
Dietary-Induced Bacterial Metabolites Reduce Inflammation and Inflammation-Associated Cancer via Vitamin D Pathway
 , , , , , , , ,
, , , , , , , ,  and add
Show full author list
and add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
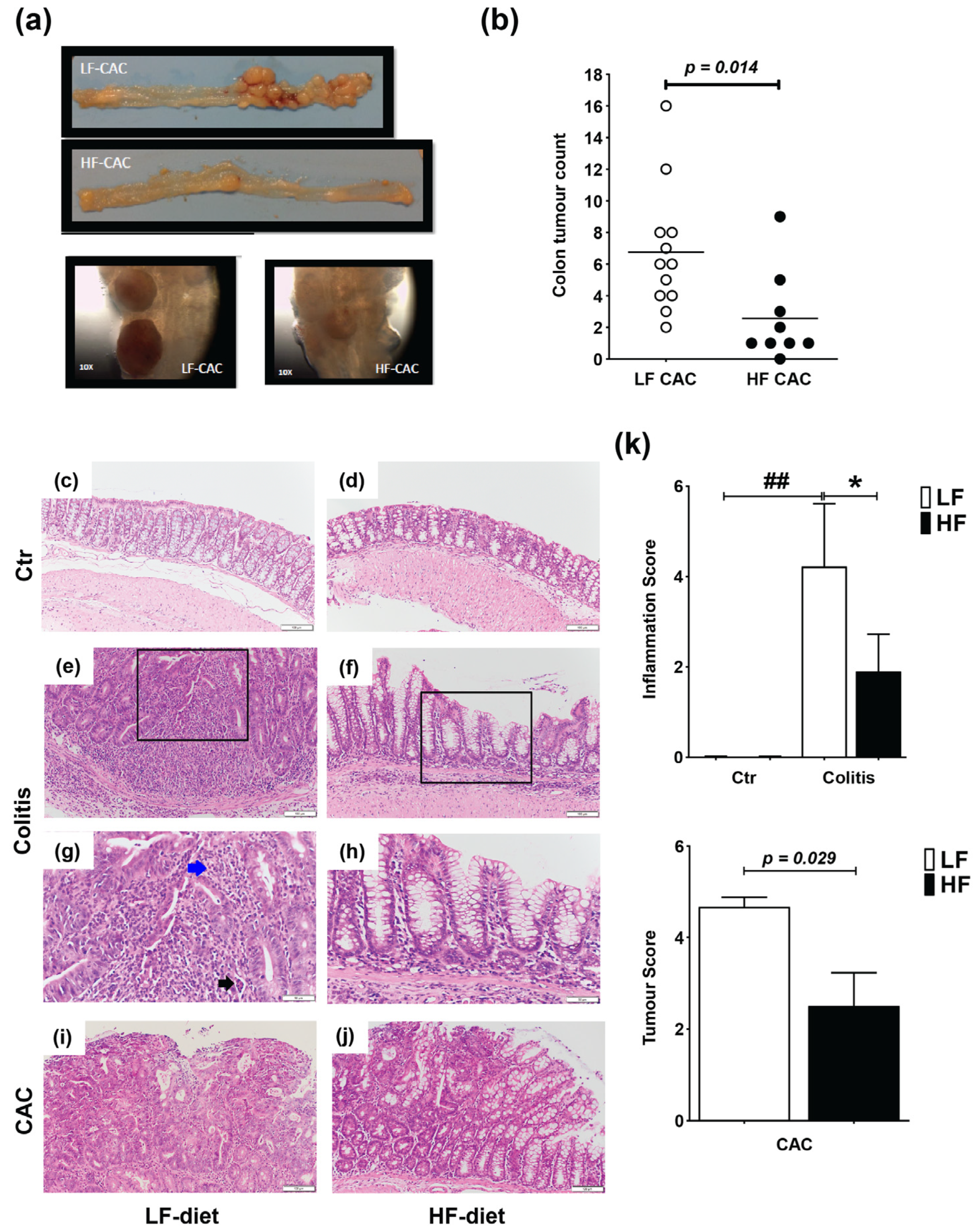
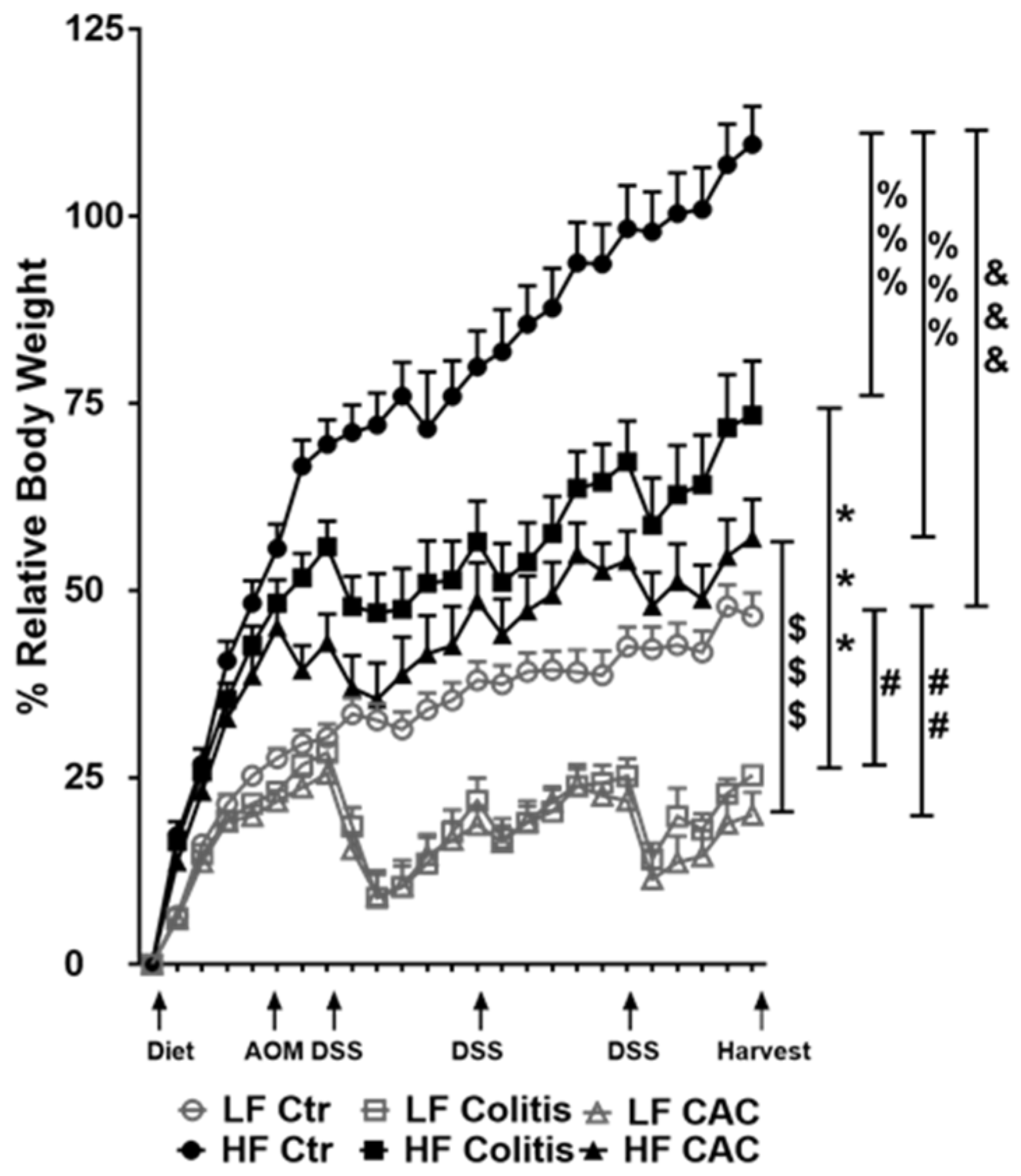
2.1. High Fat Feeding Reduces Colitis and Tumour Development
2.2. High Fat Feeding Reduces Macroscopical Signs of Inflammation
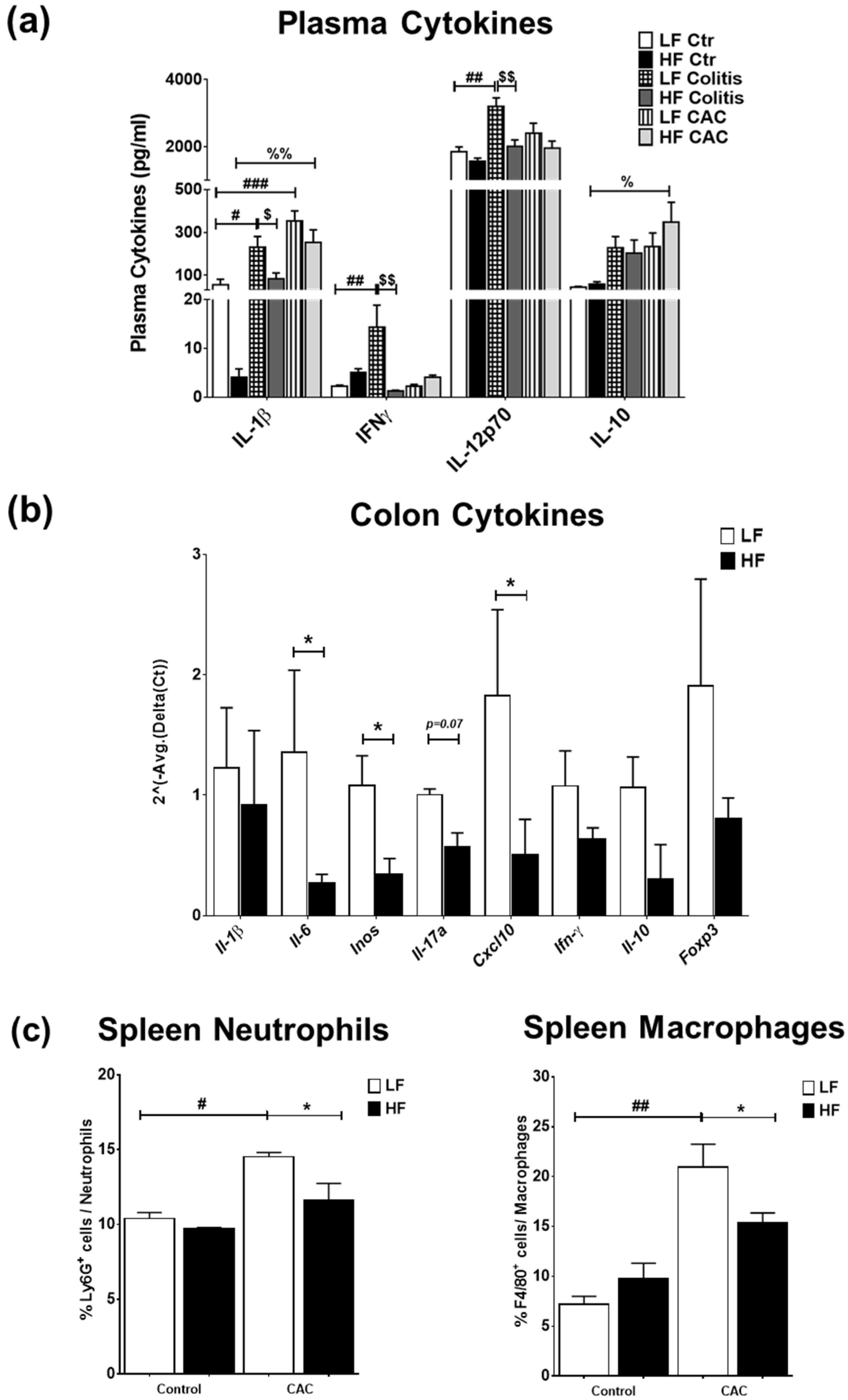
2.3. High-Fat Diet Reduces Acute Inflammatory Cell Infiltration and Tumour Malignancy and Systemic and Mucosal Inflammatory Responses
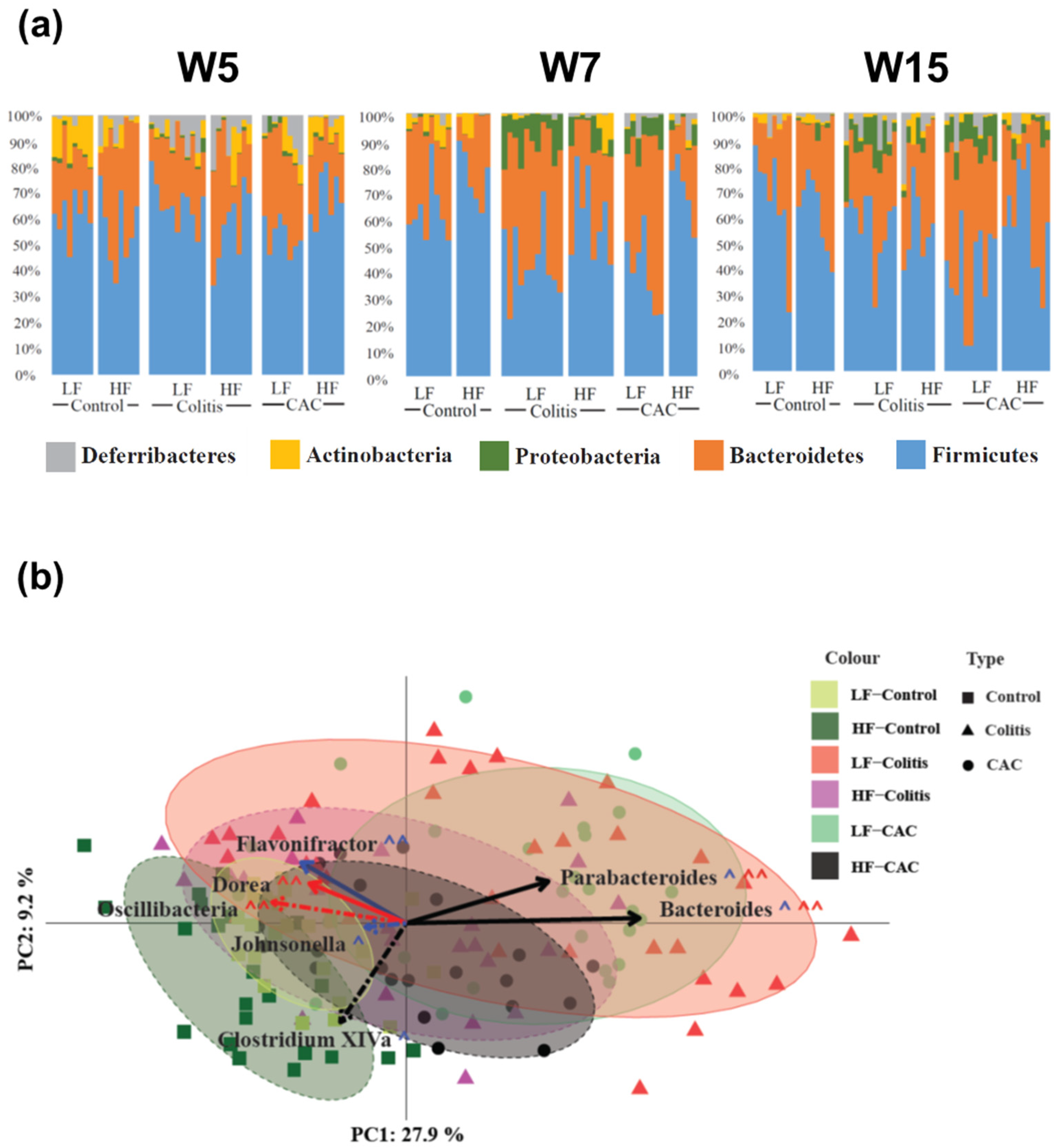
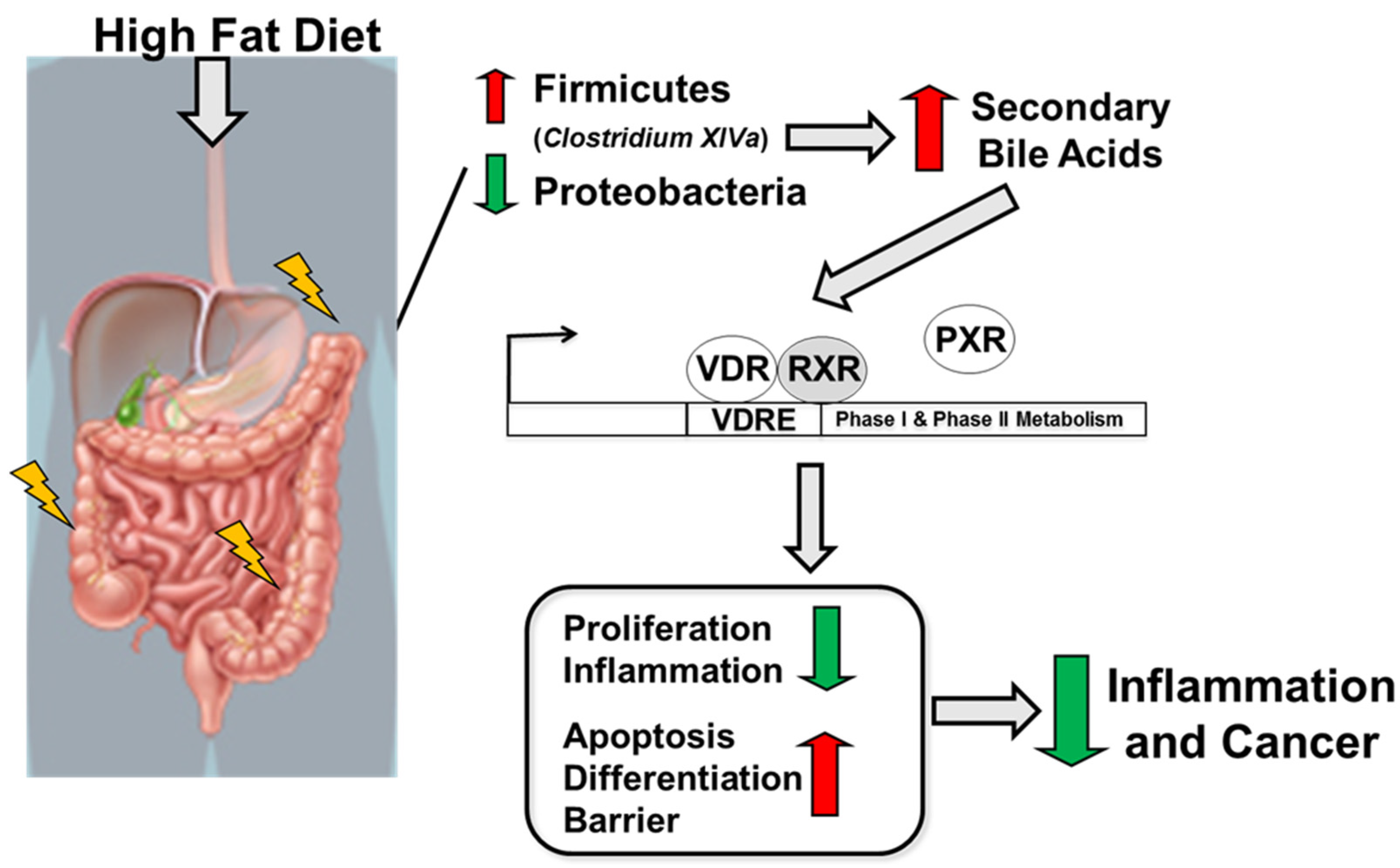
2.4. High-Fat Diet Reduces Proteobacteria and Increases Abundance of Clostridium cluster XIVa in Experimental Colitis and CAC
2.5. The Quantity of Dietary Fat Influences the Impact of Colitis and CAC on Plasma Metabolic Markers and Tissue Fatty Acids
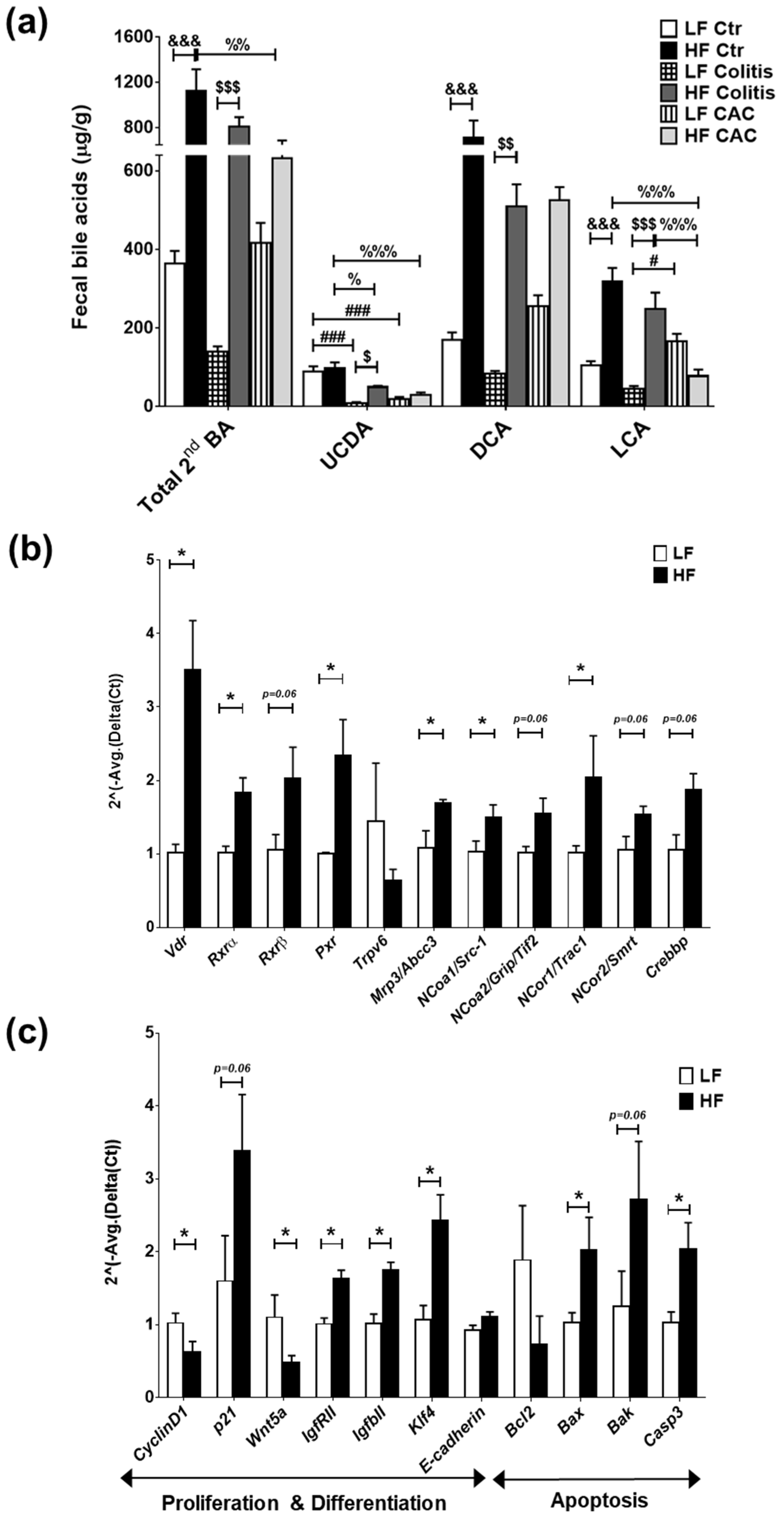
2.6. High-Fat Diet Induces High Levels of Secondary Bile Acids and Regulates the Expression of Vitamin D Receptor and Vitamin D Target Genes
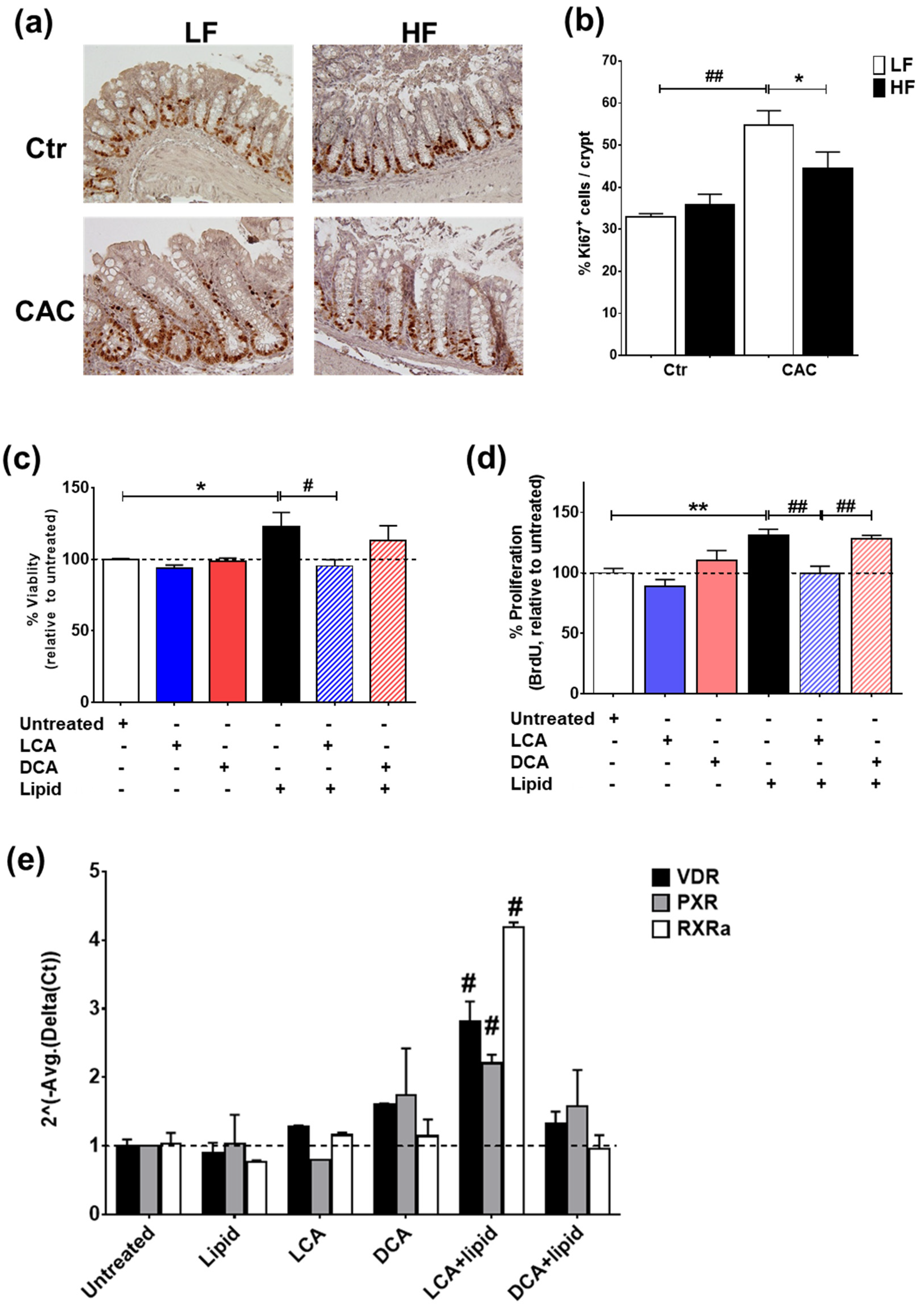
2.7. The Secondary Bile Acid LCA Reduces Proliferation and Induces the Expression of Vitamin D Regulated Genes in Lipid-Treated Epithelial Cells
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Experimental Design for Colitis and Colitis-Associated Colorectal Cancer and Diet Models
4.3. Collection of Tissue, Fluids, Serum, Caecal and Faecal Content
4.4. Isolation of Cells from Spleen and MLNs and Flow Cytometry
4.5. Cytokines/Chemokines and Metabolic Markers
4.6. RNA Extraction and PCR Analysis
4.7. Histology and Immunohistochemistry
4.8. Epithelial Cell Treatment with Saturated Lipid Mixture and Bile Acids
4.9. DNA Extraction and High-Throughput DNA Sequencing
4.10. Bioinformatics Sequence Analysis
4.11. Fatty Acid (FAA) Analysis
4.12. Short-Chain Fatty Acid (SCFA) Extraction and Analysis
4.13. Bile Acid Extractions
4.14. Chemicals for Bile Acid Analysis
4.15. Ultra Performance Liquid Chromatography–Tandem Mass Spectrometry
4.16. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Statovci, D.; Aguilera, M.; MacSharry, J.; Melgar, S. The Impact of Western Diet and Nutrients on the Microbiota and Immune Response at Mucosal Interfaces. Front. Immunol. 2017, 8, 838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ananthakrishnan, A.N.; Bernstein, C.N.; Iliopoulos, D.; Macpherson, A.; Neurath, M.F.; Raja Ali, A.R.; Vavricka, S.R.; Fiocchi, C. Environmental triggers in IBD: A review of progress and evidence. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 2017, 390, 2769–2778. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, M.; Garner, A.; Vlamakis, H.; Xavier, R.X. Microbial genes and pathways in inflammatory bowel disease. Nat. Rev. Microbiol. 2019, 17, 497–511. [Google Scholar] [CrossRef]
- Van Der Kraak, L.; Gros, P.; Beauchemin, N. Colitis-associated colon cancer: Is it in your genes? World J. Gastroenterol. 2015, 21, 11688–11699. [Google Scholar] [CrossRef]
- Hyland, N.P.; Chambers, A.P.; Keenan, C.M.; Pittman, Q.J.; Sharkey, K.A. Differential adipokine response in genetically predisposed lean and obese rats during inflammation: A role in modulating experimental colitis? Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G869–G877. [Google Scholar] [CrossRef] [Green Version]
- Kim, I.W.; Myung, S.-J.; Do, M.Y.; Ryu, Y.-M.; Kim, M.J.; Do, E.-J.; Park, S.; Yoon, S.M.; Ye, B.D.; Byeon, J.S.; et al. Western-style diets induce macrophage infiltration and contribute to colitis-associated carcinogenesis. J. Gastroenterol. Hepatol. 2010, 25, 1785–1794. [Google Scholar] [CrossRef]
- Gäbele, E.; Dostert, K.; Hofmann, C.; Wiest, R.; Schölmerich, J.; Hellerbrand, C.; Obermeier, F. DSS induced colitis increases portal LPS levels and enhances hepatic inflammation and fibrogenesis in experimental NASH. J. Hepatol. 2011, 55, 1391–1399. [Google Scholar] [CrossRef]
- Park, S.Y.; Kim, J.-S.; Seo, Y.-R.; Sung, M.-K. Effects of diet-induced obesity on colitis-associated colon tumor formation in A/J mice. Int. J. Obes. 2012, 36, 273–280. [Google Scholar] [CrossRef] [Green Version]
- Paik, J.; Fierce, Y.; Treuting, P.M.; Brabb, T.; Maggio-Price, L. High-fat diet-induced obesity exacerbates inflammatory bowel disease in genetically susceptible Mdr1a-/-male mice. J. Nutr. 2013, 143, 1240–1247. [Google Scholar] [CrossRef]
- Day, S.D.; Enos, R.T.; McClellan, J.L.; Steiner, J.L.; Velázquez, K.T.; Murphy, E.A. Linking inflammation to tumorigenesis in a mouse model of high-fat-diet-enhanced colon cancer. Cytokine 2013, 64, 454–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enos, R.T.; Velázquez, K.T.; McClellan, J.L.; Cranford, T.L.; Nagarkatti, M.; Nagarkatti, P.S.; Davis, J.M.; Murphy, E.A. High-fat diets rich in saturated fat protect against azoxymethane/dextran sulfate sodium-induced colon cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G906–G919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dermadi, D.; Valo, S.; Ollila, S.; Soliymani, R.; Sipari, N.; Pussila, M.; Sarantaus, L.; Linden, J.; Baumann, M.; Nyström, M. Western Diet Deregulates Bile Acid Homeostasis, Cell Proliferation, and Tumorigenesis in Colon. Cancer Res. 2017, 77, 3352–3363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Guo, Z.; Song, X.; Liu, L.; Dong, W.; Wang, S.; Xu, M.; Yang, C.; Wang, B.; Cao, H. High-fat diet-induced dysbiosis mediates MCP-1/CCR2 axis-dependent M2 macrophage polarization and promotes intestinal adenoma-adenocarcinoma sequence. J. Cell Mol. Med. 2020, 24, 2648–2662. [Google Scholar] [CrossRef] [Green Version]
- Devkota, S.; Wang, Y.; Musch, M.W.; Leone, V.; Fehlner-Peach, H.; Nadimpalli, A.; Antonopoulos, D.A.; Jabri, B.; Chang, E.B. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10-/-mice. Nature 2012, 487, 104–108. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, M.; Miyoshi, M.; Sakai, S.; Nishiokada, A.; Aoyama-Ishikawa, M.; Maeshige, N.; Usami, Y.; Hamada, Y.; Takahashi, M.; Usami, M. Lard-based high-fat diet increases secretory leukocyte protease inhibitor expression and attenuates the inflammatory response of acute lung injury in endotoxemic rats. Clin. Nutr. 2015, 34, 997–1009. [Google Scholar] [CrossRef] [Green Version]
- Nagy-Szakal, D.; Mir, S.A.V.; Harris, R.A.; Dowd, S.E.; Yamada, T.; Lacorazza, H.D.; Tatevian, N.; Smith, C.W.; de Zoeten, E.F.; Klein, J.; et al. Loss of n-6 fatty acid induced pediatric obesity protects against acute murine colitis. FASEB J. 2015, 29, 3151–3159. [Google Scholar] [CrossRef] [Green Version]
- Darfeuille-Michaud, A.; Neut, C.; Barnich, N.; Lederman, E.; Di Martino, P.; Desreumaux, P.; Gambiez, L.; Joly, B.; Cortot, A.; Colombel, J.F. Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn’s disease. Gastroenterology 1998, 115, 1405–1413. [Google Scholar] [CrossRef]
- Frank, D.N.; St Amand, A.L.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef] [Green Version]
- Kostic, A.D.; Chun, E.; Meyerson, M.; Garrett, W.S. Microbes and inflammation in colorectal cancer. Cancer Immunol. Res. 2013, 1, 150–157. [Google Scholar] [CrossRef]
- Prorok-Hamon, M.; Friswell, M.K.; Alswied, A.; Roberts, C.L.; Song, F.; Flanagan, P.K.; Knight, P.; Codling, C.; Marchesi, J.R.; Winstanley, C.; et al. Colonic mucosa-associated diffusely adherent afaC+ Escherichia coli expressing lpfA and pks are increased in inflammatory bowel disease and colon cancer. Gut 2014, 63, 761–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machiels, K.; Joossens, M.; Sabino, J.; De Preter, V.; Arijs, I.; Eeckhaut, V.; Ballet, V.; Claes, K.; Van Immerseel, F.; Verbeke, K.; et al. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut 2014, 63, 1275–1283. [Google Scholar] [CrossRef]
- Joyce, S.A.; Gahan, C. The gut microbiota and the metabolic health of the host. Curr. Opin. Gastroenterol. 2014, 30, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Kolho, K.L.; Korpela, K.; Jaakkola, T.; Pichai, M.V.A.; Zoetendal, E.G.; Salonen, A.; de Vos, W.M. Fecal Microbiota in Pediatric Inflammatory Bowel Disease and Its Relation to Inflammation. Am. J. Gastroenterol. 2015, 110, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Lavelle, A.; Sokol, H. Gut microbiota-derived metabolites as key actors in inflammatory bowel disease. Nat. Rev. Gastroenterol Hepatol. 2020, 17, 223–237. [Google Scholar] [CrossRef]
- Long, S.L.; Gahan, C.; Joyce, S. Interactions between gut bacteria and bile in health and disease. Mol. Aspects Med. 2017, 56, 54–65. [Google Scholar] [CrossRef]
- Li, T.; Chiang, J. Bile acids as metabolic regulators. Curr. Opin. Gastroenterol. 2015, 31, 159–165. [Google Scholar] [CrossRef] [Green Version]
- Biagioli, M.; Carino, A.; Cipriani, S.; Francisci, S.; Marchianò, S.; Scarpelli, P.; Sorcini, D.; Zampella, A.; Fiorucci, S. The Bile Acid Receptor GPBAR1 Regulates the M1/M2 Phenotype of Intestinal Macrophages and Activation of GPBAR1 Rescues Mice from Murine Colitis. J. Immunol. 2017, 199, 718–733. [Google Scholar] [CrossRef] [Green Version]
- Hang, S.; Paik, D.; Yao, L.; Kim, E.; Trinath, J.; Lu, J.; Ha, S.; Nelson, B.N.; Kelly, S.P.; Lin, W.; et al. Bile acid metabolites control T(H)17 and T(reg) cell differentiation. Nature 2019, 576, 143–148. [Google Scholar] [CrossRef]
- Song, X.; Sun, X.; Oh, S.F.; Wu, M.; Zhang, W.; Zheng, W.; Geva-Zatorsky, N.; Jupp, R.; Mathis, D.; Benoist, C.; et al. Microbial bile acid metabolites modulate gut RORγ(+) regulatory T cell homeostasis. Nature 2020, 577, 410–415. [Google Scholar] [CrossRef]
- Campbell, C.; McKenney, P.T.; Konstantinovsky, D.; Isaeva, O.I.; Schizas, M.; Verter, J.; Mai, C.; Jin, W.-B.; Guo, C.-J.; Violante, S.; et al. Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells. Nature 2020, 581, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Wirtz, S.; Neufert, C.; Weigmann, B.; Neurath, M.F. Chemically induced mouse models of intestinal inflammation. Nat. Protoc 2007, 2, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B.; Bajaj, J.S. Bile acids and the gut microbiome. Curr. Opin. Gastroenterol. 2014, 30, 332–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Yang, M.; Dong, W.; Liu, T.; Song, X.; Gu, Y.; Wang, S.; Liu, Y.; Abla, Z.; Qiao, X.; et al. Gut Dysbiosis and Abnormal Bile Acid Metabolism in Colitis-Associated Cancer. Gastroenterol. Res. Pract. 2021, 2021, 6645970. [Google Scholar] [CrossRef]
- Beyaz, S.; Mana, M.D.; Roper, J.; Kedrin, D.; Saadatpour, A.; Hong, S.-J.; Bauer-Rowe, K.E.; Xifaras, M.E.; Akkad, A.; Arias, E.; et al. High-fat diet enhances stemness and tumorigenicity of intestinal progenitors. Nature 2016, 531, 53–58. [Google Scholar] [CrossRef] [Green Version]
- Melgar, S.; Karlsson, l.; Rehnström, E.; Karlsson, A.; Utkovic, H.; Jansson, L.; Michaëlsson, E. Validation of murine dextran sulfate sodium-induced colitis using four therapeutic agents for human inflammatory bowel disease. Int. Immunopharmacol. 2008, 8, 836–844. [Google Scholar] [CrossRef]
- Lee, J.C.; Lee, H.-Y.; Kim, T.K.; Kim, M.-S.; Park, Y.M.; Kim, J.; Park, K.; Kweon, M.-N.; Kim, S.-H.; Bae, J.-W.; et al. Obesogenic diet-induced gut barrier dysfunction and pathobiont expansion aggravate experimental colitis. PLoS ONE 2017, 12, e0187515. [Google Scholar] [CrossRef] [Green Version]
- Finger, B.C.; Dinan, T.; Cryan, J. High-fat diet selectively protects against the effects of chronic social stress in the mouse. Neuroscience 2011, 192, 351–360. [Google Scholar] [CrossRef]
- de Souza, R.J.; Mente, A.; Maroleanu, A.; Cozma, A.I.; Ha, V.; Kishibe, T.; Uleryk, E.; Budylowski, P.; Schünemann, H.; Beyene, J.; et al. Intake of saturated and trans unsaturated fatty acids and risk of all cause mortality, cardiovascular disease, and type 2 diabetes: Systematic review and meta-analysis of observational studies. BMJ 2015, 351, h3978. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, A.; Redberg, R.; Meier, P. Saturated fat does not clog the arteries: Coronary heart disease is a chronic inflammatory condition, the risk of which can be effectively reduced from healthy lifestyle interventions. Br. J. Sports Med. 2017, 51, 1111–1112. [Google Scholar] [CrossRef]
- Gao, M.; Jebb, S.A.; Aveyard, P.; Ambrosini, G.L.; Perez-Cornago, A.; Carter, J.; Sun, X.; Piernas, C. Associations between dietary patterns and the incidence of total and fatal cardiovascular disease and all-cause mortality in 116,806 individuals from the UK Biobank: A prospective cohort study. BMC Med. 2021, 19, 83. [Google Scholar] [CrossRef] [PubMed]
- Hur, J.; Otegbeye, E.; Joh, H.-K.; Nimptsch, K.; Ng, K.; Ogino, S.; Meyerhardt, J.A.; Chan, A.T.; Willett, W.C.; Wu, K.; et al. Sugar-sweetened beverage intake in adulthood and adolescence and risk of early-onset colorectal cancer among women. Gut 2021, 70, 2330–2336. [Google Scholar] [CrossRef]
- Spooren, C.E.G.M.; Pierik, M.J.; Zeegers, M.P.; Feskens, E.J.M.; Masclee, A.A.M.; Jonkers, D.M.A.E. Review article: The association of diet with onset and relapse in patients with inflammatory bowel disease. Aliment. Pharmacol. Ther. 2013, 38, 1172–1187. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, J.; Garces, L.; Quintero, M.A.; Pignac-Kobinger, J.; Santander, A.M.; Fernández, I.; Ban, Y.J.; Kwon, D.; Phillips, M.C.; Knight, K.; et al. Low-Fat, High-Fiber Diet Reduces Markers of Inflammation and Dysbiosis and Improves Quality of Life in Patients With Ulcerative Colitis. Clin. Gastroenterol. Hepatol. 2021, 19, 1189–1199.e30. [Google Scholar] [CrossRef]
- Ryan, F.J.; Ahern, A.M.; Fitzgerald, R.S.; Laserna-Mendieta, E.J.; Power, E.M.; Clooney, A.G.; O’Donoghue, K.W.; McMurdie, P.J.; Iwai, S.; Crits-Christoph, A.; et al. Colonic microbiota is associated with inflammation and host epigenomic alterations in inflammatory bowel disease. Nat. Commun. 2020, 11, 1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duboc, H.; Rajca, S.; Rainteau, D.; Benarous, D.; Maubert, M.A.; Quervain, E.; Thomas, G.; Barbu, V.; Humbert, L.; Despras, G.; et al. Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases. Gut 2013, 62, 531–539. [Google Scholar] [CrossRef]
- Sinha, S.R.; Haileselassie, Y.; Nguyen, L.P.; Tropini, C.; Wang, M.; Becker, L.S.; Sim, D.; Jarr, K.; Spear, E.T.; Singh, G.; et al. Dysbiosis-Induced Secondary Bile Acid Deficiency Promotes Intestinal Inflammation. Cell Host. Microbe. 2020, 27, 659–670.e5. [Google Scholar] [CrossRef]
- Zhang, S.L.; Wang, S.; Miao, C. Influence of Microbiota on Intestinal Immune System in Ulcerative Colitis and Its Intervention. Front. Immunol. 2017, 8, 1674. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.; Xu, M.; Dong, W.; Deng, B.; Wang, S.; Zhang, Y.; Wang, S.; Luo, S.; Wang, W.; Qi, Y.; et al. Secondary bile acid-induced dysbiosis promotes intestinal carcinogenesis. Int. J. Cancer 2017, 140, 2545–2556. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Dong, W.; Wang, S.; Zhang, Y.; Liu, T.; Xie, R.; Wang, B.; Cao, H. Deoxycholic acid disrupts the intestinal mucosal barrier and promotes intestinal tumorigenesis. Food Funct. 2018, 9, 5588–5597. [Google Scholar] [CrossRef]
- Jia, W.; Xie, G.; Jia, W. Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 111–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, J.B.J.; Lajczak, N.K.; Kelly, O.B.; O’Dwyer, A.M.; Giddam, A.K.; Gabhann, J.N.; Franco, P.; Tambuwala, M.M.; Jefferies, C.A.; Keely, S.; et al. Ursodeoxycholic acid and lithocholic acid exert anti-inflammatory actions in the colon. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G550–G558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lajczak-McGinley, N.K.; Porru, E.; Fallon, C.M.; Smyth, J.; Curley, C.; McCarron, P.A.; Tambuwala, M.M.; Roda, A.; Keely, S.J. The secondary bile acids, ursodeoxycholic acid and lithocholic acid, protect against intestinal inflammation by inhibition of epithelial apoptosis. Physiol. Rep. 2020, 8, e14456. [Google Scholar] [CrossRef]
- Makishima, M.; Lu, T.T.; Xie, W.; Whitfield, G.K.; Domoto, H.; Evans, R.M.; Haussler, M.R.; Mangelsdorf, D.J. Vitamin D receptor as an intestinal bile acid sensor. Science 2002, 296, 1313–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.M.; Ong, S.S.; Chai, S.C.; Chen, T. Role of CAR and PXR in xenobiotic sensing and metabolism. Expert Opin. Drug Metab. Toxicol. 2012, 8, 803–817. [Google Scholar] [CrossRef] [Green Version]
- Pavek, P. Pregnane X Receptor (PXR)-Mediated Gene Repression and Cross-Talk of PXR with Other Nuclear Receptors via Coactivator Interactions. Front. Pharmacol. 2016, 7, 456. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.A.; Chow, E.C.; Porte, R.J.; Pang, K.S.; Groothuis, G.M. The role of lithocholic acid in the regulation of bile acid detoxication, synthesis, and transport proteins in rat and human intestine and liver slices. Toxicol. Vitr. 2011, 25, 80–90. [Google Scholar] [CrossRef]
- Song, M.; Garrett, W.; Chan, A. Nutrients, foods, and colorectal cancer prevention. Gastroenterology 2015, 148, 1244–1260. [Google Scholar] [CrossRef] [Green Version]
- Murphy, E.F.; Cotter, P.D.; Healy, S.; Marques, T.M.; O’Sullivan, O.; Fouhy, F.; Clarke, S.F.; O’Toole, P.W.; Quigley, E.M.; Stanton, C.; et al. Composition and energy harvesting capacity of the gut microbiota: Relationship to diet, obesity and time in mouse models. Gut 2010, 59, 1635–1642. [Google Scholar] [CrossRef]
- O’Mahony, C.; van der Kleij, H.; Bienenstock, J.; Shanahan, F.; O’Mahony, L. Loss of vagal anti-inflammatory effect: In Vivo visualization and adoptive transfer. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R1118–R1126. [Google Scholar] [CrossRef]
- Hall, L.J.; Faivre, E.; Quinlan, A.; Shanahan, F.; Nally, K.; Melgar, S. Induction and activation of adaptive immune populations during acute and chronic phases of a murine model of experimental colitis. Dig. Dis. Sci. 2011, 56, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.T.; Moloney, G.; Hall, L.J.; Quinlan, A.; Faivre, E.; Casey, P.; Shanahan, F.; Melgar, S.; Nally, K. Use of bioluminescence imaging to track neutrophil migration and its inhibition in experimental colitis. Clin. Exp. Immunol. 2010, 162, 188–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, C.T.; Hall, L.J.; Hurley, G.; Quinlan, A.; MacSharry, J.; Shanahan, F.; Nally, K.; Melgar, S. The sphingosine-1-phosphate analogue FTY720 impairs mucosal immunity and clearance of the enteric pathogen Citrobacter rodentium. Infect. Immun. 2012, 80, 2712–2723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Dieleman, L.A.; Palmen, M.J.; Akol, H.; Bloemena, E.; Peña, A.S.; Meuwissen, S.G.; Van Rees, E.P. Chronic experimental colitis induced by dextran sulphate sodium (DSS) is characterized by Th1 and Th2 cytokines. Clin. Exp. Immunol. 1998, 114, 385–391. [Google Scholar] [CrossRef]
- O’Connell Motherway, M.; Houston, A.; O’Callaghan, G.; Reunanen, J.; O’Brien, F.; O’Driscoll, T.; Casey, P.G.; de Vos, G.M.; van Sinderen, D.; Shanahan, F. A Bifidobacterial pilus-associated protein promotes colonic epithelial proliferation. Mol. Microbiol. 2019, 111, 287–301. [Google Scholar] [CrossRef] [Green Version]
- Clarke, S.F.; Murphy, E.F.; O’Sullivan, O.; Lucey, A.J.; Humphreys, M.; Hogan, A.; Hayes, P.; O’Reilly, M.; Jeffery, I.B.; Wood-Martin, R.; et al. Exercise and associated dietary extremes impact on gut microbial diversity. Gut 2014, 63, 1913–1920. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef]
- Schloss, P.D. A high-throughput DNA sequence aligner for microbial ecology studies. PLoS ONE 2009, 4, e8230. [Google Scholar] [CrossRef] [PubMed]
- Allard, G.; Ryan, F.J.; Jeffery, I.B.; Claesson, M.J. SPINGO: A rapid species-classifier for microbial amplicon sequences. BMC Bioinform. 2015, 16, 324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques, T.M.; Wall, R.; O’Sullivan, O.; Fitzgerald, G.F.; Shanahan, F.; Quigley, E.M.; Cotter, P.D.; Cryan, J.F.; Dinan, T.G.; Ross, R.P.; et al. Dietary trans-10, cis-12-conjugated linoleic acid alters fatty acid metabolism and microbiota composition in mice. Br. J. Nutr. 2015, 14, 728–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joyce, S.A.; MacSharry, J.; Casey, P.G.; Kinsella, M.; Murphy, E.F.; Shanahan, F.; Hill, C.; Gahan, C.G.M. Regulation of host weight gain and lipid metabolism by bacterial bile acid modification in the gut. Proc. Natl. Acad. Sci. USA 2014, 111, 7421–7426. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
O’Mahony, C.; Clooney, A.; Clarke, S.F.; Aguilera, M.; Gavin, A.; Simnica, D.; Ahern, M.; Fanning, A.; Stanley, M.; Rubio, R.C.; et al. Dietary-Induced Bacterial Metabolites Reduce Inflammation and Inflammation-Associated Cancer via Vitamin D Pathway. Int. J. Mol. Sci. 2023, 24, 1864. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24031864
O’Mahony C, Clooney A, Clarke SF, Aguilera M, Gavin A, Simnica D, Ahern M, Fanning A, Stanley M, Rubio RC, et al. Dietary-Induced Bacterial Metabolites Reduce Inflammation and Inflammation-Associated Cancer via Vitamin D Pathway. International Journal of Molecular Sciences. 2023; 24(3):1864. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24031864
Chicago/Turabian StyleO’Mahony, Caitlin, Adam Clooney, Siobhan F. Clarke, Mònica Aguilera, Aisling Gavin, Donjete Simnica, Mary Ahern, Aine Fanning, Maurice Stanley, Raul Cabrera Rubio, and et al. 2023. "Dietary-Induced Bacterial Metabolites Reduce Inflammation and Inflammation-Associated Cancer via Vitamin D Pathway" International Journal of Molecular Sciences 24, no. 3: 1864. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24031864