.X.; Logothetis, D.E.; Cui, M. The molecular mechanism by which PIP2 opens the intracellular G-loop gate of a GIRK1 channel. Biophys. J. 2012, 102, 2049–2059. [Google Scholar] [CrossRef]
- Hopkins, C.W.; Le Grand, S.; Walker, R.C.; Roitberg, A.E. Long-time-step molecular dynamics through hydrogen mass repartitioning. J. Chem. Theory Comput. 2015, 11, 1864–1874. [Google Scholar] [CrossRef] [PubMed]
- Mezei, M. Simulaid: A simulation facilitator and analysis program. J. Comp. Chem. 2010, 31, 2658–2668. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Grant, B.J.; Rodrigues, A.P.D.C.; Elsawy, K.M.; Mccammon, A.J.; Caves, L.S.D. Bio3d: An R Package for the Comparative Analysis of Protein Structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yen, J.Y. Finding the K Shortest Loopless Paths in a Network. Manag. Sci. 1971, 17, 712–716. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gazgalis, D.; Cantwell, L.; Xu, Y.; Thakur, G.A.; Cui, M.; Guarnieri, F.; Logothetis, D.E. Use of a Molecular Switch Probe to Activate or Inhibit GIRK1 Heteromers In Silico Reveals a Novel Gating Mechanism. Int. J. Mol. Sci. 2022, 23, 10820. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231810820
Gazgalis D, Cantwell L, Xu Y, Thakur GA, Cui M, Guarnieri F, Logothetis DE. Use of a Molecular Switch Probe to Activate or Inhibit GIRK1 Heteromers In Silico Reveals a Novel Gating Mechanism. International Journal of Molecular Sciences. 2022; 23(18):10820. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231810820
Chicago/Turabian StyleGazgalis, Dimitrios, Lucas Cantwell, Yu Xu, Ganesh A. Thakur, Meng Cui, Frank Guarnieri, and Diomedes E. Logothetis. 2022. "Use of a Molecular Switch Probe to Activate or Inhibit GIRK1 Heteromers In Silico Reveals a Novel Gating Mechanism" International Journal of Molecular Sciences 23, no. 18: 10820. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231810820