
Genomic and Non-Genomic Regulatory Mechanisms of the Cardiac Sodium Channel in Cardiac Arrhythmias



Abstract
:1. Introduction
2. Genomic Regulation of the Cardiac Sodium Channel
2.1. Genetic Code of SCN5A
2.2. Regulation of SCN5A Transcription
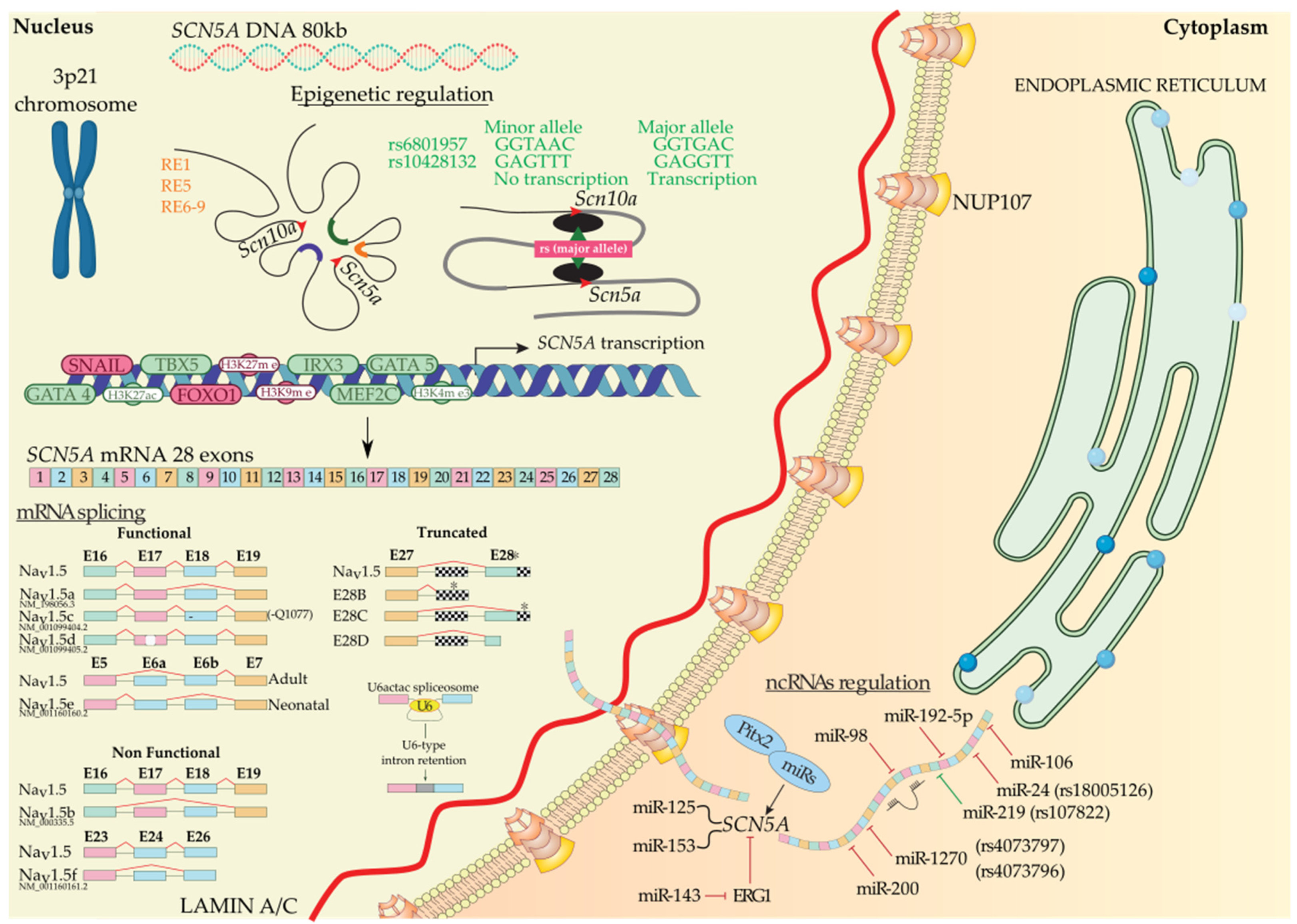
2.2.1. Epigenetic Regulation of SCN5A
Regulation of SCN5A by Distinct Regulatory Elements and Histones
Regulation of SCN5A by Transcription Factors
2.2.2. Post-Transcriptional Regulation of SCN5A
Regulation of SCN5A by Alternative Splicing
Regulation of SCN5A by Non-Coding RNAs
3. Non-Genomic Regulation of the Cardiac Sodium Channel
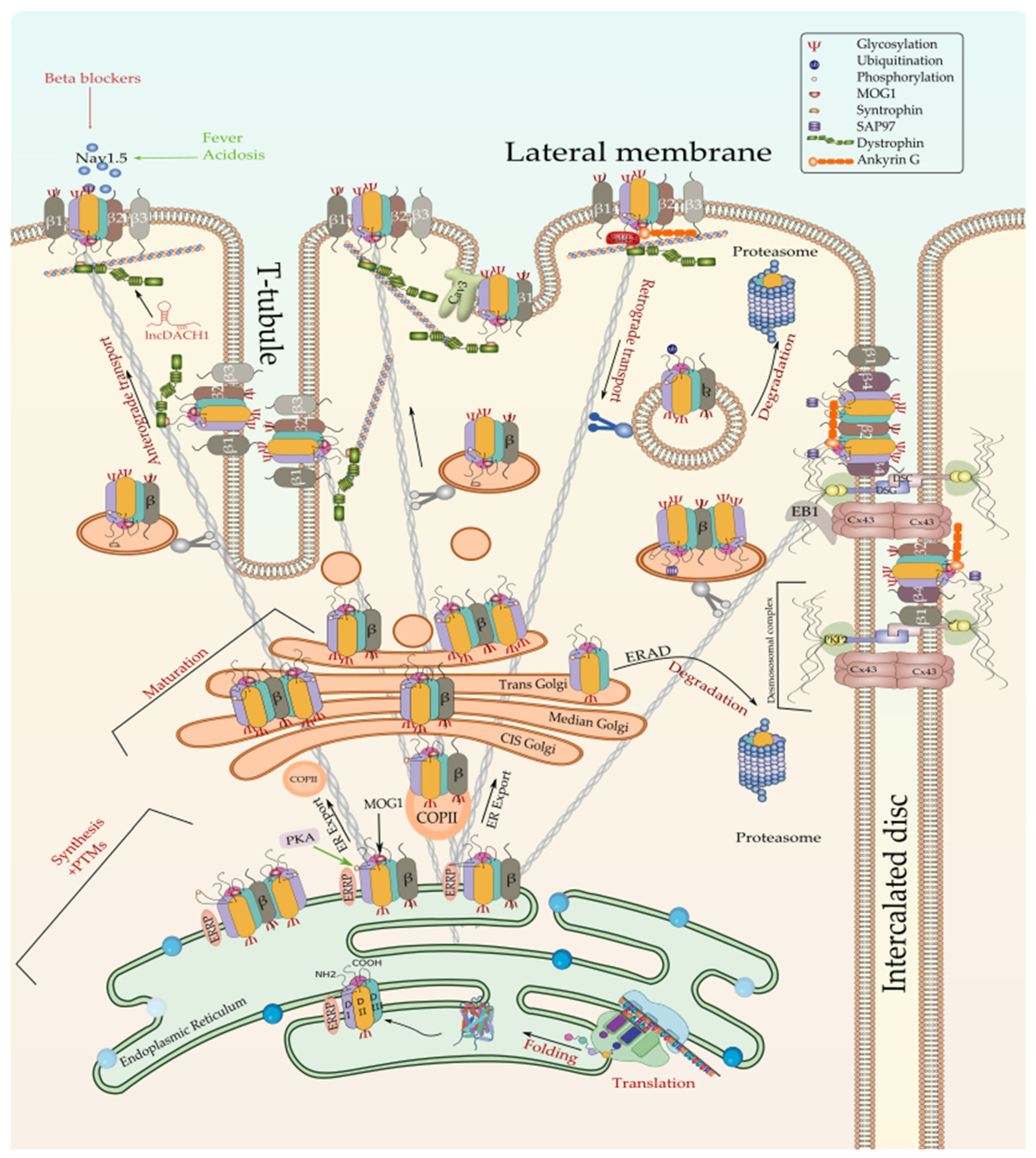
3.1. Regulation of Nav1.5 Biosynthesis and Post-Translational Modifications
3.1.1. Regulation of Nav1.5 Translation and ER Retention
3.1.2. Co-Translational and Post-Translational Regulation of Nav1.5
N-Linked Glycosylation of Nascent Nav1.5
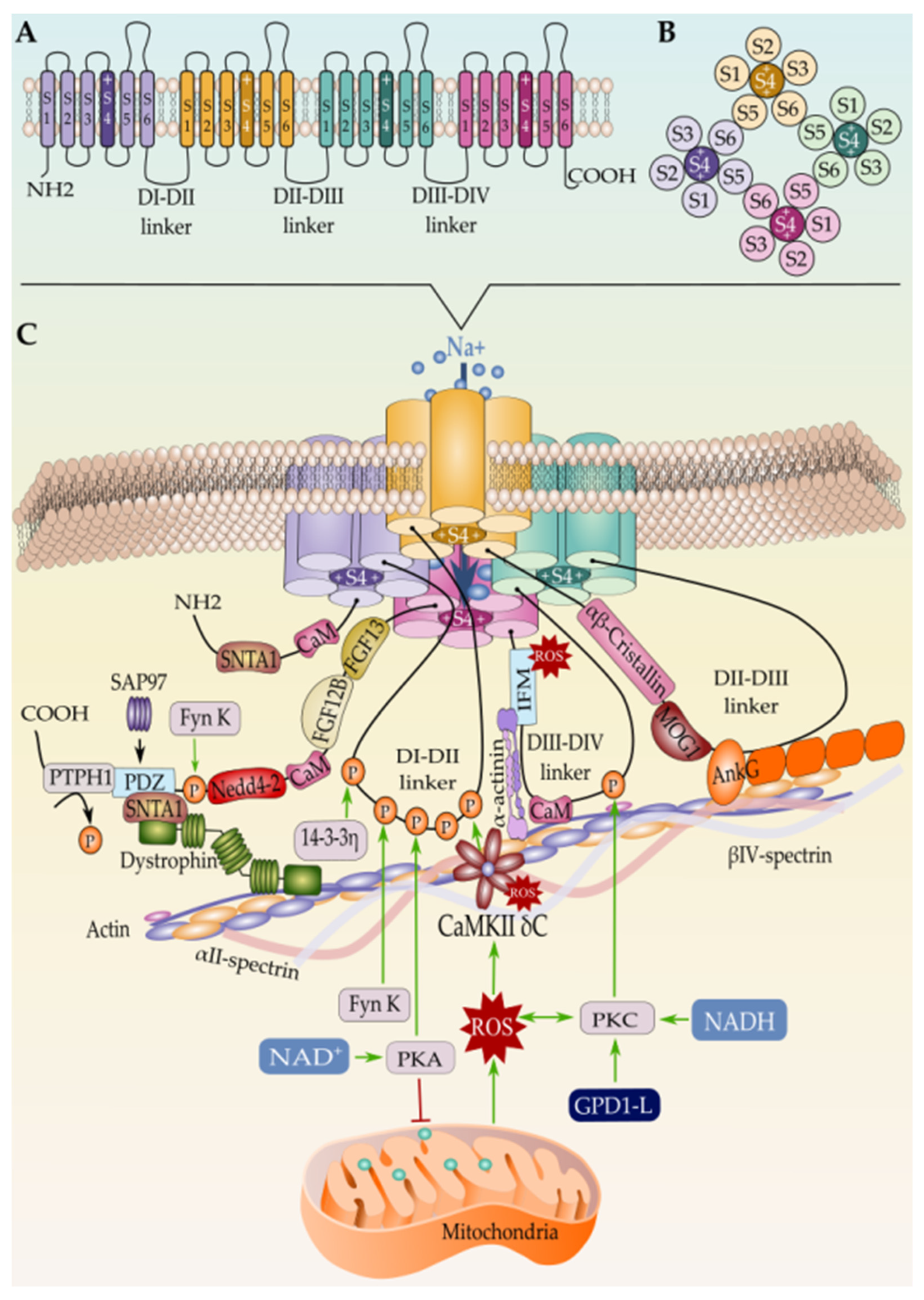
Phosphorylation and Dephosphorylation of Nav1.5
Arginine Methylation
N-Terminal and Lysine Acetylation
SUMOylation
S-Nitrosylation
Lipoxidation
Methionine Oxidation
Palmitoylation
3.1.3. Regulation of the ER-to-Golgi Trafficking
3.1.4. Regulation of Nav1.5 Maturation and Golgi Export
3.1.5. Regulation of the Nav1.5 Targeting to the Cell Membrane
Regulation of Nav1.5 by β-Subunits
The Nav1.5 and the Intercalated Disc Interactome
Nav1.5 and the Lateral Membrane’s Interactome
The Caveolar Nav1.5
3.1.6. Regulation of Nav1.5 Degradation
3.1.7. Effect of Gonadal Hormones on Nav1.5 Expression and Function
3.1.8. Effect of Temperature and pH on Nav1.5 Expression and Function
4. Cardiac Sodium Channelopathies
4.1. Long QT Syndrome
4.2. Brugada Syndrome
4.3. Atrial Fibrillation
4.4. Ventricular Fibrillation
4.5. Sick Sinus Syndrome
4.6. Sudden Infant Death Syndrome
4.7. Other Channelopathies

{kind=link}
{kind=link}
{kind=link}
| Altered Gene/Alteration | Effect on INa Current | Mode of Action | References |
|---|---|---|---|
| Long QT syndrome | |||
| SCN5A | Increased INaL, increased window current | NA | [14,336,338,339,340,341] |
| SCN1B | Increased INaL | Ancillary subunit | [342] |
| SCN4B | Undetermined | Ancillary subunit | [231,343] |
| SNTA1 | Increased INaL | Activation of nNOS–SCN5A macromolecular complex | [186,187,344,345,346,347] |
| Cav3 | Increased INaL | nNOS-dependent S-nitrosylation of SCN5A | [187] |
| BAG3 | Undetermined | Undetermined | [348] |
| ANK B | Undetermined | Undetermined | [258] |
| ACTN1 | Undetermined | Undetermined | [349] |
| TBX5 | Increased INaL | Transactivation impairment | [350,351] |
| Brugada syndrome | |||
| SCN5A | Decreased INa | NA | [24,25,26,361,362,363,364,365] |
| SCN1B | Decreased INa | Ancillary subunit; cellular trafficking defects | [232,356,359,366,367,368,369,370] |
| SCN2B | Decreased INa | Ancillary subunit; cellular trafficking defects | [229] |
| SCN3B | Decreased INa | Ancillary subunit; cellular trafficking defects | [356] |
| PKP2 | Decreased INa | INa deficit | [260,371] |
| MOG1 | Decreased INa | INa deficit; subcellular trafficking | [9,201,372,373,374,375] |
| FGF13 | Decreased INa | Enhanced Nav1.5 inactivation | [376] |
| SNTA1 | Decreased INa | Defective Nav1.5 protein interaction | [377] |
| TMEM168 | Decreased INa | Reduced Nav1-5 expression | [378,379] |
| TCAP | Decreased INa | Defective Nav1.5 protein interaction | [285,286] |
| SAP97 | Undetermined | Undetermined | [380,381] |
| TBX5 | Undetermined | Transactivation impairment | [351] |
| GATA4 | Undetermined | Undetermined | [384] |
| IRX3 | Undetermined | Undetermined | [385] |
| SCN5A promoter methylation | Undetermined | Decreased SCN5A promoter methylation | [387] |
| SCN5A promoter mutations | Undetermined | Transactivation impairment | [384] |
| Nav1.5 phosphorylation | Reduced peak INa density | Impaired PKA stimulation | [389] |
| Atrial fibrillation | |||
| SCN5A | Undetermined | NA | [384,390] |
| SCN1B | Reduced INa | Ancillary subunit; altered channel gating | [395] |
| SCN2B | Reduced INa | Ancillary subunit; altered channel gating | [395] |
| SCN3B | Undetermined | Ancillary subunit | [398] |
| SCN4B | Undetermined | Ancillary subunit | [396,397] |
| MOG1 | Reduced INa | Undetermined | [375,399] |
| ANK | Undetermined | Undetermined | [400,401] |
| ACTN1 | Undetermined | Undetermined | [402] |
| Cav3 | Undetermined | Undetermined | [403] |
| Ventricular fibrillation | |||
| SCN5A | Reduced INa | NA | [404,405,406] |
| SCN3B | Reduced peak INa | Ancillary subunit; impaired trafficking | [421] |
| CaM | Undetermined | Undetermined | [426,427] |
| ACTN1 | Undetermined | Undetermined | [428] |
| IRX3 | Undetermined | Impaired transactivation | [65] |
| Sick sinus syndrome | |||
| SCN5A | Reduced INa | NA | [29,429,430,431,432,433,434,435,436] |
| Sudden infant death syndrome | |||
| SCN5A | Increased INaL | Impaired Nav1.5 inactivation | [438,439,440,441,442,443,444] |
| SCN1B | Decreased INa density | Ancillary subunit | [447] |
| SCN3B | Decreased INa, Increased INaL | Ancillary subunit | [446,448] |
| SCN4B | Decreased INa, Increased INaL | Ancillary subunit | [446] |
| CaM | Undetermined | Undetermined | [383,449] |
| SNTA1 | Increased peak INa and window current | Undetermined | [186,346,450] |
| Cav3 | Suppression of INaL | Inhibiting nNOS-dependent S-nitrosylation of Nav1.5 | [187,451] |
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| AP | Action potential |
| VS | Voltage-sensing module |
| PM | Pore module |
| PUFA | Polyunsaturated fatty acids |
| ER | Endoplasmic reticulum |
| RE | Regulatory elements |
| UPR | Unfolded protein response |
| ERAD | ER-associated degradation pathway |
| PTMs | Post-translational modifications |
| ID | Intercalated discs |
| LM | Lateral membrane |
| Iks | Slow component of the delayed rectifier potassium current |
| Ito | Transient outward potassium current |
References
- Catterall, W.A.; Maier, S. Voltage-Gated Sodium Channels and Electrical Excitability of the Heart. In Cardiac Electrophysiology: From Cell to Bedside, 7th ed.; WB Saunders: Philadelphia, PA, USA, 2015; pp. 1–11. [Google Scholar] [CrossRef]
- Fozzard, H.A.; Hanck, D.A. Structure and function of voltage-dependent sodium channels: Comparison of brain II and cardiac isoforms. Physiol. Rev. 1996, 76, 887–926. [Google Scholar] [CrossRef]
- Gavillet, B.; Rougier, J.S.; Domenighetti, A.A.; Behar, R.; Boixel, C.; Ruchat, P.; Lehr, H.A.; Pedrazzini, T.; Abriel, H. Cardiac sodium channel Nav1.5 is regulated by a multiprotein complex composed of syntrophins and dystrophin. Circ. Res. 2006, 99, 407–414. [Google Scholar] [CrossRef] [Green Version]
- Gellens, M.E.; George, A.L.; Chen, L.; Chahine, M.; Horn, R.; Barchi, R.L.; Kallen, R.G. Primary structure and functional expression of the human cardiac tetrodotoxin-insensitive voltage-dependent sodium channel. Proc. Natl. Acad. Sci. USA 1992, 89, 554–558. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.; Shi, H.; Tonggu, L.; Gamal El-Din, T.M.; Lenaeus, M.J.; Zhao, Y.; Yoshioka, C.; Zheng, N.; Catterall, W.A. Structure of the Cardiac Sodium Channel. Cell 2020, 180, 122–134.e10. [Google Scholar] [CrossRef]
- Bohannon, B.M.; de la Cruz, A.; Wu, X.; Jowais, J.J.; Perez, M.E.; Dykxhoorn, D.M.; Liin, S.I.; Larsson, H.P. Erratum: Correction: Polyunsaturated fatty acid analogues differentially affect cardiac NaV, CaV, and KV channels through unique mechanisms. eLife 2020, 9, e60141. [Google Scholar] [CrossRef]
- Kang, J.X.; Leaf, A. Evidence that free polyunsaturated fatty acids modify Na+ channels by directly binding to the channel proteins. Proc. Natl. Acad. Sci. USA 1996, 93, 3542–3546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, Y.; Gao, X.; Tang, Q.; Huang, L.; Gao, S.; Yu, S.; Huang, J.; Li, J.; Zhou, D.; Zhang, Y.; et al. Nucleoporin 107 facilitates the nuclear export of Scn5a mRNA to regulate cardiac bioelectricity. J. Cell. Mol. Med. 2019, 23, 1448–1457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Liu, Y.; Qin, J.; Wang, Z.; Hu, Y.; Wang, F.; Li, Y.; Chakrabarti, S.; Chen, Q.; Wang, Q.K. Mechanistic insights into the interaction of the MOG1 protein with the cardiac sodium channel Nav1.5 clarify the molecular basis of Brugada syndrome. J. Biol. Chem. 2018, 293, 18207–18217. [Google Scholar] [CrossRef] [Green Version]
- Kurokawa, K.; Nakano, A. The ER exit sites are specialized ER zones for the transport of cargo proteins from the ER to the Golgi apparatus. J. Biochem. 2019, 165, 109–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, C.; Wang, Y.; Ma, A.; Wang, T. Life Cycle of the Cardiac Voltage-Gated Sodium Channel Nav1.5. Front. Physiol. 2020, 11, 609733. [Google Scholar] [CrossRef]
- Clark, K.A.; McElhinny, A.S.; Beckerle, M.C.; Gregorio, C.C. Striated muscle cytoarchitecture: An intricate web of form and function. Annu. Rev. Cell Dev. Biol. 2002, 18, 637–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balse, E.; Eichel, C. The Cardiac Sodium Channel and Its Protein Partners. In Handbook of Experimental Pharmacology; Springer: Cham, Switzerland, 2017; pp. 251–263. [Google Scholar] [CrossRef]
- Song, W.; Shou, W. Cardiac sodium channel Nav1.5 mutations and cardiac arrhythmia. Pediatr. Cardiol. 2012, 33, 943–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balser, J.R. The cardiac sodium channel: Gating function and molecular pharmacology. J. Mol. Cell. Cardiol. 2001, 33, 599–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chahine, M.; George, A.L.; Zhou, M.; Ji, S.; Sun, W.; Barchi, R.L.; Horn, R. Sodium channel mutations in paramyotonia congenita uncouple inactivation from activation. Neuron 1994, 12, 281–294. [Google Scholar] [CrossRef]
- Yang, N.; Horn, R. Evidence for voltage-dependent S4 movement in sodium channels. Neuron 1995, 15, 213–218. [Google Scholar] [CrossRef] [Green Version]
- Albert, C.; Ruben, P.C.; George, A.L.; Fujimoto, E.; Bezanilla, F. Voltage sensors in domains III and IV, but not I and II, are immobilized by Na+ channel fast inactivation. Neuron 1999, 22, 73–87. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, C.M.; Bezanilla, F. Inactivation of the sodium channel: II. gating current experiments. J. Gen. Physiol. 1977, 70, 567–590. [Google Scholar] [CrossRef]
- Armstrong, C.M.; Wm Gilly, F. Fast and slow steps in the activation of sodium channels. J. Gen. Physiol. 1979, 74, 691–711. [Google Scholar] [CrossRef] [Green Version]
- Hodgkin, A.L.; Huxley, A.F. Currents carried by sodium and potassium ions through the membrane of the giant axon of Loligo. J. Physiol. 1952, 116, 449–472. [Google Scholar] [CrossRef]
- Kapplinger, J.D.; Tester, D.J.; Alders, M.; Benito, B.; Berthet, M.; Brugada, J.; Brugada, P.; Fressart, V.; Guerchicoff, A.; Harris-Kerr, C.; et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm 2010, 7, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Kroncke, B.M.; Glazer, A.M.; Smith, D.K.; Blume, J.D.; Roden, D.M. SCN5A (Nav1.5) Variant Functional Perturbation and Clinical Presentation: Variants of a Certain Significance. Circ. Genom. Precis. Med. 2018, 11, e002095. [Google Scholar] [CrossRef] [Green Version]
- Remme, C.A.; Verkerk, A.O.; Nuyens, D.; van Ginneken, A.C.G.; van Brunschot, S.; Belterman, C.N.W.; Wilders, R.; van Roon, M.A.; Tan, H.L.; Wilde, A.A.M.; et al. Overlap Syndrome of Cardiac Sodium Channel Disease in Mice Carrying the Equivalent Mutation of Human SCN5A -1795insD. Circulation 2006, 114, 2584–2594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Kirsch, G.E.; Zhang, D.; Brugada, R.; Brugada, J.; Brugada, P.; Potenza, D.; Moya, A.; Borggrefe, M.; Breithardt, G.; et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 1998, 392, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Bezzina, C.; Veldkamp, M.W.; van den Berg, M.P.; Postma, A.V.; Rook, M.B.; Viersma, J.-W.; van Langen, I.M.; Tan-Sindhunata, G.; Bink-Boelkens, M.T.E.; van der Hout, A.H.; et al. A Single Na+ Channel Mutation Causing Both Long-QT and Brugada Syndromes. Circ. Res. 1999, 85, 1206–1213. [Google Scholar] [CrossRef] [Green Version]
- Schott, J.; Alshinawi, C.; Kyndt, F.; Probst, V.; Hoorntje, T.M.; Hulsbeek, M.; Wilde, A.A.M.; Escande, D.; Mannens, M.M.A.M.; Le Marec, H. Cardiac conduction defects associate with mutations in SCN5A. Nat. Genet. 1999, 23, 20–21. [Google Scholar] [CrossRef]
- Tan, H.L.; Bink-Boelkens, M.T.E.; Bezzina, C.R.; Viswanathan, P.C.; Beaufort-Krol, G.C.M.; Van Tintelen, P.J.; Van Den Berg, M.P.; Wilde, A.A.M.; Balser, J.R. A sodium-channel mutation causes isolated cardiac conduction disease. Nature 2001, 409, 1043–1047. [Google Scholar] [CrossRef]
- Benson, D.W.; Wang, D.W.; Dyment, M.; Knilans, T.K.; Fish, F.A.; Strieper, M.J.; Rhodes, T.H.; George, A.L. Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A). J. Clin. Investig. 2003, 112, 1019–1028. [Google Scholar] [CrossRef] [Green Version]
- Smits, J.P.P.; Koopmann, T.T.; Wilders, R.; Veldkamp, M.W.; Opthof, T.; Bhuiyan, Z.A.; Mannens, M.M.A.M.; Balser, J.R.; Tan, H.L.; Bezzina, C.R.; et al. A mutation in the human cardiac sodium channel (E161K) contributes to sick sinus syndrome, conduction disease and Brugada syndrome in two families. J. Mol. Cell. Cardiol. 2005, 38, 969–981. [Google Scholar] [CrossRef]
- Wang, Q.; Shen, J.; Splawski, I.; Atkinson, D.; Li, Z.; Robinson, J.L.; Moss, A.J.; Towbin, J.A.; Keating, M.T. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell 1995, 80, 805–811. [Google Scholar] [CrossRef] [Green Version]
- Mann, S.A.; Castro, M.L.; Ohanian, M.; Guo, G.; Zodgekar, P.; Sheu, A.; Stockhammer, K.; Thompson, T.; Playford, D.; Subbiah, R.; et al. R222Q SCN5A mutation is associated with reversible ventricular ectopy and dilated cardiomyopathy. J. Am. Coll. Cardiol. 2012, 60, 1566–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swan, H.; Amarouch, M.Y.; Leinonen, J.; Marjamaa, A.; Kucera, J.P.; Laitinen-Forsblom, P.J.; Lahtinen, A.M.; Palotie, A.; Kontula, K.; Toivonen, L.; et al. Gain-of-function mutation of the SCN5A gene causes exercise-induced polymorphic ventricular arrhythmias. Circ. Cardiovasc. Genet. 2014, 7, 771–781. [Google Scholar] [CrossRef] [Green Version]
- Laurent, G.; Saal, S.; Amarouch, M.Y.; Béziau, D.M.; Marsman, R.F.J.; Faivre, L.; Barc, J.; Dina, C.; Bertaux, G.; Barthez, O.; et al. Multifocal ectopic Purkinje-related premature contractions: A new SCN5A-related cardiac channelopathy. J. Am. Coll. Cardiol. 2012, 60, 144–156. [Google Scholar] [CrossRef]
- Makiyama, T.; Akao, M.; Shizuta, S.; Doi, T.; Nishiyama, K.; Oka, Y.; Ohno, S.; Nishio, Y.; Tsuji, K.; Itoh, H.; et al. A novel SCN5A gain-of-function mutation M1875T associated with familial atrial fibrillation. J. Am. Coll. Cardiol. 2008, 52, 1326–1334. [Google Scholar] [CrossRef] [Green Version]
- Olson, T.M.; Michels, V.V.; Ballew, J.D.; Reyna, S.P.; Karst, M.L.; Herron, K.J.; Horton, S.C.; Rodeheffer, R.J.; Anderson, J.L. Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA 2005, 293, 447–454. [Google Scholar] [CrossRef]
- McNair, W.P.; Ku, L.; Taylor, M.R.G.; Fain, P.R.; Dao, D.; Wolfel, E.; Mestroni, L. SCN5A mutation associated with dilated cardiomyopathy, conduction disorder, and arrhythmia. Circulation 2004, 110, 2163–2167. [Google Scholar] [CrossRef] [Green Version]
- Bezzina, C.R.; Rook, M.B.; Groenewegen, W.A.; Herfst, L.J.; Van der Wal, A.C.; Lam, J.; Jongsma, H.J.; Wilde, A.A.M.; Mannens, M.M.A.M. Compound heterozygosity for mutations (W156X and R225W) in SCN5A associated with severe cardiac conduction disturbances and degenerative changes in the conduction system. Circ. Res. 2003, 92, 159–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clatot, J.; Zheng, Y.; Girardeau, A.; Liu, H.; Laurita, K.R.; Marionneau, C.; Deschênes, I. Mutant voltage-gated Na+ channels can exert a dominant negative effect through coupled gating. Am. J. Physiol.—Heart Circ. Physiol. 2018, 315, H1250–H1257. [Google Scholar] [CrossRef] [PubMed]
- Keller, D.I.; Kucera, J.P.; Benammar, N. Brugada syndrome and fever: Genetic and molecular characterization of patients carrying SCN5A mutations. Cardiovasc. Res. 2005, 67, 510–519. [Google Scholar] [CrossRef] [Green Version]
- Doisne, N.; Grauso, M.; Mougenot, N.; Clergue, M.; Souil, C.; Coulombe, A.; Guicheney, P.; Neyroud, N. In vivo Dominant-Negative Effect of an SCN5A Brugada Syndrome Variant. Front. Physiol. 2021, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Neill, M.J.O.; Muhammad, A.; Li, B.; Wada, Y.; Hall, L.; Solus, J.F.; Short, L.; Roden, D.M.; Glazer, A.M. Dominant negative effects of SCN5A missense variants. bioRxiv 2021. [Google Scholar] [CrossRef]
- Galleano, I.; Harms, H.; Choudhury, K.; Khoo, K.; Delemotte, L.; Pless, S.A. Functional cross-talk between phosphorylation and disease-causing mutations in the cardiac sodium channel Nav1.5. Proc. Natl. Acad. Sci. USA 2021, 118, e2025320118. [Google Scholar] [CrossRef]
- Van der Harst, P.; van Setten, J.; Verweij, N.; Vogler, G.; Franke, L.; Maurano, M.T.; Wang, X.; Mateo Leach, I.; Eijgelsheim, M.; Sotoodehnia, N.; et al. 52 Genetic Loci Influencing Myocardial Mass A. J. Am. Coll. Cardiol. 2016, 68, 1435–1448. [Google Scholar] [CrossRef]
- Van Den Boogaard, M.; Smemo, S.; Burnicka-Turek, O.; Arnolds, D.E.; Van De Werken, H.J.G.; Klous, P.; McKean, D.; Muehlschlegel, J.D.; Moosmann, J.; Toka, O.; et al. A common genetic variant within SCN10A modulates cardiac SCN5A expression. J. Clin. Investig. 2014, 124, 1844–1852. [Google Scholar] [CrossRef] [Green Version]
- Van Den Boogaard, M.; Barnett, P.; Vincent, M.; Van Den Boogaard, M.; Wong, L.Y.E.; Tessadori, F.; Bakker, M.L. Genetic variation in T-box binding element functionally affects SCN5A/SCN10A enhancer Find the latest version: Genetic variation in T-box binding element functionally affects SCN5A/SCN10A enhancer. J. Clin. Investig. 2012, 122, 2519–2530. [Google Scholar] [CrossRef] [Green Version]
- Verweij, N.; Leach, I.M.; Van Den Boogaard, M.; Van Veldhuisen, D.J.; Christoffels, V.M.; Hillege, H.L.; Van Gilst, W.H.; Barnett, P.; De Boer, R.A.; Van Der Harst, P. Genetic Determinants of P Wave Duration and PR Segment. Circ. Cardiovasc. Genet. 2014, 7, 475–481. [Google Scholar] [CrossRef] [Green Version]
- van Setten, J.; Brody, J.A.; Jamshidi, Y.; Swenson, B.R.; Butler, A.M.; Campbell, H.; Del Greco, F.M.; Evans, D.S.; Gibson, Q.; Gudbjartsson, D.F.; et al. PR interval genome-wide association meta-analysis identifies 50 loci associated with atrial and atrioventricular electrical activity. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Man, J.C.; Mohan, R.A.; van den Boogaard, M.; Hilvering, C.R.; Jenkins, C.; Wakker, V.; Bianchi, V.; de Laat, W.; Barnett, P.; Boukens, B.J.; et al. An enhancer cluster controls gene activity and topology of the SCN5A-SCN10A locus in vivo. Nat. Commun. 2019, 10, 4943. [Google Scholar] [CrossRef] [Green Version]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Postma, A.V.; Bezzina, C.R.; Christoffels, V.M. Genetics of congenital heart disease: The contribution of the noncoding regulatory genome. J. Hum. Genet. 2016, 61, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Bezzina, C.R.; Barc, J.; Mizusawa, Y.; Remme, C.A.; Gourraud, J.B.; Simonet, F.; Verkerk, A.O.; Schwartz, P.J.; Crotti, L.; Dagradi, F.; et al. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat Genet. 2013, 45, 1044–1049. [Google Scholar] [CrossRef] [PubMed]
- Gilsbach, R.; Preissl, S.; Grüning, B.A.; Schnick, T.; Burger, L.; Benes, V.; Würch, A.; Bönisch, U.; Günther, S.; Backofen, R.; et al. Dynamic DNA methylation orchestrates cardiomyocyte development, maturation and disease. Nat. Commun. 2014, 5, 5288. [Google Scholar] [CrossRef] [Green Version]
- Carreras, D.; Martinez-Moreno, R.; Pinsach-Abuin, M.L.; Santafe, M.M.; Gomà, P.; Brugada, R.; Scornik, F.S.; Pérez, G.J.; Pagans, S. Epigenetic changes governing Scn5a expression in denervated skeletal muscle. Int. J. Mol. Sci. 2021, 22, 2755. [Google Scholar] [CrossRef]
- Salvarani, N.; Crasto, S.; Miragoli, M.; Bertero, A.; Paulis, M.; Kunderfranco, P.; Serio, S.; Forni, A.; Lucarelli, C.; Dal Ferro, M.; et al. The K219T-Lamin mutation induces conduction defects through epigenetic inhibition of SCN5A in human cardiac laminopathy. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Arnolds, D.E.; Liu, F.; Fahrenbach, J.P.; Kim, G.H.; Schillinger, K.J.; Smemo, S.; McNally, E.M.; Nobrega, M.A.; Patel, V.V.; Moskowitz, I.P. TBX5 drives Scn5a expression to regulate cardiac conduction system function. J. Clin. Investig. 2012, 122, 2509–2518. [Google Scholar] [CrossRef] [Green Version]
- Moskowitz, I.P.G.; Pizard, A.; Patel, V.V.; Bruneau, B.G.; Kim, J.B.; Kupershmidt, S.; Roden, D.; Berul, C.I.; Seidman, C.E.; Seidman, J.G. The T-Box transcription factor Tbx5 is required for the patterning and maturation of the murine cardiac conduction system. Development 2004, 131, 4107–4116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steimle, J.D.; Moskowitz, I.P. TBX5: A Key Regulator of Heart Development. Curr. Top. Dev. Biol. 2017, 122, 195–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brewer, A.; Pizzey, J. GATA factors in vertebrate heart development and disease. Expert Rev. Mol. Med. 2006, 8, 1–20. [Google Scholar] [CrossRef]
- Garg, V.; Kathiriya, I.S.; Barnes, R.; Schluterman, M.K.; King, I.N.; Butler, C.A.; Rothrock, C.R.; Eapen, R.S.; Hirayama-Yamada, K.; Joo, K.; et al. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature 2003, 424, 443–447. [Google Scholar] [CrossRef]
- Tarradas, A.; Pinsach-Abuin, M.; Mackintosh, C.; Llora-Batlle, O.; Perez-Serra, A.; Batlle, M.; Pérez-Villa, F.; Zimmer, T.; Garcia-Bassets, I.; Brugada, R.; et al. Transcriptional regulation of the sodium channel gene (SCN5A) by GATA4 in human heart. J. Mol. Cell. Cardiol. 2017, 102, 74–82. [Google Scholar] [CrossRef] [Green Version]
- Zhou, A.; Shi, G.; Kang, G.J.; Xie, A.; Liu, H.; Jiang, N.; Liu, M.; Jeong, E.M.; Dudley, S.C. RNA binding protein, HuR, regulates SCN5A expression through stabilizing MEF2C transcription factor mRNA. J. Am. Heart Assoc. 2018, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhou, A.; Xie, A.; Kim, T.; Liu, H.; Shi, G.; Kang, G.; Jiang, N.; Liu, M.; Jeong, E.; Choi, B.; et al. HuR-mediated SCN5A mRNA stability reduces arrhythmic risk in heart failure. Heart Rhythm 2018, 15, 1072–1080. [Google Scholar] [CrossRef]
- Gaborit, N.; Sakuma, R.; Wylie, J.N.; Kim, K.H.; Zhang, S.S.; Hui, C.C.; Bruneau, B.G. Cooperative and antagonistic roles for Irx3 and Irx5 in cardiac morphogenesis and postnatal physiology. Development 2012, 139, 4007–4019. [Google Scholar] [CrossRef] [Green Version]
- Koizumi, A.; Sasano, T.; Kimura, W.; Miyamoto, Y.; Aiba, T.; Ishikawa, T.; Nogami, A.; Fukamizu, S.; Sakurada, H.; Takahashi, Y.; et al. Genetic defects in a His-Purkinje system transcription factor, IRX3, cause lethal cardiac arrhythmias. Eur. Heart J. 2016, 37, 1469–1475. [Google Scholar] [CrossRef] [Green Version]
- Cai, B.; Wang, N.; Mao, W.; You, T.; Lu, Y.; Li, X.; Ye, B.; Li, F.; Xu, H. Deletion of FoxO1 leads to shortening of QRS by increasing Na+ channel activity through enhanced expression of both cardiac Nav1.5 and β3 subunit. J. Mol. Cell. Cardiol. 2014, 74, 297–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, W.; You, T.; Ye, B.; Li, X.; Dong, H.H.; Hill, J.A.; Li, F.; Xu, H. Reactive oxygen species suppress cardiac Nav1.5 expression through Foxo1. PLoS ONE 2012, 7, e32738. [Google Scholar] [CrossRef] [Green Version]
- Atack, T.C.; Stroud, D.M.; Watanabe, H.; Yang, T.; Hall, L.; Susan, B.; Lowe, J.S.; Leake, B.; Magnuson, M.A.; Yang, P.; et al. Informatic and Functional Approaches to Identifying a Regulatory Region for the Cardiac Sodium Channel. Circ. Res. 2012, 109, 38–46. [Google Scholar] [CrossRef] [Green Version]
- Hesse, M.; Kondo, C.S.; Clark, R.B.; Su, L.; Allen, F.L.; Geary-Joo, C.T.M.; Kunnathu, S.; Severson, D.L.; Nygren, A.; Giles, W.R.; et al. Dilated cardiomyopathy is associated with reduced expression of the cardiac sodium channel Scn5a. Cardiovasc. Res. 2007, 75, 498–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieto, M.A. The snail superfamily of zinc-finger transcription factors. Nat. Rev. Mol. Cell Biol. 2002, 3, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Schroeter, A.; Walzik, S.; Blechschmidt, S.; Haufe, V.; Benndorf, K.; Zimmer, T. Structure and function of splice variants of the cardiac voltage-gated sodium channel Nav1.5. J. Mol. Cell. Cardiol. 2010, 49, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Makielski, J.C.; Ye, B.; Valdivia, C.R.; Pagel, M.D.; Pu, J.; Tester, D.J.; Ackerman, M.J. A Ubiquitous Splice Variant and a Common Polymorphism Affect Heterologous Expression of Recombinant Human SCN5A Heart Sodium Channels. Circ. Res. 2003, 93, 821–828. [Google Scholar] [CrossRef] [Green Version]
- Camacho, J.A.; Hensellek, S.; Rougier, J.S.; Blechschmidt, S.; Abriel, H.; Benndorf, K.; Zimmer, T. Modulation of Nav1.5 channel function by an alternatively spliced sequence in the DII/DIII linker region. J. Biol. Chem. 2006, 281, 9498–9506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blechschmidt, S.; Haufe, V.; Benndorf, K.; Zimmer, T. Voltage-gated Na+ channel transcript patterns in the mammalian heart are species-dependent. Prog. Biophys. Mol. Biol. 2008, 98, 309–318. [Google Scholar] [CrossRef]
- Chioni, A.M.; Fraser, S.P.; Pani, F.; Foran, P.; Wilkin, G.P.; Diss, J.K.J.; Djamgoz, M.B.A. A novel polyclonal antibody specific for the Nav1.5 voltage-gated Na+ channel “neonatal” splice form. J. Neurosci. Methods 2005, 147, 88–98. [Google Scholar] [CrossRef]
- Onkal, R.; Mattis, J.H.; Fraser, S.P.; Diss, J.K.J.; Shao, D.; Okuse, K.; Djamgoz, M.B.A. Alternative splicing of Nav1.5: An electrophysiological comparison of “neonatal” and “adult” isoforms and critical involvement of a lysine residue. J. Cell. Physiol. 2008, 216, 716–726. [Google Scholar] [CrossRef]
- Zimmer, T.; Bollensdorff, C.; Haufe, V.; Birch-Hirschfeld, E.; Benndorf, K. Mouse heart Na+ channels: Primary structure and function of two isoforms and alternatively spliced variants. Am. J. Physiol.—Heart Circ. Physiol. 2002, 282, 1007–1017. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Ou, S.W.; Wang, Y.J.; Zong, Z.H.; Lin, L.; Kameyama, M.; Kameyama, A. New variants of Nav1.5/SCN5A encode Na+ channels in the brain. J. Neurogenet. 2008, 22, 57–75. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ou, S.W.; Wang, Y.J.; Kameyama, M.; Kameyama, A.; Zong, Z.H. Analysis of four novel variants of Nav1.5/SCN5A cloned from the brain. Neurosci. Res. 2009, 64, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Shang, L.L.; Pfahnl, A.E.; Sanyal, S.; Jiao, Z.; Allen, J.; Banach, K.; Fahrenbach, J.; Weiss, D.; Taylor, W.R.; Zafari, A.M.; et al. Human Heart Failure Is Associated With Abnormal C-Terminal Splicing Variants in the Cardiac Sodium Channel. Physiology 2007, 101, 1146–1154. [Google Scholar] [CrossRef] [Green Version]
- Montañés-Agudo, P.; Casini, S.; Aufiero, S.; Ernault, A.C.; van der Made, I.; Pinto, Y.M.; Remme, C.A.; Creemers, E.E. Inhibition of minor intron splicing reduces Na+ and Ca2+ channel expression and function in cardiomyocytes. J. Cell Sci. 2021, 135, 259191. [Google Scholar] [CrossRef]
- Wang, Z.; Luo, X.; Lu, Y.; Yang, B. miRNAs at the heart of the matter. J. Mol. Med. 2008, 86, 771–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Lu, Y.; Wang, Z. Control of cardiac excitability by microRNAs. Cardiovasc. Res. 2008, 79, 571–580. [Google Scholar] [CrossRef] [Green Version]
- Remme, C.A.; Bezzina, C.R. Sodium channel (Dys)function and cardiac arrhythmias. Cardiovasc. Ther. 2010, 28, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Boštjančič, E.; Zidar, N.; Štajer, D.; Glavač, D. MicroRNAs miR-1, miR-133a, miR-133b and miR-208 are dysregulated in human myocardial infarction. Cardiology 2010, 115, 163–169. [Google Scholar] [CrossRef]
- Daimi, H.; Lozano-Velasco, E.; Haj Khelil, A.; Chibani, J.B.E.; Barana, A.; Amorós, I.; González De La Fuente, M.; Caballero, R.; Aranega, A.; Franco, D. Regulation of SCN5A by microRNAs: MiR-219 modulates SCN5A transcript expression and the effects of flecainide intoxication in mice. Heart Rhythm 2015, 12, 1333–1342. [Google Scholar] [CrossRef] [PubMed]
- Lozano-Velasco, E.; Hernández-Torres, F.; Daimi, H.; Serra, S.A.S.A.; Herraiz, A.; Hove-Madsen, L.; Aránega, A.; Franco, D. Pitx2 impairs calcium handling in a dose-dependent manner by modulating Wnt signalling. Cardiovasc. Res. 2016, 109, 55–66. [Google Scholar] [CrossRef] [Green Version]
- Lozano-Velasco, E.; Wangensteen, R.; Quesada, A.; Garcia-Padilla, C.; Osorio, J.A.; Ruiz-Torres, M.D.; Aranega, A.; Franco, D. Hyperthyroidism, but not hypertension, impairs PITX2 expression leading to Wnt-microRNA-ion channel remodeling. PLoS ONE 2017, 12, e0188473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinchilla, A.; Daimi, H.; Lozano-velasco, E.; Dominguez, J.N.; Caballero, R.; Delpo, E.; Tamargo, J.; Cinca, J.; Hove-madsen, L.; Aranega, A.E.; et al. PITX2 Insufficiency Leads to Atrial Electrical and Structural Remodeling Linked to Arrhythmogenesis. Circ. Cardiovasc. Genet. 2011, 4, 269–279. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Huang, Y.; Li, W.; Wang, Z.; Zhan, S.; Zhou, M.; Yao, Y.; Zeng, Z.; Hou, Y.; Chen, Q.; et al. Post-transcriptional regulation of cardiac sodium channel gene SCN5A expression and function by miR-192-5p. Biochim. Biophys. Acta—Mol. Basis Dis. 2015, 1852, 2024–2034. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Xu, C.; Liu, Y.; Li, Y.; Du, S.; Zhang, R.; Sun, Y.; Zhang, R.; Wang, Y.; Xue, H.; et al. Fibroblast growth factor 21 inhibited ischemic arrhythmias via targeting miR-143/EGR1 axis. Basic Res. Cardiol. 2020, 115, 1–17. [Google Scholar] [CrossRef]
- Zhang, X.; Yoon, J.Y.; Morley, M.; McLendon, J.M.; Mapuskar, K.A.; Gutmann, R.; Mehdi, H.; Bloom, H.L.; Dudley, S.C.; Ellinor, P.T.; et al. A common variant alters SCN5A-miR-24 interaction and associates with heart failure mortality. J. Clin. Investig. 2018, 128, 1154–1163. [Google Scholar] [CrossRef] [Green Version]
- Daimi, H.; Khelil, A.H.; Neji, A.; Ben Hamda, K.; Maaoui, S.; Aranega, A.; Chibani, J.B.; Franco, D. Role of SCN5A coding and non-coding sequences in Brugada syndrome onset: What’s behind the scenes? Biomed. J. 2019, 42, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.J.; Chang, H.Y. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016, 17, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Bär, C.; Chatterjee, S.; Thum, T. Long Noncoding RNAs in Cardiovascular Pathology, Diagnosis, and Therapy. Circulation 2016, 134, 1484–1499. [Google Scholar] [CrossRef]
- Long, Q.Q.; Wang, H.; Gao, W.; Fan, Y.; Li, Y.F.; Ma, Y.; Yang, Y.; Shi, H.J.; Chen, B.R.; Meng, H.Y.; et al. Long noncoding RNA Kcna2 antisense RNA contributes to ventricular arrhythmias via silencing Kcna2 in rats with congestive heart failure. J. Am. Heart Assoc. 2017, 6, e005965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, P.; Yang, M.; Ren, H.; Shen, G.; Chen, J.; Zhang, J.; Liu, J.; Sun, C. Long noncoding RNA MALAT1 downregulates cardiac transient outward potassium current by regulating miR-200c/HMGB1 pathway. J. Cell. Biochem. 2018, 119, 10239–10249. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z. Binding of LncRNA-DACH1 to dystrophin impairs the membrane tra cking of Nav1.5 protein and increases ventricular arrhythmia susceptibility. Preprint 2021, 1–17. [Google Scholar] [CrossRef]
- Schmidt, J.W.; Catterall, W.A. Palmitylation, sulfation, and glycosylation of the alpha subunit of the sodium channel. Role of post-translational modifications in channel assembly. J. Biol. Chem. 1987, 262, 13713–13723. [Google Scholar] [CrossRef]
- Marionneau, C.; Abriel, H. Regulation of the cardiac Na+ channel Nav1.5 by post-translational modifications. J. Mol. Cell. Cardiol. 2015, 82, 36–47. [Google Scholar] [CrossRef]
- Abriel, H. Cardiac sodium channel Nav1.5 and interacting proteins: Physiology and pathophysiology. J. Mol. Cell. Cardiol. 2010, 48, 2–11. [Google Scholar] [CrossRef]
- Delisle, B.P.; Anson, B.D.; Rajamani, S.; January, C.T. Biology of Cardiac Arrhythmias Ion Channel Protein Trafficking. Circ. Res. 2004, 94, 1418–1428. [Google Scholar] [CrossRef] [Green Version]
- Jan, C.H.; Williams, C.C.; Weissman, J.S. Principles of ER cotranslational translocation revealed by proximity-specific ribosome profiling. Science 2014, 346, 716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Shin, H.G.; Yi, J.; Shen, W.; Williams, C.P.; Murray, K.T. Phosphorylation and putative ER retention signals are required for protein kinase A-mediated potentiation of cardiac sodium current. Circ. Res. 2002, 91, 540–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abriel, H. Roles and regulation of the cardiac sodium channel Nav1.5: Recent insights from experimental studies. Cardiovasc. Res. 2007, 76, 381–389. [Google Scholar] [CrossRef] [Green Version]
- Fiona, S.; Cusdin, J.J.C.; Jackson, A.P. Trafficking and Cellular Distribution of Voltage-Gated Sodium Channels. Traffic 2008, 9, 17–26. [Google Scholar] [CrossRef]
- Herfst, L.J.; Rook, M.B.; Jongsma, H.J. Trafficking and functional expression of cardiac Na+ channels. J. Mol. Cell. Cardiol. 2004, 36, 185–193. [Google Scholar] [CrossRef]
- Ranek, M.J.; Stachowski, M.J.; Kirk, J.A.; Willis, M.S. The role of heat shock proteins and co-chaperones in heart failure. Phil. Trans. R. Soc. B 2017, 373, 20160530. [Google Scholar] [CrossRef] [Green Version]
- Remme, C.A.; Wilde, A.A. Targeting sodium channels in cardiac arrhythmia. Curr. Opin. Pharmacol. 2014, 15, 53–60. [Google Scholar] [CrossRef]
- Abriel, H.; Kass, R.S. Regulation of the voltage-gated cardiac sodium channel Nav1.5 by interacting proteins. Trends Cardiovasc. Med. 2005, 15, 35–40. [Google Scholar] [CrossRef]
- Shinlapawittayatorn, K.; Dudash, L.A.; Du, X.X.; Heller, L.; Poelzing, S.; Ficker, E.; Deschênes, I. A novel strategy using cardiac sodium channel polymorphic fragments to rescue trafficking-deficient SCN5A Mutations. Circ. Cardiovasc. Genet. 2011, 4, 500–509. [Google Scholar] [CrossRef] [Green Version]
- Moreau, A.; Keller, D.I.; Huang, H.; Fressart, V.; Schmied, C.; Timour, Q.; Chahine, M. Mexiletine differentially restores the trafficking defects caused by two Brugada syndrome mutations. Front. Pharmacol. 2012, 3, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Mercier, A.; Clément, R.; Harnois, T.; Bourmeyster, N.; Bois, P.; Chatelier, A. Nav1.5 channels can reach the plasma membrane through distinct N-glycosylation states. Biochim. Biophys. Acta—Gen. Subj. 2015, 1850, 1215–1223. [Google Scholar] [CrossRef]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Young, J.C. The role of the cytosolic HSP70 chaperone system in diseases caused by misfolding and aberrant trafficking of ion channels. DMM Dis. Models Mech. 2014, 7, 319–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, K.; Jiang, Q.; Bai, X.; Yang, Y.F.; Ruan, M.Y.; Cai, S.Q. Tetrameric Assembly of K+ Channels Requires ER-Located Chaperone Proteins. Mol. Cell 2017, 65, 52–65. [Google Scholar] [CrossRef] [Green Version]
- Tarone, G.; Brancaccio, M. Keep your heart in shape: Molecular chaperone networks for treating heart disease. Cardiovasc. Res. 2014, 102, 346–361. [Google Scholar] [CrossRef] [Green Version]
- Penna, C.; Sorge, M.; Femminò, S.; Pagliaro, P.; Brancaccio, M. Redox aspects of chaperones in cardiac function. Front. Physiol. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Poelzing, S.; Forleo, C.; Samodell, M.; Dudash, L.; Sorrentino, S.; Anaclerio, M.; Troccoli, R.; Iacoviello, M.; Romito, R.; Guida, P.; et al. SCN5A polymorphism restores trafficking of a Brugada syndrome mutation on a separate gene. Circulation 2006, 114, 368–376. [Google Scholar] [CrossRef] [Green Version]
- Valdivia, C.R.; Tester, D.J.; Rok, B.A.; Porter, C.-B.J.; Munger, T.M.; Jahangir, A.; Makielski, J.C.; Ackerman, M.J. A trafficking defective, Brugada syndrome-causing SCN5A mutation rescued by drugs. Cardiovasc. Res. 2004, 62, 53–62. [Google Scholar] [CrossRef]
- Ulloa-Aguirre, A.; Janovick, J.A.; Brothers, S.P.; Conn, P.M. Pharmacologic rescue of conformationally-defective proteins: Implications for the treatment of human disease. Traffic 2004, 5, 821–837. [Google Scholar] [CrossRef]
- Cohen, S.A.; Levitt, L.K. Partial characterization of the rH1 sodium channel protein from rat heart using subtype-specific antibodies. Circ. Res. 1993, 73, 735–742. [Google Scholar] [CrossRef] [Green Version]
- Caramelo, J.J.; Parodi, A.J. A sweet code for glycoprotein folding. FEBS Lett. 2015, 589, 3379–3387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verges, M. Trafficking of Cardiac Ion Channels; MDPI AG: Basel, Switzerland, 2021; ISBN 9783039434725. [Google Scholar]
- Bennett, E.S. Isoform-specific effects of sialic acid on voltage-dependent Na+ channel gating: Functional sialic acids are localized to the S5–S6 loop of domain I. J. Physiol. 2002, 538, 675–690. [Google Scholar] [CrossRef] [PubMed]
- Stocker, P.J.; Bennett, E.S. Differential sialylation modulates voltage-gated Na+ channel gating throughout the developing myocardium. J. Gen. Physiol. 2006, 127, 253–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arakel, E.C.; Brandenburg, S.; Uchida, K.; Zhang, H.; Lin, Y.W.; Kohl, T.; Schrul, B.; Sulkin, M.S.; Efimov, I.R.; Nichols, C.G.; et al. Tuning the electrical properties of the heart by differential trafficking of KATP ion channel complexes. J. Cell Sci. 2014, 127, 2106–2119. [Google Scholar] [CrossRef] [Green Version]
- Roth, J.; Zuber, C.; Park, S.; Jang, I.; Lee, Y.; Kysela, K.G.; Le Fourn, V.; Santimaria, R.; Guhl, B.; Cho, J.W. Protein N-glycosylation, protein folding and protein quality control. Mol. Cells 2010, 30, 497–506. [Google Scholar] [CrossRef]
- Xiao, X.; Chen, C.; Yu, T.M.; Ou, J.; Rui, M.; Zhai, Y.; He, Y.; Xue, L.; Ho, M.S. Molecular chaperone calnexin regulates the function of Drosophila sodium channel paralytic. Front. Mol. Neurosci. 2017, 10, 57. [Google Scholar] [CrossRef] [Green Version]
- Khanna, R.; Lee, E.J.; Papazian, D.M. Transient calnexin interaction confers long-term stability on folded K+ channel protein in the ER. J. Cell Sci. 2004, 117, 2897–2908. [Google Scholar] [CrossRef] [Green Version]
- Proft, J.; Rzhepetskyy, Y.; Lazniewska, J.; Zhang, F.X.; Cain, S.M.; Snutch, T.P.; Zamponi, G.W.; Weiss, N. The Cacna1h mutation in the GAERS model of absence epilepsy enhances T-type Ca2+ currents by altering calnexin-dependent trafficking of Cav3.2 channels. Sci. Rep. 2017, 7, 11513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zumhagen, S.; Veldkamp, M.W.; Stallmeyer, B.; Baartscheer, A.; Eckardt, L.; Paul, M.; Remme, C.A.; Bhuiyan, Z.A.; Bezzina, C.R.; Schulze-Bahr, E. A Heterozygous Deletion Mutation in the Cardiac Sodium Channel Gene SCN5A with Loss- and Gain-of-Function Characteristics Manifests as Isolated Conduction Disease, without Signs of Brugada or Long QT Syndrome. PLoS ONE 2013, 8, e67963. [Google Scholar] [CrossRef]
- Casini, S.; Tan, H.L.; Demirayak, I.; Remme, C.A.; Amin, A.S.; Scicluna, B.P.; Chatyan, H.; Ruijter, J.M.; Bezzina, C.R.; Van Ginneken, A.C.G.; et al. Tubulin polymerization modifies cardiac sodium channel expression and gating. Cardiovasc. Res. 2010, 85, 691–700. [Google Scholar] [CrossRef] [Green Version]
- Abriel, H.; Sottas, V. Unexpected α-α interactions with nav1.5 genetic variants in brugada syndrome. Circ. Cardiovasc. Genet. 2014, 7, 97–99. [Google Scholar] [CrossRef] [Green Version]
- Gao, G.; Xie, A.; Zhang, J.; Herman, A.M.; Jeong, E.M.; Gu, L.; Liu, M.; Yang, K.C.; Kamp, T.J.; Dudley, S.C. Unfolded protein response regulates cardiac sodium current in systolic human heart failure. Circ. Arrhythmia Electrophysiol. 2013, 6, 1018–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, J.; Bi, Y.; Sowers, J.R.; Hetz, C.; Zhang, Y. Endoplasmic reticulum stress and unfolded protein response in cardiovascular diseases. Nat. Rev. Cardiol. 2021, 18, 499–521. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, S.M.; Lemmens-Gruber, R. Phosphorylation of cardiac voltage-gated sodium channel: Potential players with multiple dimensions. Acta Physiol. 2019, 225, 1–18. [Google Scholar] [CrossRef]
- Schubert, B.; Vandongen, A.M.J.; Kirsch, G.E.; Brown, A.M. Inhibition of cardiac Na+ currents by isoproterenol. Am. J. Physiol.—Heart Circ. Physiol. 1990, 258. [Google Scholar] [CrossRef] [Green Version]
- Frohnwieser, B.; Chen, L.Q.; Schreibmayer, W.; Kallen, R.G. Modulation of the human cardiac sodium channel α-subunit by cAMP-dependent protein kinase and the responsible sequence domain. J. Physiol. 1997, 498, 309–318. [Google Scholar] [CrossRef] [Green Version]
- Murphy, B.J.; Rogers, J.; Perdichizzi, A.P.; Colvin, A.A.; Catterall, W.A. cAMP-dependent phosphorylation of two sites in the α subunit of the cardiac sodium channel. J. Biol. Chem. 1996, 271, 28837–28843. [Google Scholar] [CrossRef] [Green Version]
- Tateyama, M.; Rivolta, I.; Clancy, C.E.; Kass, R.S. Modulation of Cardiac Sodium Channel Gating by Protein Kinase A Can Be Altered by Disease-linked Mutation. J. Biol. Chem. 2003, 278, 46718–46726. [Google Scholar] [CrossRef] [Green Version]
- Rook, M.B.; Evers, M.M.; Vos, M.A.; Bierhuizen, M.F. Biology of cardiac sodium channel Nav1.5 expression. Cardiovasc. Res. 2012, 93, 12–23. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Yi, J.; Hu, N.N.; George, A.L.; Murray, K.T. Activation of protein kinase A modulates trafficking of the human cardiac sodium channel in Xenopus oocytes. Circ. Res. 2000, 87, 33–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, D.B.; Blanpied, T.A.; Ehlers, M.D. Coordinated PKA and PKC phosphorylation suppresses RXR-mediated ER retention and regulates the surface delivery of NMDA receptors. Neuropharmacology 2003, 45, 755–767. [Google Scholar] [CrossRef]
- Hallaqa, H.; Yanga, Z.; Viswanathanb, P.C.; Fukudab, K.; Shena, W.; Wanga, D.W.; Wellsc, K.S.; Zhoua, J.; Yia, J.; Hallaqa, K.T.M.; et al. Quantitation of protein kinase a mediated trafficking of cardiac sodium channels in living cells. Cardiovasc. Res. 2006, 72, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Rogers, J.; Tanada, T.; Scheuer, T.; Catterall, W.A. Modulation of cardiac Na+ channels expressed in a mammalian cell line and in ventricular myocytes by protein kinase C. Proc. Natl. Acad. Sci. USA 1994, 91, 3289–3293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, H.G.; Murray, K.T. Conventional protein kinase C isoforms and cross-activation of protein kinase A regulate cardiac Na+ current. FEBS Lett. 2001, 495, 154–158. [Google Scholar] [CrossRef] [Green Version]
- Xiao, G.Q.; Qu, Y.; Sun, Z.Q.; Mochly-Rosen, D.; Boutjdir, M. Evidence for functional role of εPKC isozyme in the regulation of cardiac Na+ channels. Am. J. Physiol.—Cell Physiol. 2001, 281, C1477–C1486. [Google Scholar] [CrossRef]
- Ferreira, J.C.B.; Mochly-Rosen, D.; Boutjdir, M. Regulation of cardiac excitability by protein kinase C isozymes. Front. Biosci.—Sch. 2012, 4, 532–546. [Google Scholar] [CrossRef]
- Liu, M.; Shi, G.; Yang, K.C.; Gu, L.; Kanthasamy, A.G.; Anantharam, V.; Dudley, S.C., Jr. The role of protein kinase C in the metabolic regulation of the cardiac Na+ channel. Heart Rhythm 2017, 14, 440–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathieu, S.; El Khoury, N.; Rivard, K.; Gélinas, R.; Goyette, P.; Paradis, P.; Nemer, M.; Fiset, C. Reduction in Na+ current by angiotensin II is mediated by PKCα in mouse and human-induced pluripotent stem cell-derived cardiomyocytes. Heart Rhythm 2016, 13, 1346–1354. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Rogers, J.C.; Tanada, T.N.; Catterall, W.A.; Scheuer, T. Phosphorylation of S1505 in the cardiac Na+ channel inactivation gate is required for modulation by protein kinase C. J. Gen. Physiol. 1996, 108, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Murray, K.T.; Hu, N.; Daw, J.R.; Shin, H.G.; Watson, M.T.; Mashburn, A.B.; George, A.L. Functional effects of protein kinase C activation on the human cardiac Na+ channel. Circ. Res. 1997, 80, 370–376. [Google Scholar] [CrossRef]
- Liu, M.; Sanyal, S.; Gao, G.; Gurung, I.S.; Zhu, X.; Gaconnet, G.; Kerchner, L.J.; Shang, L.L.; Huang, C.L.H.; Grace, A.; et al. Cardiac Na+ Current regulation by pyridine nucleotides. Circ. Res. 2009, 105, 737–745. [Google Scholar] [CrossRef] [Green Version]
- Strickland, M.; Yacoubi-Loueslati, B.; Bouhaouala-Zahar, B.; Pender, S.L.F.; Larbi, A. Relationships between ion channels, mitochondrial functions and inflammation in human aging. Front. Physiol. 2019, 10, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.F.; Wu, C.H.; Lai, T.C.; Chang, Y.C.; Hwang, M.J.; Chang, T.Y.; Weng, C.H.; Chang, P.M.H.; Chen, C.H.; Mochly-Rosen, D.; et al. ALDH2 deficiency induces atrial fibrillation through dysregulated cardiac sodium channel and mitochondrial bioenergetics: A multi-omics analysis. Biochim. Biophys. Acta—Mol. Basis Dis. 2021, 1867, 166088. [Google Scholar] [CrossRef] [PubMed]
- Valdivia, C.R.; Ueda, K.; Ackerman, M.J.; Makielski, J.C. GPD1L links redox state to cardiac excitability by PKC-dependentphosphorylation of the sodium channel SCN5A. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1446–H1452. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Liu, H.; Dudley, S.C. Reactive oxygen species originating from mitochondria regulate the cardiac sodium channel. Circ. Res. 2010, 107, 967–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Gu, L.; Sulkin, M.S.; Liu, H.; Jeong, E.M.; Greener, I.; Xie, A.; Efimov, I.R.; Dudley, S.C. Mitochondrial dysfunction causing cardiac sodium channel downregulation in cardiomyopathy. J. Mol. Cell. Cardiol. 2013, 54, 25–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matasic, D.S.; Yoon, J.Y.; McLendon, J.M.; Mehdi, H.; Schmidt, M.S.; Greiner, A.M.; Quinones, P.; Morgan, G.M.; Boudreau, R.L.; Irani, K.; et al. Modulation of the cardiac sodium channel Nav1.5 peak and late currents by NAD+ precursors. J. Mol. Cell. Cardiol. 2020, 141, 70–81. [Google Scholar] [CrossRef]
- Fouda, M.A.; Ghovanloo, M.R.; Ruben, P.C. Cannabidiol protects against high glucose-induced oxidative stress and cytotoxicity in cardiac voltage-gated sodium channels. Br. J. Pharmacol. 2020, 177, 2932–2946. [Google Scholar] [CrossRef]
- Fouda, M.A.; Ruben, P.C. Protein kinases mediate anti-inflammatory effects of cannabidiol and estradiol against high glucose in cardiac sodium channels. Front. Pharmacol. 2021, 12, 1–17. [Google Scholar] [CrossRef]
- Ashpole, N.M.; Herren, A.W.; Ginsburg, K.S.; Brogan, J.D.; Johnson, D.E.; Cummins, T.R.; Bers, D.M.; Hudmon, A. Ca2+/calmodulin-dependent protein kinase II (CaMKII) regulates cardiac sodium channel Nav1.5 gating by multiple phosphorylation sites. J. Biol. Chem. 2012, 287, 19856–19869. [Google Scholar] [CrossRef] [Green Version]
- Herren, A.W.; Weber, D.M.; Rigor, R.R.; Margulies, K.B.; Phinney, B.S.; Bers, D.M. CaMKII Phosphorylation of Nav1.5: Novel in Vitro Sites Identified by Mass Spectrometry and Reduced S516 Phosphorylation in Human Heart Failure. J. Proteome Res. 2015, 14, 2298–2311. [Google Scholar] [CrossRef] [Green Version]
- Burel, S.; Coyan, F.C.; Lorenzini, M.; Meyer, M.R.; Lichti, C.F.; Brown, J.H.; Loussouarn, G.; Charpentier, F.; Nerbonne, J.M.; Townsend, R.R.; et al. C-terminal phosphorylation of Nav1.5 impairs FGF13-dependent regulation of channel inactivation. J. Biol. Chem. 2017, 292, 17431–17448. [Google Scholar] [CrossRef] [Green Version]
- Wagner, S.; Dybkova, N.; Rasenack, E.C.L.; Jacobshagen, C.; Fabritz, L.; Kirchhof, P.; Maier, S.K.G.; Zhang, T.; Hasenfuss, G.; Brown, J.H.; et al. Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J. Clin. Investig. 2006, 116, 3127–3138. [Google Scholar] [CrossRef] [PubMed]
- El Refaey, M.; Musa, H.; Murphy, N.P.; Lubbers, E.R.; Skaf, M.; Han, M.; Cavus, O.; Koenig, S.N.; Wallace, M.J.; Gratz, D.; et al. Protein Phosphatase 2A Regulates Cardiac Na+ Channels. Circ. Res. 2019, 124, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Deschênes, I.; Neyroud, N.; DiSilvestre, D.; Marbán, E.; Yue, D.T.; Tomaselli, G.F. Isoform-specific modulation of voltage-gated Na+ channels by calmodulin. Circ. Res. 2002, 90, 49–57. [Google Scholar] [CrossRef] [Green Version]
- Ahern, C.A.; Zhang, J.F.; Wookalis, M.J.; Horn, R. Modulation of the cardiac sodium channel Nav1.5 by Fyn, a Src family tyrosine kinase. Circ. Res. 2005, 96, 991–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razani, B.; Woodman, S.E.; Lisanti, M.P. Caveolae: From cell biology to animal physiology. Pharmacol. Rev. 2002, 54, 431–467. [Google Scholar] [CrossRef]
- Maier, S.K.; Westenbroek, R.E.; Schenkman, K.A.; Feigl, E.O.; Scheuer, T.; Catterall, W.A. An unexpected role for brain type sodium channels in coupling of cell surface depolarization to contraction in the heart. Proc. Natl. Acad. Sci. USA 2002, 99, 4073–4078. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, S.M.; Andavan, G.S.B.; Lemmens-Gruber, R. Differential modulation of fast inactivation in cardiac sodium channel splice variants by fyn tyrosine kinase. Cell. Physiol. Biochem. 2015, 37, 825–837. [Google Scholar] [CrossRef]
- Iqbal, S.M.; Aufy, M.; Shabbir, W.; Lemmens-Gruber, R. Identification of phosphorylation sites and binding pockets for modulation of Nav1.5 channel by Fyn tyrosine kinase. FEBS J. 2018, 285, 2520–2530. [Google Scholar] [CrossRef] [Green Version]
- Sarhan, M.F.; Van Petegem, F.; Ahern, C.A. A double tyrosine motif in the cardiac sodium channel domain III-IV linker couples calcium-dependent calmodulin binding to inactivation gating. J. Biol. Chem. 2009, 284, 33265–33274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jespersen, T.; Gavillet, B.; van Bemmelen, M.X.; Cordonier, S.; Thomas, M.A.; Staub, O.; Abriel, H. Cardiac sodium channel Nav1.5 interacts with and is regulated by the protein tyrosine phosphatase PTPH1. Biochem. Biophys. Res. Commun. 2006, 348, 1455–1462. [Google Scholar] [CrossRef] [PubMed]
- Beltran-Alvarez, P.; Pagans, S.; Brugada, R. The cardiac sodium channel is post-translationally modified by arginine methylation. J. Proteome Res. 2011, 10, 3712–3719. [Google Scholar] [CrossRef] [PubMed]
- Beltran-Alvarez, P.; Espejo, A.; Schmauder, R.; Beltran, C.; Mrowka, R.; Linke, T.; Batlle, M.; Pérez-Villa, F.; Pérez, G.J.; Scornik, F.S.; et al. Protein arginine methyl transferases-3 and -5 increase cell surface expression of cardiac sodium channel. FEBS Lett. 2013, 587, 3159–3165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beltran-Alvarez, P.; Tarradas, A.; Chiva, C.; Pérez-Serra, A.; Batlle, M.; Pérez-Villa, F.; Schulte, U.; Sabidó, E.; Brugada, R.; Pagans, S. Identification of N-terminal protein acetylation and arginine methylation of the voltage-gated sodium channel in end-stage heart failure human heart. Curr. Ther. Res.—Clin. Exp. 2014, 76, 126–129. [Google Scholar] [CrossRef]
- Yoon, J.Y.; Vikram, A.; London, B.; Irani, K. Reversible lysine acetylation: Another layer of post-translational regulation of the cardiac sodium channel. Channels 2017, 11, 360–361. [Google Scholar] [CrossRef] [Green Version]
- Vikram, A.; Lewarchik, C.M.; Yoon, J.-Y.; Naqvi, A.; Kumar, S.; Morgan, G.M.; Jacobs, J.S.; Li, Q.; Kim, Y.-R.; Kassan, M.; et al. Sirtuin 1 regulates cardiac electrical activity by deacetylating the cardiac sodium channel. Nat. Med. 2017, 23, 361–370. [Google Scholar] [CrossRef]
- Henley, J.M.; Craig, T.J.; Wilkinson, K.A. Neuronal Sumoylation: Mechanisms, Dysfunction. Physiol. Rev. 2014, 94, 1249–1285. [Google Scholar] [CrossRef]
- Plant, L.D.; Xiong, D.; Romero, J.; Dai, H.; Goldstein, S.A.N. Hypoxia Produces Pro-arrhythmic Late Sodium Current in Cardiac Myocytes by SUMOylation of Nav1.5 Channels. Cell Rep. 2020, 30, 2225–2236.e4. [Google Scholar] [CrossRef] [Green Version]
- Hess, D.T.; Matsumoto, A.; Kim, S.O.; Marshall, H.E.; Stamler, J.S. Protein S-nitrosylation: Purview and parameters. Nat. Rev. Mol. Cell Biol. 2005, 6, 150–166. [Google Scholar] [CrossRef]
- Nathan, C.; Xie, Q. wen Nitric oxide synthases: Roles, tolls, and controls. Cell 1994, 78, 915–918. [Google Scholar] [CrossRef]
- Ahern, G.P.; Hsu, S.F.; Klyachko, V.A.; Jackson, M.B. Induction of persistent sodium current by exogenous and endogenous nitric oxide. J. Biol. Chem. 2000, 275, 28810–28815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.; Van Norstrand, D.W.; Medeiros-Domingo, A.; Valdivia, C.; Tan, B.; Ye, B.; Kroboth, S.; Vatta, M.; Tester, D.J.; January, C.T.; et al. α1-Syntrophin Mutations Identified in Sudden Infant Death Syndrome Cause an Increase in Late Cardiac Sodium Current. Circ. Arrhythmia Electrophysiol. 2009, 2, 667–676. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Valdivia, C.R.; Vaidyanathan, R.; Balijepalli, R.C.; Ackerman, M.J.; Makielski, J.C. Caveolin-3 suppresses late sodium current by inhibiting nNOS-dependent S-nitrosylation of SCN5A. J. Mol. Cell. Cardiol. 2013, 61, 102–110. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Wei, M.; Zhu, X.; Liu, Y.; Yoshimura, K.; Zheng, M.; Liu, G.; Kume, S.; Morishima, M.; Kurokawa, T.; et al. Nitric oxide down-regulates voltage-gated Na+ channel in cardiomyocytes possibly through S-nitrosylation-mediated signaling. Sci. Rep. 2021, 11, 11273. [Google Scholar] [CrossRef] [PubMed]
- Krüger, E.; Hahn, K. Lipoxidation products as biomarkers of oxidative damage to proteins during lipid peroxidation reactions. Nephrol.—Dial.—Transplant. 2010, 11, 1–191. [Google Scholar]
- Herren, A.W.; Bers, D.M.; Grandi, E. Post-translational modifications of the cardiac Na channel: Contribution of CaMKII-dependent phosphorylation to acquired arrhythmias. Am. J. Physiol.—Heart Circ. Physiol. 2013, 305, H431–H445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakajima, T.; Davies, S.S.; Matafonova, E.; Potet, F.; Amarnath, V.; Tallman, K.A.; Serwa, R.A.; Porter, N.A.; Balser, J.R.; Kupershmidt, S.; et al. Selective γ-ketoaldehyde scavengers protect Nav1.5 from oxidant-induced inactivation. J. Mol. Cell. Cardiol. 2010, 48, 352–359. [Google Scholar] [CrossRef] [Green Version]
- Quiñonez, M.; DiFranco, M.; González, F. Involvement of methionine residues in the fast inactivation mechanism of the sodium current from toad skeletal muscle fibers. J. Membr. Biol. 1999, 169, 83–90. [Google Scholar] [CrossRef]
- Kassmann, M.; Hansel, A.; Leipold, E.; Birkenbeil, J.; Lu, S.Q.; Hoshi, T.; Heinemann, S.H. Oxidation of multiple methionine residues impairs rapidsodium channel inactivation. Pflugers Arch.—Eur. J. Physiol. 2008, 456, 1085–1095. [Google Scholar] [CrossRef] [Green Version]
- Erickson, J.R.; Mei-ling, A.J.; Guan, X.; Kutschke, W.; Yang, J.; Oddis, C.V.; Bartlett, R.K.; Lowe, J.S.; O’Donnell, S.E.; Aykin-Burns, N.; et al. A Dynamic Pathway for Calcium-Independent Activation of CaMKII by Methionine Oxidation. Cell 2008, 133, 462–474. [Google Scholar] [CrossRef] [Green Version]
- Chamberlain, L.H.; Shipston, M.J. The physiology of protein s-acylation. Physiol. Rev. 2015, 95, 341–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Bi, D.; Tian, L.; McClafferty, H.; Steeb, F.; Ruth, P.; Knaus, H.G.; Shipston, M.J. Palmitoylation of the β4-subunit regulates surface expression of large conductance calcium-activated potassium channel splice variants. J. Biol. Chem. 2013, 288, 13136–13144. [Google Scholar] [CrossRef] [Green Version]
- Pei, Z.; Xiao, Y.; Meng, J.; Hudmon, A.; Cummins, T.R. Cardiac sodium channel palmitoylation regulates channel availability and myocyte excitability with implications for arrhythmia generation. Nat. Commun. 2016, 7, 12035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahl, G. Protein truncation test (PTT; protein truncation assay, PTA). Dict. Genom. Transcr. Proteom. 2015, 1. [Google Scholar] [CrossRef]
- Zimmer, T.; Biskup, C.; Dugarmaa, S.; Vogel, F.; Steinbis, M.; Böhle, T.; Wu, Y.S.; Dumaine, R.; Benndorf, K. Functional expression of GFP-linked human heart sodium channel (hH1) and subcellular localization of the α subunit in HEK293 cells and dog cardiac myocytes. J. Membr. Biol. 2002, 186, 1–12. [Google Scholar] [CrossRef]
- Wu, L.; Yong, S.L.; Fan, C.; Ni, Y.; Yoo, S.; Zhang, T.; Zhang, X.; Obejero-Paz, C.A.; Rho, H.J.; Ke, T.; et al. Identification of a new co-factor, MOG1, required for the full function of cardiac sodium channel Nav1.5. J. Biol. Chem. 2008, 283, 6968–6978. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, S.; Wu, X.; Yang, Z.; Wu, L.; Yong, S.L.; Zhang, C.; Hu, K.; Wang, Q.K.; Chen, Q. MOG1 Rescues Defective Trafficking of Nav1.5 Mutations in Brugada Syndrome and Sick Sinus Syndrome. Circ. Arrhythmia Electrophysiol. 2013, 6, 392–401. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Yu, G.; Liu, Y.; Liu, S.; Aridor, M.; Huang, Y.; Hu, Y.; Wang, L.; Li, S.; Xiong, H.; et al. Small GTPases SAR1A and SAR1B regulate the trafficking of the cardiac sodium channel Nav1.5. Biochim. Biophys. Acta—Mol. Basis Dis. 2018, 1864, 3672–3684. [Google Scholar] [CrossRef]
- Chatin, B.; Colombier, P.; Gamblin, A.L.; Allouis, M.; Le Bouffant, F. Dynamitin affects cell-surface expression of voltage-gated sodium channel Nav1.5. Biochem. J. 2014, 463, 339–349. [Google Scholar] [CrossRef]
- Ednie, A.R.; Horton, K.K.; Wu, J.; Bennett, E.S. Expression of the sialyltransferase, ST3Gal4, impacts cardiac voltage-gated sodium channel activity, refractory period and ventricular conduction. J. Mol. Cell. Cardiol. 2013, 59, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Baroni, D.; Picco, C.; Moran, O. A mutation of SCN1B associated with GEFS+ causes functional and maturation defects of the voltage-dependent sodium channel. Hum. Mutat. 2018, 39, 1402–1415. [Google Scholar] [CrossRef] [PubMed]
- Rosnoblet, C.; Peanne, R.; Legrand, D.; Foulquier, F. Glycosylation disorders of membrane trafficking. Glycoconj. J. 2013, 30, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Bennett, E.; Urcan, M.S.; Tinkle, S.S.; Koszowski, A.G.; Levinson, S.R. Contribution of sialic acid to the voltage dependence of sodium channel gating: A possible electrostatic mechanism. J. Gen. Physiol. 1997, 109, 327–343. [Google Scholar] [CrossRef] [Green Version]
- Ufret-Vincenty, C.A.; Baro, D.J.; Lederer, W.J.; Rockman, H.A.; Quiñones, L.E.; Santana, L.F. Role of Sodium Channel Deglycosylation in the Genesis of Cardiac Arrhythmias in Heart Failure. J. Biol. Chem. 2001, 276, 28197–28203. [Google Scholar] [CrossRef] [Green Version]
- Ednie, A.R.; Bennett, E.S. Modulation of voltage-gated ion channels by sialylation. Compr. Physiol. 2012, 2, 1269–1301. [Google Scholar] [CrossRef]
- Gu, F.; Crump, C.M.; Thomas, G. Trans-golgi network sorting. Cell. Mol. Life Sci. 2001, 58, 1067–1084. [Google Scholar] [CrossRef] [Green Version]
- Ponce-Balbuena, D.; Guerrero-Serna, G.; Valdivia, C.R.; Caballero, R.; Diez-Guerra, F.J.; Jiménez-Vázquez, E.N.; Ramírez, R.J.; Monteiro Da Rocha, A.; Herron, T.J.; Campbell, K.F.; et al. Cardiac Kir2.1 and Nav1.5 Channels Traffic Together to the Sarcolemma to Control Excitability. Circ. Res. 2018, 122, 1501–1516. [Google Scholar] [CrossRef]
- Willis, B.C.; Ponce-Balbuena, D.; Jalife, J. Protein assemblies of sodium and inward rectifier potassium channels control cardiac excitability and arrhythmogenesis. Am. J. Physiol.—Heart Circ. Physiol. 2015, 308, H1463–H1473. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.-Q.; Perez-Hernandez, M.; Sanchez-Alonso, J.; Shevchuk, A.; Gorelik, J.; Rothenberg, E.; Delmar, M.; Coetzee, W.A. Ankyrin-G mediates targeting of both Na+ and K ATP channels to the rat cardiac intercalated disc. Elife 2020, 9, 1–23. [Google Scholar] [CrossRef]
- Shy, D.; Gillet, L.; Abriel, H. Targeting the sodium channel Nav1.5 to specific membrane compartments of cardiac cells not a simple task! Circ. Res. 2014, 115, 901–903. [Google Scholar] [CrossRef] [Green Version]
- Maier, S.K.G.; Westenbroek, R.E.; McCormick, K.A.; Curtis, R.; Scheuer, T.; Catterall, W.A. Distinct Subcellular Localization of Different Sodium Channel α and β Subunits in Single Ventricular Myocytes from Mouse Heart. Circulation 2004, 109, 1421–1427. [Google Scholar] [CrossRef] [Green Version]
- Domínguez, J.N.; De La Rosa, Á.; Navarro, F.; Franco, D.; Aránega, A.E. Tissue distribution and subcellular localization of the cardiac sodium channel during mouse heart development. Cardiovasc. Res. 2008, 78, 45–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, S.A. Immunocytochemical localization of rh1 sodium channel in adult rat heart atria and ventricle: Presence in terminal intercalated disks. Circulation 1996, 94, 3083–3086. [Google Scholar] [CrossRef] [PubMed]
- Shy, D.; Gillet, L.; Abriel, H. Cardiac sodium channel Nav1.5 distribution in myocytes via interacting proteins: The multiple pool model. Biochim. Biophys. Acta—Mol. Cell Res. 2013, 1833, 886–894. [Google Scholar] [CrossRef] [Green Version]
- Peeters, U.; Scornik, F.; Riuró, H.; Pérez, G.; Komurcu-Bayrak, E.; Van Malderen, S.; Pappaert, G.; Tarradas, A.; Pagans, S.; Daneels, D.; et al. Contribution of cardiac sodium channel β-subunit variants to brugada syndrome. Circ. J. 2015, 79, 2118–2129. [Google Scholar] [CrossRef] [Green Version]
- Salvage, S.C.; Rees, J.S.; McStea, A.; Hirsch, M.; Wang, L.; Tynan, C.J.; Reed, M.W.; Irons, J.R.; Butler, R.; Thompson, A.J.; et al. Supramolecular clustering of the cardiac sodium channel Nav1.5 in HEK293F cells, with and without the auxiliary β3-subunit. FASEB J. 2020, 34, 3537–3553. [Google Scholar] [CrossRef]
- Catterall, W.A. Voltage-gated sodium channels at 60_ structure, function and pathophysiology. J. Physiol. 2012, 590, 2577–2589. [Google Scholar] [CrossRef]
- Chen, C.; Calhoun, J.D.; Zhang, Y.; Lopez-Santiago, L.; Zhou, N.; Davis, T.H.; Salzer, J.L.; Isom, L.L. Identification of the cysteine residue responsible for disulfide linkage of Na+ channel alpha and beta2. J. Biol. Chem. 2012, 287, 39061–39069. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.H.; Westenbroek, R.E.; Silos-santiago, I.; Mccormick, K.A.; Lawson, D.; Ge, P.; Ferriera, H.; Lilly, J.; Distefano, P.S.; Catterall, W.A.; et al. Sodium Channel β4, a New Disulfide-Linked Auxiliary Subunit with Similarity to β2. J. Neurosci. 2003, 23, 7577–7585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, W.; Voelker, T.L.; Varga, Z.; Schubert, A.R.; Nerbonne, J.M.; Silva, J.R. Mechanisms of noncovalent β subunit regulation of Nav channel gating. J. Gen. Physiol. 2017, 149, 813–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malhotra, J.D.; Kazen-Gillespie, K.; Hortsch, M.; Isom, L.L. Sodium channel β subunits mediate homophilic cell adhesion and recruit ankyrin to points of cell-cell contact. J. Biol. Chem. 2000, 275, 11383–11388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domínguez, J.N.; Navarro, F.; Franco, D.; Thompson, R.P.; Aránega, A.E. Temporal and spatial expression pattern ofh1 sodium channel subunitduring heart development. Cardiovasc. Res. 2004, 65, 842–850. [Google Scholar] [CrossRef] [Green Version]
- Zimmer, T.; Biskup, C.; Bollensdorff, C.; Benndorf, K. The β1 Subunit but not the β2 Subunit Colocalizes with the Human Heart Na+ Channel (hHl) already within the Endoplasmic Reticulum. J. Membr. Biol. 2002, 186, 13–21. [Google Scholar] [CrossRef]
- Mercier, A.; Clement, R.; Harnois, T.; Bourmeyster, N.; Faivre, J.F.; Findlay, I.; Chahine, M.; Bois, P.; Chatelier, A. The β1-Subunit of Nav1.5 Cardiac Sodium Channel Is Required for a Dominant Negative Effect through α-α Interaction. PLoS ONE 2012, 7, e48690. [Google Scholar]
- Dulsat, G.; Palomeras, S.; Cortada, E.; Riuró, H.; Brugada, R.; Vergés, M. Trafficking and localisation to the plasma membrane of Na v 1.5 promoted by the β2 subunit is defective due to a β2 mutation associated with Brugada syndrome. Biol. Cell 2017, 109, 273–291. [Google Scholar] [CrossRef]
- Namadurai, S.; Balasuriya, D.; Rajappa, R.; Wiemhöfer, M.; Stott, K.; Klingauf, J.; Edwardson, J.M.; Chirgadze, D.Y.; Jackson, A.P. Crystal structure and molecular imaging of the Nav channelβ3 subunit indicates a trimeric assembly. J. Biol. Chem. 2014, 289, 10797–10811. [Google Scholar] [CrossRef] [Green Version]
- Medeiros-Domingo, A.; Kaku, T.; Tester, D.J.; Iturralde-Torres, P.; Itty, A.; Ye, B.; Valdivia, C.; Ueda, K.; Canizales-Quinteros, S.; Tusié-Luna, M.T.; et al. SCN4B-Encoded Sodium Channel β4 Subunit in Congenital Long-QT Syndrome. Circulation 2007, 116, 134–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, H.; Koopmann, T.T.; Le Scouarnec, S.; Yang, T.; Ingram, C.R.; Schott, J.-J.; Demolombe, S.; Probst, V.; Anselme, F.; Escande, D.; et al. Sodium channel β1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J. Clin. Investig. 2008, 118, 2260–2268. [Google Scholar] [CrossRef] [Green Version]
- Wilde, A.A.M.; Brugada, R. Phenotypical manifestations of mutations in the genes encoding subunits of the cardiac sodium channel. Circ. Res. 2011, 108, 884–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Malley, H.A.; Isom, L.L. Sodium channel β subunits: Emerging targets in channelopathies. Annu. Rev. Physiol. 2015, 77, 481–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, Y.; Curtis, R.; Lawson, D.; Gilbride, K.; Ge, P.; DiStefano, P.S.; Silos-Santiago, I.; Catterall, W.A.; Scheuer, T. Differential Modulation of Sodium Channel Gating and Persistent Sodium Currents by the β1, β2, and β3 Subunits. Mol. Cell. Neurosci. 2001, 18, 570–580. [Google Scholar] [CrossRef] [PubMed]
- Morgan, K.; Stevens, E.B.; Shah, B.; Cox, P.J.; Dixon, A.K.; Lee, K.; Pinnock, R.D.; Hughes, J.; Richardson, P.J.; Mizuguchi, K.; et al. β3: An additional auxiliary subunit of the voltage-sensitive sodium channel that modulates channel gating with distinct kinetics. Proc. Natl. Acad. Sci. USA 2000, 97, 2308–2313. [Google Scholar] [CrossRef] [Green Version]
- Delmar, M. Connexin43 regulates sodium current; Ankyrin-G modulates gap junctions: The intercalated disc exchanger. Cardiovasc. Res. 2012, 93, 220–222. [Google Scholar] [CrossRef]
- Abriel, H.; Rougier, J.S.; Jalife, J. Ion Channel Macromolecular Complexes in Cardiomyocytes: Roles in Sudden Cardiac Death. Circ. Res. 2015, 116, 1971–1988. [Google Scholar] [CrossRef] [Green Version]
- Godreau, D.; Vranckx, R.; Maguy, A.; Goyenvalle, C.; Hatem, S.N. Different Isoforms of Synapse-associated Protein, SAP97, Are Expressed in the Heart and Have Distinct Effects on the Voltage-gated K+ Channel Kv1.5. J. Biol. Chem. 2003, 278, 47046–47052. [Google Scholar] [CrossRef] [Green Version]
- Milstein, M.L.; Musa, H.; Balbuena, D.P.; Anumonwo, J.M.B.; Auerbach, D.S.; Furspan, P.B.; Hou, L.; Hu, B.; Schumacher, S.M.; Vaidyanathan, R.; et al. Dynamic reciprocity of sodium and potassium channel expression in a macromolecular complex controls cardiac excitability and arrhythmia. Proc. Natl. Acad. Sci. USA 2012, 109, E2134–E2143. [Google Scholar] [CrossRef] [Green Version]
- Petitprez, S.; Zmoos, A.F.; Ogrodnik, J.; Balse, E.; Raad, N.; El-Haou, S.; Albesa, M.; Bittihn, P.; Luther, S.; Lehnart, S.E.; et al. SAP97 and dystrophin macromolecular complexes determine two pools of cardiac sodium channels Nav1.5 in cardiomyocytes. Circ. Res. 2011, 108, 294–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillet, L.; Rougier, J.S.; Shy, D.; Sonntag, S.; Mougenot, N.; Essers, M.; Shmerling, D.; Balse, E.; Hatem, S.N.; Abriel, H. Cardiac-specific ablation of synapse-associated protein SAP97 in mice decreases potassium currents but not sodium current. Heart Rhythm 2015, 12, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Mohler, P.J. Editorial: Sodium channel traffic on the cardiac microtubule highway. Cardiovasc. Res. 2010, 85, 645–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agullo-Pascual, E.; Lin, X.; Leo-Macias, A.; Zhang, M.; Liang, F.; Li, Z.; Pfenniger, A.; Bkemeier, I.L.; Keegan, S.; Fenyo, D.; et al. Super-resolution imaging reveals that loss of theC-terminus of connexin43 limits microtubuleplus-end capture and Nav1.5 localization at theintercalated disc. Cardiovasc. Res. 2014, 104, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Marchal, G.A.; Jouni, M.; Chiang, D.Y.; Pérez-Hernández, M.; Podliesna, S.; Yu, N.; Casini, S.; Potet, F.; Veerman, C.C.; Klerk, M.; et al. Targeting the Microtubule EB1-CLASP2 Complex Modulates Nav1.5 at Intercalated Discs. Circ. Res. 2021, 129, 349–365. [Google Scholar] [CrossRef] [PubMed]
- Mimori-Kiyosue, Y.; Grigoriev, I.; Lansbergen, G.; Sasaki, H.; Matsui, C.; Severin, F.; Galjart, N.; Grosveld, F.; Vorobjev, I.; Tsukita, S.; et al. CLASP1 and CLASP2 bind to EB1 and regulate microtubule plus-end dynamics at the cell cortex. J. Cell Biol. 2005, 168, 141–153. [Google Scholar] [CrossRef]
- Desplantez, T. Cardiac Cx43, Cx40 and Cx45 co-assembling: Involvement of connexins epitopes in formation of hemichannels and Gap junction channels. BMC Cell Biol. 2017, 18, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Hunter, A.W.; Barker, R.J.; Zhu, C.; Gourdie, R.G. Zonula occludens-1 alters connexin43 gap junction size and organization by influencing channel accretion. Mol. Biol. Cell 2005, 16, 5686–5698. [Google Scholar] [CrossRef] [Green Version]
- Rhett, J.M.; Ongstad, E.L.; Jourdan, J.; Gourdie, R.G. Cx43 Associates with Nav1.5 in the Cardiomyocyte Perinexus. J. Membr. Biol. 2012, 245, 411–422. [Google Scholar]
- Noorman, M.; Hakim, S.; Kessler, E.; Groeneweg, J.; Cox, M.G.P.J.; Asimaki, A.; van Rijen, H.V.M.; van Stuijvenberg, L.; Chkourko, H.; van der Heyden, M.A.G.; et al. Remodeling of the cardiac sodium channel, Connexin43 andPlakoglobin at the intercalated disk in patients witharrhythmogenic cardiomyopathy. Heart Rhythm 2013, 10, 412–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhett, J.M.; Veeraraghavan, R.; Poelzing, S.; Gourdie, R.G. The perinexus: Sign-post on the path to a new model of cardiac conduction? Trends Cardiovasc. Med. 2013, 23, 222–228. [Google Scholar] [CrossRef] [Green Version]
- Cunha, S.R.; Mohler, P.J. Cardiac ankyrins: Essential components for development and maintenance of excitable membrane domains in heart. Cardiovasc. Res. 2006, 71, 22–29. [Google Scholar] [CrossRef] [Green Version]
- Hashemi, S.M.; Hund, T.J.; Mohler, P.J. Cardiac ankyrins in health and disease. J. Mol. Cell. Cardiol. 2009, 47, 203–209. [Google Scholar] [CrossRef] [Green Version]
- Makara, M.A.; Curran, J.; Little, S.C.; Musa, H.; Polina, I.; Smith, S.A.; Wright, P.J.; Unudurthi, S.D.; Snyder, J.; Bennett, V.; et al. Ankyrin-G Coordinates Intercalated Disc Signaling Platform to Regulate Cardiac Excitability In Vivo. Circ Res. 2014, 115, 929–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hund, T.J.; Koval, O.M.; Li, J.; Wright, P.J.; Qian, L.; Snyder, J.S.; Gudmundsson, H.; Kline, C.F.; Davidson, N.P.; Cardona, N.; et al. A β IV -spectrin/CaMKII signaling complex is essential for membrane excitability in mice. J. Clin. Investig. 2010, 120, 3508–3519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, P.Y.; Coombs, W.; Lin, X.; Nekrasova, O.; Green, K.J.; Isom, L.L.; Taffet, S.M.; Delmar, M. Interactions Between Ankyrin-G, Plakophilin-2, and Connexin43 at the Cardiac Intercalated Disc. Circ Res. 2011, 109, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Makara, M.A.; Curran, J.; Lubbers, E.R.; Murphy, N.P.; Little, S.C.; Musa, H.; Smith, S.A.; Unudurthi, S.D.; Rajaram, M.V.S.; Janssen, P.M.L.; et al. Novel Mechanistic Roles for Ankyrin-G in Cardiac Remodeling and Heart Failure. JACC Basic Transl. Sci. 2018, 3, 675–689. [Google Scholar] [CrossRef] [PubMed]
- Mohler, P.J.; Rivolta, I.; Napolitano, C.; Lemaillet, G.; Lambert, S.; Priori, S.G.; Bennett, V. Nav1.5 E1053K mutation causing Brugada syndrome blocks binding to ankyrin-G and expression of Nav1.5 on the surface of cardiomyocytes. Proc. Natl. Acad. Sci. USA 2004, 101, 17533–17538. [Google Scholar] [CrossRef] [Green Version]
- Cavus, O.; Williams, J.; Musa, H.; el Refaey, M.; Gratz, D.; Shaheen, R.; Schwieterman, N.A.; Koenig, S.; Antwi-Boasiako, S.; Young, L.J.; et al. Giant ankyrin-G regulates cardiac function. J. Biol. Chem. 2021, 296, 1–13. [Google Scholar] [CrossRef]
- Cerrone, M.; Delmar, M. Desmosomes and the sodium channel complex: Implications for arrhythmogenic cardiomyopathy and Brugada syndrome. Trends Cardiovasc. Med. 2014, 24, 184–190. [Google Scholar] [CrossRef] [Green Version]
- Cerrone, M.; Lin, X.; Zhang, M.; Agullo-Pascual, E.; Pfenniger, A.; Chkourko Gusky, H.; Novelli, V.; Kim, C.; Tirasawadichai, T.; Judge, D.P.; et al. Missense mutations in plakophilin-2 cause sodium current deficit and associate with a brugada syndrome phenotype. Circulation 2014, 129, 1092–1103. [Google Scholar] [CrossRef] [Green Version]
- Cerrone, M.; Lin, X.; Zhang, M.; Agullo-Pascual, E.; Pfenniger, A.; Chkourko Gusky, H.; Novelli, V.; Kim, C.; Tirasawadischai, T.; Judge, D.P.; et al. Missense Mutations in Plakophilin-2 Can Cause Brugada Syndrome Phenotype By Decreasing Sodium Current and Nav1.5 Membrane Localization. Heart Rhythm 2013, 10, 1743. [Google Scholar] [CrossRef]
- Sato, P.Y.; Musa, H.; Coombs, W.; Guerrero-Serna, G.; Patiño, G.A.; Taffet, S.M.; Isom, L.L.; Delmar, M. Loss of plakophilin-2 expression leads to decreased sodium current and slower conduction velocity in cultured cardiac myocytes. Circ. Res. 2009, 105, 523–526. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Wong, J.; Wen, J.; Wang, S.; Wang, C.; Spiering, S.; Kan, N.G.; Forcales, S.; Puri, P.L.; Leone, T.C.; et al. Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature 2013, 494, 105–110. [Google Scholar] [CrossRef] [Green Version]
- Rizzo, S.; Lodder, E.M.; Verkerk, A.O.; Wolswinkel, R.; Beekman, L.; Pilichou, K.; Basso, C.; Remme, C.A.; Thiene, G.; Bezzina, C.R. Intercalated disc abnormalities, reduced Na+ current density, and conduction slowing in desmoglein-2 mutant mice prior to cardiomyopathic changes. Cardiovasc. Res. 2012, 95, 409–418. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Deng, C.; Rao, F.; Modi, R.M.; Zhu, J.; Liu, X.; Mai, L.; Tan, H.; Yu, X.; Lin, Q.; et al. Silencing of desmoplakin decreases connexin43/Nav1.5 expression and sodium current in HL-1 cardiomyocytes. Mol. Med. Rep. 2013, 8, 780–786. [Google Scholar] [CrossRef]
- Cunha, S.R.; Mohler, P.J. Ankyrin protein networks in membrane formation and stabilization. J. Cell. Mol. Med. 2009, 13, 4364–4376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nassal, D.; Yu, J.; Min, D.; Lane, C.; Shaheen, R.; Gratz, D.; Hund, T.J. Regulation of cardiac conduction and arrhythmias by ankyrin/spectrin-based macromolecular complexes. J. Cardiovasc. Dev. Dis. 2021, 8, 48. [Google Scholar] [CrossRef] [PubMed]
- Bennett, V.; Chen, L. Ankyrins and cellular targeting of diverse membrane proteins to physiological sites. Curr. Opin. Cell Biol. 2001, 13, 61–67. [Google Scholar] [CrossRef]
- Glynn, P.; Musa, H.; Wu, X.; Unudurthi, S.D.; Little, S.; Qian, L.; Wright, P.J.; Radwanski, P.B.; Gyorke, S.; Mohler, P.J.; et al. Voltage-Gated Sodium Channel Phosphorylation at Ser571 Regulates Late Current, Arrhythmia, and Cardiac Function in Vivo. Circulation 2015, 132, 567–577. [Google Scholar] [CrossRef] [Green Version]
- Greer-Short, A.; Musa, H.; Alsina, K.M.; Ni, L.; Word, T.A.; Reynolds, J.O.; Gratz, D.; Lane, C.; El-Refaey, M.; Unudurthi, S.; et al. Calmodulin kinase II regulates atrial myocyte late sodium current, calcium handling, and atrial arrhythmia. Heart Rhythm 2020, 17, 503–511. [Google Scholar] [CrossRef]
- Marsman, R.F.; Bezzina, C.R.; Freiberg, F.; Verkerk, A.O.; Adriaens, M.E.; Podliesna, S.; Chen, C.; Purfürst, B.; Spallek, B.; Koopmann, T.T.; et al. Coxsackie and adenovirus receptor (CAR) is a modifier of cardiac conduction and arrhythmia vulnerability in the setting of myocardial ischemia. J. Am. Coll. Cardiol. 2015, 63, 549–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noutsias, M.; Fechner, H.; De Jonge, H.; Wang, X.; Dekkers, D.; Houtsmuller, A.B.; Pauschinger, M.; Bergelson, J.; Warraich, R.; Yacoub, M.; et al. Human Coxsackie-Adenovirus Receptor Is Colocalized With Integrins avb3 and avb5 on the Cardiomyocyte Sarcolemma and Upregulated in Dilated Cardiomyopathy Implications for Cardiotropic Viral Infections. Heart 2001, 104, 275–280. [Google Scholar]
- Clatot, J.; Ziyadeh-Isleem, A.; Maugenre, S.; Denjoy, I.; Liu, H.; Dilanian, G.; Hatem, S.N.; Deschenes, I.; Coulombe, A.; Guicheney, P.; et al. Dominant-negative effect of SCN5A N-terminal mutations through the interaction of Nav1.5 alpha subunits. Cardiovasc. Res. 2012, 96, 53–56. [Google Scholar] [CrossRef] [Green Version]
- Clatot, J.; Hoshi, M.; Wan, X.; Liu, H.; Jain, A.; Shinlapawittayatorn, K.; Marionneau, C.; Ficker, E.; Ha, T.; Deschênes, I. Voltage-gated sodium channels assemble and gate as dimers. Nat. Commun. 2017, 8, 2077. [Google Scholar] [CrossRef] [Green Version]
- Allouis, M.; Le Bouffant, F.; Wilders, R.; Péroz, D.; Schott, J.J.; Noireaud, J.; Le Marec, H.; Mérot, J.; Escande, D.; Baró, I. 14-3-3 Is a regulator of the cardiac voltage-gated sodium channel Nav1.5. Circ. Res. 2006, 98, 1538–1546. [Google Scholar] [CrossRef] [Green Version]
- Rougier, J.S.; Essers, M.C.; Gillet, L.; Guichard, S.; Sonntag, S.; Shmerling, D.; Abriel, H. A Distinct Pool of Nav1.5 Channels at the Lateral Membrane of Murine Ventricular Cardiomyocytes. Front. Physiol. 2019, 10, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Gee, S.H.; Madhavan, R.; Levinson, S.R.; Caldwell, J.H.; Sealock, R.; Froehner, S.C. Interaction of muscle and brain sodium channels with multiple members of the syntrophin family of dystrophin-associated proteins. J. Neurosci. 1998, 18, 128–137. [Google Scholar] [CrossRef]
- Ou, Y.; Strege, P.; Miller, S.M.; Makielski, J.; Ackerman, M.; Gibbons, S.J.; Farrugia, G. Syntrophin γ2 regulates SCN5A gating by a PDZ domain-mediated interaction. J. Biol. Chem. 2003, 278, 1915–1923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, J.; Hoffmuller, U.; Krause, G.; Ashurst, J.; Macias, M.J.; Schmieder, P.; Schneider-Mergener, J.; Oschkinat, H. Specific interactions between the syntrophin PDZ domain and voltagegated sodium channels. Nat. Struct. Biol. 1998, 5, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Matamoros, M.; Perez-Hernandez, M.; Guerrero-Serna, G.; Amoros, I.; Barana, A.; Nunez, M.; Ponce-Balbuena, D.; Sacristan, S.; Gomez, R.; Tamargo, J.; et al. Nav1.5 N-terminal domain binding to alpha1 syntrophin incraeses membrane density of human Kir2.1 Kir2.2 and Nav1.5. Cardiovasc. Res. 2016, 110, 279–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-hernández, M.; Caballero, R.; Delpón, E.; Pérez-hernández, M.; Matamoros, M.; Alfayate, S.; Nieto-marín, P.; Utrilla, R.G.; Tinaquero, D.; De Andrés, R.; et al. channels can trap cardiac Kir2.1/2.2 channels. JCI Insights 2018, 3, e96291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichel, C.A.; Beuriot, A.; Chevalier, M.Y.E.; Rougier, J.S.; Louault, F.; Dilanian, G.; Amour, J.; Coulombe, A.; Abriel, H.; Hatem, S.N.; et al. Lateral Membrane-Specific MAGUK CASK Down-Regulates Nav1.5 Channel in Cardiac Myocytes. Circ. Res. 2016, 119, 544–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziane, R.; Huang, H.; Moghadaszadeh, B.; Beggs, A.H.; Levesque, G.; Chahine, M. Cell membrane expression of cardiac sodium channel Nav1.5 is modulated by α-actinin-2 interaction. Biochemistry 2010, 49, 166–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzone, A.; Strege, P.R.; Tester, D.J.; Bernard, C.E.; Faulkner, G.; De Giorgio, R.; Makielski, J.C.; Stanghellini, V.; Gibbons, S.J.; Ackerman, M.J.; et al. A Mutation in Telethonin Alters Nav1.5 Function. J. Biol. Chem. 2008, 283, 16537–16544. [Google Scholar] [CrossRef] [Green Version]
- Turker, I.; Makiyama, T.; Ueyama, T.; Shimizu, A.; Yamakawa, M.; Chen, P.S.; Vatta, M.; Horie, M.; Ai, T. Telethonin Variants Found in Brugada Syndrome, J-Wave Pattern ECG, and ARVC Reduce Peak Nav1.5 Currents in HEK-293 Cells. PACE—Pacing Clin. Electrophysiol. 2020, 43, 838–846. [Google Scholar] [CrossRef]
- Goldfarb, M. Fibroblast growth factor homologous factors evolution, structure, and function. Cytokine Growth Factor Rev. 2005, 16, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Rush, A.M.; Wittmack, E.K.; Tyrrell, L.; Black, J.A.; Dib-hajj, S.D.; Waxman, S.G. Differential modulation of sodium channel Nav1.6 by two members of the fibroblast growth factor homologous factor 2 subfamily. Eur. J. Neurosci. 2006, 23, 2551–2562. [Google Scholar] [CrossRef]
- Liu, C.; Dib-Hajj, S.D.; Renganathan, M.; Cummins, T.R.; Waxman, S.G. Modulation of the cardiac sodium channel Nav1.5 by fibroblast growth factor homologous factor 1B. J. Biol. Chem. 2003, 278, 1029–1036. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Hennessey, J.A.; Kirkton, R.D.; Wang, C.; Graham, V.; Puranam, R.S.; Rosenberg, P.B.; Bursac, N.; Pitt, G.S. Fibroblast Growth Factor Homologous Factor 13 Regulates Na+ Channels and Conduction Velocity in Murine Hearts. Circ. Res. 2011, 109, 775–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Wang, C.; Hoch, E.G.; Pitt, G.S. Identification of Novel Interaction Sites that Determine Specificity between Fibroblast Growth Factor Homologous Factors and Voltage-gated Sodium Channels. J. Biol. Chem. 2011, 286, 24253–24263. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Wang, Z.; Sinden, D.S.; Wang, X.; Shan, B.; Yu, X.; Zhang, H.; Pitt, G.S.; Wang, C. FGF13 modulates the gating properties of the cardiac sodium channel Nav1.5 in an isoform-specific maner. Channels 2016, 10, 410–420. [Google Scholar] [CrossRef] [Green Version]
- Lou, J.Y.; Laezza, F.; Gerber, B.R.; Xiao, M.; Yamada, K.A.; Hartmann, H.; Craig, A.M.; Nerbonne, J.M.; Ornitz, D.M. Fibroblast growth factor 14 is an intracellular modulator of voltage-gated sodium channels. J. Physiol. 2005, 569, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Mohler, P.J.; Splawski, I.; Napolitano, C.; Bottelli, G.; Sharpe, L.; Timothy, K.; Priori, S.G.; Keating, M.T.; Bennett, V. A cardiac arrhythmia syndrome caused by loss of ankyrin-B function. Proc. Natl. Acad. Sci. USA 2004, 101, 9137–9142. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Chung, B.C.; Yan, H.; Wang, H.G.; Lee, S.Y.; Pitt, G.S. Structural analyses of Ca2+/CaM interaction with NaV channel C-termini reveal mechanisms of calcium-dependent regulation. Nat. Commun. 2014, 5, 4896. [Google Scholar] [CrossRef] [Green Version]
- Urbauer, J.L. Direct Sodium Channel Regulation by Calmodulin. Structure 2018, 26, 677–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, P.W.; Chakouri, N.; Diaz, J.; Tomaselli, G.F.; Yue, D.T.; Ben-Johny, M. Elementary mechanisms of calmodulin regulation of Nav1.5 producing divergent arrhythmogenic phenotypes. Proc. Natl. Acad. Sci. USA 2021, 118, e2025085118. [Google Scholar] [CrossRef] [PubMed]
- Turnow, K.; Metzner, K.; Cotella, D.; Morales, M.J.; Schaefer, M.; Christ, T.; Ravens, U.; Wettwer, E.; Kämmerer, S. Interaction of DPP10a with Kv4.3 channel complex results in a sustained current component of human transient outward current Ito. Basic Res. Cardiol. 2015, 110, 5. [Google Scholar] [CrossRef]
- Belau, F.; Metzner, K.; Christ, T.; Ravens, U.; Schaefer, M.; Künzel, S.; Li, W.; Wettwer, E.; Dobrev, D.; El-Armouche, A.; et al. DPP10 is a new regulator of Nav1.5 channels in human heart. Int. J. Cardiol. 2019, 284, 68–73. [Google Scholar] [CrossRef]
- Yarbrough, T.L.; Lu, T.; Lee, H.-C.; Shibata, E.F. Localization of Cardiac Sodium Channels in Caveolin-Rich Membrane Domains. Circ. Res. 2002, 90, 443–449. [Google Scholar] [CrossRef] [Green Version]
- Balijepalli, R.C.; Kamp, T.J. Caveolae, Ion Channels and Cardiac Arrhythmias. Prog. Biophys. Mol. Biol. 2008, 98, 149–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Y.F.; Kang, J.X.; Morgan, J.P.; Leaf, A. Blocking effects of polyunsaturated fatty acids on Na+ channels of neonatal rat ventricular myocytes. Proc. Natl. Acad. Sci. USA 1995, 92, 11000–11004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, J.X.; Xiao, Y.F.; Leaf, A. Free, long-chain, polyunsaturated fatty acids reduce membrane electrical excitability in neonatal rat cardiac myocytes. Proc. Natl. Acad. Sci. USA 1995, 92, 3997–4001. [Google Scholar] [CrossRef] [Green Version]
- Shibata, E.F.; Brown, T.L.Y.; Washburn, Z.W.; Bai, J.; Revak, T.J.; Butters, C.A. Autonomic Regulation of Voltage-Gated Cardiac Ion Channels. J. Cardiovasc. Electrophysiol. 2006, 17, 34–42. [Google Scholar] [CrossRef]
- Vatta, M.; Ackerman, M.J.; Ye, B.; Makielski, J.C.; Ughanze, E.E.; Taylor, E.W.; Tester, D.J.; Balijepalli, R.C.; Foell, J.D.; Li, Z.; et al. Mutant Caveolin-3 Induces Persistent Late Sodium Current and Is Associated With Long-QT Syndrome. Circulation 2006, 114, 2104–2112. [Google Scholar] [CrossRef] [Green Version]
- Vaidyanathan, R.; Reilly, L.; Eckhardt, L.L. Caveolin-3 Microdomain: Arrhythmia Implications for Potassium Inward Rectifier and Cardiac Sodium Channel. Front. Physiol. 2018, 9, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrlich, J.R. Taking ion channel degradation to heart. Cardiovasc. Res. 2007, 74, 6–7. [Google Scholar] [CrossRef]
- Abriel, H.; Staub, O. Ubiquitylation of ion channels. Physiology 2005, 20, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chen, Z.; Han, Z.; Liu, Y.; Wu, X.; Peng, Y.; Di, W.; Lan, R.; Sun, B.; Xu, B.; et al. AMPK-mediated degradation of Nav1.5 through autophagy. FASEB J. 2019, 33, 5366–5376. [Google Scholar] [CrossRef] [PubMed]
- Van Bemmelen, M.X.; Rougier, J.S.; Gavillet, B.; Apothéloz, F.; Daidié, D.; Tateyama, M.; Rivolta, I.; Thomas, M.A.; Kass, R.S.; Staub, O.; et al. Cardiac voltage-gated sodium channel Nav1.5 is regulated by Nedd4-2 mediated ubiquitination. Circ. Res. 2004, 95, 284–291. [Google Scholar] [CrossRef] [Green Version]
- Rougier, J.S.; Van Bemmelen, M.X.; Bruce, M.C.; Jespersen, T.; Gavillet, B.; Apothéloz, F.; Cordonier, S.; Staub, O.; Rotin, D.; Abriel, H. Molecular determinants of voltage-gated sodium channel regulation by the Nedd4/Nedd4-like proteins. Am. J. Physiol.—Cell Physiol. 2005, 288, 692–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Wang, Z.; Liu, Y.; Xiong, H.; Zhao, Y.; Wu, L.; Yuan, C.; Wang, L.; Hou, Y.; Yu, G.; et al. αB-crystallin interacts with Nav1.5 and regulates ubiquitination and internalization of cell surface Nav1.5. J. Biol. Chem. 2016, 291, 11030–11041. [Google Scholar] [CrossRef] [Green Version]
- Boehmer, C.; Wilhelm, V.; Palmada, M.; Wallisch, S.; Henke, G.; Brinkmeier, H.; Cohen, P.; Pieske, B.; Lang, F. Serum and glucocorticoid inducible kinases in the regulation of the cardiac sodium channel SCN5A. Cardiovasc. Res. 2003, 57, 1079–1084. [Google Scholar] [CrossRef] [Green Version]
- Tang, B.; Hu, Y.; Wang, Z.; Cheng, C.; Wang, P.; Liang, L.; Xiong, H.; Luo, C.; Xu, C.; Chen, Q.; et al. UBC9 regulates cardiac sodium channel Nav1.5 ubiquitination, degradation and sodium current density. J. Mol. Cell Cardiol. 2019, 129, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ge, J.; Chen, C.; Shen, Y.; Xie, J.; Zhu, X.; Liu, M.; Hu, J.; Chen, L.; Guo, L.; et al. FAT10 protects against ischemia-induced ventricular arrhythmia by decreasing Nedd4-2_Nav1.5 complex formation. Cell Death Dis. 2021, 12, 1–12. [Google Scholar]
- Bhalla, V.; Oyster, N.M.; Fitch, A.C.; Wijngaarden, M.A.; Neumann, D.; Schlattner, U.; Pearce, D.; Hallows, K.R. AMP-activated kinase inhibits the epithelial Na+ channel through functional regulation of the ubiquitin ligase Nedd4-2. J. Biol. Chem. 2006, 281, 26159–26169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tadros, R.; Ton, A.T.; Fiset, C.; Nattel, S. Sex differences in cardiac electrophysiology and clinical arrhythmias: Epidemiology, therapeutics, and mechanisms. Can. J. Cardiol. 2014, 30, 783–792. [Google Scholar] [CrossRef]
- Costa, S.; Saguner, A.M.; Gasperetti, A.; Akdis, D.; Brunckhorst, C.; Duru, F. The Link Between Sex Hormones and Susceptibility to Cardiac Arrhythmias: From Molecular Basis to Clinical Implications. Front. Cardiovasc. Med. 2021, 8, 85. [Google Scholar] [CrossRef]
- Gaborit, N.; Varro, A.; Le Bouter, S.; Szuts, V.; Escande, D.; Nattel, S.; Demolombe, S. Gender-related differences in ion-channel and transporter subunit expression in non-diseased human hearts. J. Mol. Cell. Cardiol. 2010, 49, 639–646. [Google Scholar] [CrossRef]
- Furukawa, T.; Kurokawa, J. Regulation of cardiac ion channels via non-genomic action of sex steroid hormones: Implication for the gender difference in cardiac arrhythmias. Pharmacol. Ther. 2007, 115, 106–115. [Google Scholar] [CrossRef]
- Clemens Moller, R.N. Effects of estradiol on cardiac ion channel currents. Eur. J. Pharmacol. 2006, 532, 44–49. [Google Scholar] [CrossRef]
- Yang, G.; Liu, J.; Wang, Y.; Du, Y.; Ma, A.; Wang, T. Lack of influence of sex hormones on Brugada syndrome-associated mutant Nav1.5 sodium channel. J. Electrocardiol. 2019, 52, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Peto, K.; Miko, I.; Ba, T.; To, I.B. Effects of sex hormones on ECG parameters and expression of cardiac ion channels in dogs. Acta Physiol. 2006, 188, 163–171. [Google Scholar] [CrossRef]
- Hu, X.; Fu, L.; Zhao, M.; Wang, D.; Zhang, H.; Gong, Z.; Ma, T.; Zhang, Y.; Machuki, J.; Pan, X.; et al. Sex hormones ameliorated sodium channel dysfunction induced by β-adrenergic overstimulation: The role of estrogen and G protein-coupled estrogen receptor. Authorea Prepr. 2020, 1–12. [Google Scholar] [CrossRef]
- Mangold, K.E.; Brumback, B.D.; Angsutararux, P.; Voelker, T.L.; Zhu, W.; Kang, P.W.; Moreno, J.D.; Silva, J.R. Mechanisms and models of cardiac sodium channel inactivation. Channels 2017, 11, 517–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nassal, M.M.J.; Wan, X.; Dale, Z.; Deschênes, I.; Wilson, L.D.; Piktel, J.S. Mild hypothermia preserves myocardial conduction during ischemia by maintaining gap junction intracellular communication and Na+ channel function. Am. J. Physiol. Heart Circ. Physiol. 2021, 312, 886–895. [Google Scholar] [CrossRef] [Green Version]
- Porres, J.M.; Brugada, J.; Urbistondo, V.; García, F.; Reviejo, K.; Marco, P. Fever unmasking the Brugada syndrome. Pacing Clin. Electrophysiol. 2002, 25, 1646–1648. [Google Scholar] [CrossRef] [PubMed]
- Abdelsayed, M.; Peters, C.H.; Ruben, P.C. Differential thermosensitivity in mixed syndrome cardiac sodium channel mutants. J. Physiol. 2015, 593, 4201–4223. [Google Scholar] [CrossRef]
- Dumaine, R.; Towbin, J.A.; Brugada, P.; Vatta, M.; Nesterenko, D.V.; Nesterenko, V.V.; Brugada, J.; Brugada, R.; Antzelevitch, C. Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are temperature dependent. Circ. Res. 1999, 85, 803–809. [Google Scholar] [CrossRef] [Green Version]
- Peters, C.H.; Abdelsayed, M.; Ruben, P.C. Triggers for arrhythmogenesis in the Brugada and long QT 3 syndromes. Prog. Biophys. Mol. Biol. 2016, 120, 77–88. [Google Scholar] [CrossRef]
- Kazutaka Gima and Yoram Rudy Ionic current basis of electrocardiographic waveforms: A model study. Circ. Res. 2007, 90, 889–896.
- Park, D.S.; Shekhar, A.; Marra, C.; Lin, X.; Vasquez, C.; Solinas, S.; Kelley, K.; Morley, G.; Goldfarb, M.; Fishman, G.I. Fhf2 gene deletion causes temperature-sensitive cardiac conduction failure. Nat. Commun. 2016, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.K.; Peters, C.H.; Tolhurst, S.A.; Claydon, T.W.; Ruben, P.C. Extracellular proton modulation of the cardiac voltage-gated sodium channel, Nav1.5. Biophys. J. 2011, 101, 2147–2156. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.K.; Claydon, T.W.; Ruben, P.C. Extracellular protons inhibit charge immobilization in the cardiac voltage-gated sodium channel. Biophys. J. 2013, 105, 101–107. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.K.; Peters, C.H.; Allard, C.R.; Claydon, T.W.; Ruben, P.C. Proton Sensors in the Pore Domain of the Cardiac Voltage-gated Sodium Channel. J. Biol. Chem. 2013, 288, 4782–4791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Shen, J.; Li, Z.; Timothy, K.; Vincent, G.M.; Priori, S.G.; Schwartz, P.J.; Keating, M.T. Cardiac sodium channel mutations in patients with long QT syndrome, an inherited cardiac arrhythmia. Hum. Mol. Genet. 1995, 4, 1603–1607. [Google Scholar] [CrossRef]
- Wang, D.W.; Yazawa, K.; George, A.L.; Bennett, P.B. Characterization of human cardiac Na+ channel mutations in the congenital long QT syndrome. Proc. Natl. Acad. Sci. USA 1996, 93, 13200–13205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilde, A.A.M.; Amin, A.S. Clinical Spectrum of SCN5A Mutations. JACC Clin. Electrophysiol. 2018, 4, 569–579. [Google Scholar] [CrossRef]
- Tester, D.J.; Ackerman, M.J. Genetics of Long QT Syndrome. Methodist Debakey Cardiovasc. J. 2014, 10, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Riera, A.R.; Barbosa-Barros, R.; Daminello Raimundo, R.; da Costa de Rezende Barbosa, M.P.; Esposito Sorpreso, I.C.; de Abreu, L.C. The congenital long QT syndrome Type 3: An update. Indian Pacing Electrophysiol. J. 2018, 18, 25–35. [Google Scholar] [CrossRef]
- Remme, C.A. Cardiac sodium channelopathy associated with SCN5A mutations: Electrophysiological, molecular and genetic aspects. J. Physiol. 2013, 591, 4099–4116. [Google Scholar] [CrossRef]
- Riuró, H.; Campuzano, O.; Arbelo, E.; Iglesias, A.; Batlle, M.; Pérez-Villa, F.; Brugada, J.; Pérez, G.J.; Scornik, F.S.; Brugada, R. A missense mutation in the sodium channel β1b subunit reveals SCN1B as a susceptibility gene underlying long QT syndrome. Heart Rhythm 2014, 11, 1202–1209. [Google Scholar] [CrossRef]
- Adler, A.; Novelli, V.; Amin, A.S.; Abiusi, E.; Care, M.; Nannenberg, E.A.; Feilotter, H.; Amenta, S.; Mazza, D.; Bikker, H.; et al. An International, Multicentered, Evidence-Based Reappraisal of Genes Reported to Cause Congenital Long QT Syndrome. Circulation 2020, 141, 418–428. [Google Scholar] [CrossRef]
- Choi, J.-I.; Wang, C.; Thomas, M.J.; Pitt, G.S. α1-Syntrophin Variant Identified in Drug-Induced Long QT Syndrome Increases Late Sodium Current. PLoS ONE 2016, 11, e0152355. [Google Scholar] [CrossRef]
- Ueda, K.; Valdivia, C.; Medeiros-Domingo, A.; Tester, D.J.; Vatta, M.; Farrugia, G.; Ackerman, M.J.; Makielski, J.C. Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex. Proc. Natl. Acad. Sci. USA 2008, 105, 9355–9360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.; Ai, T.; Kim, J.J.; Mohapatra, B.; Xi, Y.; Li, Z.; Abbasi, S.; Purevjav, E.; Samani, K.; Ackerman, M.J.; et al. α-1-Syntrophin Mutation and the Long-QT Syndrome. Circ. Arrhythmia Electrophysiol. 2008, 1, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Hu, R.-M.; Tan, B.-H.; Orland, K.M.; Valdivia, C.R.; Peterson, A.; Pu, J.; Makielski, J.C. Digenic inheritance novel mutations in SCN5A and SNTA1 increase late INa contributing to LQT syndrome. Am. J. Physiol. Circ. Physiol. 2013, 304, H994–H1001. [Google Scholar] [CrossRef] [Green Version]
- Kostera-Pruszczyk, A.; Suszek, M.; Płoski, R.; Franaszczyk, M.; Potulska-Chromik, A.; Pruszczyk, P.; Sadurska, E.; Karolczak, J.; Kamińska, A.M.; Rędowicz, M.J. BAG3-related myopathy, polyneuropathy and cardiomyopathy with long QT syndrome. J. Muscle Res. Cell Motil. 2015, 36, 423–432. [Google Scholar] [CrossRef] [Green Version]
- Prondzynski, M.; Lemoine, M.D.; Zech, A.T.; Horváth, A.; Di Mauro, V.; Koivumäki, J.T.; Kresin, N.; Busch, J.; Krause, T.; Krämer, E.; et al. Disease modeling of a mutation in α-actinin 2 guides clinical therapy in hypertrophic cardiomyopathy. EMBO Mol. Med. 2019, 11, e11115. [Google Scholar] [CrossRef]
- Markunas, A.M.; Manivannan, P.K.R.; Ezekian, J.E.; Agarwal, A.; Eisner, W.; Alsina, K.; Allen, H.D.; Wray, G.A.; Kim, J.J.; Wehrens, X.H.T.; et al. TBX5-encoded T-box transcription factor 5 variant T223M is associated with long QT syndrome and pediatric sudden cardiac death. Am. J. Med. Genet. Part A 2021, 185, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Marín, P.; Tinaquero, D.; Utrilla, R.G.; Cebrián, J.; González-Guerra, A.; Crespo-García, T.; Cámara-Checa, A.; Rubio-Alarcón, M.; Dago, M.; Alfayate, S.; et al. Tbx5 variants disrupt Nav1.5 function differently in patients diagnosed with Brugada or Long QT Syndrome. Cardiovasc. Res. 2021, cvab045. [Google Scholar] [CrossRef]
- Splawski, I.; Shen, J.; Timothy, K.W.; Lehmann, M.H.; Priori, S.; Robinson, J.L.; Moss, A.J.; Schwartz, P.J.; Towbin, J.A.; Vincent, G.M.; et al. Spectrum of Mutations in Long-QT Syndrome Genes. Circulation 2000, 102, 1178–1185. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.-K.; MacCormick, J.M.; McCulley, C.H.; Crawford, J.; Eddy, C.-A.; Mitchell, E.A.; Shelling, A.N.; French, J.K.; Skinner, J.R.; Rees, M.I. Long QT and Brugada syndrome gene mutations in New Zealand. Heart Rhythm 2007, 4, 1306–1314. [Google Scholar] [CrossRef]
- Chang, R.-K.R.; Lan, Y.-T.; Silka, M.J.; Morrow, H.; Kwong, A.; Smith-Lang, J.; Wallerstein, R.; Lin, H.J. Genetic Variants for Long QT Syndrome among Infants and Children from a Statewide Newborn Hearing Screening Program Cohort. J. Pediatr. 2014, 164, 590–595.e3. [Google Scholar] [CrossRef] [Green Version]
- Murphy, L.L.; Moon-Grady, A.J.; Cuneo, B.F.; Wakai, R.T.; Yu, S.; Kunic, J.D.; Benson, D.W.; George, A.L. Developmentally regulated SCN5A splice variant potentiates dysfunction of a novel mutation associated with severe fetal arrhythmia. Heart Rhythm 2012, 9, 590–597. [Google Scholar] [CrossRef] [Green Version]
- Hu, D.; Barajas-Martínez, H.; Medeiros-Domingo, A.; Crotti, L.; Veltmann, C.; Schimpf, R.; Urrutia, J.; Alday, A.; Casis, O.; Pfeiffer, R.; et al. A novel rare variant in SCN1Bb linked to Brugada syndrome and SIDS by combined modulation of Nav1.5 and Kv4.3 channel currents. Heart Rhythm 2012, 9, 760–769. [Google Scholar] [CrossRef] [Green Version]
- Rossenbacker, T. Unconventional intronic splice site mutation in SCN5A associates with cardiac sodium channelopathy. J. Med. Genet. 2005, 42, e29. [Google Scholar] [CrossRef] [Green Version]
- Casini, S.; Albesa, M.; Wang, Z.; Portero, V.; Ross-Kaschitza, D.; Rougier, J.-S.; Marchal, G.A.; Chung, W.K.; Bezzina, C.R.; Abriel, H.; et al. Functional Consequences of the SCN5A-p.Y1977N Mutation within the PY Ubiquitylation Motif: Discrepancy between HEK293 Cells and Transgenic Mice. Int. J. Mol. Sci. 2019, 20, 5033. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Han, Z.; Dai, J.; Cao, K. Brugada Syndrome Caused by Sodium Channel Dysfunction Associated with a SCN1B Variant A197V. Arch. Med. Res. 2020, 51, 245–253. [Google Scholar] [CrossRef]
- Liu, L.; Hayashi, K.; Kaneda, T.; Ino, H.; Fujino, N.; Uchiyama, K.; Konno, T.; Tsuda, T.; Kawashiri, M.; Ueda, K.; et al. A novel mutation in the transmembrane nonpore region of the KCNH2 gene causes severe clinical manifestations of long QT syndrome. Heart Rhythm 2013, 10, 61–67. [Google Scholar] [CrossRef]
- Rivolta, I.; Abriel, H.; Tateyama, M.; Liu, H.; Memmi, M.; Vardas, P.; Napolitano, C.; Priori, S.G.; Kass, R.S. Inherited Brugada and Long QT-3 Syndrome Mutations of a Single Residue of the Cardiac Sodium Channel Confer Distinct Channel and Clinical Phenotypes. J. Biol. Chem. 2001, 276, 30623–30630. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Tateyama, M.; Clancy, C.E.; Abriel, H.; Kass, R.S. Channel Openings Are Necessary but not Sufficient for Use-dependent Block of Cardiac Na+ Channels by Flecainide. J. Gen. Physiol. 2002, 120, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Davis, R.P.; Casini, S.; van den Berg, C.W.; Hoekstra, M.; Remme, C.A.; Dambrot, C.; Salvatori, D.; Oostwaard, D.W.; Wilde, A.A.M.; Bezzina, C.R.; et al. Cardiomyocytes Derived From Pluripotent Stem Cells Recapitulate Electrophysiological Characteristics of an Overlap Syndrome of Cardiac Sodium Channel Disease. Circulation 2012, 125, 3079–3091. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, Y.; Yang, J.; Xu, B.; Wen, Y.; Xiang, G.; Wei, G.; Zhu, C.; Xing, Y.; Li, Y. Electrophysiological and trafficking defects of the SCN5A T353I mutation in Brugada syndrome are rescued by alpha-allocryptopine. Eur. J. Pharmacol. 2015, 746, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Bonnin, B.; Rinné, S.; Moss, R.; Streit, A.K.; Scharf, M.; Richter, K.; Stöber, A.; Pfeufer, A.; Seemann, G.; Kääb, S.; et al. Electrophysiological characterization of a large set of novel variants in the SCN5A-gene: Identification of novel LQTS3 and BrS mutations. Pflügers Arch.—Eur. J. Physiol. 2016, 468, 1375–1387. [Google Scholar] [CrossRef]
- El-Battrawy, I.; Müller, J.; Zhao, Z.; Cyganek, L.; Zhong, R.; Zhang, F.; Kleinsorge, M.; Lan, H.; Li, X.; Xu, Q.; et al. Studying Brugada Syndrome With an SCN1B Variants in Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Front. Cell Dev. Biol. 2019, 7, 261. [Google Scholar] [CrossRef] [PubMed]
- Gray, B.; Hasdemir, C.; Ingles, J.; Aiba, T.; Makita, N.; Probst, V.; Wilde, A.A.M.; Newbury-Ecob, R.; Sheppard, M.N.; Semsarian, C.; et al. Lack of genotype-phenotype correlation in Brugada Syndrome and Sudden Arrhythmic Death Syndrome families with reported pathogenic SCN1B variants. Heart Rhythm 2018, 15, 1051–1057. [Google Scholar] [CrossRef] [Green Version]
- Ricci, M.T.; Menegon, S.; Vatrano, S.; Mandrile, G.; Cerrato, N.; Carvalho, P.; De Marchi, M.; Gaita, F.; Giustetto, C.; Giachino, D.F. SCN1B gene variants in Brugada Syndrome: A study of 145 SCN5A-negative patients. Sci. Rep. 2015, 4, 6470. [Google Scholar] [CrossRef]
- Ogawa, R.; Kishi, R.; Takagi, A.; Sakaue, I.; Takahashi, H.; Matsumoto, N.; Masuhara, K.; Nakazawa, K.; Kobayashi, S.; Miyake, F.; et al. A novel microsatellite polymorphism of sodium channel beta1-subunit gene (SCN1B) may underlie abnormal cardiac excitation manifested by coved-type ST-elevation compatible with Brugada syndrome in Japanese. Int. J. Clin. Pharmacol. Ther. 2010, 48, 109–119. [Google Scholar] [CrossRef]
- Olesen, M.S.; Holst, A.G.; Svendsen, J.H.; Haunsø, S.; Tfelt-Hansen, J. SCN1Bb R214Q found in 3 patients: 1 with Brugada syndrome and 2 with lone atrial fibrillation. Heart Rhythm 2012, 9, 770–773. [Google Scholar] [CrossRef]
- Peters, S. Arrhythmogenic cardiomyopathy and provocable Brugada ECG in a patient caused by missense mutation in plakophilin-2. Int. J. Cardiol. 2014, 173, 317–318. [Google Scholar] [CrossRef]
- Forkmann, M.; Tomala, J.; Huo, Y.; Mayer, J.; Christoph, M.; Wunderlich, C.; Salmas, J.; Gaspar, T.; Piorkowski, C. Epicardial Ventricular Tachycardia Ablation in a Patient With Brugada ECG Pattern and Mutation of PKP2 and DSP Genes. Circ. Arrhythmia Electrophysiol. 2015, 8, 505–507. [Google Scholar] [CrossRef] [Green Version]
- Kattygnarath, D.; Maugenre, S.; Neyroud, N.; Balse, E.; Ichai, C.; Denjoy, I.; Dilanian, G.; Martins, R.P.; Fressart, V.; Berthet, M.; et al. MOG1. Circ. Cardiovasc. Genet. 2011, 4, 261–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Wang, L.; Zuo, M.; Wang, X.; Ahmed, A.S.I.; Chen, Q.; Wang, Q.K. Cardiac sodium channel regulator MOG1 regulates cardiac morphogenesis and rhythm. Sci. Rep. 2016, 6, 21538. [Google Scholar] [CrossRef] [Green Version]
- Olesen, M.S.; Jensen, N.F.; Holst, A.G.; Nielsen, J.B.; Tfelt-Hansen, J.; Jespersen, T.; Sajadieh, A.; Haunsø, S.; Lund, J.T.; Calloe, K.; et al. A Novel Nonsense Variant in Nav1.5 Cofactor MOG1 Eliminates Its Sodium Current Increasing Effect and May Increase the Risk of Arrhythmias. Can. J. Cardiol. 2011, 27, 523.e17–523.e23. [Google Scholar] [CrossRef] [PubMed]
- Park, D.S.; Shekhar, A.; Santucci, J.; Redel-Traub, G.; Solinas, S.; Mintz, S.; Lin, X.; Chang, E.W.; Narke, D.; Xia, Y.; et al. Ionic Mechanisms of Impulse Propagation Failure in the FHF2-Deficient Heart. Circ. Res. 2020, 127, 1536–1548. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C.; Wiedmann, F.; El-Battrawy, I.; Fritz, M.; Ratte, A.; Beller, C.J.; Lang, S.; Rudic, B.; Schimpf, R.; Akin, I.; et al. Reduced Na+ Current in Native Cardiomyocytes of a Brugada Syndrome Patient Associated With β-2-Syntrophin Mutation. Circ. Genom. Precis. Med. 2018, 11, e002263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, A.; Zankov, D.P.; Sato, A.; Komeno, M.; Toyoda, F.; Yamazaki, S.; Makita, T.; Noda, T.; Ikawa, M.; Asano, Y.; et al. Identification of transmembrane protein 168 mutation in familial Brugada syndrome. FASEB J. 2020, 34, 6399–6417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.K.C.; Shimizu, A.; Soh, J.E.C.; Komeno, M.; Sato, A.; Ogita, H. Transmembrane protein 168 mutation reduces cardiomyocyte cell surface expression of Nav1.5 through αB-crystallin intracellular dynamics. J. Biochem. 2021, 170, mvab066. [Google Scholar] [CrossRef]
- Di Resta, C.; Pietrelli, A.; Sala, S.; Della Bella, P.; De Bellis, G.; Ferrari, M.; Bordoni, R.; Benedetti, S. High-throughput genetic characterization of a cohort of Brugada syndrome patients. Hum. Mol. Genet. 2015, 24, 5828–5835. [Google Scholar] [CrossRef] [Green Version]
- Musa, H.; Marcou, C.A.; Herron, T.J.; Makara, M.A.; Tester, D.J.; O’Connell, R.P.; Rosinski, B.; Guerrero-Serna, G.; Milstein, M.L.; Monteiro da Rocha, A.; et al. Abnormal myocardial expression of SAP97 is associated with arrhythmogenic risk. Am. J. Physiol. Circ. Physiol. 2020, 318, H1357–H1370. [Google Scholar] [CrossRef]
- Wang, Z.; Vermij, S.H.; Sottas, V.; Shestak, A.; Ross-Kaschitza, D.; Zaklyazminskaya, E.V.; Hudmon, A.; Pitt, G.S.; Rougier, J.S.; Abriel, H. Calmodulin binds to the N-terminal domain of the cardiac sodium channel Nav1.5. Channels 2020, 14, 268–286. [Google Scholar] [CrossRef]
- Jiménez-Jáimez, J.; Palomino Doza, J.; Ortega, Á.; Macías-Ruiz, R.; Perin, F.; Rodríguez-Vázquez del Rey, M.M.; Ortiz-Genga, M.; Monserrat, L.; Barriales-Villa, R.; Blanca, E.; et al. Calmodulin 2 Mutation N98S Is Associated with Unexplained Cardiac Arrest in Infants Due to Low Clinical Penetrance Electrical Disorders. PLoS ONE 2016, 11, e0153851. [Google Scholar] [CrossRef] [PubMed]
- Yagihara, N.; Watanabe, H.; Barnett, P.; Duboscq-Bidot, L.; Thomas, A.C.; Yang, P.; Ohno, S.; Hasegawa, K.; Kuwano, R.; Chatel, S.; et al. Variants in the SCN5A Promoter Associated with Various Arrhythmia Phenotypes. J. Am. Heart Assoc. 2016, 5, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Kimura, Y.; Aiba, T.; Sasano, T.; Furukawa, T.; Kusano, K.; Shimizu, W. IRX3 variant as a modifier of Brugada syndrome with frequent ventricular fibrillation. Heart Case Rep. 2016, 2, 465–468. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.; Koopmann, T.T.; Pfeufer, A.; Jalilzadeh, S.; Schulze-Bahr, E.; Kääb, S.; Wilde, A.A.; Roden, D.M.; Bezzina, C.R. Polymorphisms in the cardiac sodium channel promoter displaying variant in vitro expression activity. Eur. J. Hum. Genet. 2008, 16, 350–357. [Google Scholar] [CrossRef] [Green Version]
- Matsumura, H.; Nakano, Y.; Ochi, H.; Onohara, Y.; Sairaku, A.; Tokuyama, T.; Tomomori, S.; Motoda, C.; Amioka, M.; Hironobe, N.; et al. H558R, a common SCN5A polymorphism, modifies the clinical phenotype of Brugada syndrome by modulating DNA methylation of SCN5A promoters. J. Biomed. Sci. 2017, 24, 91. [Google Scholar] [CrossRef]
- Scumaci, D.; Oliva, A.; Concolino, A.; Curcio, A.; Fiumara, C.V.; Tammè, L.; Campuzano, O.; Pascali, V.L.; Coll, M.; Iglesias, A.; et al. Integration of “Omics” Strategies for Biomarkers Discovery and for the Elucidation of Molecular Mechanisms Underlying Brugada Syndrome. Proteom.—Clin. Appl. 2018, 12, 1800065. [Google Scholar] [CrossRef]
- Aiba, T.; Farinelli, F.; Kostecki, G.; Hesketh, G.G.; Edwards, D.; Biswas, S.; Tung, L.; Tomaselli, G.F. A Mutation Causing Brugada Syndrome Identifies a Mechanism for Altered Autonomic and Oxidant Regulation of Cardiac Sodium Currents. Circ. Cardiovasc. Genet. 2014, 7, 249–256. [Google Scholar] [CrossRef] [Green Version]
- Darbar, D.; Kannankeril, P.J.; Donahue, B.S.; Kucera, G.; Stubblefield, T.; Haines, J.L.; George, A.L.J.; Roden, D.M. Cardiac sodium channel (SCN5A) variants associated with atrial fibrillation. Circulation 2008, 117, 1927–1935. [Google Scholar] [CrossRef] [Green Version]
- Takehara, N.; Makita, N.; Kawabe, J.; Sato, N.; Kawamura, Y.; Kitabatake, A.; Kikuchi, K. A cardiac sodium channel mutation identified in Brugada syndrome associated with atrial standstill. J. Intern. Med. 2004, 255, 137–142. [Google Scholar] [CrossRef] [Green Version]
- Laitinen-Forsblom, P.J.; Makynen, P.; Makynen, H.; Yli-Mayry, S.; Virtanen, V.; Kontula, K.; Aalto-Setala, K. SCN5A Mutation Associated with Cardiac Conduction Defect and Atrial Arrhythmias. J. Cardiovasc. Electrophysiol. 2006, 17, 480–485. [Google Scholar] [CrossRef]
- Benito, B.; Brugada, R.; Perich, R.M.; Lizotte, E.; Cinca, J.; Mont, L.; Berruezo, A.; Tolosana, J.M.; Freixa, X.; Brugada, P.; et al. A mutation in the sodium channel is responsible for the association of long QT syndrome and familial atrial fibrillation. Heart Rhythm 2008, 5, 1434–1440. [Google Scholar] [CrossRef]
- Rossenbacker, T.; Carroll, S.J.; Liu, H.; Kuipéri, C.; de Ravel, T.J.L.; Devriendt, K.; Carmeliet, P.; Kass, R.S.; Heidbüchel, H. Novel pore mutation in SCN5A manifests as a spectrum of phenotypes ranging from atrial flutter, conduction disease, and Brugada syndrome to sudden cardiac death. Heart Rhythm 2004, 1, 610–615. [Google Scholar] [CrossRef]
- Watanabe, H.; Darbar, D.; Kaiser, D.W.; Jiramongkolchai, K.; Chopra, S.; Donahue, B.S.; Kannankeril, P.J.; Roden, D.M. Mutations in sodium channel β1- and β2-subunits associated with atrial fibrillation. Circ. Arrhythmia Electrophysiol. 2009, 2, 268–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, H.; Yang, Q.; Zhang, X.; Wang, P.; Chen, F.; Liu, Y.; Wang, P.; Zhao, Y.; Li, S.; Huang, Y.; et al. Significant association of rare variant p.Gly8Ser in cardiac sodium channel β4-subunit SCN4B with atrial fibrillation. Ann. Hum. Genet. 2019, 83, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Li, R.-G.; Wang, Q.; Xu, Y.-J.; Zhang, M.; Qu, X.-K.; Liu, X.; Fang, W.-Y.; Yang, Y.-Q. Mutations of the SCN4B-encoded sodium channel β4 subunit in familial atrial fibrillation. Int. J. Mol. Med. 2013, 32, 144–150. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Yang, Q.; Wu, X.; Yang, Y.; Shi, L.; Wang, C.; Wu, G.; Xia, Y.; Yang, B.; Zhang, R.; et al. Functional dominant-negative mutation of sodium channel subunit gene SCN3B associated with atrial fibrillation in a Chinese GeneID population. Biochem. Biophys. Res. Commun. 2010, 398, 98–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Husser, D.; Ueberham, L.; Hindricks, G.; Büttner, P.; Ingram, C.; Weeke, P.; Shoemaker, M.B.; Adams, V.; Arya, A.; Sommer, P.; et al. Rare variants in genes encoding the cardiac sodium channel and associated compounds and their impact on outcome of catheter ablation of atrial fibrillation. PLoS ONE 2017, 12, e0183690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunha, S.R.; Hund, T.J.; Hashemi, S.; Voigt, N.; Li, N.; Wright, P.; Koval, O.; Li, J.; Gudmundsson, H.; Gumina, R.J.; et al. Defects in Ankyrin-Based Membrane Protein Targeting Pathways Underlie Atrial Fibrillation. Circulation 2011, 124, 1212–1222. [Google Scholar] [CrossRef] [Green Version]
- Robaei, D.; Ford, T.; Ooi, S.-Y. Ankyrin-B Syndrome: A Case of Sinus Node Dysfunction, Atrial Fibrillation and Prolonged QT in a Young Adult. Heart Lung Circ. 2015, 24, e31–e34. [Google Scholar] [CrossRef]
- Girolami, F.; Iascone, M.; Tomberli, B.; Bardi, S.; Benelli, M.; Marseglia, G.; Pescucci, C.; Pezzoli, L.; Sana, M.E.; Basso, C.; et al. Novel α-Actinin 2 Variant Associated With Familial Hypertrophic Cardiomyopathy and Juvenile Atrial Arrhythmias. Circ. Cardiovasc. Genet. 2014, 7, 741–750. [Google Scholar] [CrossRef]
- LIU, Y.; NI, B.; LIN, Y.; CHEN, X.-G.; CHEN, M.; HU, Z.; ZHANG, F. The rs3807989 G/A Polymorphism in CAV1 is Associated with the Risk of Atrial Fibrillation in Chinese Han Populations. Pacing Clin. Electrophysiol. 2015, 38, 164–170. [Google Scholar] [CrossRef]
- Watanabe, H.; Nogami, A.; Ohkubo, K.; Kawata, H.; Hayashi, Y.; Ishikawa, T.; Makiyama, T.; Nagao, S.; Yagihara, N.; Takehara, N.; et al. Electrocardiographic Characteristics and SCN5A Mutations in Idiopathic Ventricular Fibrillation Associated with Early Repolarization. Circ. Arrhythmia Electrophysiol. 2011, 4, 874–881. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, H.; Ohkubo, K.; Watanabe, I.; Matsuyama, T.; Ishibashi-Ueda, H.; Yagihara, N.; Shimizu, W.; Horie, M.; Minamino, T.; Makita, N. SCN5A mutation associated with ventricular fibrillation, early repolarization, and concealed myocardial abnormalities. Int. J. Cardiol. 2013, 165, e21–e23. [Google Scholar] [CrossRef]
- Reithmann, C.; Beckmann, B.-M.; Kääb, S. Purkinje-related ventricular fibrillation associated with a homozygous H558R polymorphism in the sodium channel SCN5A gene. Europace 2016, 18, 896. [Google Scholar] [CrossRef] [PubMed]
- Vilin, Y.; Fujimoto, E.; Ruben, P. A novel mechanism associated with idiopathic ventricular fibrillation (IVF) mutations R1232W and T1620M in human cardiac sodium channels. Pflügers Arch. Eur. J. Physiol. 2001, 442, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Akai, J.; Makita, N.; Sakurada, H.; Shirai, N.; Ueda, K.; Kitabatake, A.; Nakazawa, K.; Kimura, A.; Hiraoka, M. A novel SCN5A mutation associated with idiopathic ventricular fibrillation without typical ECG findings of Brugada syndrome. FEBS Lett. 2000, 479, 29–34. [Google Scholar] [CrossRef] [Green Version]
- Wan, X.; Chen, S.; Sadeghpour, A.; Wang, Q.; Kirsch, G.E. Accelerated inactivation in a mutant Na+ channel associated with idiopathic ventricular fibrillation. Am. J. Physiol.—Heart Circ. Physiol. 2001, 280, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Raharjo, S.B.; Maulana, R.; Maghfirah, I.; Alzahra, F.; Putrinarita, A.D.; Hanafy, D.A.; Yuniadi, Y. SCN 5A gene mutations and the risk of ventricular fibrillation and syncope in Brugada syndrome patients: A meta-analysis. J. Arrhythmia 2018, 34, 473–477. [Google Scholar] [CrossRef]
- Nishii, N.; Ogawa, M.; Morita, H.; Nakamura, K.; Banba, K.; Miura, D.; Kumagai, N.; Matsunaga, A.; Kawamura, H.; Urakawa, S.; et al. SCN5A Mutation Is Associated With Early and Frequent Recurrence of Ventricular Fibrillation in Patients With Brugada Syndrome. Circ. J. 2010, 74, 2572–2578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- KELLER, D.; HUANG, H.; ZHAO, J.; FRANK, R.; SUAREZ, V.; DELACRETAZ, E.; BRINK, M.; OSSWALD, S.; SCHWICK, N.; CHAHINE, M. A novel SCN5A mutation, F1344S, identified in a patient with Brugada syndrome and fever-induced ventricular fibrillation. Cardiovasc. Res. 2006, 70, 521–529. [Google Scholar] [CrossRef] [Green Version]
- Kawamura, M.; Ozawa, T.; Yao, T.; Ashihara, T.; Sugimoto, Y.; Yagi, T.; Itoh, H.; Ito, M.; Makiyama, T.; Horie, M. Dynamic Change in ST-Segment and Spontaneous Occurrence of Ventricular Fibrillation in Brugada Syndrome With a Novel Nonsense Mutation in the SCN5A Gene During Long-Term Follow-up. Circ. J. 2009, 73, 584–588. [Google Scholar] [CrossRef] [Green Version]
- Lieve, K.V.; Verkerk, A.O.; Podliesna, S.; van der Werf, C.; Tanck, M.W.; Hofman, N.; van Bergen, P.F.; Beekman, L.; Bezzina, C.R.; Wilde, A.A.M.; et al. Gain-of-function mutation in SCN5A causes ventricular arrhythmias and early onset atrial fibrillation. Int. J. Cardiol. 2017, 236, 187–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boehringer, T.; Bugert, P.; Borggrefe, M.; Elmas, E. SCN5A mutations and polymorphisms in patients with ventricular fibrillation during acute myocardial infarction. Mol. Med. Rep. 2014, 10, 2039–2044. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Liu, Y.; Liao, H.; Xue, Y.; Zhan, X.; Fang, X.; Liang, Y.; Wei, W.; Rao, F.; Zhang, Q.; et al. Genetic Variants on SCN5A, KCNQ1, and KCNH2 in Patients with Ventricular Arrhythmias during Acute Myocardial Infarction in a Chinese Population. Cardiology 2020, 145, 38–45. [Google Scholar] [CrossRef]
- Hu, D.; Viskin, S.; Oliva, A.; Carrier, T.; Cordeiro, J.M.; Barajas-Martinez, H.; Wu, Y.; Burashnikov, E.; Sicouri, S.; Brugada, R.; et al. Novel mutation in the SCN5A gene associated with arrhythmic storm development during acute myocardial infarction. Heart Rhythm 2007, 4, 1072–1080. [Google Scholar] [CrossRef] [Green Version]
- Ter Bekke, R.M.A.; Isaacs, A.; Barysenka, A.; Hoos, M.B.; Jongbloed, J.D.H.; Hoorntje, J.C.A.; Patelski, A.S.M.; Helderman-van den Enden, A.T.J.M.; van den Wijngaard, A.; Stoll, M.; et al. Heritability in a SCN5A -mutation founder population with increased female susceptibility to non-nocturnal ventricular tachyarrhythmia and sudden cardiac death. Heart Rhythm 2017, 14, 1873–1881. [Google Scholar] [CrossRef] [PubMed]
- Son, M.K.; Ki, C.-S.; Park, S.-J.; Huh, J.; Kim, J.S.; On, Y.K. Genetic Mutation in Korean Patients of Sudden Cardiac Arrest as a Surrogating Marker of Idiopathic Ventricular Arrhythmia. J. Korean Med. Sci. 2013, 28, 1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stawiarski, K.; Clarke, J.-R.D.; Pollack, A.; Winslow, R.; Majumdar, S. Ventricular fibrillation in Graves disease reveals a rare SCN5A mutation with W1191X variant associated with Brugada syndrome. Heart Case Rep. 2021, 7, 95–99. [Google Scholar] [CrossRef]
- Valdivia, C.R.; Medeiros-Domingo, A.; Ye, B.; Shen, W.K.; Algiers, T.J.; Ackerman, M.J.; Makielski, J.C. Loss-of-function mutation of the SCN3B-encoded sodium channel 3 subunit associated with a case of idiopathic ventricular fibrillation. Cardiovasc. Res. 2010, 86, 392–400. [Google Scholar] [CrossRef] [Green Version]
- Lasam, G.; Oaks, J.B. A Rare Desmoglein-2 Gene Mutation in Arrhythmogenic Right Ventricular Cardiomyopathy Inciting Incessant Ventricular Fibrillation. Cureus 2018, 10, e3388. [Google Scholar] [CrossRef] [Green Version]
- Rigato, I.; Bauce, B.; Rampazzo, A.; Zorzi, A.; Pilichou, K.; Mazzotti, E.; Migliore, F.; Marra, M.P.; Lorenzon, A.; De Bortoli, M.; et al. Compound and Digenic Heterozygosity Predicts Lifetime Arrhythmic Outcome and Sudden Cardiac Death in Desmosomal Gene–Related Arrhythmogenic Right Ventricular Cardiomyopathy. Circ. Cardiovasc. Genet. 2013, 6, 533–542. [Google Scholar] [CrossRef] [Green Version]
- Bhonsale, A.; Groeneweg, J.A.; James, C.A.; Dooijes, D.; Tichnell, C.; Jongbloed, J.D.H.; Murray, B.; te Riele, A.S.J.M.; van den Berg, M.P.; Bikker, H.; et al. Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated mutation carriers. Eur. Heart J. 2015, 36, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Cox, M.G.P.J.; van der Zwaag, P.A.; van der Werf, C.; van der Smagt, J.J.; Noorman, M.; Bhuiyan, Z.A.; Wiesfeld, A.C.P.; Volders, P.G.A.; van Langen, I.M.; Atsma, D.E.; et al. Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. Circulation 2011, 123, 2690–2700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsman, R.F.; Barc, J.; Beekman, L.; Alders, M.; Dooijes, D.; van den Wijngaard, A.; Ratbi, I.; Sefiani, A.; Bhuiyan, Z.A.; Wilde, A.A.M.; et al. A Mutation in CALM1 Encoding Calmodulin in Familial Idiopathic Ventricular Fibrillation in Childhood and Adolescence. J. Am. Coll. Cardiol. 2014, 63, 259–266. [Google Scholar] [CrossRef] [Green Version]
- Nomikos, M.; Thanassoulas, A.; Beck, K.; Vassilakopoulou, V.; Hu, H.; Calver, B.L.; Theodoridou, M.; Kashir, J.; Blayney, L.; Livaniou, E.; et al. Altered RyR2 regulation by the calmodulin F90L mutation associated with idiopathic ventricular fibrillation and early sudden cardiac death. FEBS Lett. 2014, 588, 2898–2902. [Google Scholar] [CrossRef] [Green Version]
- Bagnall, R.D.; Molloy, L.K.; Kalman, J.M.; Semsarian, C. Exome sequencing identifies a mutation in the ACTN2 gene in a family with idiopathic ventricular fibrillation, left ventricular noncompaction, and sudden death. BMC Med. Genet. 2014, 15, 99. [Google Scholar] [CrossRef] [Green Version]
- Butters, T.D.; Aslanidi, O.V.; Inada, S.; Boyett, M.R.; Hancox, J.C.; Lei, M.; Zhang, H. Mechanistic Links Between Na+ Channel (SCN5A) Mutations and Impaired Cardiac Pacemaking in Sick Sinus Syndrome. Circ. Res. 2010, 107, 126–137. [Google Scholar] [CrossRef] [Green Version]
- GUI, J.; WANG, T.; TRUMP, D.; ZIMMER, T.; LEI, M. Mutation-Specific Effects of Polymorphism H558R in SCN5A-Related Sick Sinus Syndrome. J. Cardiovasc. Electrophysiol. 2010, 21, 564–573. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Machida, T.; Sumitomo, N.; Yamamoto, H.; Ohkubo, K.; Watanabe, I.; Makiyama, T.; Fukae, S.; Kohno, M.; Harrell, D.T.; et al. Sodium Channelopathy Underlying Familial Sick Sinus Syndrome With Early Onset and Predominantly Male Characteristics. Circ. Arrhythmia Electrophysiol. 2014, 7, 511–517. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, S.; Makiyama, T.; Hanazawa, K.; Kaitani, K.; Amano, M.; Hayama, Y.; Onishi, N.; Tamaki, Y.; Miyake, M.; Tamura, T.; et al. A Novel SCN5A Mutation Demonstrating a Variety of Clinical Phenotypes in Familial Sick Sinus Syndrome. Intern. Med. 2013, 52, 1805–1808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, B.; Iturraldetorres, P.; Medeirosdomingo, A.; Nava, S.; Tester, D.; Valdivia, C.; Tusieluna, T.; Ackerman, M.; Makielski, J. A novel C-terminal truncation SCN5A mutation from a patient with sick sinus syndrome, conduction disorder and ventricular tachycardia. Cardiovasc. Res. 2007, 76, 409–417. [Google Scholar] [CrossRef] [Green Version]
- Kodama, T.; Serio, A.; Disertori, M.; Bronzetti, G.; Diegoli, M.; Narula, N.; Grasso, M.; Mazzola, S.; Arbustini, E. Autosomal recessive paediatric sick sinus syndrome associated with novel compound mutations in SCN5A. Int. J. Cardiol. 2013, 167, 3078–3080. [Google Scholar] [CrossRef] [PubMed]
- Holst, A.G.; Liang, B.; Jespersen, T.; Bundgaard, H.; Haunso, S.; Svendsen, J.H.; Tfelt-Hansen, J. Sick Sinus Syndrome, Progressive Cardiac Conduction Disease, Atrial Flutter and Ventricular Tachycardia Caused by a Novel SCN5A Mutation. Cardiology 2010, 115, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Alkorashy, M.; Al-Ghamdi, B.; Tulbah, S.; Al-Numair, N.S.; Alhadeq, F.; Takroni, S.A.; Al-Hassnan, Z.N. A novel homozygous SCN5A variant detected in sick sinus syndrome. Pacing Clin. Electrophysiol. 2021, 44, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Ziyadeh-Isleem, A.; Clatot, J.; Duchatelet, S.; Gandjbakhch, E.; Denjoy, I.; Hidden-Lucet, F.; Hatem, S.; Deschênes, I.; Coulombe, A.; Neyroud, N.; et al. A truncating SCN5A mutation combined with genetic variability causes sick sinus syndrome and early atrial fibrillation. Heart Rhythm 2014, 11, 1015–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ackerman, M.J. Postmortem Molecular Analysis of SCN5A Defects in Sudden Infant Death Syndrome. JAMA 2001, 286, 2264. [Google Scholar] [CrossRef]
- Wang, D.W.; Desai, R.R.; Crotti, L.; Arnestad, M.; Insolia, R.; Pedrazzini, M.; Ferrandi, C.; Vege, A.; Rognum, T.; Schwartz, P.J.; et al. Cardiac Sodium Channel Dysfunction in Sudden Infant Death Syndrome. Circulation 2007, 115, 368–376. [Google Scholar] [CrossRef] [Green Version]
- Turillazzi, E.; La Rocca, G.; Anzalone, R.; Corrao, S.; Neri, M.; Pomara, C.; Riezzo, I.; Karch, S.B.; Fineschi, V. Heterozygous nonsense SCN5A mutation W822X explains a simultaneous sudden infant death syndrome. Virchows Arch. 2008, 453, 209–216. [Google Scholar] [CrossRef]
- Van Norstrand, D.W.; Tester, D.J.; Ackerman, M.J. Overrepresentation of the proarrhythmic, sudden death predisposing sodium channel polymorphism S1103Y in a population-based cohort of African-American sudden infant death syndrome. Heart Rhythm 2008, 5, 712–715. [Google Scholar] [CrossRef] [Green Version]
- Wedekind, H.; Smits, J.P.P.; Schulze-Bahr, E.; Arnold, R.; Veldkamp, M.W.; Bajanowski, T.; Borggrefe, M.; Brinkmann, B.; Warnecke, I.; Funke, H.; et al. De Novo Mutation in the SCN5A Gene Associated With Early Onset of Sudden Infant Death. Circulation 2001, 104, 1158–1164. [Google Scholar] [CrossRef] [Green Version]
- Plant, L.D. A common cardiac sodium channel variant associated with sudden infant death in African Americans, SCN5A S1103Y. J. Clin. Investig. 2006, 116, 430–435. [Google Scholar] [CrossRef] [Green Version]
- Todd, S.J.; Campbell, M.J.; Roden, D.M.; Kannankeril, P.J. Novel Brugada SCN5A mutation causing sudden death in children. Heart Rhythm 2005, 2, 540–543. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, J.; Rougier, J.-S.; Abriel, H.; Haas, C. Functional implications of a rare variant in the sodium channel β1B subunit (SCN1B) in a 5-month-old male sudden infant death syndrome case. Heart Case Rep. 2018, 4, 187–190. [Google Scholar] [CrossRef] [Green Version]
- Tan, B.-H.; Pundi, K.N.; Van Norstrand, D.W.; Valdivia, C.R.; Tester, D.J.; Medeiros-Domingo, A.; Makielski, J.C.; Ackerman, M.J. Sudden infant death syndrome–associated mutations in the sodium channel beta subunits. Heart Rhythm 2010, 7, 771–778. [Google Scholar] [CrossRef] [Green Version]
- Denti, F.; Bentzen, B.H.; Wojciak, J.; Thomsen, N.M.; Scheinman, M.; Schmitt, N. Multiple genetic variations in sodium channel subunits in a case of sudden infant death syndrome. Pacing Clin. Electrophysiol. 2018, 41, 620–626. [Google Scholar] [CrossRef]
- Winkel, B.G.; Yuan, L.; Olesen, M.S.; Sadjadieh, G.; Wang, Y.; Risgaard, B.; Jabbari, R.; Haunsø, S.; Holst, A.G.; Hollegaard, M.V.; et al. The role of the sodium current complex in a nonreferred nationwide cohort of sudden infant death syndrome. Heart Rhythm 2015, 12, 1241–1249. [Google Scholar] [CrossRef]
- Makita, N.; Yagihara, N.; Crotti, L.; Johnson, C.N.; Beckmann, B.-M.; Roh, M.S.; Shigemizu, D.; Lichtner, P.; Ishikawa, T.; Aiba, T.; et al. Novel Calmodulin Mutations Associated With Congenital Arrhythmia Susceptibility. Circ. Cardiovasc. Genet. 2014, 7, 466–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.; Van Norstrand, D.W.; Medeiros-Domingo, A.; Tester, D.J.; Valdivia, C.R.; Tan, B.-H.; Vatta, M.; Makielski, J.C.; Ackerman, M.J. LQTS-associated mutation A257G in α1-syntrophin interacts with the intragenic variant P74L to modify its biophysical phenotype. Cardiogenetics 2011, 1, 13. [Google Scholar] [CrossRef] [PubMed]
- Cronk, L.B.; Ye, B.; Kaku, T.; Tester, D.J.; Vatta, M.; Makielski, J.C.; Ackerman, M.J. Novel mechanism for sudden infant death syndrome: Persistent late sodium current secondary to mutations in caveolin-3. Heart Rhythm 2007, 4, 161–166. [Google Scholar] [CrossRef] [Green Version]
- Veltmann, C.; Barajas-Martinez, H.; Wolpert, C.; Borggrefe, M.; Schimpf, R.; Pfeiffer, R.; Cáceres, G.; Burashnikov, E.; Antzelevitch, C.; Hu, D. Further Insights in the Most Common SCN5A Mutation Causing Overlapping Phenotype of Long QT Syndrome, Brugada Syndrome, and Conduction Defect. J. Am. Heart Assoc. 2016, 5, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyndt, F.; Probst, V.; Potet, F.; Demolombe, S.; Chevallier, J.C.; Baro, I.; Moisan, J.P.; Boisseau, P.; Schott, J.J.; Escande, D.; et al. Novel SCN5A mutation leading either to isolated cardiac conduction defect or Brugada syndrome in a large French family. Circulation 2001, 104, 3081–3086. [Google Scholar] [CrossRef] [Green Version]
- Hothi, S.S.; Ara, F.; Timperley, J. p.Y1449C SCN5A Mutation Associated with Overlap Disorder Comprising Conduction Disease, Brugada Syndrome, and Atrial Flutter. J. Cardiovasc. Electrophysiol. 2015, 26, 93–97. [Google Scholar] [CrossRef]
- Chen, S. SNP S1103Y in the cardiac sodium channel gene SCN5A is associated with cardiac arrhythmias and sudden death in a white family. J. Med. Genet. 2002, 39, 913–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Márquez, M.F.; Cruz-Robles, D.; Inés-Real, S.; Gallardo, G.J.; Gonzlez-Hermosillo, A.; Cárdenas, M.; Vargas-Alarcón, G. A novel SCN5A deletion mutation in a child with ventricular tachycardia, recurrent aborted sudden death, and Brugada electrocardiographic pattern. Arch. Cardiol. Mex. 2007, 77, 284–287. [Google Scholar] [PubMed]
- Patel, C.; Silcock, L.; McMullan, D.; Brueton, L.; Cox, H. TBX5 intragenic duplication: A family with an atypical Holt–Oram syndrome phenotype. Eur. J. Hum. Genet. 2012, 20, 863–869. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daimi, H.; Lozano-Velasco, E.; Aranega, A.; Franco, D. Genomic and Non-Genomic Regulatory Mechanisms of the Cardiac Sodium Channel in Cardiac Arrhythmias. Int. J. Mol. Sci. 2022, 23, 1381. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23031381
Daimi H, Lozano-Velasco E, Aranega A, Franco D. Genomic and Non-Genomic Regulatory Mechanisms of the Cardiac Sodium Channel in Cardiac Arrhythmias. International Journal of Molecular Sciences. 2022; 23(3):1381. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23031381
Chicago/Turabian StyleDaimi, Houria, Estefanía Lozano-Velasco, Amelia Aranega, and Diego Franco. 2022. "Genomic and Non-Genomic Regulatory Mechanisms of the Cardiac Sodium Channel in Cardiac Arrhythmias" International Journal of Molecular Sciences 23, no. 3: 1381. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23031381