
A Hybrid Machine Learning and Network Analysis Approach Reveals Two Parkinson’s Disease Subtypes from 115 RNA-Seq Post-Mortem Brain Samples

 , , , , ,
, , , , ,
Abstract
:
1. Introduction
2. Results
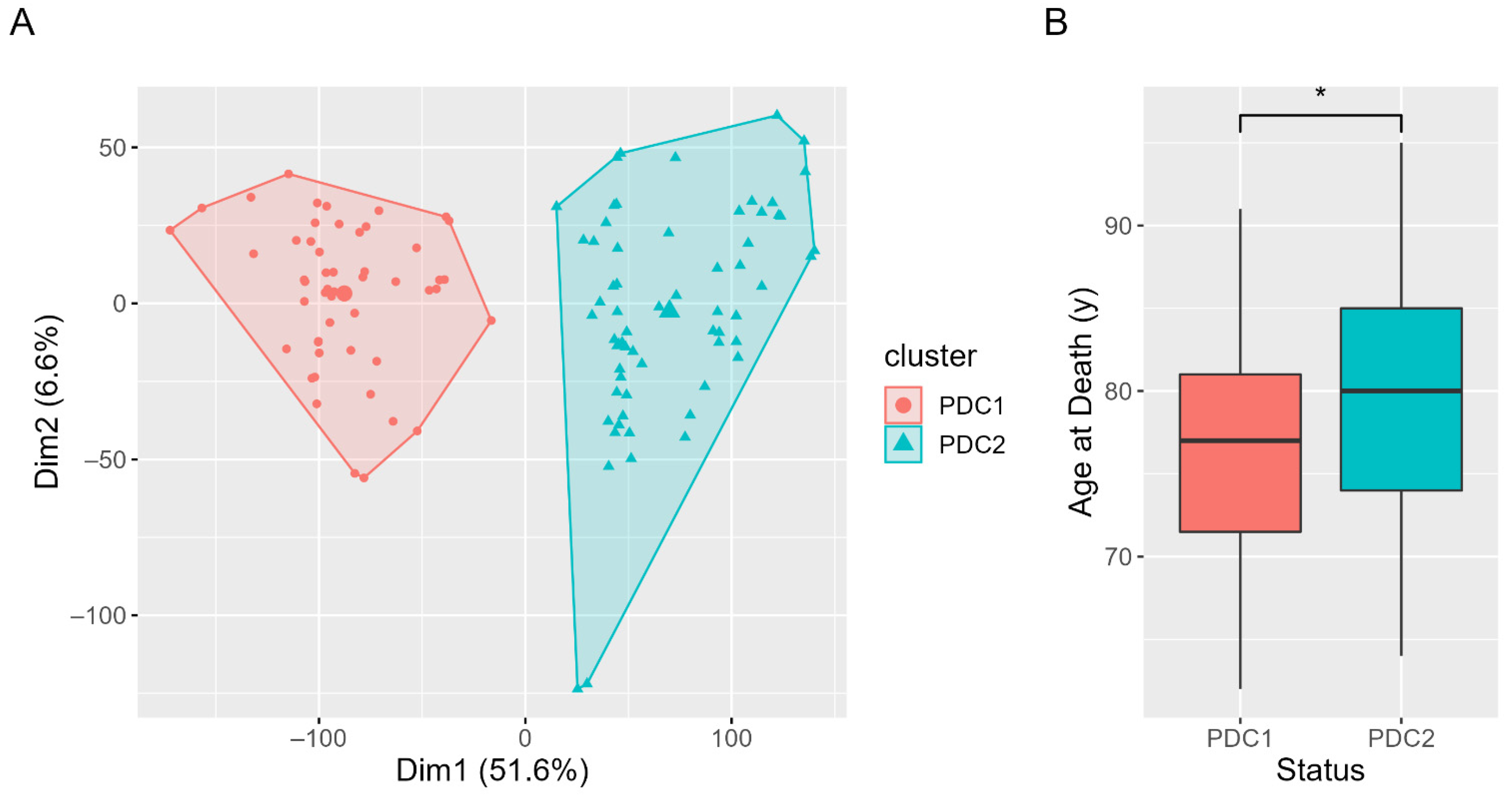
2.1. Clustering Results and Clinical Characterization
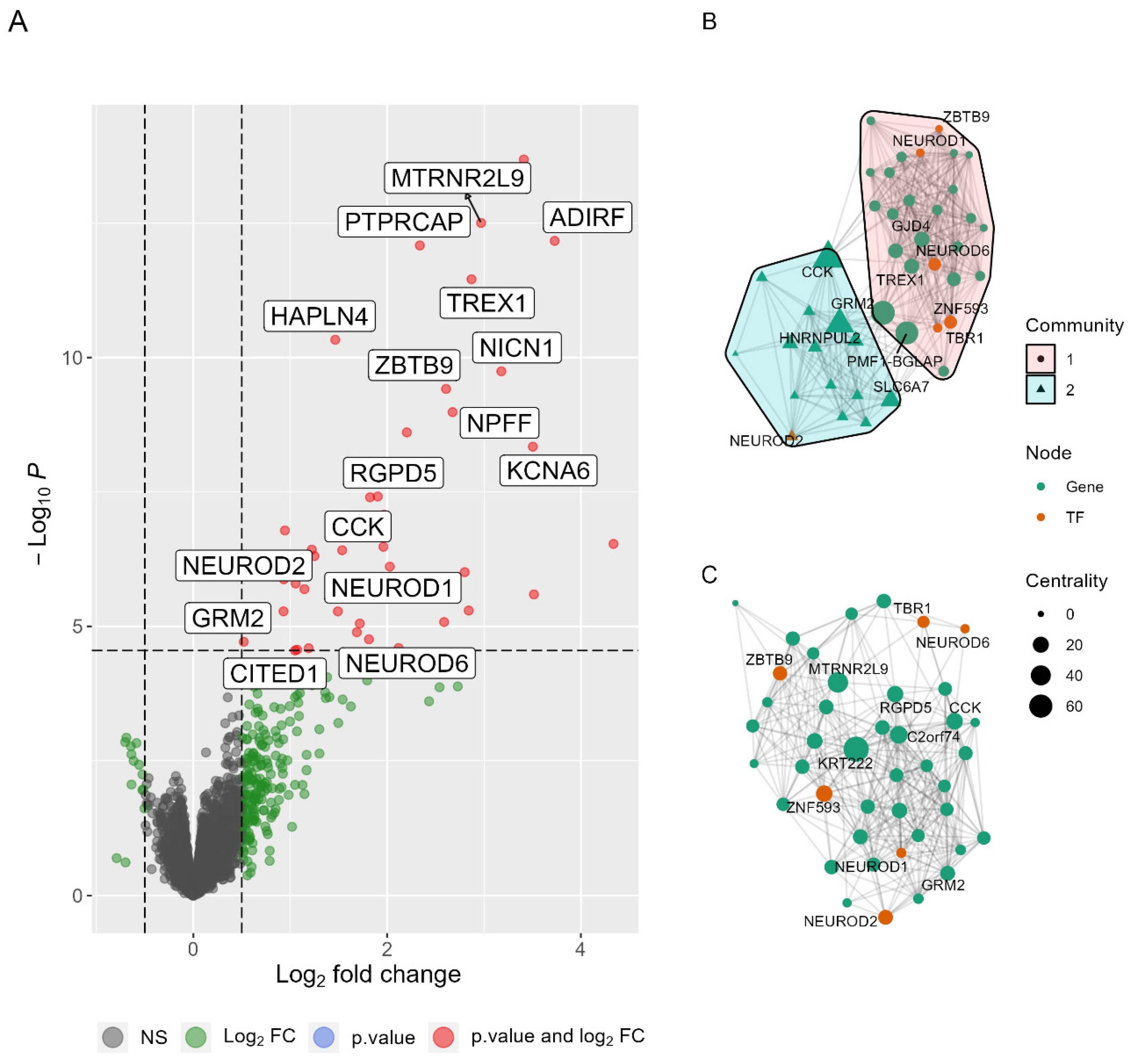
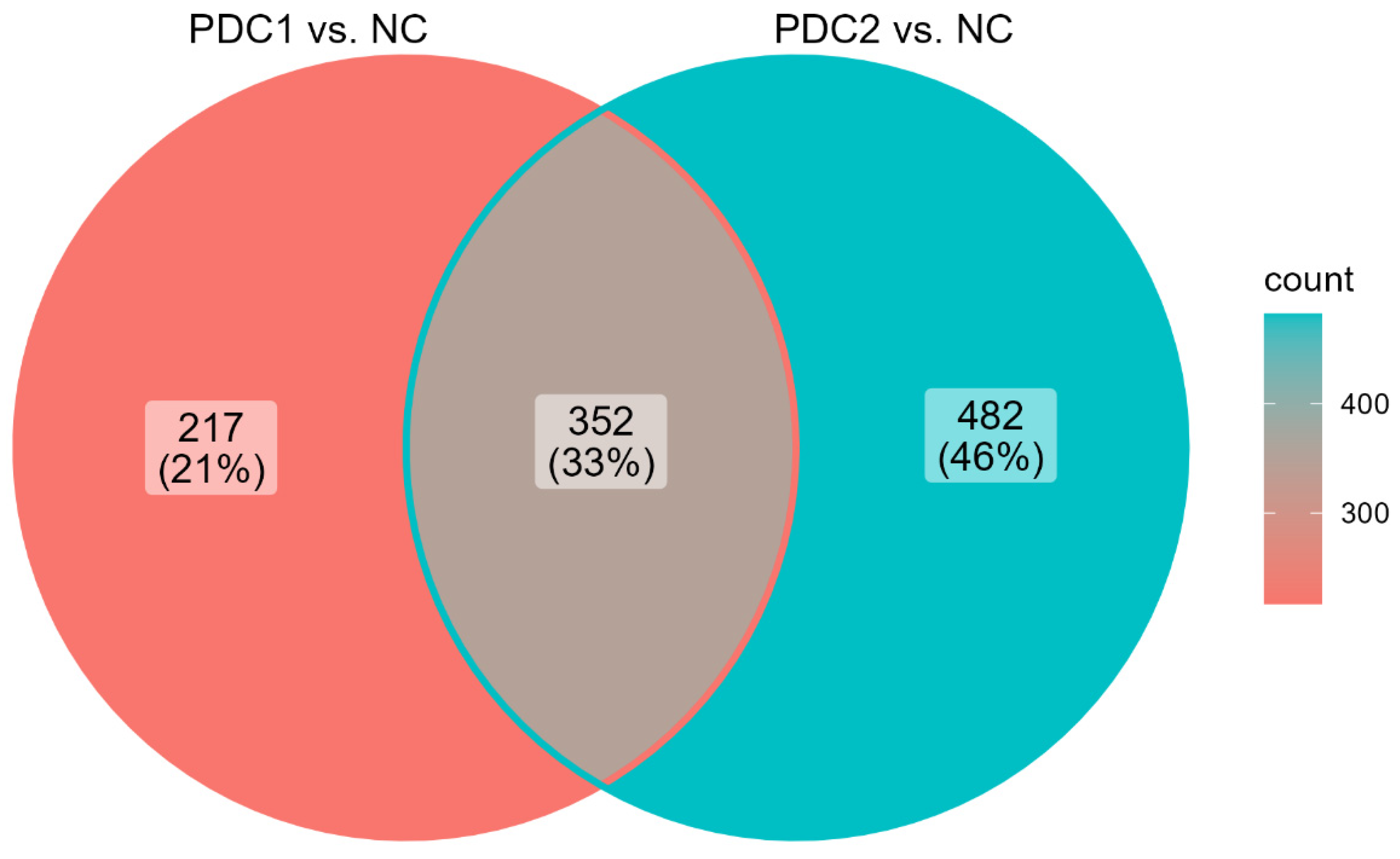
2.2. Differentially Expressed Genes in PDC1 vs. PDC2
2.3. PDC1 and PDC2 Networks Differ for Neuroprotective Pathways
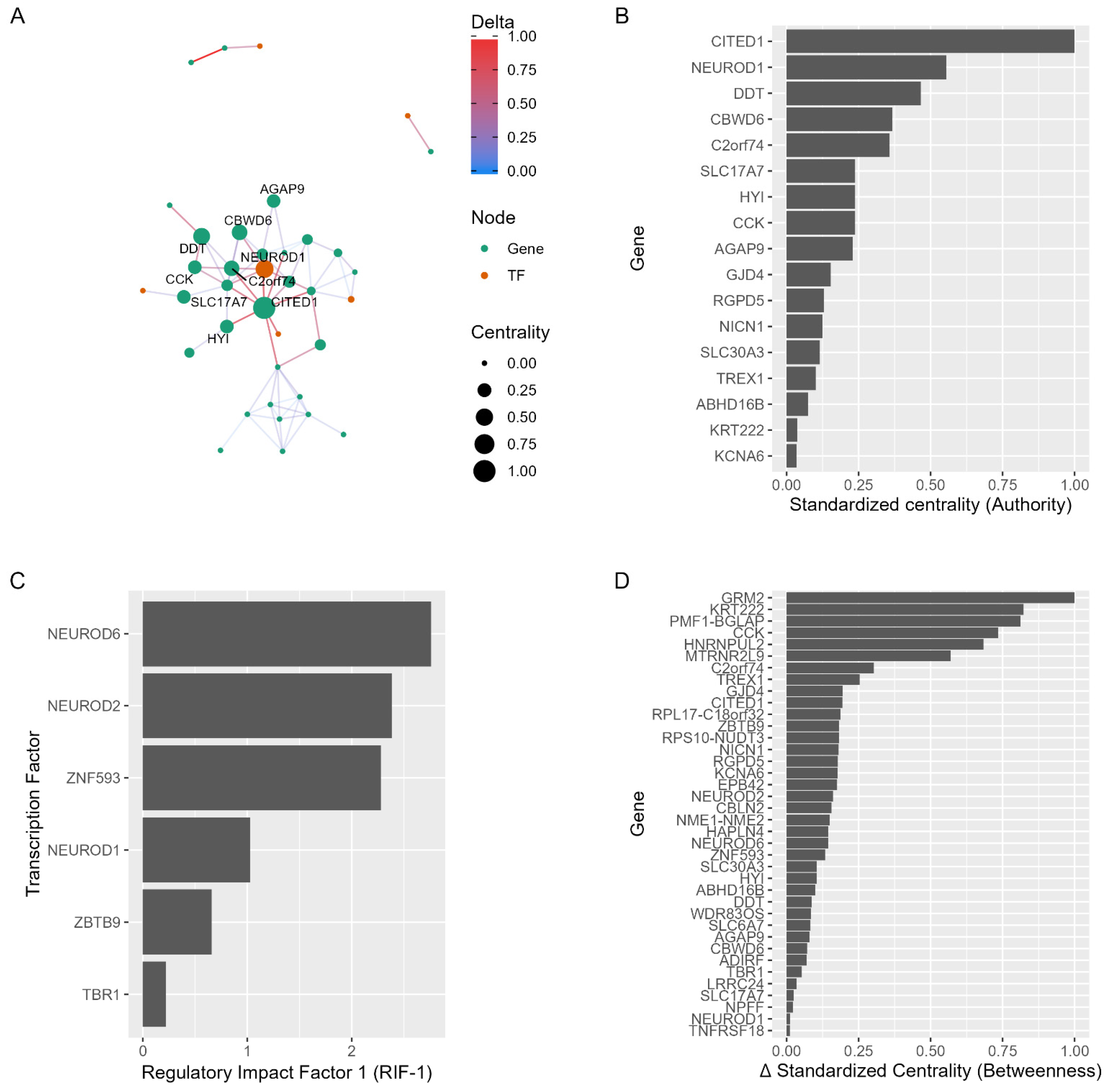
2.4. Network Regulators as Drug Repurposing Candidates
2.5. Drug Repurposing Pipeline
2.6. Molecular Characterization of PDC1 or PDC2 vs. NC
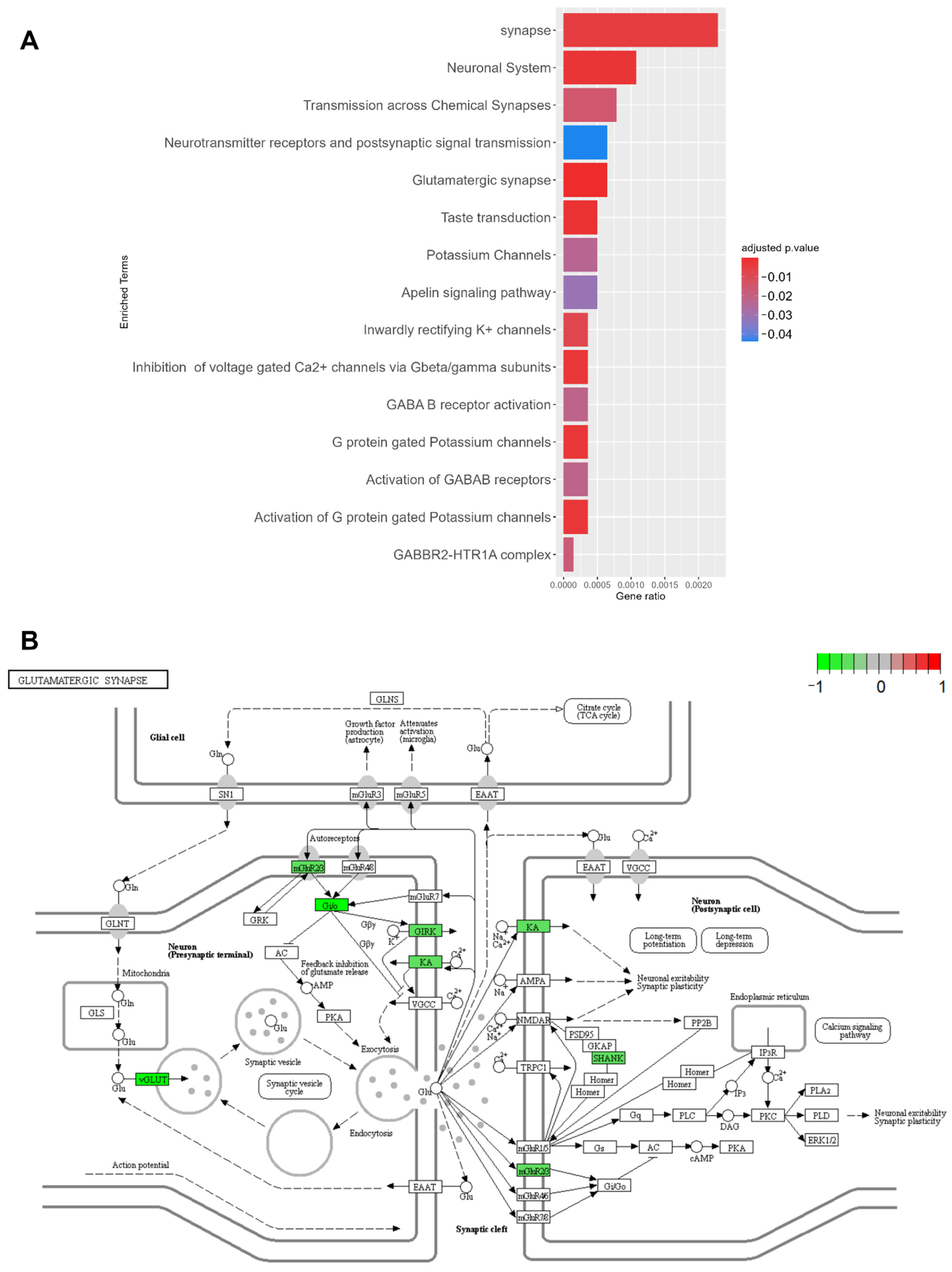
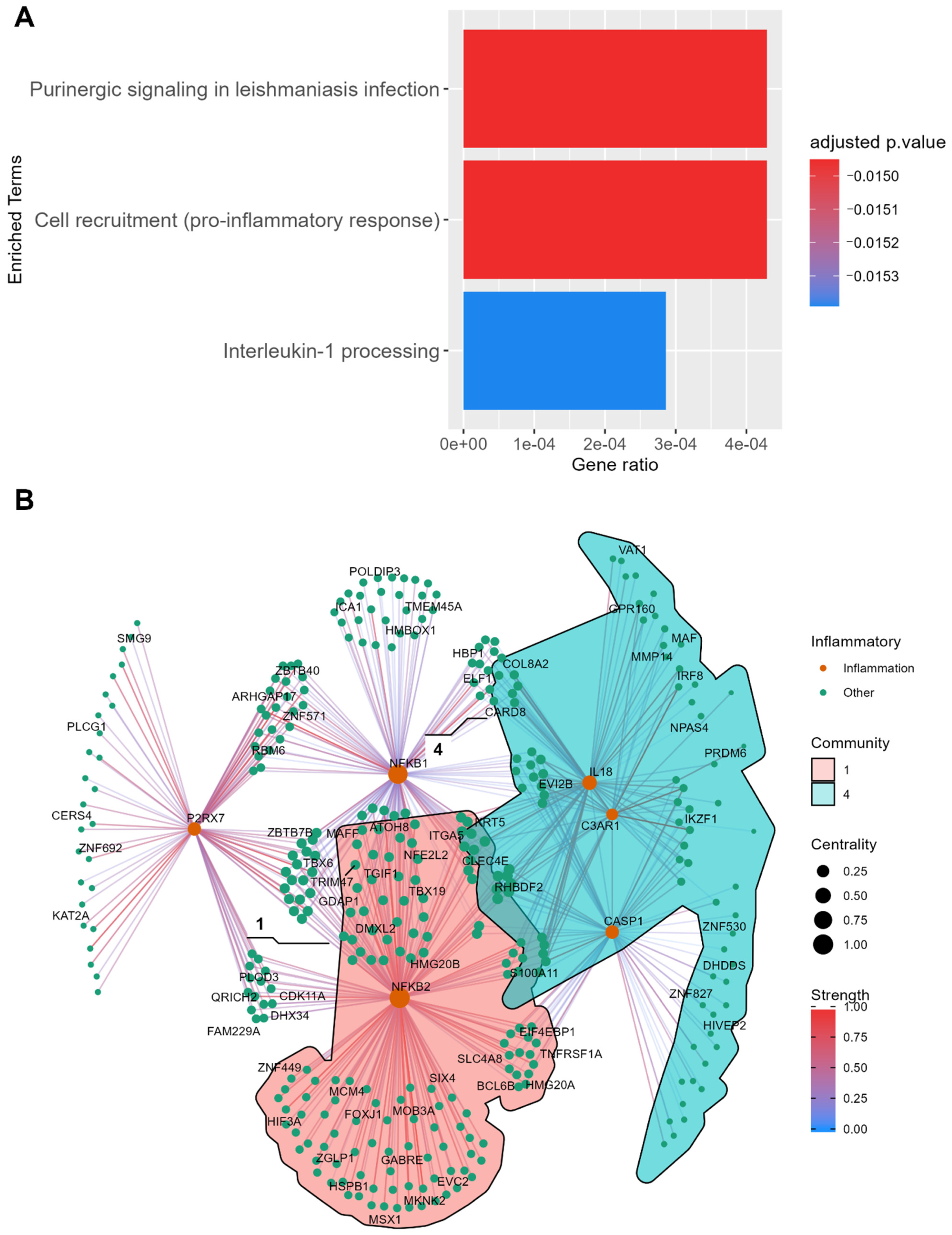
2.6.1. Disease Mechanism of PDC1
2.6.2. Disease Mechanism of PDC2
3. Discussion
4. Materials and Methods
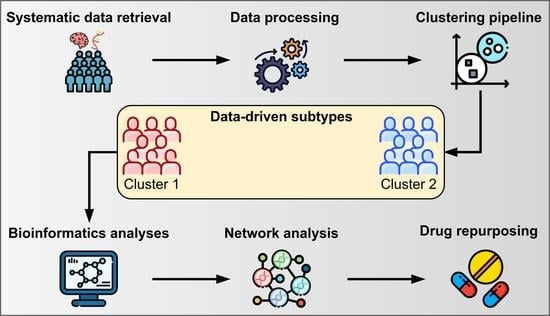
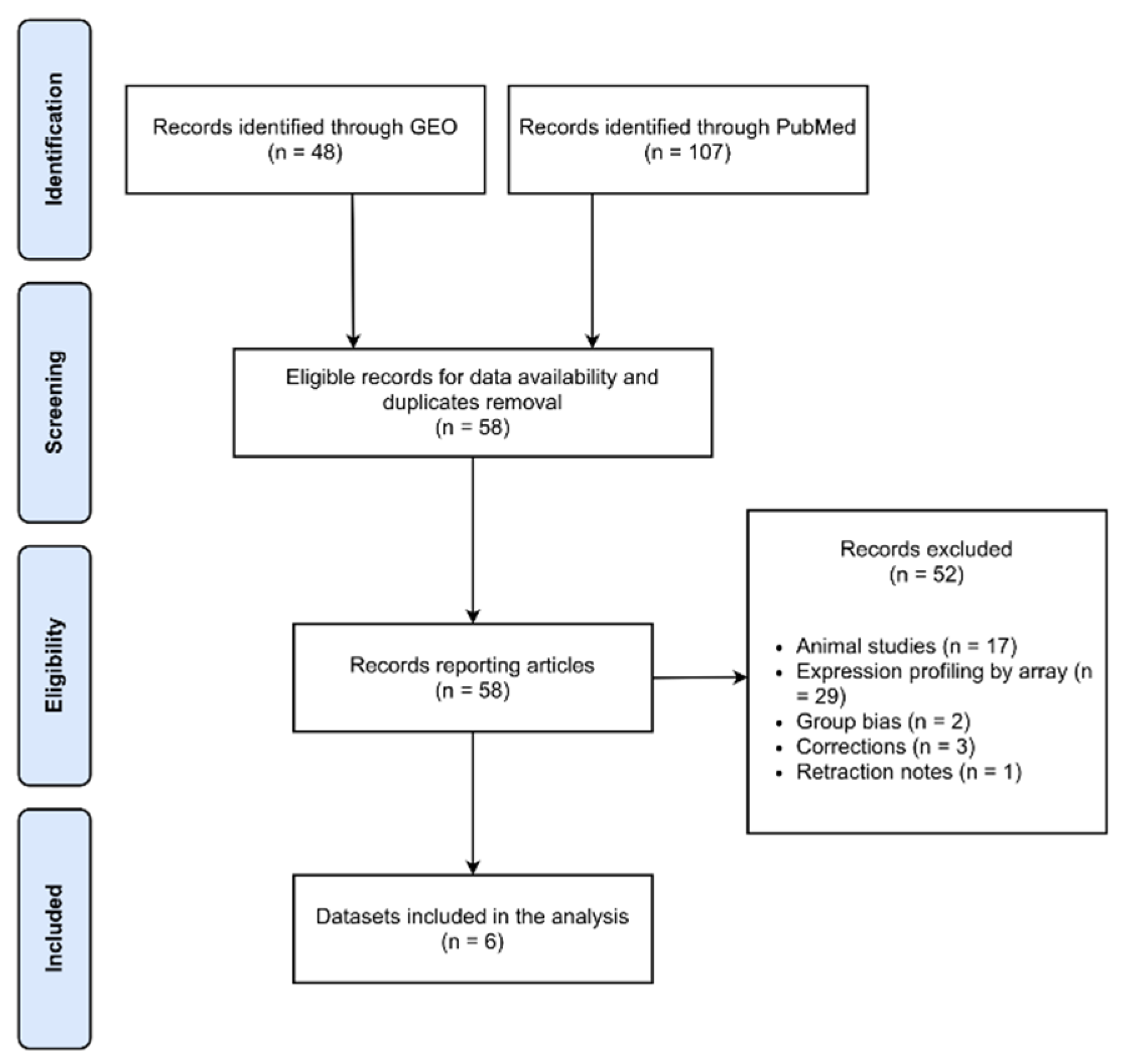
4.1. Systematic Data Retrieval
4.2. Raw Counts Preprocessing
4.3. Clustering Pipeline
4.4. Statistical Analyses
4.5. Differential Expression Analysis
4.6. Gene Network Analysis
4.7. Drug Repurposing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s Disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef]
- Deuschl, G.; Beghi, E.; Fazekas, F.; Varga, T.; Christoforidi, K.A.; Sipido, E.; Bassetti, C.L.; Vos, T.; Feigin, V.L. The Burden of Neurological Diseases in Europe: An Analysis for the Global Burden of Disease Study 2017. Lancet Public Health 2020, 5, e551–e567. [Google Scholar] [CrossRef]
- Dorsey, E.R.; Elbaz, A.; Nichols, E.; Abd-Allah, F.; Abdelalim, A.; Adsuar, J.C.; Ansha, M.G.; Brayne, C.; Choi, J.-Y.J.; Collado-Mateo, D.; et al. Global, Regional, and National Burden of Parkinson’s Disease, 1990–2016: A Systematic Analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef] [Green Version]
- Lang, A.E.; Espay, A.J. Disease Modification in Parkinson’s Disease: Current Approaches, Challenges, and Future Considerations. Mov. Disord. Off. J. Mov. Disord. Soc. 2018, 33, 660–677. [Google Scholar] [CrossRef] [PubMed]
- Paolini Paoletti, F.; Gaetani, L.; Parnetti, L. The Challenge of Disease-Modifying Therapies in Parkinson’s Disease: Role of CSF Biomarkers. Biomolecules 2020, 10, 335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, A.; Stacy, M. Disease-Modifying Drugs in Parkinson’s Disease. Drugs 2015, 75, 2065–2071. [Google Scholar] [CrossRef]
- Blauwendraat, C.; Nalls, M.A.; Singleton, A.B. The Genetic Architecture of Parkinson’s Disease. Lancet Neurol. 2020, 19, 170–178. [Google Scholar] [CrossRef]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Identification of Novel Risk Loci, Causal Insights, and Heritable Risk for Parkinson’s Disease: A Meta-Analysis of Genome-Wide Association Studies. Lancet Neurol. 2019, 18, 1091–1102. [Google Scholar] [CrossRef]
- Chang, D.; Nalls, M.A.; Hallgrímsdóttir, I.B.; Hunkapiller, J.; van der Brug, M.; Cai, F.; International Parkinson’s Disease Genomics Consortium; 23andMe Research Team; Kerchner, G.A.; Ayalon, G.; et al. A Meta-Analysis of Genome-Wide Association Studies Identifies 17 New Parkinson’s Disease Risk Loci. Nat. Genet. 2017, 49, 1511–1516. [Google Scholar] [CrossRef]
- International Parkinson Disease Genomics Consortium; Nalls, M.A.; Plagnol, V.; Hernandez, D.G.; Sharma, M.; Sheerin, U.-M.; Saad, M.; Simón-Sánchez, J.; Schulte, C.; Lesage, S.; et al. Imputation of Sequence Variants for Identification of Genetic Risks for Parkinson’s Disease: A Meta-Analysis of Genome-Wide Association Studies. Lancet 2011, 377, 641–649. [Google Scholar] [CrossRef] [Green Version]
- Fereshtehnejad, S.-M.; Romenets, S.R.; Anang, J.B.M.; Latreille, V.; Gagnon, J.-F.; Postuma, R.B. New Clinical Subtypes of Parkinson Disease and Their Longitudinal Progression: A Prospective Cohort Comparison With Other Phenotypes. JAMA Neurol. 2015, 72, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Greenland, J.C.; Williams-Gray, C.H.; Barker, R.A. The Clinical Heterogeneity of Parkinson’s Disease and Its Therapeutic Implications. Eur. J. Neurosci. 2019, 49, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Riggare, S.; Hägglund, M. Precision Medicine in Parkinson’s Disease—Exploring Patient-Initiated Self-Tracking. J. Park. Dis. 2018, 8, 441–446. [Google Scholar] [CrossRef] [Green Version]
- Severson, K.A.; Chahine, L.M.; Smolensky, L.A.; Dhuliawala, M.; Frasier, M.; Ng, K.; Ghosh, S.; Hu, J. Discovery of Parkinson’s Disease States and Disease Progression Modelling: A Longitudinal Data Study Using Machine Learning. Lancet Digit. Health 2021, 3, e555–e564. [Google Scholar] [CrossRef]
- Fabrizio, C.; Termine, A.; Caltagirone, C.; Sancesario, G. Artificial Intelligence for Alzheimer’s Disease: Promise or Challenge? Diagnostics 2021, 11, 1473. [Google Scholar] [CrossRef] [PubMed]
- Qian, E.; Huang, Y. Subtyping of Parkinson’s Disease—Where Are We Up To? Aging Dis. 2019, 10, 1130–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertucci Filho, D.; Teive, H.A.G.; Werneck, L.C. Early-Onset Parkinson’s Disease and Depression. Arq. Neuropsiquiatr. 2007, 65, 5–10. [Google Scholar] [CrossRef] [Green Version]
- Wickremaratchi, M.M.; Knipe, M.D.W.; Sastry, B.S.D.; Morgan, E.; Jones, A.; Salmon, R.; Weiser, R.; Moran, M.; Davies, D.; Ebenezer, L.; et al. The Motor Phenotype of Parkinson’s Disease in Relation to Age at Onset. Mov. Disord. Off. J. Mov. Disord. Soc. 2011, 26, 457–463. [Google Scholar] [CrossRef]
- Dienstmann, R.; Vermeulen, L.; Guinney, J.; Kopetz, S.; Tejpar, S.; Tabernero, J. Consensus Molecular Subtypes and the Evolution of Precision Medicine in Colorectal Cancer. Nat. Rev. Cancer 2017, 17, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Neff, R.A.; Wang, M.; Vatansever, S.; Guo, L.; Ming, C.; Wang, Q.; Wang, E.; Horgusluoglu-Moloch, E.; Song, W.; Li, A. Molecular Subtyping of Alzheimer’s Disease Using RNA Sequencing Data Reveals Novel Mechanisms and Targets. Sci. Adv. 2021, 7, eabb5398. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, Y.; Wang, M.; Song, W.-M.; Shen, Q.; McKenzie, A.; Choi, I.; Zhou, X.; Pan, P.-Y.; Yue, Z.; et al. The Landscape of Multiscale Transcriptomic Networks and Key Regulators in Parkinson’s Disease. Nat. Commun. 2019, 10, 5234. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.-X.; Yin, J.-C.; Chen, G. Transcriptome Analysis of Small Molecule–Mediated Astrocyte-to-Neuron Reprogramming. Front. Cell Dev. Biol. 2019, 7, 82. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Tutukova, S.; Tarabykin, V.; Hernandez-Miranda, L.R. The Role of Neurod Genes in Brain Development, Function, and Disease. Front. Mol. Neurosci. 2021, 14, 109. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, B. Wnt/β-Catenin Signaling Pathway Governs a Full Program for Dopaminergic Neuron Survival, Neurorescue and Regeneration in the MPTP Mouse Model of Parkinson’s Disease. Int. J. Mol. Sci. 2018, 19, 3743. [Google Scholar] [CrossRef] [Green Version]
- Marchetti, B.; Tirolo, C.; L’Episcopo, F.; Caniglia, S.; Testa, N.; Smith, J.A.; Pluchino, S.; Serapide, M.F. Parkinson’s Disease, Aging and Adult Neurogenesis: Wnt/β-Catenin Signalling as the Key to Unlock the Mystery of Endogenous Brain Repair. Aging Cell 2020, 19, e13101. [Google Scholar] [CrossRef]
- Valor, L.M.; Viosca, J.; Lopez-Atalaya, J.P.; Barco, A. Lysine Acetyltransferases CBP and P300 as Therapeutic Targets in Cognitive and Neurodegenerative Disorders. Curr. Pharm. Des. 2013, 19, 5051–5064. [Google Scholar] [CrossRef] [Green Version]
- Ono, M.; Lai, K.K.Y.; Wu, K.; Nguyen, C.; Lin, D.P.; Murali, R.; Kahn, M. Nuclear Receptor/Wnt Beta-Catenin Interactions Are Regulated via Differential CBP/P300 Coactivator Usage. PLoS ONE 2018, 13, e0200714. [Google Scholar] [CrossRef]
- Fu, M.; Wang, C.; Li, Z.; Sakamaki, T.; Pestell, R.G. Minireview: Cyclin D1: Normal and Abnormal Functions. Endocrinology 2004, 145, 5439–5447. [Google Scholar] [CrossRef]
- Hagey, D.W.; Topcic, D.; Kee, N.; Reynaud, F.; Bergsland, M.; Perlmann, T.; Muhr, J. CYCLIN-B1/2 and -D1 Act in Opposition to Coordinate Cortical Progenitor Self-Renewal and Lineage Commitment. Nat. Commun. 2020, 11, 2898. [Google Scholar] [CrossRef]
- Xu, D.; Hou, K.; Li, F.; Chen, S.; Fang, W.; Li, Y. XQ-1H Alleviates Cerebral Ischemia in Mice through Inhibition of Apoptosis and Promotion of Neurogenesis in a Wnt/β-Catenin Signaling Dependent Way. Life Sci. 2019, 235, 116844. [Google Scholar] [CrossRef] [PubMed]
- Brulet, R.; Matsuda, T.; Zhang, L.; Miranda, C.; Giacca, M.; Kaspar, B.K.; Nakashima, K.; Hsieh, J. NEUROD1 Instructs Neuronal Conversion in Non-Reactive Astrocytes. Stem Cell Rep. 2017, 8, 1506–1515. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.-H.; Tsai, P.-H.; Wang, K.-Y.; Wei, Y.-T.; Chiou, S.-H.; Mou, C.-Y. Generation of Functional Dopaminergic Neurons from Reprogramming Fibroblasts by Nonviral-Based Mesoporous Silica Nanoparticles. Sci. Rep. 2018, 8, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.; Piao, X.; Hu, S.; Gao, J.; Bao, M. LncRNA H19 Diminishes Dopaminergic Neuron Loss by Mediating MicroRNA-301b-3p in Parkinson’s Disease via the HPRT1-Mediated Wnt/β-Catenin Signaling Pathway. Aging 2020, 12, 8820–8836. [Google Scholar] [CrossRef] [PubMed]
- Parmar, M.; Björklund, A.; Björklund, T. In Vivo Conversion of Dopamine Neurons in Mouse Models of Parkinson’s Disease—A Future Approach for Regenerative Therapy? Curr. Opin. Genet. Dev. 2021, 70, 76–82. [Google Scholar] [CrossRef]
- Rivetti di Val Cervo, P.; Romanov, R.A.; Spigolon, G.; Masini, D.; Martín-Montañez, E.; Toledo, E.M.; La Manno, G.; Feyder, M.; Pifl, C.; Ng, Y.-H.; et al. Induction of Functional Dopamine Neurons from Human Astrocytes in Vitro and Mouse Astrocytes in a Parkinson’s Disease Model. Nat. Biotechnol. 2017, 35, 444–452. [Google Scholar] [CrossRef]
- Gao, Z.; Ure, K.; Ables, J.L.; Lagace, D.C.; Nave, K.-A.; Goebbels, S.; Eisch, A.J.; Hsieh, J. Neurod1 Is Essential for the Survival and Maturation of Adult-Born Neurons. Nat. Neurosci. 2009, 12, 1090–1092. [Google Scholar] [CrossRef] [Green Version]
- Pataskar, A.; Jung, J.; Smialowski, P.; Noack, F.; Calegari, F.; Straub, T.; Tiwari, V.K. NeuroD1 Reprograms Chromatin and Transcription Factor Landscapes to Induce the Neuronal Program. EMBO J. 2016, 35, 24–45. [Google Scholar] [CrossRef]
- Dickerson, J.W.; Conn, P.J. Therapeutic Potential of Targeting Metabotropic Glutamate Receptors for Parkinson’s Disease. Neurodegener. Dis. Manag. 2012, 2, 221–232. [Google Scholar] [CrossRef] [Green Version]
- Xicoy, H.; Brouwers, J.F.; Wieringa, B.; Martens, G.J.M. Explorative Combined Lipid and Transcriptomic Profiling of Substantia Nigra and Putamen in Parkinson’s Disease. Cells 2020, 9, 1966. [Google Scholar] [CrossRef]
- Crupi, R.; Impellizzeri, D.; Cuzzocrea, S. Role of Metabotropic Glutamate Receptors in Neurological Disorders. Front. Mol. Neurosci. 2019, 12, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, T.K.; Messenger, M.J.; Ward, M.A.; Woodhouse, S.; Osborne, D.J.; Duty, S.; O’Neill, M.J. Evaluation of the MGluR2/3 Agonist LY379268 in Rodent Models of Parkinson’s Disease. Pharmacol. Biochem. Behav. 2002, 73, 455–466. [Google Scholar] [CrossRef]
- Nickols, H.H.; Conn, P.J. Development of Allosteric Modulators of GPCRs for Treatment of CNS Disorders. Neurobiol. Dis. 2014, 61, 55–71. [Google Scholar] [CrossRef] [Green Version]
- Bettio, L.E.B.; Gil-Mohapel, J.; Patten, A.R.; O’Rourke, N.F.; Hanley, R.P.; Gopalakrishnan, K.; Wulff, J.E.; Christie, B.R. Effects of Isx-9 and Stress on Adult Hippocampal Neurogenesis: Experimental Considerations and Future Perspectives. Neurogenesis 2017, 4, e1317692. [Google Scholar] [CrossRef] [Green Version]
- Petersen, H.V.; Jensen, J.N.; Stein, R.; Serup, P. Glucose Induced MAPK Signalling Influences NeuroD1-Mediated Activation and Nuclear Localization. FEBS Lett. 2002, 528, 241–245. [Google Scholar] [CrossRef] [Green Version]
- Rui, Y.; Sun, Z.; Gu, J.; Sheng, Z.; He, X.; Xie, Z. MEK Inhibitor PD98059 Acutely Inhibits Synchronized Spontaneous Ca2+ Oscillations in Cultured Hippocampal Networks. Acta Pharmacol. Sin. 2006, 27, 869–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohush, A.; Niewiadomska, G.; Filipek, A. Role of Mitogen Activated Protein Kinase Signaling in Parkinson’s Disease. Int. J. Mol. Sci. 2018, 19, 2973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Nie, S.; Han, C.; Ma, K.; Xu, Y.; Zhang, Z.; Papa, S.M.; Cao, X. Antidyskinetic Effects of MEK Inhibitor Are Associated with Multiple Neurochemical Alterations in the Striatum of Hemiparkinsonian Rats. Front. Neurosci. 2017, 11, 112. [Google Scholar] [CrossRef] [Green Version]
- Kosyakovsky, J.; Fine, J.M.; Frey, W.H.; Hanson, L.R. Mechanisms of Intranasal Deferoxamine in Neurodegenerative and Neurovascular Disease. Pharmaceuticals 2021, 14, 95. [Google Scholar] [CrossRef]
- Farr, A.C.; Xiong, M.P. Challenges and Opportunities of Deferoxamine Delivery for Treatment of Alzheimer’s Disease, Parkinson’s Disease, and Intracerebral Hemorrhage. Mol. Pharm. 2021, 18, 593–609. [Google Scholar] [CrossRef]
- Mounsey, R.B.; Teismann, P. Chelators in the Treatment of Iron Accumulation in Parkinson’s Disease. Int. J. Cell Biol. 2012, 2012, e983245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Febbraro, F.; Andersen, K.J.; Sanchez-Guajardo, V.; Tentillier, N.; Romero-Ramos, M. Chronic Intranasal Deferoxamine Ameliorates Motor Defects and Pathology in the α-Synuclein RAAV Parkinson’s Model. Exp. Neurol. 2013, 247, 45–58. [Google Scholar] [CrossRef]
- Kim, H.-J.; Hida, H.; Jung, C.-G.; Miura, Y.; Nishino, H. Treatment with Deferoxamine Increases Neurons from Neural Stem/Progenitor Cells. Brain Res. 2006, 1092, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Mursaleen, L.; Somavarapu, S.; Zariwala, M.G. Deferoxamine and Curcumin Loaded Nanocarriers Protect Against Rotenone-Induced Neurotoxicity. J. Parkinson Dis. 2020, 10, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.J.; Dexter, D.T.; Martin-Bastida, A.; Crichton, R.R. Is Chelation Therapy a Potential Treatment for Parkinson’s Disease? Int. J. Mol. Sci. 2021, 22, 3338. [Google Scholar] [CrossRef] [PubMed]
- Foster, D.J.; Conn, P.J. Allosteric Modulation of GPCRs: New Insights and Potential Utility for Treatment of Schizophrenia and Other CNS Disorders. Neuron 2017, 94, 431–446. [Google Scholar] [CrossRef] [Green Version]
- Shamseer, L.; Moher, D.; Clarke, M.; Ghersi, D.; Liberati, A.; Petticrew, M.; Shekelle, P.; Stewart, L.A. Preferred Reporting Items for Systematic Review and Meta-Analysis Protocols (PRISMA-P) 2015: Elaboration and Explanation. BMJ 2015, 349, g7647. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Ensink, E.; Lang, S.; Marshall, L.; Schilthuis, M.; Lamp, J.; Vega, I.; Labrie, V. Hemispheric Asymmetry in the Human Brain and in Parkinson’s Disease Is Linked to Divergent Epigenetic Patterns in Neurons. Genome Biol. 2020, 21, 61. [Google Scholar] [CrossRef] [Green Version]
- Schulze, M.; Sommer, A.; Plötz, S.; Farrell, M.; Winner, B.; Grosch, J.; Winkler, J.; Riemenschneider, M.J. Sporadic Parkinson’s Disease Derived Neuronal Cells Show Disease-Specific MRNA and Small RNA Signatures with Abundant Deregulation of PiRNAs. Acta Neuropathol. Commun. 2018, 6, 58. [Google Scholar] [CrossRef] [Green Version]
- Dumitriu, A.; Golji, J.; Labadorf, A.T.; Gao, B.; Beach, T.G.; Myers, R.H.; Longo, K.A.; Latourelle, J.C. Integrative Analyses of Proteomics and RNA Transcriptomics Implicate Mitochondrial Processes, Protein Folding Pathways and GWAS Loci in Parkinson Disease. BMC Med. Genom. 2016, 9, 5. [Google Scholar] [CrossRef] [Green Version]
- Nido, G.S.; Dick, F.; Toker, L.; Petersen, K.; Alves, G.; Tysnes, O.-B.; Jonassen, I.; Haugarvoll, K.; Tzoulis, C. Common Gene Expression Signatures in Parkinson’s Disease Are Driven by Changes in Cell Composition. Acta Neuropathol. Commun. 2020, 8, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leek, J.T. Svaseq: Removing Batch Effects and Other Unwanted Noise from Sequencing Data. Nucleic Acids Res. 2014, 42, e161. [Google Scholar] [CrossRef] [PubMed]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The Sva Package for Removing Batch Effects and Other Unwanted Variation in High-Throughput Experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Parmigiani, G.; Johnson, W.E. ComBat-Seq: Batch Effect Adjustment for RNA-Seq Count Data. NAR Genom. Bioinform. 2020, 2, lqaa078. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Dave, R.N. Validating clusters using the Hopkins statistic. In Proceedings of the IEEE International Conference on Fuzzy Systems, Budapest, Hungary, 25–29 July 2004; Volume 1, pp. 149–153. [Google Scholar]
- Charrad, M.; Ghazzali, N.; Boiteau, V.; Niknafs, A. NbClust: An R Package for Determining the Relevant Number of Clusters in a Data Set. J. Stat. Softw. 2014, 61, 1–36. [Google Scholar] [CrossRef] [Green Version]
- Kassambara, A.; Mundt, F. Package ‘Factoextra’. Extract Visual Results Multivariative Data Anallysis. Available online: https://cran.r-project.org/web/packages/factoextra/index.html (accessed on 10 May 2021).
- Rousseeuw, P.J. Silhouettes: A Graphical Aid to the Interpretation and Validation of Cluster Analysis. J. Comput. Appl. Math. 1987, 20, 53–65. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Li, Z.; Xiong, H.; Gao, X.; Wu, J. Understanding of internal clustering validation measures. In Proceedings of the IEEE International Conference on Data Mining, Sydney, Australia, 13–17 December 2010; Institute of Electrical and Electronics Engineers (IEEE): Piscataway, NJ, USA, 2010; pp. 911–916. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Hollander, M.; Wolfe, D.A.; Chicken, E. Nonparametric Statistical Methods; John Wiley & Sons: Hoboken, NJ, USA, 2013; Volume 751, ISBN 1118553292. [Google Scholar]
- Agresti, A. An Introduction to Categorical Data Analysis; John Wiley & Sons: Hoboken, NJ, USA, 2018; ISBN 1119405262. [Google Scholar]
- Smyth, G.K. Limma: Linear models for microarray Data. In Bioinformatics and Computational Biology Solutions Using R and Bioconductor; Springer: Cham, Switzerland, 2005; pp. 397–420. [Google Scholar]
- Benjamini, Y. Discovering the False Discovery Rate. J. R. Stat. Soc. Ser. B Stat. Methodol. 2010, 72, 405–416. [Google Scholar] [CrossRef]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A Web Server for Functional Enrichment Analysis and Conversions of Gene Lists (2019 Update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [Green Version]
- Piñero, J.; Ramírez-Anguita, J.M.; Saüch-Pitarch, J.; Ronzano, F.; Centeno, E.; Sanz, F.; Furlong, L.I. The DisGeNET Knowledge Platform for Disease Genomics: 2019 Update. Nucleic Acids Res. 2020, 48, D845–D855. [Google Scholar] [CrossRef] [Green Version]
- Fujita, K.A.; Ostaszewski, M.; Matsuoka, Y.; Ghosh, S.; Glaab, E.; Trefois, C.; Crespo, I.; Perumal, T.M.; Jurkowski, W.; Antony, P.M.A.; et al. Integrating Pathways of Parkinson’s Disease in a Molecular Interaction Map. Mol. Neurobiol. 2014, 49, 88–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, W.; Brouwer, C. Pathview: An R/Bioconductor Package for Pathway-Based Data Integration and Visualization. Bioinformatics 2013, 29, 1830–1831. [Google Scholar] [CrossRef] [Green Version]
- Oliveira de Biagi, C.A.; Nociti, R.P.; Brotto, D.B.; Funicheli, B.O.; de Cássia Ruy, P.; Bianchi Ximenez, J.P.; Alves Figueiredo, D.L.; Araújo Silva, W. CeTF: An R/Bioconductor Package for Transcription Factor Co-Expression Networks Using Regulatory Impact Factors (RIF) and Partial Correlation and Information (PCIT) Analysis. BMC Genom. 2021, 22, 624. [Google Scholar] [CrossRef] [PubMed]
- Reverter, A.; Hudson, N.J.; Nagaraj, S.H.; Pérez-Enciso, M.; Dalrymple, B.P. Regulatory Impact Factors: Unraveling the Transcriptional Regulation of Complex Traits from Expression Data. Bioinformatics 2010, 26, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, T.L. Tidygraph: A Tidy API for Graph Manipulation. Available online: https://cran.r-project.org/web/packages/tidygraph/index.html (accessed on 10 May 2021).
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING V11: Protein–Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Van Borkulo, C.D.; Boschloo, L.; Kossakowski, J.; Tio, P.; Schoevers, R.A.; Borsboom, D.; Waldorp, L.J. Comparing Network Structures on Three Aspects: A Permutation Test. J. Stat. Softw. 2017, 10. [Google Scholar] [CrossRef]
- Corsello, S.M.; Bittker, J.A.; Liu, Z.; Gould, J.; McCarren, P.; Hirschman, J.E.; Johnston, S.E.; Vrcic, A.; Wong, B.; Khan, M.; et al. The Drug Repurposing Hub: A next-Generation Drug Library and Information Resource. Nat. Med. 2017, 23, 405–408. [Google Scholar] [CrossRef] [Green Version]
- Freshour, S.L.; Kiwala, S.; Cotto, K.C.; Coffman, A.C.; McMichael, J.F.; Song, J.J.; Griffith, M.; Griffith, O.L.; Wagner, A.H. Integration of the Drug-Gene Interaction Database (DGIdb 4.0) with Open Crowdsource Efforts. Nucleic Acids Res. 2021, 49, D1144–D1151. [Google Scholar] [CrossRef] [PubMed]
- Zarin, D.A.; Tse, T.; Williams, R.J.; Califf, R.M.; Ide, N.C. The ClinicalTrials.Gov Results Database—Update and Key Issues. N. Engl. J. Med. 2011, 364, 852–860. [Google Scholar] [CrossRef] [Green Version]
- National Research Council (US) Committee on A Framework for Developing a New Taxonomy of Disease. Toward Precision Medicine: Building a Knowledge Network for Biomedical Research and a New Taxonomy of Disease; National Academies Press: Washington, DC, USA, 2011; ISBN 9780309222259.
- Strafella, C.; Caputo, V.; Termine, A.; Fabrizio, C.; Ruffo, P.; Potenza, S.; Cusumano, A.; Ricci, F.; Caltagirone, C.; Giardina, E. Genetic Determinants Highlight the Existence of Shared Etiopathogenetic Mechanisms Characterizing Age-Related Macular Degeneration and Neurodegenerative Disorders. Front. Neurol. 2021, 12, 626066. [Google Scholar] [CrossRef]
- Termine, A.; Fabrizio, C.; Strafella, C.; Caputo, V.; Petrosini, L.; Caltagirone, C.; Giardina, E.; Cascella, R. Multi-Layer Picture of Neurodegenerative Diseases: Lessons from the Use of Big Data through Artificial Intelligence. J. Pers. Med. 2021, 11, 280. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Drug | ChEMBL-ID | Phase | Modality of Action | PMID |
|---|---|---|---|---|---|
| NEUROD1 | PD98059 | CHEMBL35482 | Preclinical | ERK1⁄2 pathway inhibitor | 12297313 |
| 28337120 | |||||
| 30274251 | |||||
| 16787571 | |||||
| NEUROD1 | DEFEROXAMINE | CHEMBL556 | Launched | hexadentate iron chelator | 16697980 |
| 32926630 | |||||
| 23531432 | |||||
| 22754573 | |||||
| 31868679 | |||||
| 33513737 | |||||
| 33805195 | |||||
| NEUROD1 | ISX9 | CHEMBL1222381 | Preclinical | neural stem cell inducer | 29311646 |
| 18552832 | |||||
| 26407349 | |||||
| 28656155 | |||||
| 22542682 | |||||
| 28216149 | |||||
| GRM2 | JNJ-40411813 | CHEMBL3337527 | Phase 2 | glutamate receptor positive allosteric modulator | 25462291 |
| 25586401 | |||||
| 25735992 | |||||
| GRM2 | LY2979165 | CHEMBL3544939 | Phase 2 | glutamate receptor positive allosteric modulator | 32052375 |
| 33071070 | |||||
| 29564482 | |||||
| GRM2 | LY2969822 | CHEMBL3545270 | Phase 1 | glutamate receptor agonist | 28177520 |
| 31306647 | |||||
| 30934533 | |||||
| GRM2 | LY404039 | CHEMBL375611 | Phase 1 | glutamate receptor agonist | 32403118 |
| 32403118 | |||||
| GRM2 | BINA | CHEMBL593013 | Preclinical | glutamate receptor positive allosteric modulator | 16046122 |
| 16608916 | |||||
| 17526600 | |||||
| 24076101 | |||||
| 28472649 | |||||
| GRM2 | CBiPES | CHEMBL4303163 | Preclinical | glutamate receptor positive allosteric modulator | 15717213 |
| 19951716 | |||||
| 22659090 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Termine, A.; Fabrizio, C.; Strafella, C.; Caputo, V.; Petrosini, L.; Caltagirone, C.; Cascella, R.; Giardina, E. A Hybrid Machine Learning and Network Analysis Approach Reveals Two Parkinson’s Disease Subtypes from 115 RNA-Seq Post-Mortem Brain Samples. Int. J. Mol. Sci. 2022, 23, 2557. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23052557
Termine A, Fabrizio C, Strafella C, Caputo V, Petrosini L, Caltagirone C, Cascella R, Giardina E. A Hybrid Machine Learning and Network Analysis Approach Reveals Two Parkinson’s Disease Subtypes from 115 RNA-Seq Post-Mortem Brain Samples. International Journal of Molecular Sciences. 2022; 23(5):2557. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23052557
Chicago/Turabian StyleTermine, Andrea, Carlo Fabrizio, Claudia Strafella, Valerio Caputo, Laura Petrosini, Carlo Caltagirone, Raffaella Cascella, and Emiliano Giardina. 2022. "A Hybrid Machine Learning and Network Analysis Approach Reveals Two Parkinson’s Disease Subtypes from 115 RNA-Seq Post-Mortem Brain Samples" International Journal of Molecular Sciences 23, no. 5: 2557. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23052557