

Growth Hormone Improves Adipose Tissue Browning and Muscle Wasting in Mice with Chronic Kidney Disease-Associated Cachexia
Abstract
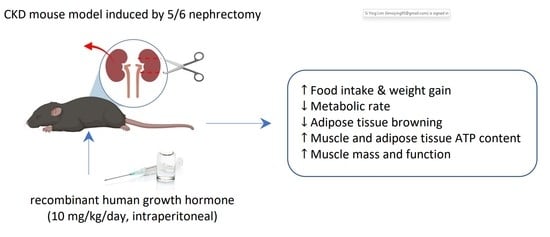
:
1. Introduction
2. Results
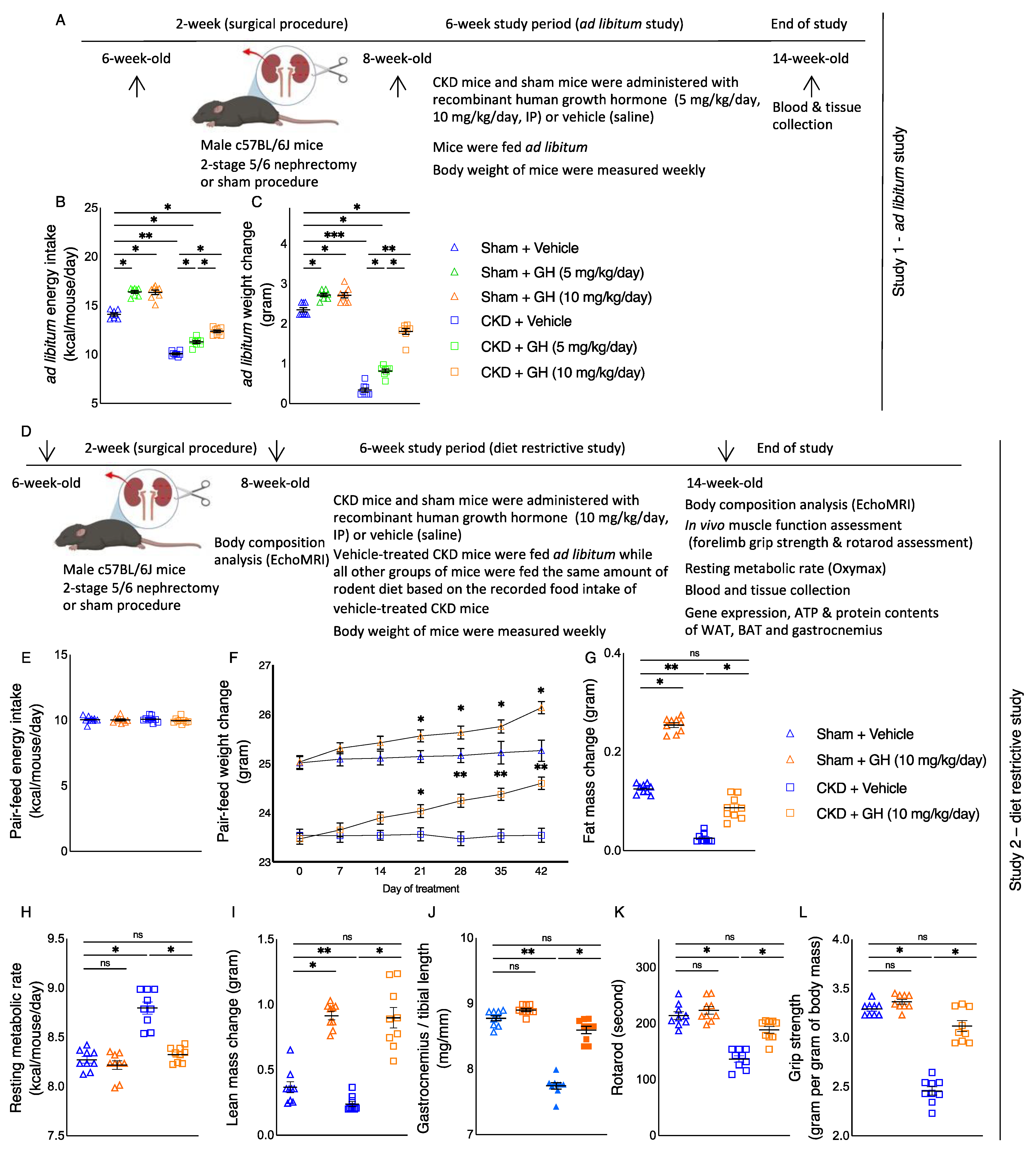
2.1. GH Stimulates Food Intake and Increases Body Weight in CKD Mice
2.2. GH Improves Energy Homeostasis in CKD Mice
2.3. GH Improves Skeletal Muscle and Adipose Tissue Energy Homeostasis in CKD Mice
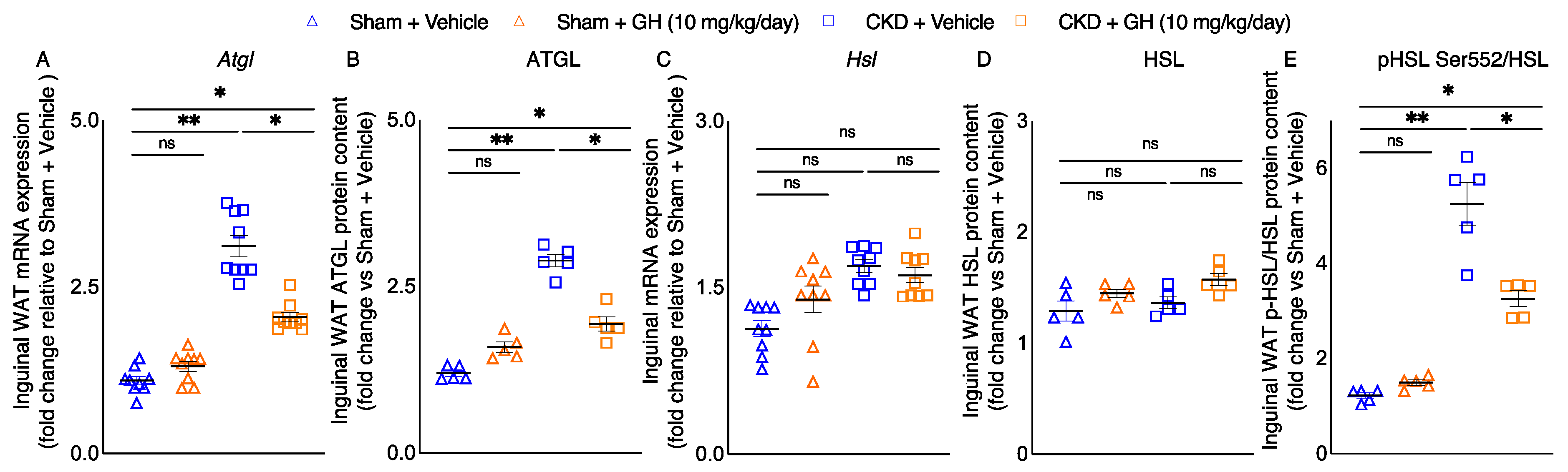
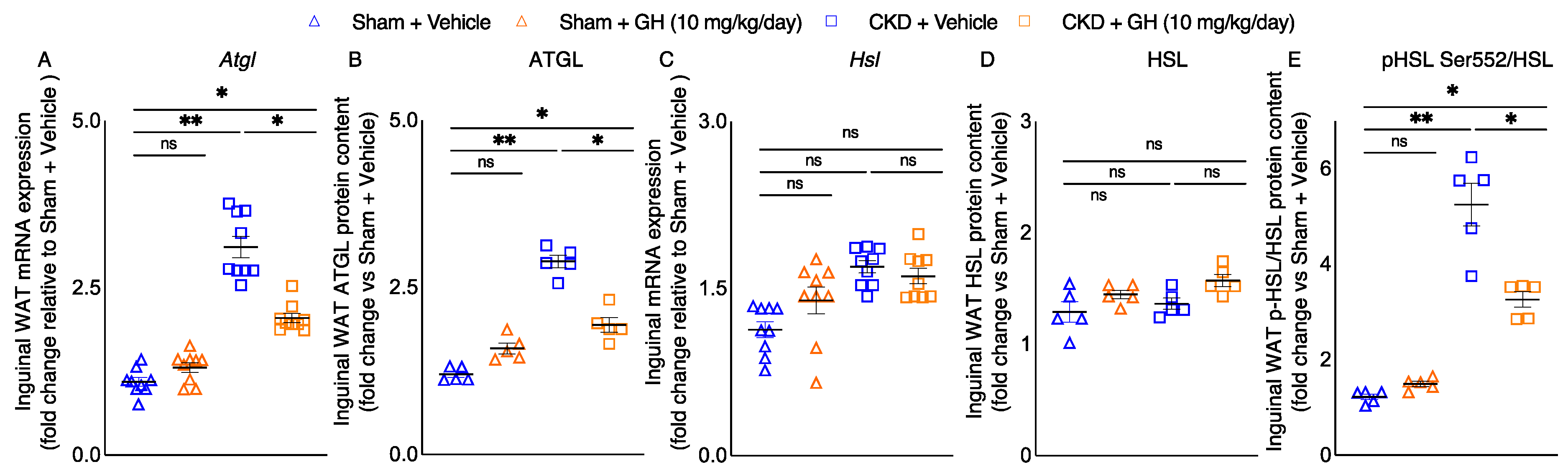
2.4. GH Mitigates Lipolytic Enzymes in CKD Mice
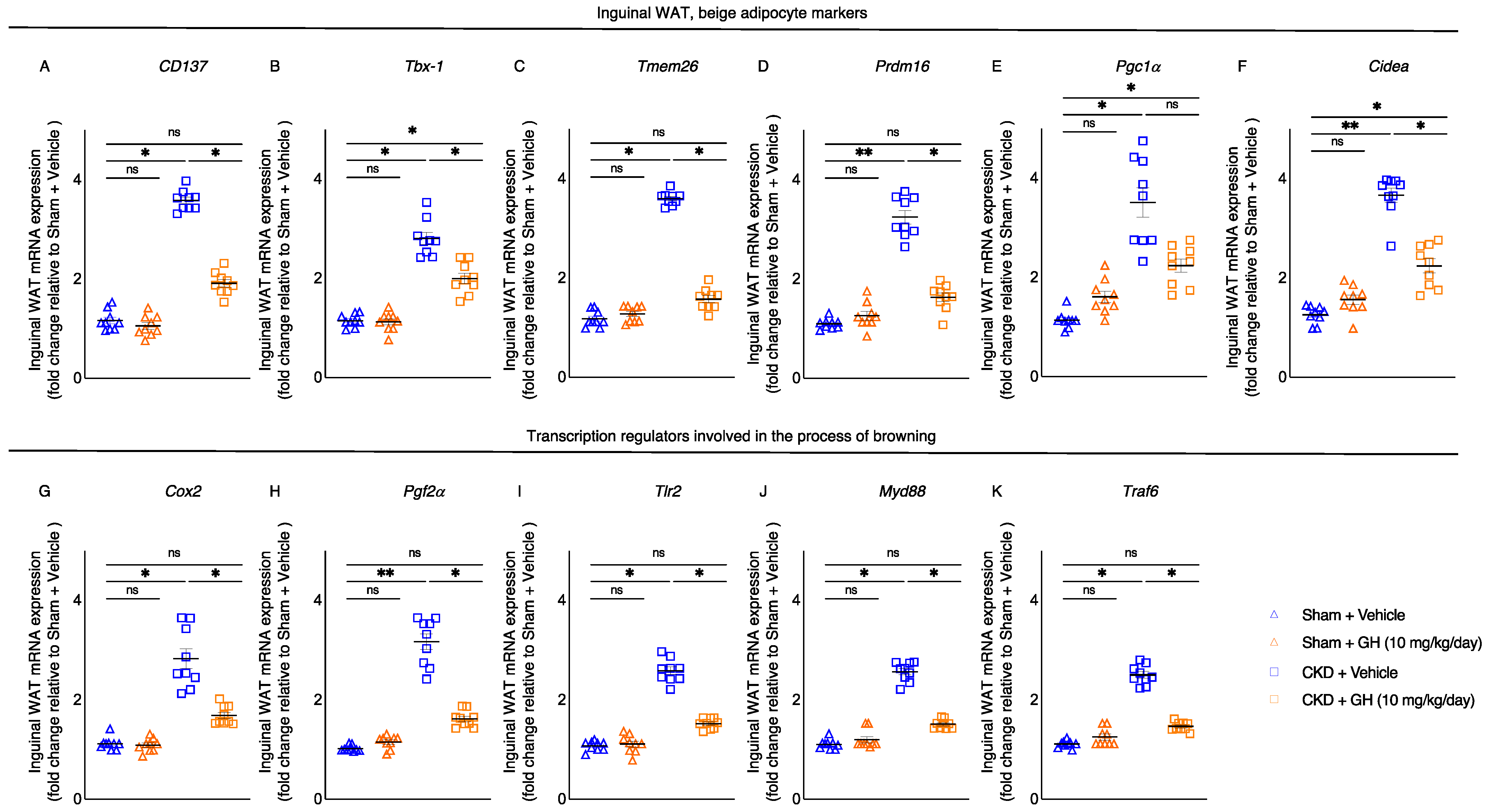
2.5. GH Mitigates White Adipose Tissue Browning in CKD Mice
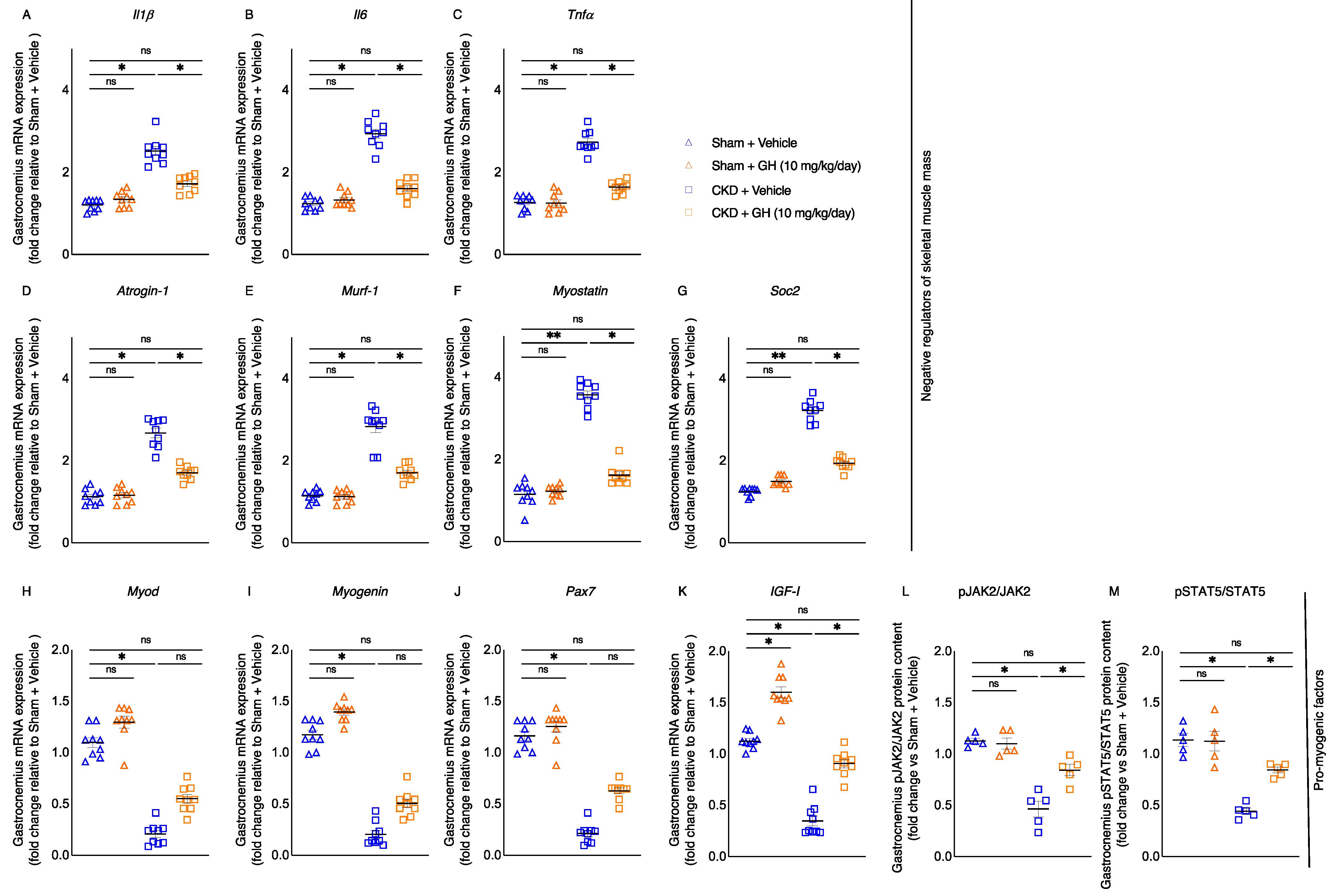
2.6. GH Attenuates Muscle-Wasting Signaling and GH Resistance Pathways in CKD Mice
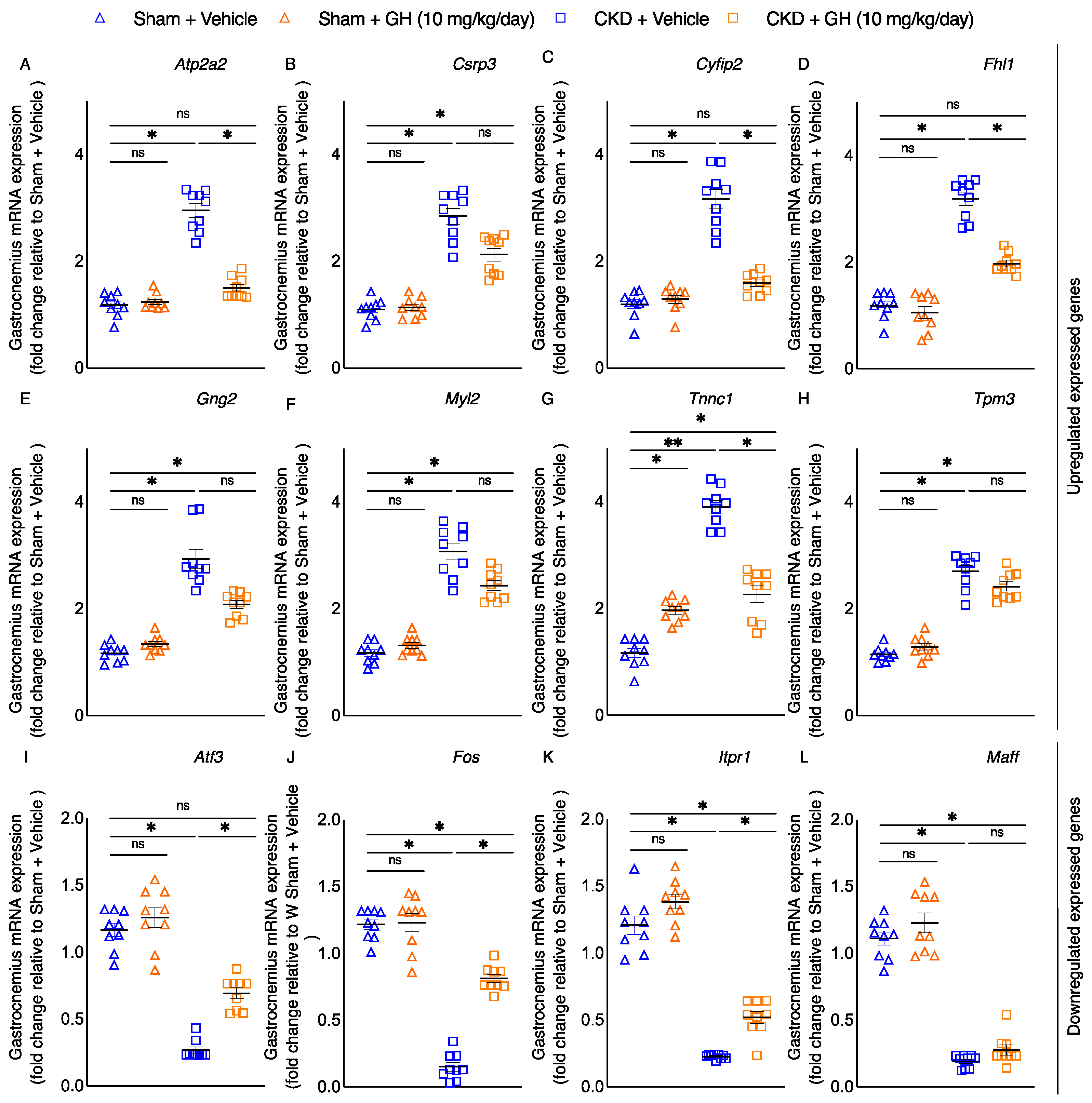
2.7. Molecular Mechanism of GH on Muscle Function by RNAseq Analysis
3. Discussion
4. Materials and Methods
4.1. Study Design
4.2. Body Composition, Metabolic Rate, and In Vivo Muscle Function
4.3. Serum and Blood Chemistry
4.4. Protein Assay for Muscle and Adipose Tissue
4.5. Muscle RNAseq Analysis
4.6. Quantative Real-Time PCR
4.7. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mak, R.H.; Ikizler, A.T.; Kovesdy, C.P.; Raj, D.S. Wasting in Chronic Kidney Disease. J. Cachexia Sarcopenia Muscle 2011, 2, 9–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koppe, L.; Fouque, D.; Kalantar-Zadeh, K. Kidney Cachexia or Protein-Energy Wasting in CKD: Facts and Numbers. J. Cachexia Sarcopenia Muscle 2019, 10, 479–484. [Google Scholar] [CrossRef] [Green Version]
- Gungor, O.; Ulu, S.; Hasbal, N.B.; Anker, S.D. Effects of Hormonal Changes on Sarcopenia in Chronic Kidney Disease: Where are we now and what can we do? J. Cachexia Sarcopenia Muscle 2021, 12, 1380–1392. [Google Scholar] [CrossRef] [PubMed]
- Oliveria, E.A.; Carter, C.E.; Mak, R.H. The Role of Growth Hormone in Chronic Kidney Disease. Semin. Nephrol. 2021, 41, 144–155. [Google Scholar] [CrossRef] [PubMed]
- Tavares, A.B.W.; Micmacher, E.; Biesek, S.; Assumpcao, R. Effects of Growth Hormone Administration on Muscle Strength in Men over 50 Years Old. Int. J. Endocrinol. 2013, 2013, 942030. [Google Scholar] [CrossRef] [Green Version]
- Gabribotto, G.; Barreca, A.; Russo, R.; Sofia, A. Effects of Recombinant Human Growth Hormone on Muscle Protein Turnover in Malnourished Hemodialysis Patients. J. Clin. Investig. 1997, 99, 97–105. [Google Scholar] [CrossRef] [Green Version]
- Hansen, T.B.; Gram, J.; Jensen, P.B.; Kristiansen, J.H. Influence of Growth hormone on Whole Body and Regional Soft Tissue Composition in Adult Patients on Hemodialysis. A Double-Blind, Randomized, Placebo-Controlled Study. Clin. Nephrol. 2000, 53, 99–107. [Google Scholar]
- Feldt-Rasmussen, B.; Lange, M.; Sulowicz, W.; Gafter, U. Growth Hormone Treatment during Hemodialysis in a Randomized Trial Improves Nutrition, Quality of Life, and Cardiovascular Risk. J. Am. Soc. Nephrol. 2007, 18, 2161–2171. [Google Scholar] [CrossRef] [Green Version]
- Nienczyk, S.; Sikorsk, H.; Wiecek, A.; Zukowska-Szczechowska, E. A Super-Agonist of Growth Hormone-Releasing Hormone Causes Rapid Improvement of Nutritional Status in Patients with Chronic Kidney Disease. Kidney Int. 2010, 77, 450–458. [Google Scholar] [CrossRef] [Green Version]
- Mendias, C.L.; Sibilsky Enselman, E.R.; Olszewski, A.M.; Gumucio, J.P. The Use of Recombinant Human Growth Hormone to Protect Against Muscle Weakness in Patients Undergoing Anterior Cruciate Ligament Reconstruction: A Pilot, Randomized Placebo-Controlled Trial. Am. J. Sports Med. 2020, 48, 1916–1928. [Google Scholar] [CrossRef]
- Tavoian, D.; Ampomah, K.; Amano, S.; Law, T.D. Changes in DXA-Derived Lean Mass and MRI-Derived Cross-Sectional Area of the Thigh are Modestly Associated. Sci. Rep. 2019, 9, 10028. [Google Scholar] [CrossRef] [PubMed]
- Sabatino, A.; D’Alessandro, C.D.; Regolisti, G.; di Mario, F. Muscle Mass Assessment in Renal Disease: The Role of Imaging Techniques. Quant. Imaging Med. Surg. 2020, 10, 1672–1686. [Google Scholar] [CrossRef] [PubMed]
- Cheung, W.W.; Ding, W.; Gunta, S.S.; Gu, Y.; Mak, R.H. A Pegylated Leptin Antagonist Ameliorates CKD-Associated Cachexia in Mice. J. Am. Soc. Nephrol. 2014, 25, 119–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, D.F.; Chen, Y.; Rabkin, R. Work-Induced Changes in Skeletal Muscle IGF-1 and Myostatin Gene Expression in Uremia. Kidney Int. 2006, 70, 453–459. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, F.; Chen, Y.; Tsao, T.; Nouri, P. Impaired JAK-STAT Signal Transduction Contributes to Growth Hormone Resistance in Chronic Uremia. J. Clin. Investig. 2001, 108, 467–475. [Google Scholar] [CrossRef]
- Choe, S.S.; Huh, J.Y.; Hwang, I.J.; Kim, J.I. Adipose Tissue Remodeling: Its Role in Energy Metabolism and Metabolic Disorders. Front. Endocrinol. 2016, 7, 30. [Google Scholar] [CrossRef] [Green Version]
- Kir, S.; White, J.P.; Kleiner, S.; Kazak, L. Tumor-Derived PTHrP Triggers Adipose Tissue Browning and Cancer Cachexia. Nature 2014, 513, 100–104. [Google Scholar] [CrossRef] [Green Version]
- Kir, S.; Komaba, H.; Garcia, A.P.; Economopoulos, K.P. PTH/PTHrP Receptor Mediates Cachexia in Models of Kidney Failure and Cancer. Cell Metab. 2016, 23, 315–323. [Google Scholar] [CrossRef] [Green Version]
- Berryman, D.E.; List, E.O. Growth Hormone’s Effect on Adipose Tissue: Quality versus Quantity. Int. J. Mol. Sci. 2017, 18, 1621. [Google Scholar] [CrossRef] [Green Version]
- Kopchick, J.J.; Berryman, D.E.; Puri, V.; Lee, K.Y. The Effects of Growth Hormone on Adipose Tissue: Old Observations, New Mechanisms. Nat. Rev. Endocrinol. 2020, 16, 135–146. [Google Scholar] [CrossRef]
- Kliewer, K.L.; Ke, J.Y.; Tian, M.; Cole, R.M.; Andridge, R.R.; Belury, M.A. Adipose tissue lipolysis and energy metabolism in early cancer cachexia mice. Cancer Biol. Ther. 2015, 16, 886–897. [Google Scholar] [CrossRef] [PubMed]
- Vegiopoulos, A.; Müller-Decker, K.; Strzoda, D.; Schmitt, I. Cyclooxygenase-2 Controls Energy Homeostasis in Mice by de Novo Recruitment of Brown Adipocytes. Science 2010, 328, 1158–1161. [Google Scholar] [CrossRef] [PubMed]
- Cheung, W.W.; Ding, W.; Hoffman, H.M.; Wang, Z.; Mak, R.H. Vitamin D Ameliorates Adipose Browning in Chronic Kidney Disease Cachexia. Sci. Rep. 2020, 10, 14175. [Google Scholar] [CrossRef] [PubMed]
- Pupim, L.B.; Flakoll, P.J.; Yu, C.; Alp Ikizer, T. Recombinant Human Growth Hormone Improves Muscle Amino Acid Uptake and Whole-Body Protein Metabolism in Chronic Hemodialysis Patients. Am. J. Clin. Nutr. 2005, 82, 1235–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricquier, D.; Bouillaud, F. Mitochondrial uncoupling proteins: From mitochondria to the regulation of energy balance. J. Physiol. 2000, 529, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Demine, S.; Renard, P.; Arnould, T. Mitochondrial uncoupling: A key controller of biological processes in physiology and diseases. Cells 2019, 8, 795. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Wray, C.; Tian, X.; Hasselgren, P.O.; Lu, J. Expression of uncoupling protein is upregulated in skeletal muscle during sepsis. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E512–E520. [Google Scholar] [CrossRef]
- Pohl, E.E.; Rupprecht, A.; Macher, G.; Hilse, K.E. Important trends in UCP3 investigation. Front. Physiol. 2019, 10, 470. [Google Scholar] [CrossRef] [Green Version]
- Minnaard, R.; Schrauwen, P.; Schaart, G.; Hesselink, M.K.C. UCP3 in muscle wasting, a role in modulating lipotoxicity? FEBS Lett. 2006, 580, 5172–5176. [Google Scholar] [CrossRef] [Green Version]
- Petruzzelli, M.; Schweiger, M.; Schreiber, R.; Campos-Olivas, R.; Tsoli, M.; Allen, J.; Swarbrick, M.; Rose-John, S.; Rincon, M.; Robertson, G.; et al. A switch from white to brown fat increased energy expenditure in cancer-associated cachexia. Cell Metab. 2014, 20, 443–447. [Google Scholar] [CrossRef] [Green Version]
- Rohm, M.; Schafer, M.; Laurent, V.; Ustunel, B.K.; Niopek, K.; Algrire, C.; Hautzinger, O.; Sijmonsma, T.P.; Zota, A.; Medrikova, D.; et al. An AMP-activated protein kinase–stabilizing peptide ameliorates adipose tissue wasting in cancer cachexia in mice. Nat. Med. 2016, 22, 1120–1130. [Google Scholar] [CrossRef] [PubMed]
- Sanders, K.J.C.; Wierts, R.; Lichtenbelt, W.D.V.M.; Vos-Geelen, J.D.; Plasqui, G.; Kelders, M.C.J.M.; Schrauwen-Hinderling, V.B.; Bucerius, J.; Dingemans, A.M.C.; Mottaghy, F.M.; et al. Brown adipose tissue activation is not related to hypermetabolism in emphysematous chronic obstructive pulmonary disease patients. J. Cachexia Sarcopenia Muscle 2022, 13, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Wijers, S.L.J.; Schrauwen, P.; van Baak, M.A.; Saris, W.H.M.; Lichtenbelt, W.D.V.M. β-Adrenergic receptor blockade does not inhibit cold-induced thermogenesis in humans: Possible involvement of brown adipose tissue. J. Clin. Endocrinol. Metab. 2011, 96, E598–E605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ootsuka, Y.; Kulasekara, K.; de Menezes, R.C.; Blessing, W.M. SR59230A, a beta-3 adrenergic antagonist, inhibit ultradian brown adipose tissue thermogenesis and interrupts associated episodic brain and body heating. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R987–R994. [Google Scholar] [CrossRef] [Green Version]
- Zhai, M.; Yang, D.; Yi, W.; Sun, W. Involvement of calcium channel in the regulation of adipogenesis. Adipocyte 2020, 9, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Mao, Y.; Xiao, L.; Sun, Z.; Liu, J.; Zhou, D.; Xu, Z.; Liu, L.; Fu, T.; Ding, C.; et al. FNIPI regulates adipocyte browning and systemic glucose homeostasis in mice by shaping intracellular calcium dynamics. J. Exp. Med. 2022, 219, e20212491. [Google Scholar] [CrossRef]
- Murphy, R.A.; Wilke, M.S.; Perrine, M.; Pawlowicz, M.; Mourtzakis, M.; Lieffers, J.R.; Maneshgar, M.; Bruera, E.; Clandinin, M.T.; Baracos, V.E.; et al. Loss of adipose tissue and plasma phospholipids: Relationship to survival in advanced cancer patients. Clin. Nutr. 2010, 29, 482–487. [Google Scholar] [CrossRef]
- Dalal, S.; Hui, D.; Bidaut, L.; Lem, K.; Del Fabbro, E.; Crane, C.; Reyes-Gibby, C.C.; Bedi, D.; Bruera, E. Relationships among body mass index, longitudinal body composition alterations, and survival in patients with locally advanced pancreatic cancer receiving chemoradiation: A pilot study. J. Pain Symptom Manag. 2012, 44, 181–191. [Google Scholar] [CrossRef]
- Di Sebastiano, K.M.; Yang, L.; Zbuk, K.; Wong, R.K.; Chow, T.; Koff, D.; Moran, G.R.; Mourtzakis, M. Accelerated muscle and adipose tissue loss may predict survival in pancreatic cancer patients: The relationship with diabetes and anaemia. Br. J. Nutr. 2013, 109, 302–312. [Google Scholar] [CrossRef] [Green Version]
- Miyoshi, H.; Perfield, J.W.; Obin, M.S.; Greenberg, A.S. Adipose triglyceride lipase regulates basal lipolysis and lipid droplet size in adipocytes. J. Cell. Biochem. 2008, 105, 1430–1436. [Google Scholar] [CrossRef] [Green Version]
- Das, S.K.; Eder, S.; Schauer, S.; Diwoky, C.; Temmel, H.; Guertl, B.; Gorkiewicz, G.; Tamilarasan, K.P.; Kumari, P.; Trauner, M.; et al. Adipose triglyceride lipase contributes to cancer-associated cachexia. Science 2011, 33, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Tsoli, M.; Schweiger, M.; Vanniasinghe, A.S.; Painter, A.; Zechner, R.; Clarke, S.; Robertson, G. Depletion of white adipose tissue in cancer cachexia syndrome is associated with inflammatory signaling and disrupted circadian regulation. PLoS ONE 2014, 9, e92966. [Google Scholar] [CrossRef] [PubMed]
- Silverio, R.; Lira, F.S.; Oyama, L.M.; do Nascimento, C.M.O.; Otoch, J.P.; Alcantara, P.S.M.; Batista Jr, M.L.; Seelaender, M. Lipase and lipid droplet-associated protein expression in subcutaneous white adipose tissue of cachectic patients with cancer. Lipids Health Dis. 2017, 16, 159–169. [Google Scholar] [CrossRef]
- Wu, J.; Dong, J.; Verzolaa, D.; Hruska, K.; Garibotto, G.; Hu, Z.; Mithc, W.E.; Thomas, S.S. Signal regulatory protein alpha initiates cachexia through muscle to adipose tissue crosstalk. J. Cachexia Sarcopenia Muscle 2019, 10, 1210–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ran, L.; Wang, X.; Mi, A.; Liu, Y. Loss of Adipose Growth Hormone Receptor in Mice Enhances Local Fatty Acid Tapping and Impairs Brown Adipose Tissue Thermogenesis. iSciences 2019, 16, 106–121. [Google Scholar] [CrossRef] [Green Version]
- Witkowska-Sedek, E.; Pyrzak, B. Chronic inflammation and the growth hormone/insulin-like growth factor-1 axis. Cent. Eur. J. Immunol. 2020, 45, 469–475. [Google Scholar] [CrossRef]
- Han, J.; Meng, Q.; Shen, L.; Wu, G. Interleukin-6 induces fat loss in cancer cachexia by promoting white adipose tissue lipolysis and browning. Lipids Health Dis. 2018, 17, 14. [Google Scholar] [CrossRef] [Green Version]
- Bozzola, M.; De Amici, M.; Zecca, M.; Schimpff, R.M.; Rapaport, R. Modulating effect of human growth hormone on tumour necrosis factor-alpha and interleukin-1beta. Eur. J. Endocrinol. 1998, 138, 640–643. [Google Scholar] [CrossRef] [Green Version]
- Buckingham, M.; Relaix, F. PAX3 and PAX7 as Upstream Regulators of Myogenesis. Semin. Cell Dev. Biol. 2015, 44, 115–125. [Google Scholar] [CrossRef]
- Wang, Y.X.; Rudnicki, M. Satellite Cells, the Engines of Muscle Repair. Nat. Rev. Mol. Cell Biol. 2011, 13, 127–133. [Google Scholar] [CrossRef]
- Foster, B.J.; Kalkwarf, H.J.; Shults, J.; Zemel, B.S. Association of Chronic Kidney Disease with Muscle Deficits in Children. J. Am. Soc. Nephrol. 2011, 22, 377–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziegler, T.R.; Lazarus, J.M.; Young, L.S.; Hakim, R. Effects of Recombinant Human Growth Hormone in Adults Receiving Maintenance Gemodialysis. J. Am. Soc. Nephrol. 1991, 2, 1130–1135. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, P.; Diez, J.J.; Fernandez-Reyes, M.J.; Aguilera, A. Recombinant Human Growth Hormone Therapy in Malnourished Dialysis Patients: A Randomized Controlled Study. Am. J. Kidney Dis. 1998, 32, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Kotzmann, H.; Yilmaz, N.; Lercher, P.; Riedl, M. Differential Effects of Growth Hormone Therapy in Malnourished Hemodialysis Patients. Kidney Int. 2001, 60, 1578–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florini, J.R.; Ewton, D.Z.; Coolican, S.A. Growth Hormone and the Insulin-Like Growth Factor System in Myogenesis. Endocr. Rev. 1996, 15, 481–517. [Google Scholar]
- Lopez, J.; Quan, A.; Budihardjo, J.; Xiang, S. Growth Hormone Improves Nerve Regeneration, Muscle Re-innervation and Functional Outcomes after Chronic Denervation Injury. Sci. Rep. 2019, 9, 3117. [Google Scholar] [CrossRef] [Green Version]
- Gautsch, T.A.; Kandl, S.M.; Donovan, S.M.; Layman, D.K. Growth Hormone Promotes Somatic and Skeletal Muscle Growth Recovery in Rats Following Chronic-Energy Malnutrition. J. Nutr. 1999, 129, 828–837. [Google Scholar] [CrossRef] [Green Version]
- Sotiropoulos, A.; Ohanna, M.; Kedzia, C.; Menon, R.K. Growth Hormone Promotes Skeletal Muscle Cell Fusion Independent of Insulin-Like Growth Factor 1 Upregulation. Proc. Natl. Acad. Sci. USA 2006, 103, 7315–7320. [Google Scholar] [CrossRef] [Green Version]
- Mahan, J.D.; Warady, B.A.; Consensus Committee. Assessment and treatment of short stature in pediatric patients with chronic kidney disease: A consensus statement. Pediatr. Nephrol. 2006, 21, 917–930. [Google Scholar] [CrossRef]
- Bielohuby, M.; Schaab, M.; Kummann, M.; Sawitzky, M.; Gebhardt, R.; Binder, G.; Frystyk, J.; Bjerre, M.; Hoeflich, A.; Kratzsch, J.; et al. Serum IGF-I is not a reliable pharmacodynamic marker of exogenous growth hormone activity in mice. Endocrinology 2011, 152, 4764–4776. [Google Scholar] [CrossRef]
- Ho, K.K.Y.; 2007 GH Deficiency Consensus Workshop Participants. Consensus guidelines for the diagnosis and treatment of adults with GH deficiency II: A statement of the GH Research Society in association with the European Society for Pediatric Endocrinology, Lawson Wilkins Society, European Society of Endocrinology, Japan Endocrine Society, and Endocrine Society of Australia. Eur. J. Endocrinol. 2007, 157, 695–700. [Google Scholar] [PubMed] [Green Version]
- Sjogren, K.; Liu, J.L.; Blad, K.; Skrtic, S.; Vidal, O.; Wallenius, V.; LeRoith, D.; Tornell, J.; Isaksson, O.G.; Jansson, J.O.; et al. Liver-derived insulin-like growth factor I (IGF-I) is the principal source of IGF-I in blood but is not required for postnatal body growth in mice. Proc. Natl. Acad. Sci. USA 1999, 96, 7088–7092. [Google Scholar] [CrossRef] [PubMed]
- List, E.O.; Palmer, A.J.; Berryman, D.E.; Bower, B.; Kelder, B.; Kopchick, J.J. Growth hormone improves body composition, fasting blood glucose, glucose tolerance and liver triacylglycerol in a mouse model of diet-induced obesity and type 2 diabetes. Diabetologia 2009, 52, 1647–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, K.C.; Doyle, N.; Ballesteros, M.; Waters, M.J.; Ho, K.K. Insulin regulation of human hepatic growth hormone receptors: Divergent effects on biosynthesis and surface translocation. J. Clin. Endocrinol. Metab. 2000, 85, 4712–4720. [Google Scholar] [CrossRef]
- Baxter, R.C.; Turtle, J.R. Regulation of hepatic growth hormone receptors by insulin. Biochem. Biophys. Res. Commun. 1978, 84, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Carnieli, D.S.; Yoshioka, E.; Silva, L.F.F.; Lamcas, T.; Arantes, F.M.; Perini, A.; Martins, M.A.; Saldiva, P.H.N.; Dolhnikoff, M.; Mauad, T. Inflammation and remodeling in infantile, juvenile, and adult allergic sensitized mice. Pediatr. Pulmonol. 2011, 46, 650–665. [Google Scholar] [CrossRef] [PubMed]
- Astori, M.; Finke, D.; Karapetian, O.; Acha-Orbea, H. Development of T–B cell collaboration in neonatal mice. Int. Immunol. 1999, 11, 445–451. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Rusznak, Z.; Herculano-Houzel, S.; Watson, C.; Paxinos, G. Cellular composition characterizing postnatal development and maturation of the mouse brain and spinal cord. Brain Struct. Funct. 2013, 218, 1337–1354. [Google Scholar] [CrossRef] [Green Version]
- Somerville, J.M.; Aspden, R.M.; Armour, K.E.; Armour, K.J.; Reid, D.M. Growth of C57BL/6 mice and the material and mechanical properties of cortical bone from the tibia. Calcif. Tissue Int. 2004, 74, 469–475. [Google Scholar] [CrossRef]
- Brodt, M.D.; Ellis, C.B.; Silva, M.J. Growing C57BL/6 mice increase whole bone mechanical properties by increasing geometric and material properties. J. Bone Miner. Res. 1999, 14, 2159–2166. [Google Scholar] [CrossRef]
- Halloran, B.P.; Ferguson, V.L.; Simske, S.J.; Burghardt, A.; Venton, L.L.; Majumdar, S. Changes in bone structure and mass with advancing age in the male C57BL/6J mouse. J. Bone Miner. Res. 2002, 17, 1044–1050. [Google Scholar] [CrossRef] [PubMed]
- Gargiolo, S.; Gramanzini, M.; Megna, R.; Greco, A.; Albanese, S.; Manfredi, C.; Brunetti, A. Evaluation of growth patterns and body composition in c57BL/6J mice using dual energy- X-ray absorptiometry. Biomed Res. Int. 2014, 2014, 253067. [Google Scholar] [CrossRef] [PubMed]
- Loboda, A.; Kraft, W.K.; Fine, B.; Joseph, J.; Nebozhyn, M.; Zhang, C.; He, Y.; Yang, X.; Wright, C.; Morris, M.; et al. Diurnal variation of the human adipose transcriptome and the link to metabolic disease. BMC Med. Genom. 2009, 2, 7. [Google Scholar] [CrossRef] [Green Version]
- Zvonic, S.; Ptitsyn, A.A.; Conrad, S.A.; Scott, L.K.; Floyd, Z.E.; Kilroy, G.; Wu, X.; Goh, B.C.; Mynatt, R.L.; Gimble, J.M. Characterization of peripheral circadian clocks in adipose tissues. Diabetes 2006, 55, 962–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serin, Y.; Acar, T.N. Effect of circadian rhythm on metabolic processes and the regulation of energy balance. Ann. Nutr. Metab. 2019, 74, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Nadal-Casellas, A.; Proenza, A.M.; Llado, I.; Gianotti, M. Effects of ovariectomy and 17-b edstradiol replacement on rat brown adipose tissue mitochondrial function. Steroids 2011, 76, 1051–1056. [Google Scholar] [CrossRef]
- Abdulnour, J.; Doucet, E.; Brochu, M.; Lavoie, J.M.; Streychar, I.; Rabasa-Lhoret, R.; Orud’homme, D. The effect of the menopausal transition on body composition and cardiometabolic risk factors: A Montreal-Ottawa New Emerging Team group study. Menopause 2012, 19, 760–767. [Google Scholar] [CrossRef]
- Wade, G.N.; Gray, J.M. Cytoplasmic 17 beta-[3H] estradiol binding in rat adipose tissues. Endocrinology 1978, 103, 1695–1701. [Google Scholar] [CrossRef]
- Velickovic, K.; Cvoro, A.; Srdic, B.; Stokic, E.; Markelic, M.; Golic, I.; Otasevic, V.; Stancic, A.; Jankovic, A.; Vucetic, M.; et al. Expression and subcellular localization of estrogen receptors α and β in human fetal brown adipose tissue. J. Clin. Endocrinol. Metab. 2014, 99, 151–159. [Google Scholar] [CrossRef] [Green Version]
- de Morentin, P.B.M.; Gonzalez-Garcia, I.; Martins, L.; Lage, R.; Fernandez-Mallo, D.; Martinez-Sanchez, N.; Ruiz-Pino, F.; Liu, J.; Morgan, D.A.; Pinilla, L.; et al. Estradiol regulates brown adipose tissue thermogenesis via hypothalamic AMPK. Cell Metab. 2014, 20, 41–53. [Google Scholar]
- Liu, P.; Ji, Y.; Yuen, T.; Rendina-Ruedy, E.; DeMambro, V.E.; Dhawan, S.; Abu-Amer, W.; Izadmehr, S.; Zhou, B.; Shin, A.C.; et al. Blocking FSH induces thermogenic adipose tissue and reduces body fat. Nature 2017, 546, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Dieudonne, M.; Pecquery, R.; Leneveu, M.C.; Giudicelli, Y. Opposite Effects of Androgens and Estrogens on Adipogenesis in Rat Preadipocytes: Evidence for Sex and Site-Related Specificities and Possible Involvement of Insulin-Like Growth Factor 1 Receptor and Peroxisome Proliferator-Activated Receptorγ 2. Endocrinology 2000, 141, 649–656. [Google Scholar] [CrossRef] [PubMed]
- O’Mara, A.E.; Johnson, J.W.; Limderman, J.D.; Brychta, R.J.; McGehee, S.; Fletcher, L.A.; Fink, Y.A.; Kapuria, D.; Cassimatis, T.M.; Kelsey, N.; et al. Chronic mirabegron treatment increases human brown fat, HDL cholesterol, and insulin activity. J. Clin. Investig. 2020, 130, 2209–2219. [Google Scholar] [CrossRef] [Green Version]
- Carpentier, A.C.; Blondin, D.P.; Haman, F.; Richard, D. Brown adipose tissue—A translational perspective. Endocr. Rev. 2022, bnac015. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sham + Vehicle (n = 9) | Sham + GH (5 mg/kg/day) (n = 9) | Sham + GH (10 mg/kg/day) (n = 9) | CKD + Vehicle (n = 9) | CKD + GH (5 mg/kg/day) (n = 9) | CKD + GH (10 mg/kg/day) (n = 9) | |
|---|---|---|---|---|---|---|
| BUN (mg/dL) | 34.5 ± 3.5 | 36.7 ± 4.6 | 32.6 ± 3.7 | 65.8 ± 6.9 a | 75.6 ± 8.1 a | 65.9 ± 5.8 a |
| Creatinine (mg/dL) | 0.32 ± 0.11 | 0.35 ± 0.14 | 0.28 ± 0.09 | 0.57 ± 0.15 a | 0.65 ± 0.13 a | 0.75 ± 0.13 a |
| Sham + Vehicle (n = 9) | Sham + GH (10 mg/kg/day) (n = 9) | CKD + Vehicle (n = 9) | CKD + GH (10 mg/kg/day) (n = 9) | |
|---|---|---|---|---|
| BUN (mg/dL) | 36.5 ± 5.8 | 26.7 ± 4.7 | 59.8 ± 7.4 a | 72.8 ± 11.5 a |
| Creatinine (mg/dL) | 0.25 ± 0.06 | 0.31 ± 0.13 | 0.63 ± 0.21 a | 0.75 ± 0.25 a |
| Human GH (µg/L) | - | 364.6 ± 76.4 | - | 325.3 ± 65.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mak, R.H.; Gunta, S.; Oliveira, E.A.; Cheung, W.W. Growth Hormone Improves Adipose Tissue Browning and Muscle Wasting in Mice with Chronic Kidney Disease-Associated Cachexia. Int. J. Mol. Sci. 2022, 23, 15310. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232315310
Mak RH, Gunta S, Oliveira EA, Cheung WW. Growth Hormone Improves Adipose Tissue Browning and Muscle Wasting in Mice with Chronic Kidney Disease-Associated Cachexia. International Journal of Molecular Sciences. 2022; 23(23):15310. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232315310
Chicago/Turabian StyleMak, Robert H., Sujana Gunta, Eduardo A. Oliveira, and Wai W. Cheung. 2022. "Growth Hormone Improves Adipose Tissue Browning and Muscle Wasting in Mice with Chronic Kidney Disease-Associated Cachexia" International Journal of Molecular Sciences 23, no. 23: 15310. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232315310