
Veratridine Can Bind to a Site at the Mouth of the Channel Pore at Human Cardiac Sodium Channel NaV1.5

 ,
,  , and
, and
Abstract
:1. Introduction
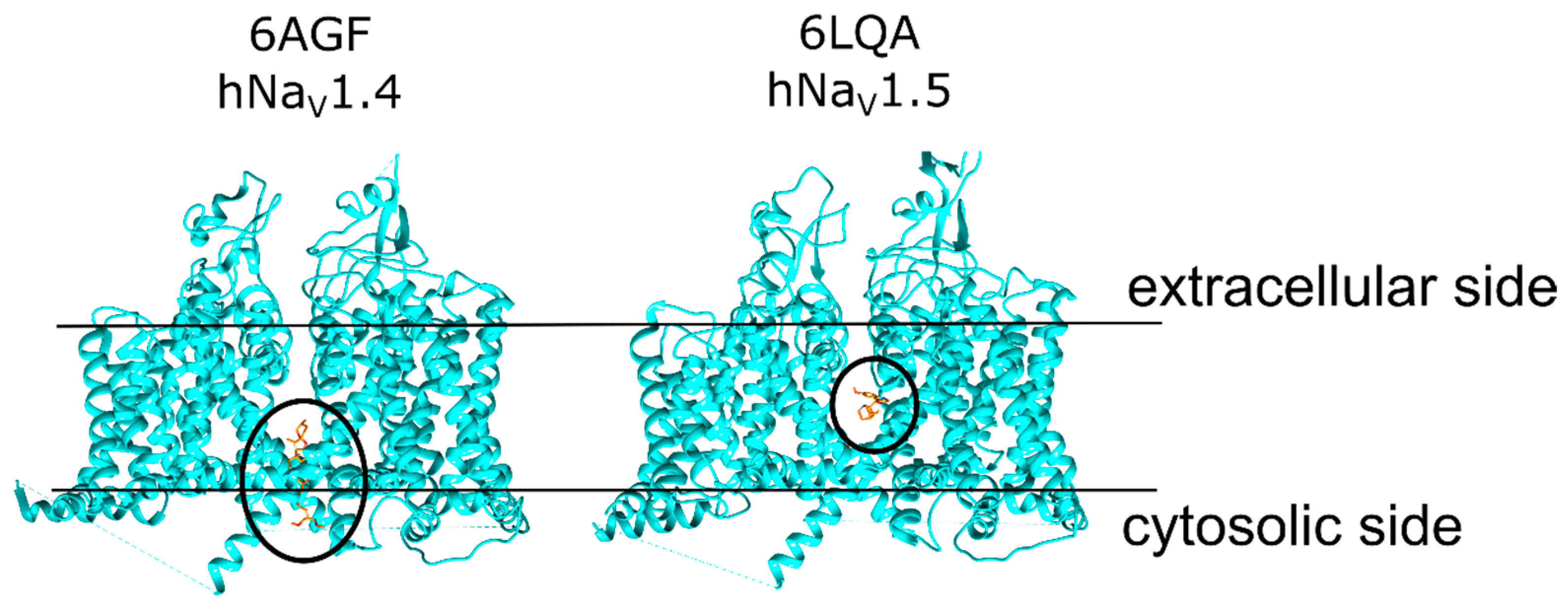
1.1. NaV Structure and Function
1.2. NaV1.5 Activity Is Modulated by Multiple Classes of Drugs
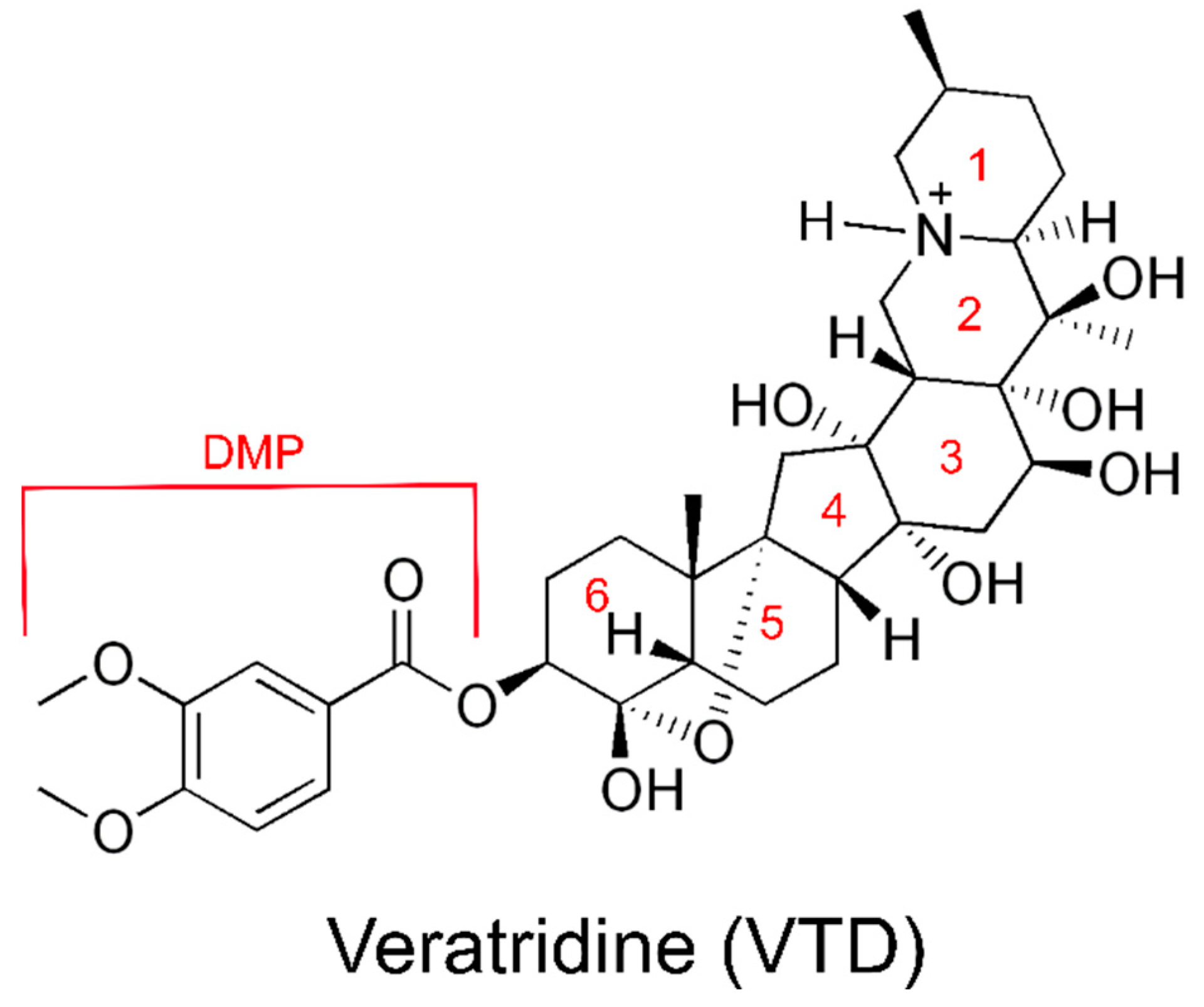
1.3. Veratridine Is a Plant Alkaloid That Delays Channel Desensitization
1.4. VTD May Bind to Multiple Sites on NaV1.5
2. Results and Discussion
2.1. Veratridine Was Docked to the Mouth Site and the Pore Site
2.2. Three Pose Clusters Were Identified to Bind Close to the Mouth of the Pore When the Docking Grid Was Centered at the Mouth Site
2.3. Two Pose Clusters Bound Deeper in the Pore with the Charged Nitrogen Facing Opposite Directions inside the Pore
2.4. Three VTD Poses at the Pore Site Were Buried Mostly within the Ion Channel and Intercalated the Surrounding α-Helices
2.5. Two Pore Site Poses Had Tenuous Interactions with the Pore Residues and Interacted Mostly with the α-Helices Lining the Pore
2.6. Eleven Mutations Were Selected to Probe the Effect of NaV1.5 Mutations on VTD Binding
2.7. E417 and F942 Were Designed to Selectively Affect Mouth Site Binding
2.8. Four Mutations Were Designed to Diminish Hydrophobic Interactions between NaV1.5 and VTD
2.9. Aromatic Residue Mutations at Two Sites Were Introduced to Alter Hydrogen Bonding, Hydrophobic Interactions, and π-Cation Interactions
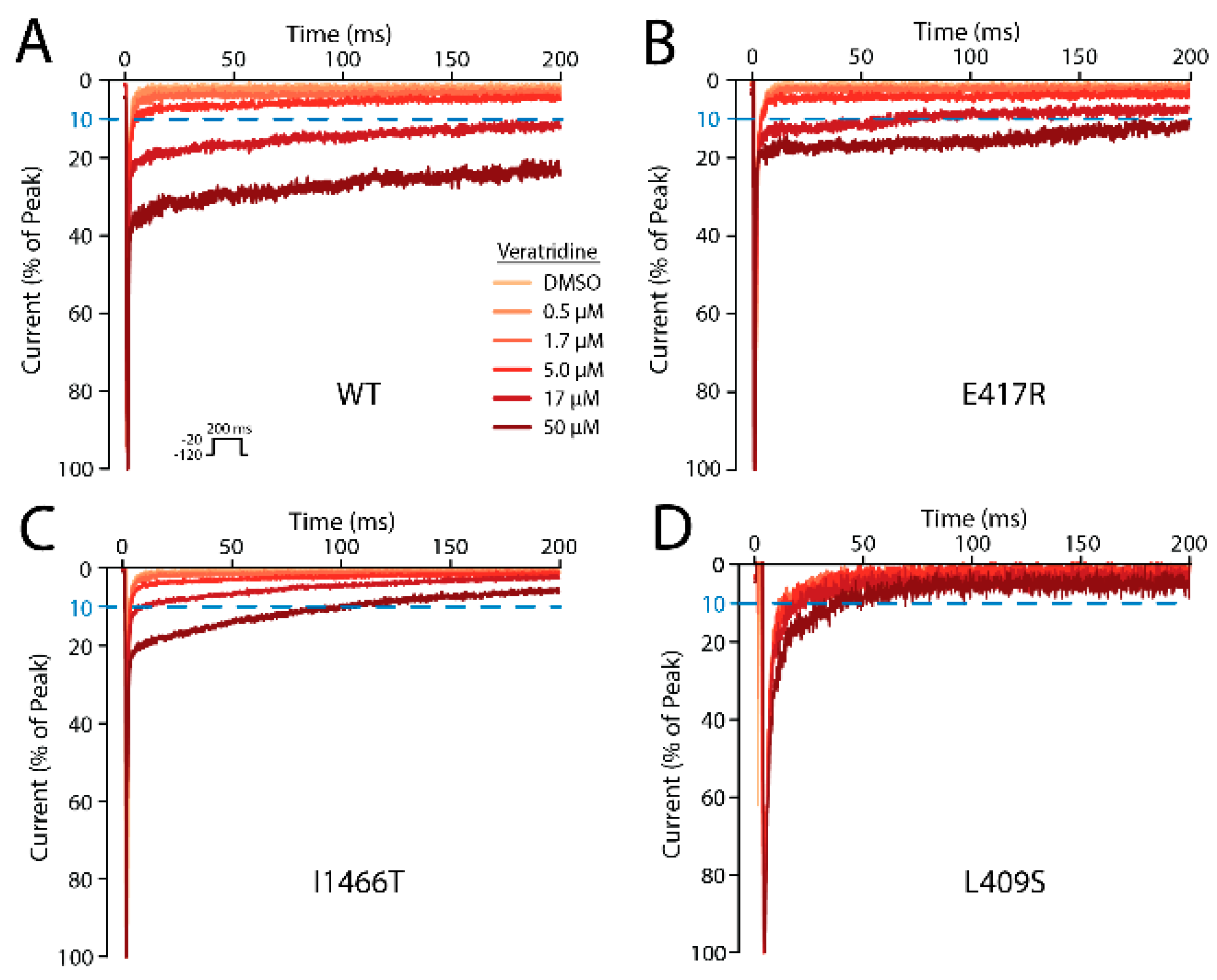
2.10. Five Mutations Had a Moderate or Large Effect on VTD Binding
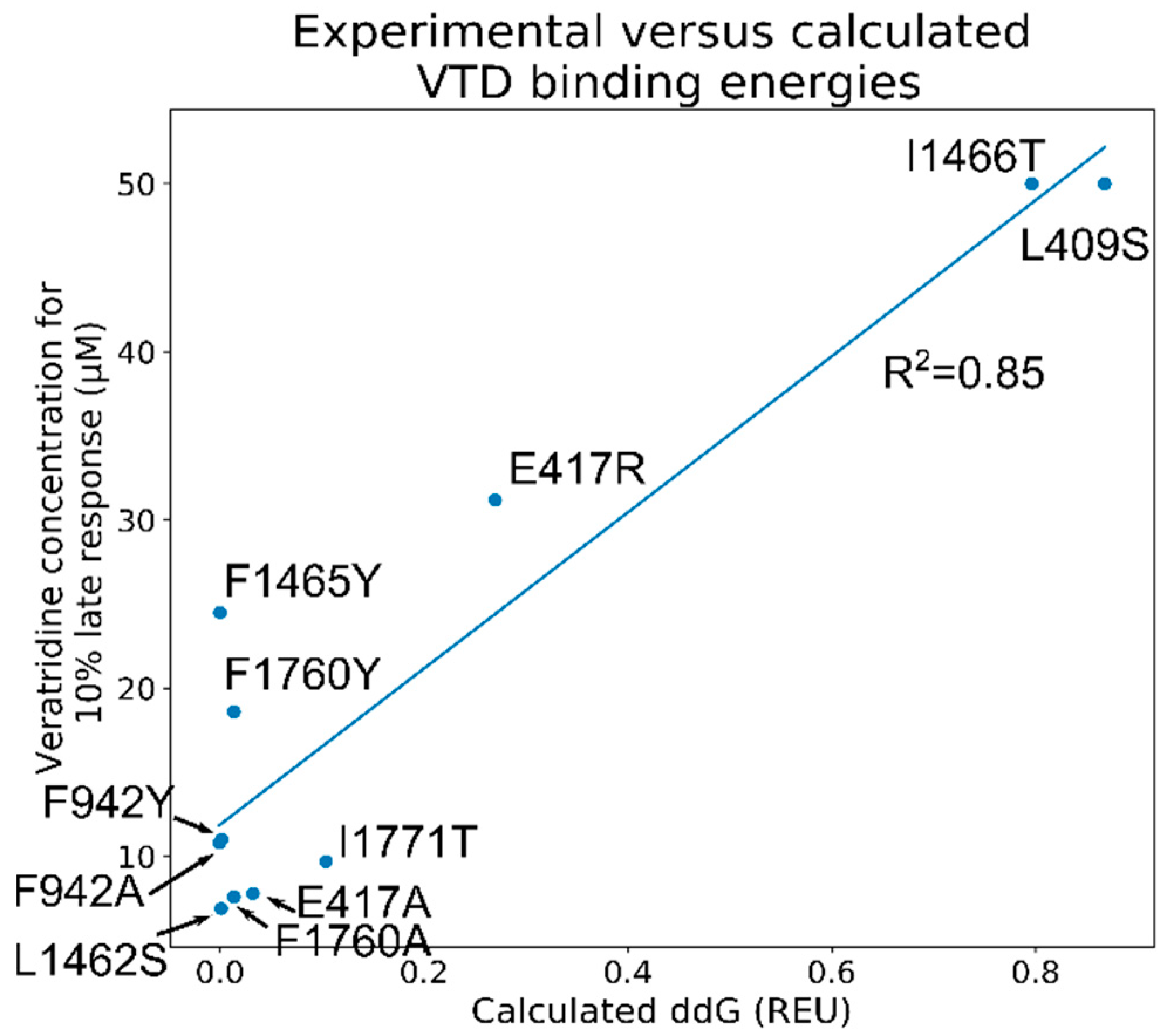
2.11. Screening of the 1000 Lowest-Scoring Poses for ΔΔG Changes Identified at the Mouth Site Pose Are More Consistent with the Electrophysiology Experiments
2.12. The VTD Pose That Showed the Best Correlation Had Interactions Consistent with the Experimental Data
2.13. Delayed Activation and Channel Block by VTD May Work through Two Distinct Mechanisms
2.14. Limitations of the Method
3. Conclusions
4. Methods and Materials
4.1. Structure Preparation and Refinement
4.2. Ligand Docking to the Mouth and Pore Sites of NaV1.5
4.3. Clustering and Analysis Based on the Interface Scores
4.4. VTD ΔΔG Calculations with WT and the Mutant NaV1.5
4.5. Mutagenesis and Stable Cell Line Generation
4.6. Veratridine Binding Measurements
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- William, A. Catterall From Ionic Currents to Molecular Review Mechanisms: The Structure and Function of Voltage-Gated Sodium Channels. Neuron 2000, 26, 13–25. [Google Scholar]
- Deuis, J.R.; Mueller, A.; Israel, M.R.; Vetter, I. The pharmacology of voltage-gated sodium channel activators. Neuropharmacology 2017, 127, 87–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remme, C.A. Cardiac sodium channelopathy associated with SCN5A mutations: Electrophysiological, molecular and genetic aspects. J. Physiol. 2013, 591, 4099–4116. [Google Scholar] [CrossRef] [PubMed]
- Abriel, H. Cardiac sodium channel Nav1.5 and interacting proteins: Physiology and pathophysiology. J. Mol. Cell. Cardiol. 2010, 48, 2–11. [Google Scholar] [CrossRef]
- Ragsdale, D.S.; McPhee, J.C.; Scheuer, T.; Catterall, W.A. Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science 1994, 265, 1724–1728. [Google Scholar] [CrossRef]
- Ragsdale, D.S.; Mcphee, J.C.; Scheuer, T.; Catterall, W.A. Common molecular determinants of local anesthetic, antiarrhythmic, and anticonvulsant block of voltage-gated Na+ channels. Proc. Natl. Acad. Sci. USA 1996, 93, 9270–9275. [Google Scholar] [CrossRef] [Green Version]
- Vaughan Williams, E.M. Classification of antiarrhythmic drugs. In Proceedings of the Symposium on Cardiac Arrhythmias, Elsinore, Denmark, 23–25 April 1970; AB Astra: Sodertalje, Sweden, 1970. [Google Scholar]
- Roden, D.M. Pharmacology and toxicology of Nav1.5-class1 antiarrhythmic drugs. Card. Electrophysiol. Clin. 2014, 6, 695–704. [Google Scholar] [CrossRef] [Green Version]
- Vickery, R.G.; Amagasu, S.M.; Chang, R.; Mai, N.; Kaufman, E.; Martin, J.; Hembrador, J.; O’Keefe, M.D.; Gee, C.; Marquess, D.; et al. Comparison of the pharmacological properties of rat Nav 1.8 with rat Nav 1.2a and human Nav 1.5 voltage-gated sodium channel subtypes using a membrane potential sensitive dye and FLIPRR. Recept. Channels 2004, 10, 11–23. [Google Scholar] [CrossRef]
- Barnes, S.; Hille, B. Veratridine modifies open sodium channels. J. Gen. Physiol. 1988, 91, 421–443. [Google Scholar] [CrossRef]
- Sutro, J.B. Kinetics of veratridine action on Na channels of skeletal muscle. J. Gen. Physiol. 1986, 87, 1–24. [Google Scholar] [CrossRef]
- Cataldi, M. Veratridine. In Hayes’ Handbook of Pesticide Toxicology; Krieger, R., Ed.; Elsevier: Amsterdam, The Netherlands, 2010; pp. 1–11. [Google Scholar]
- Denac, H.; Mevissen, M.; Scholtysik, G. Structure, function and pharmacology of voltage-gated sodium channels. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2000, 362, 453–479. [Google Scholar] [CrossRef] [PubMed]
- Craig, R.A.; Garrison, C.E.; Nguyen, P.T.; Yarov-Yarovoy, V.; Du Bois, J. Veratridine: A Janus-Faced Modulator of Voltage-Gated Sodium Ion Channels. ACS Chem. Neurosci. 2020, 11, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Li, Z.; Zhou, Q.; Shen, H.; Wu, K.; Huang, X.; Chen, J.; Zhang, J.; Zhu, X.; Lei, J.; et al. Structure of the human voltage-gated sodium channel Nav1.4 in complex with β1. Science 2018, 362, eaau2486. [Google Scholar] [CrossRef] [PubMed]
- Glazer, A.M.; Wada, Y.; Li, B.; Muhammad, A.; Kalash, O.R.; O’Neill, M.J.; Shields, T.; Hall, L.; Short, L.; Blair, M.A.; et al. High-Throughput Reclassification of SCN5A Variants. Am. J. Hum. Genet. 2020, 107, 111–123. [Google Scholar] [CrossRef]
- Kortemme, T.; Baker, D. A simple physical model for binding energy hot spots in protein-protein complexes. Proc. Natl. Acad. Sci. USA 2002, 99, 14116–14121. [Google Scholar] [CrossRef] [Green Version]
- Kortemme, T.; Kim, D.E.; Baker, D. Computational Alanine Scanning of Protein-Protein Interfaces. Sci. STKE 2004, 2004, p12. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Jin, X.; Wu, T.; Huang, G.; Wu, K.; Lei, J.; Pan, X.; Yan, N. Structural Basis for Pore Blockade of the Human Cardiac Sodium Channel Nav1.5 by the Antiarrhythmic Drug Quinidine **. Angew. Chem. 2021, 60, 11474–11480. [Google Scholar] [CrossRef]
- Tyka, M.D.; Keedy, D.A.; André, I.; Dimaio, F.; Song, Y.; Richardson, D.C.; Richardson, J.S.; Baker, D. Alternate states of proteins revealed by detailed energy landscape mapping. J. Mol. Biol. 2011, 405, 607–618. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.Y.-R.; Song, Y.; Barad, B.A.; Cheng, Y.; Fraser, J.S.; DiMaio, F. Automated structure refinement of macromolecular assemblies from cryo-EM maps using Rosetta. eLife 2016, 5, e17219. [Google Scholar] [CrossRef]
- Alford, R.F.; Leaver-Fay, A.; Jeliazkov, J.R.; O’Meara, M.J.; DiMaio, F.P.; Park, H.; Shapovalov, M.V.; Renfrew, P.D.; Mulligan, V.K.; Kappel, K.; et al. The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design. J. Chem. Theory Comput. 2017, 13, 3031–3048. [Google Scholar] [CrossRef]
- Lovell, S.C.; Davis, I.W.; Arendall, W.B.; de Bakker, P.I.W.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Cα geometry: φ,ψ and Cβ deviation. Proteins Struct. Funct. Bioinform. 2003, 50, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Kothiwale, S.; Mendenhall, J.L.; Meiler, J. BCL::Conf: Small molecule conformational sampling using a knowledge based rotamer library. J. Cheminform. 2015, 7, 47. [Google Scholar] [CrossRef] [Green Version]
- Meiler, J.; Baker, D. ROSETTALIGAND: Protein-small molecule docking with full side-chain flexibility. Proteins 2006, 65, 538–548. [Google Scholar] [CrossRef]
- Davis, I.W.; Baker, D. RosettaLigand docking with full ligand and receptor flexibility. J. Mol. Biol. 2009, 385, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Matreyek, K.A.; Stephany, J.J.; Chiasson, M.A.; Hasle, N.; Fowler, D.M. An improved platform for functional assessment of large protein libraries in mammalian cells. Nucleic Acids Res. 2020, 48, e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
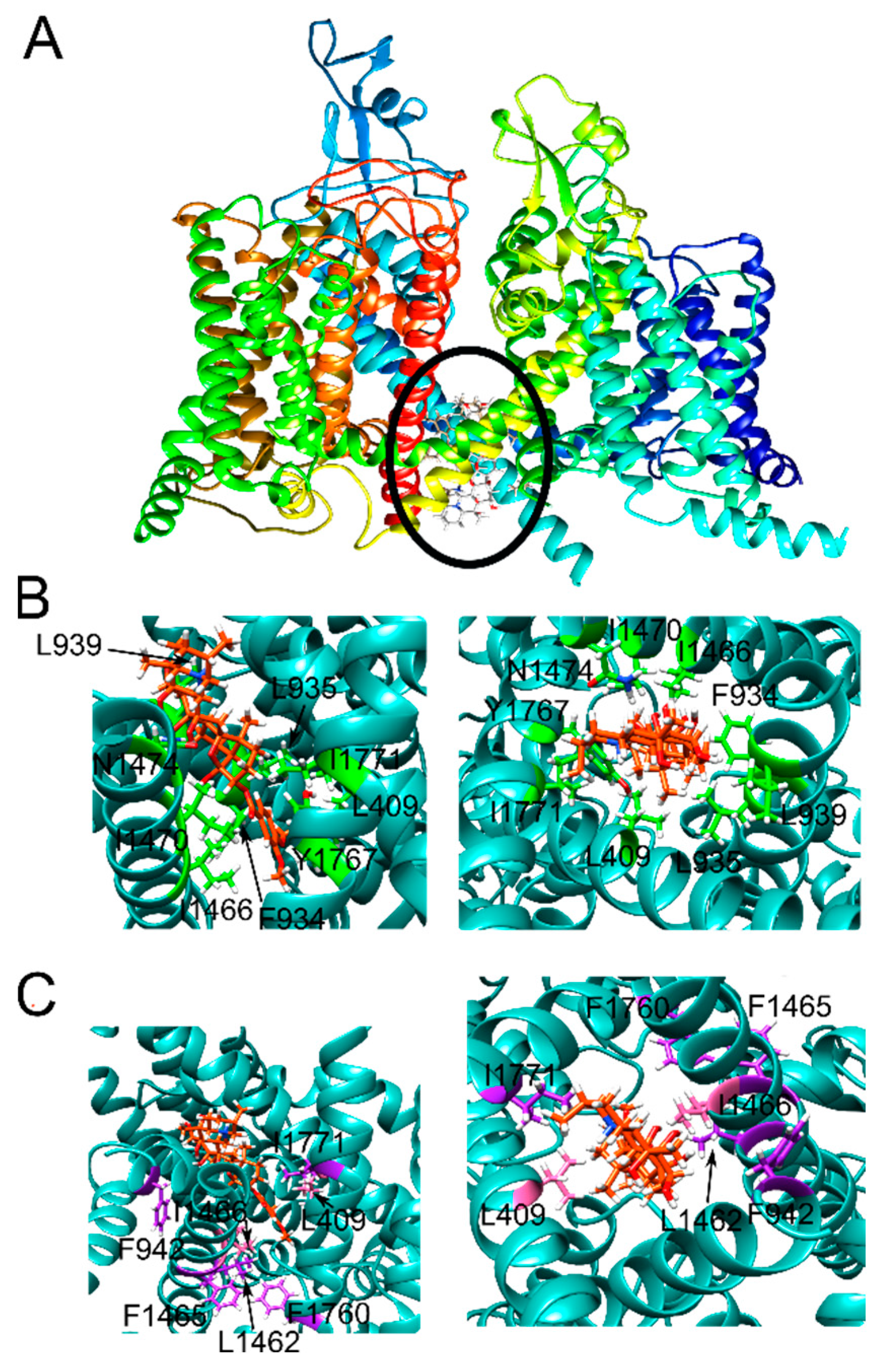{kind=link}
| Pose Number | Mouth Site Score | Pore Site Score |
|---|---|---|
| Pose 1 | −15.4 | −13.9 |
| Pose 2 | −13.4 | −15.4 |
| Pose 3 | −14.6 | −16.6 |
| Pose 4 | −14.5 | −16.0 |
| Pose 5 | −15.9 | −15.9 |
| Mutation | Peak Density (Norm. to WT) | Number of Cells | [Veratridine] for 10% Late Current (µM) ± S.E. |
|---|---|---|---|
| WT | 100 ± 6.4 | 15 | 8.2 ± 0.4 |
| E417A | 111.5 ± 14.6 | 7 | 7.8 ± 0.6 |
| E417R | 94 ± 9.6 | 9 | 31.2 ± 1.9 |
| L409S | 61.3 ±16 | 2 | >50 |
| F942A | 48.2 ± 5.1 | 6 | 10.8 ± 0.9 |
| F942Y | 88.5 ± 8 | 7 | 11.0 ± 0.8 |
| L1462S | 4.2 ± 0.7 | 1 | 6.9 |
| F1465Y | 50 ± 5.7 | 5 | 24.5 ± 2.1 |
| I1466T | 117.7 ± 12.8 | 5 | >50 |
| F1760A | 59.4 ± 8 | 6 | 7.6 ± 0.5 |
| F1760Y | 15.2 ± 1.6 | 1 | 18.6 |
| I1771T | 54.6 ± 7.3 | 8 | 9.8 ± 0.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gulsevin, A.; Glazer, A.M.; Shields, T.; Kroncke, B.M.; Roden, D.M.; Meiler, J. Veratridine Can Bind to a Site at the Mouth of the Channel Pore at Human Cardiac Sodium Channel NaV1.5. Int. J. Mol. Sci. 2022, 23, 2225. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23042225
Gulsevin A, Glazer AM, Shields T, Kroncke BM, Roden DM, Meiler J. Veratridine Can Bind to a Site at the Mouth of the Channel Pore at Human Cardiac Sodium Channel NaV1.5. International Journal of Molecular Sciences. 2022; 23(4):2225. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23042225
Chicago/Turabian StyleGulsevin, Alican, Andrew M. Glazer, Tiffany Shields, Brett M. Kroncke, Dan M. Roden, and Jens Meiler. 2022. "Veratridine Can Bind to a Site at the Mouth of the Channel Pore at Human Cardiac Sodium Channel NaV1.5" International Journal of Molecular Sciences 23, no. 4: 2225. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23042225