Potential Suppressive Effect of Nicotine on the Inflammatory Response in Oral Epithelial Cells: An In Vitro Study

 , and
, and 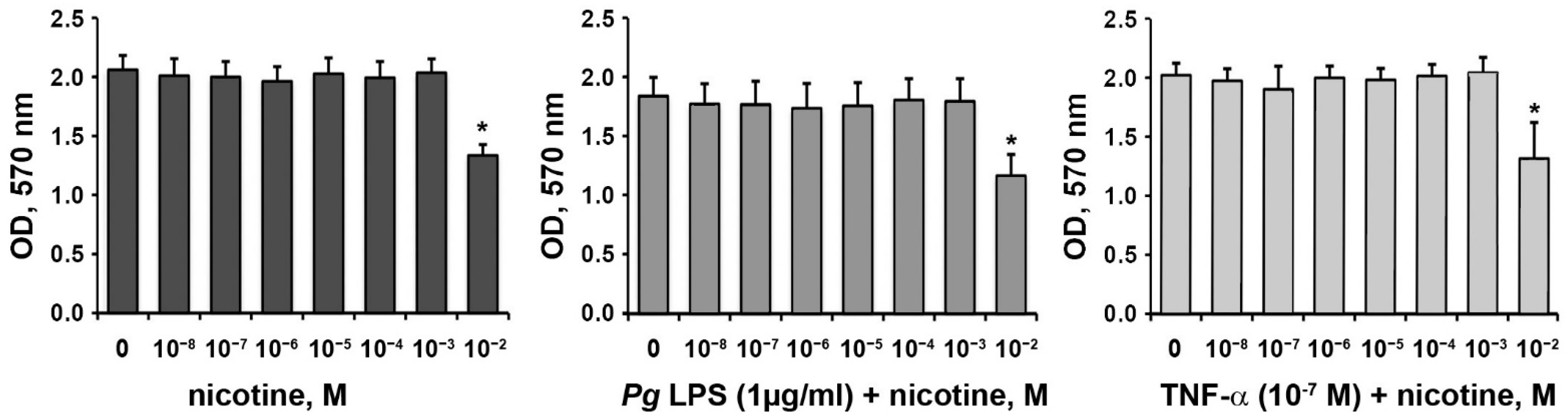{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Proliferation/Viability
2.3. Gene and Protein Expression Analyses
2.4. Statistical Analysis
3. Results
3.1. Effect of Nicotine on the Proliferation/Viability of HSCs
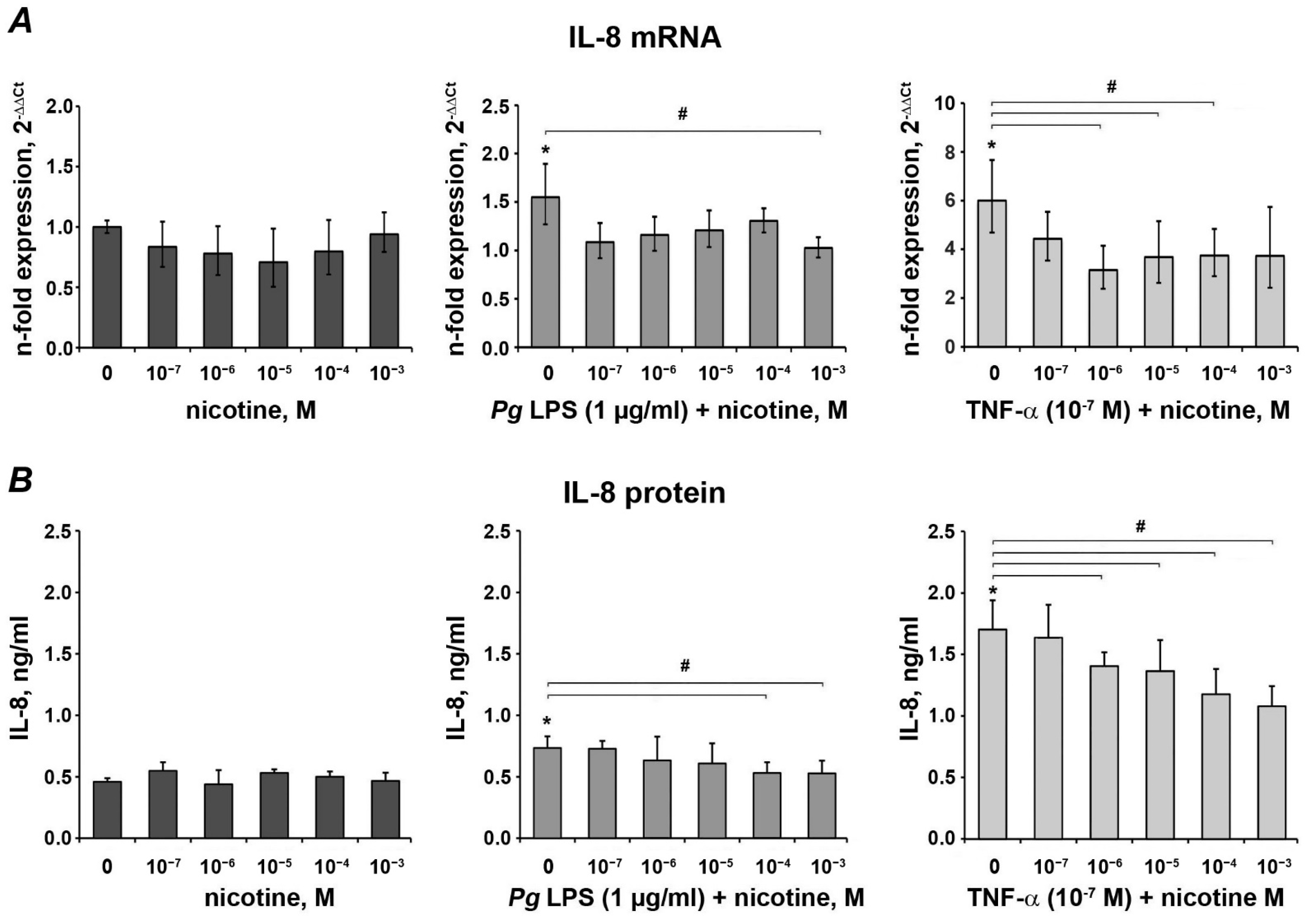
3.2. Effect of Nicotine on the Expression of IL-8
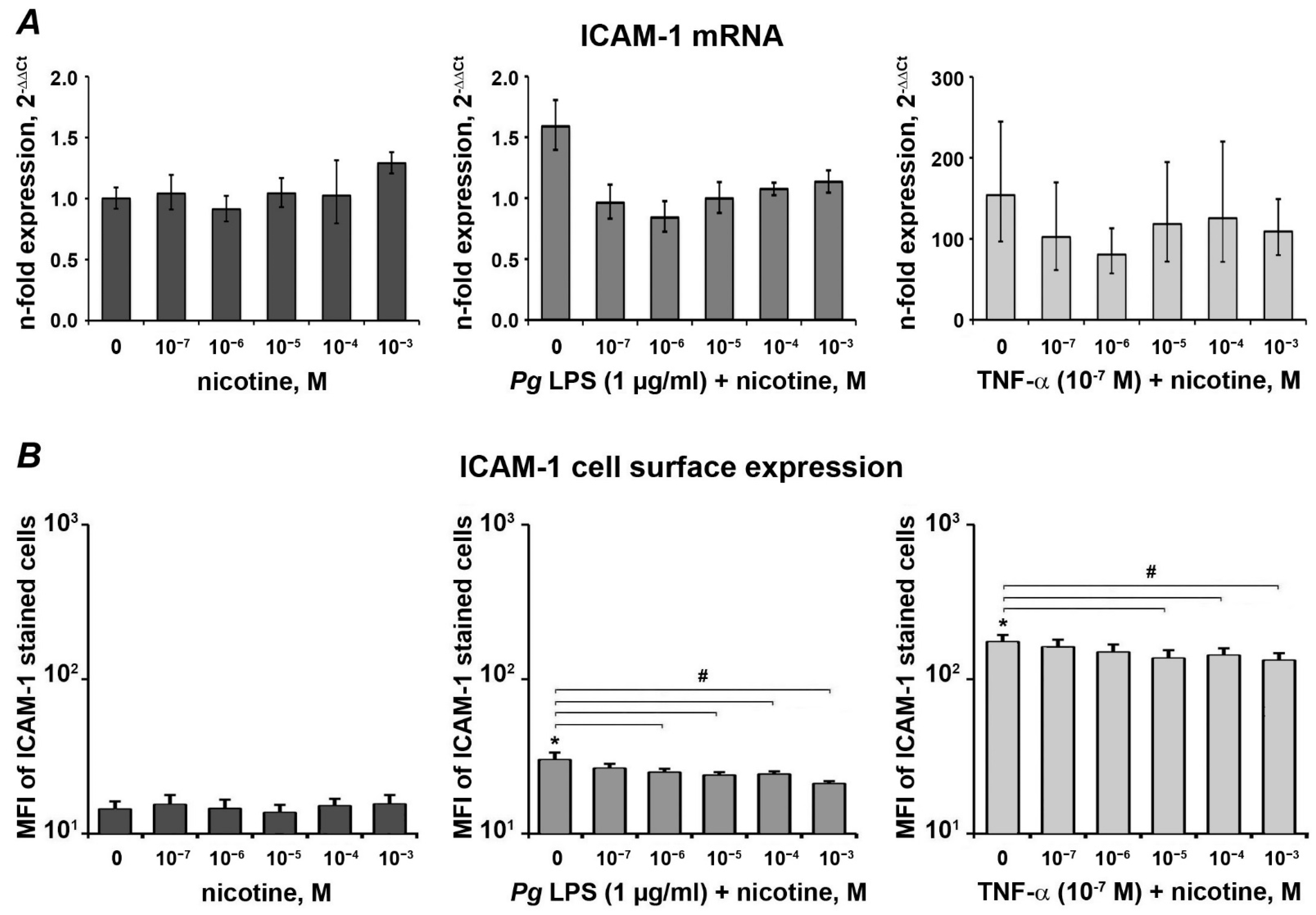
3.3. Effect of Nicotine on the Expression of ICAM-1
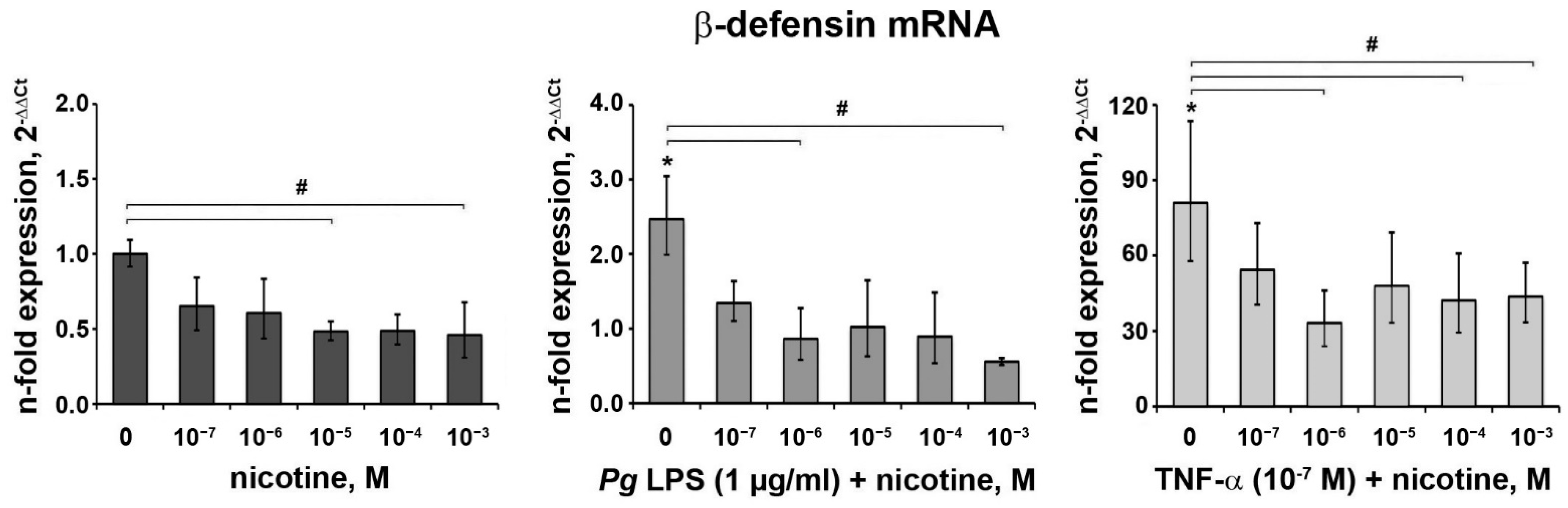
3.4. Effect of Nicotine on the Expression of β-Defensin
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, S.; Yu, N.; Arce, R.M. Periodontal inflammation: Integrating genes and dysbiosis. Periodontology 2000 2019, 82, 129–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamont, R.J.; Koo, H.; Hajishengallis, G. The oral microbiota: Dynamic communities and host interactions. Nat. Rev. Microbiol. 2018, 16, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Cekici, A.; Kantarci, A.; Hasturk, H.; Van Dyke, T.E. Inflammatory and immune pathways in the pathogenesis of periodontal disease. Periodontology 2000 2014, 64, 57–80. [Google Scholar] [CrossRef] [Green Version]
- Hajishengallis, G.; Darveau, R.P.; Curtis, M.A. The keystone-pathogen hypothesis. Nat. Rev. Microbiol. 2012, 10, 717–725. [Google Scholar] [CrossRef]
- Bostanci, N.; Belibasakis, G.N. Porphyromonas gingivalis: An invasive and evasive opportunistic oral pathogen. FEMS Microbiol. Lett. 2012, 333, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Lamont, R.J.; Chan, A.; Belton, C.M.; Izutsu, K.T.; Vasel, D.; Weinberg, A. Porphyromonas gingivalis invasion of gingival epithelial cells. Infect. Immun. 1995, 63, 3878–3885. [Google Scholar] [CrossRef] [Green Version]
- Groeger, S.; Meyle, J. Oral Mucosal Epithelial Cells. Front. Immunol. 2019, 10, 208. [Google Scholar] [CrossRef] [Green Version]
- Baggiolini, M.; Clark-Lewis, I. Interleukin-8, a chemotactic and inflammatory cytokine. FEBS Lett. 1992, 307, 97–101. [Google Scholar] [CrossRef] [Green Version]
- Long, E.O. ICAM-1: Getting a grip on leukocyte adhesion. J. Immunol. 2011, 186, 5021–5023. [Google Scholar] [CrossRef] [Green Version]
- Meade, K.G.; O’Farrelly, C. Beta-defensins: Farming the microbiome for homeostasis and health. Front. Immunol. 2018, 9, 3072. [Google Scholar] [CrossRef]
- Tonetti, M.S. Cigarette smoking and periodontal diseases: Etiology and management of disease. Ann. Periodontol. 1998, 3, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.K.; Guthmiller, J.M. The impact of cigarette smoking on periodontal disease and treatment. Periodontology 2000 2007, 44, 178–194. [Google Scholar] [CrossRef] [PubMed]
- Rose, J.E.; Salley, A.; Behm, F.M.; Bates, J.E.; Westman, E.C. Reinforcing effects of nicotine and non-nicotine components of cigarette smoke. Psychopharmacology 2010, 210, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Walley, S.C.; Wilson, K.M.; Winickoff, J.P.; Groner, J. A public health crisis: Electronic cigarettes, vape, and JUUL. Pediatrics 2019, 143. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Song, F.; Windsor, L.J. Effects of tobacco and P. gingivalis on gingival fibroblasts. J. Dent. Res. 2010, 89, 527–531. [Google Scholar] [CrossRef]
- Kim, S.Y.; Kang, K.L.; Lee, J.C.; Heo, J.S. Nicotinic acetylcholine receptor α7 and β4 subunits contribute nicotine-induced apoptosis in periodontal ligament stem cells. Mol. Cells 2012, 33, 343–350. [Google Scholar] [CrossRef] [Green Version]
- Imamura, K.; Kokubu, E.; Kita, D.; Ota, K.; Yoshikawa, K.; Ishihara, K.; Saito, A. Role of mitogen-activated protein kinase pathways in migration of gingival epithelial cells in response to stimulation by cigarette smoke condensate and infection by Porphyromonas gingivalis. J. Periodontal. Res. 2016, 51, 613–621. [Google Scholar] [CrossRef]
- Han, Y.-K.; Lee, I.S.; Lee, S.-I. JAK/STAT pathway modulates on porphyromonas gingivalis lipopolysaccharide- and nicotine-induced inflammation in osteoblasts. J. Dent. Hyg. Sci. 2017, 17, 81–86. [Google Scholar] [CrossRef] [Green Version]
- Holliday, R.S.; Campbell, J.; Preshaw, P.M. Effect of nicotine on human gingival, periodontal ligament and oral epithelial cells. A systematic review of the literature. J. Dent. 2019, 86, 81–88. [Google Scholar] [CrossRef]
- Moore, D. Is nicotine damaging to oral tissues? Evid. Based Dent. 2020, 21, 32–33. [Google Scholar] [CrossRef]
- Desjardins, J.; Grenier, D. Neutralizing effect of green tea epigallocatechin-3-gallate on nicotine-induced toxicity and chemokine (C-C motif) ligand 5 secretion in human oral epithelial cells and fibroblasts. J. Investig. Clin. Dent. 2012, 3, 189–197. [Google Scholar] [CrossRef]
- Johnson, G.K.; Guthmiller, J.M.; Joly, S.; Organ, C.C.; Dawson, D.V. Interleukin-1 and interleukin-8 in nicotine- and lipopolysaccharide-exposed gingival keratinocyte cultures. J. Periodontal. Res. 2010, 45, 583–588. [Google Scholar] [CrossRef]
- Almasri, A.; Wisithphrom, K.; Windsor, L.J.; Olson, B. Nicotine and lipopolysaccharide affect cytokine expression from gingival fibroblasts. J. Periodontol. 2007, 78, 533–541. [Google Scholar] [CrossRef]
- Kudo, Y.; Kitajjma, S.; Sato, S.; Miyauchi, M.; Ogawa, I.; Takata, T. Establishment of an oral squamous cell carcinoma cell line with high invasive and p27 degradation activities from a lymph node metastasis. Oral Oncol. 2003, 39, 515–520. [Google Scholar] [CrossRef]
- Behm, C.; Blufstein, A.; Abhari, S.Y.; Koch, C.; Gahn, J.; Schäffer, C.; Moritz, A.; Rausch-Fan, X.; Andrukhov, O. Response of human mesenchymal stromal cells from periodontal tissue to LPS depends on the purity but not on the LPS source. Mediat. Inflamm. 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Ozdemir, B.; Nguyen, P.Q.; Andrukhov, O.; Rausch-Fan, X. Methanandamide diminish the Porphyromonas gingivalis lipopolysaccharide induced response in human periodontal ligament cells. BMC Oral Health 2020, 20, 107. [Google Scholar] [CrossRef] [Green Version]
- An, N.; Andrukhov, O.; Tang, Y.; Falkensammer, F.; Bantleon, H.P.; Ouyang, X.; Rausch-Fan, X. Effect of nicotine and porphyromonas gingivalis lipopolysaccharide on endothelial cells in vitro. PLoS ONE 2014, 9, e96942. [Google Scholar] [CrossRef]
- Andrukhov, O.; Andrukhova, O.; Ozdemir, B.; Haririan, H.; Muller-Kern, M.; Moritz, A.; Rausch-Fan, X. Soluble CD14 enhances the response of periodontal ligament stem cells to P. gingivalis lipopolysaccharide. PLoS ONE 2016, 11, e0160848. [Google Scholar] [CrossRef]
- Behm, C.; Blufstein, A.; Gahn, J.; Nemec, M.; Moritz, A.; Rausch-Fan, X.; Andrukhov, O. Cytokines differently define the Immunomodulation of mesenchymal stem cells from the periodontal ligament. Cells 2020, 9, 1222. [Google Scholar] [CrossRef]
- Blufstein, A.; Behm, C.; Nguyen, P.Q.; Rausch-Fan, X.; Andrukhov, O. Human periodontal ligament cells exhibit no endotoxin tolerance upon stimulation with Porphyromonas gingivalis lipopolysaccharide. J. Periodont. Res. 2018, 53, 589–597. [Google Scholar] [CrossRef] [Green Version]
- Blufstein, A.; Behm, C.; Gahn, J.; Uitz, O.; Naumovska, I.; Moritz, A.; Rausch-Fan, X.; Andrukhov, O. Synergistic effects triggered by simultaneous Toll-like receptor-2 and -3 activation in human periodontal ligament stem cells. J. Periodontol. 2019, 90, 1190–1201. [Google Scholar] [CrossRef]
- Andrukhov, O.; Steiner, I.; Liu, S.; Bantleon, H.P.; Moritz, A.; Rausch-Fan, X. Different effects of Porphyromonas gingivalis lipopolysaccharide and TLR2 agonist Pam3CSK4 on the adhesion molecules expression in endothelial cells. Odontology 2013. [Google Scholar] [CrossRef]
- Robson, N.; Bond, A.J.; Wolff, K. Salivary nicotine and cotinine concentrations in unstimulated and stimulated saliva. Afr. J. Pharm. Pharmaco 2010, 4, 61–65. [Google Scholar]
- Feyerabend, C.; Higenbottam, T.; Russell, M.A. Nicotine concentrations in urine and saliva of smokers and non-smokers. Br. Med. J. 1982, 284, 1002–1004. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, D.; Adams, J.D. A Study of tobacco carcinogenesis 23. Carcinogenic tobacco-specific N-nitrosamines in snuff and in the saliva of snuff dippers. Cancer Res. 1981, 41, 4305–4308. [Google Scholar]
- Johannsen, A.; Susin, C.; Gustafsson, A. Smoking and inflammation: Evidence for a synergistic role in chronic disease. Periodontology 2000 2014, 64, 111–126. [Google Scholar] [CrossRef]
- Liu, Q.; Zhao, M.; Chen, W.; Xu, K.; Huang, F.; Qu, J.; Xu, Z.; Wang, X.; Wang, Y.; Zhu, Y.; et al. Mainstream cigarette smoke induces autophagy and promotes apoptosis in oral mucosal epithelial cells. Arch. Oral Biol. 2020, 111, 104646. [Google Scholar] [CrossRef]
- Deveci, B.; Ayna, B.; Tacir, I.H.; Deveci, E.; Tuncer, M.C.; Pala, A. Effects of nicotine administration in rats on MMP2 and VEGF levels in periodontal membrane. Folia Morphol. 2018, 77, 471–477. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; He, J.; He, B.; Huang, R.; Li, M. Effect of tobacco on periodontal disease and oral cancer. Tob. Induc. Dis. 2019, 17, 40. [Google Scholar] [CrossRef]
- Johnson, G.K.; Organ, C.C. Prostaglandin E2 and interleukin-1 concentrations in nicotine-exposed oral keratinocyte cultures. J. Periodont. Res. 1997, 32, 447–454. [Google Scholar] [CrossRef]
- Argentin, G.; Cicchetti, R. Genotoxic and antiapoptotic effect of nicotine on human gingival fibroblasts. Toxicol. Sci. 2004, 79, 75–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciapetti, G.; Remiddi, G.; Savioli, F.; Monaco, G.; Ori, G.; Checchi, L. In vitro testing of the responses of human gingival fibroblasts and L-929 cells to nicotine. Altern. Lab. Anim. 1999, 27, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Kraus, D.; Winter, J.; Jepsen, S.; Jager, A.; Meyer, R.; Deschner, J. Interactions of adiponectin and lipopolysaccharide from Porphyromonas gingivalis on human oral epithelial cells. PLoS ONE 2012, 7, e30716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonçalves, M.; Cappellari, Á.R.; Santos Junior, A.A.D.; Marchi, F.O.D.; Macchi, F.S.; Antunes, K.H.; Souza, A.P.D.D.; Morrone, F.B. Effect of LPS on the Viability and Proliferation of Human Oral and Esophageal Cancer Cell Lines. Braz. Arch. Biol. Technol. 2016, 59. [Google Scholar] [CrossRef] [Green Version]
- Basso, F.G.; Pansani, T.N.; Turrioni, A.P.; Soares, D.G.; de Souza Costa, C.A.; Hebling, J. Tumor necrosis factor-alpha and interleukin (IL)-1beta, IL-6, and IL-8 impair in vitro migration and induce apoptosis of gingival fibroblasts and epithelial cells, delaying wound healing. J. Periodontol. 2016, 87, 990–996. [Google Scholar] [CrossRef] [PubMed]
- Finoti, L.S.; Nepomuceno, R.; Pigossi, S.C.; Corbi, S.C.T.; Secolin, R.; Scarel-Caminaga, R.M. Association between interleukin-8 levels and chronic periodontal disease A PRISMA-compliant systematic review and meta-analysis. Medicine 2017, 96. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.A.; Shiue, L.F.; Yu, H.S.; Hsieh, T.Y.; Tsai, C.C. Interleukin-8 secretion by cultured oral epidermoid carcinoma cells induced with nicotine and/or arecoline treatments. Kaohsiung J. Med. Sci. 2000, 16, 126–133. [Google Scholar]
- Mahanonda, R.; Sa-Ard-Iam, N.; Eksomtramate, M.; Rerkyen, P.; Phairat, B.; Schaecher, K.E.; Fukuda, M.M.; Pichyangkul, S. Cigarette smoke extract modulates human beta-defensin-2 and interleukin-8 expression in human gingival epithelial cells. J. Periodont. Res. 2009, 44, 557–564. [Google Scholar] [CrossRef]
- Han, D.C.; Huang, G.T.; Lin, L.M.; Warner, N.A.; Gim, J.S.; Jewett, A. Expression of MHC Class II, CD70, CD80, CD86 and pro-inflammatory cytokines is differentially regulated in oral epithelial cells following bacterial challenge. Oral Microbiol. Immunol. 2003, 18, 350–358. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, Y.; Cardell, L.O. Nicotine exaggerates LPS-induced airway hyperreactivity via JNK-mediated up-regulation of Toll-like receptor 4. Am. J. Respir. Cell Mol. Biol. 2014, 51, 370–379. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.S.; Shin, S.I.; Kang, K.L.; Chung, J.H.; Herr, Y.; Bae, W.J.; Kim, E.C. Nicotine and lipopolysaccharide stimulate the production of MMPs and prostaglandin E2 by hypoxia-inducible factor-1alpha up-regulation in human periodontal ligament cells. J. Periodont. Res. 2012, 47, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Gotts, J.E.; Abbott, J.; Fang, X.; Yanagisawa, H.; Takasaka, N.; Nishimura, S.L.; Calfee, C.S.; Matthay, M.A. Cigarette smoke exposure worsens endotoxin-induced lung injury and pulmonary edema in mice. Nicot. Tob. Res. 2017, 19, 1033–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barua, R.S.; Sharma, M.; Dileepan, K.N. Cigarette smoke amplifies inflammatory response and atherosclerosis progression through activation of the H1R-TLR2/4-COX2 Axis. Front. Immunol. 2015, 6, 572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leite, F.R.M.; Nascimento, G.G.; Scheutz, F.; Lopez, R. Effect of smoking on periodontitis: A systematic review and meta-regression. Am. J. Prev. Med. 2018, 54, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Osborne-Hereford, A.V.; Rogers, S.W.; Gahring, L.C. Neuronal nicotinic alpha7 receptors modulate inflammatory cytokine production in the skin following ultraviolet radiation. J. Neuroimmunol. 2008, 193, 130–139. [Google Scholar] [CrossRef] [Green Version]
- Bao, J.; Liu, Y.; Yang, J.; Gao, Q.; Shi, S.Q.; Garfield, R.E.; Liu, H. Nicotine inhibits LPS-induced cytokine production and leukocyte infiltration in rat placenta. Placenta 2016, 39, 77–83. [Google Scholar] [CrossRef]
- Teng, P.; Liu, Y.; Dai, Y.; Zhang, H.; Liu, W.T.; Hu, J. Nicotine attenuates osteoarthritis pain and matrix metalloproteinase-9 expression via the alpha7 nicotinic acetylcholine receptor. J. Immunol. 2019, 203, 485–492. [Google Scholar] [CrossRef]
- Jamal Uddin, M.; Joe, Y.; Zheng, M.; Blackshear, P.J.; Ryter, S.W.; Park, J.W.; Chung, H.T. A functional link between heme oxygenase-1 and tristetraprolin in the anti-inflammatory effects of nicotine. Free Radic. Biol. Med. 2013, 65, 1331–1339. [Google Scholar] [CrossRef] [Green Version]
- Bao, J.; Zou, Y.; Liu, Y.; Yuan, L.; Garfield, R.E.; Liu, H. Nicotine protects fetus against LPS-induced fetal growth restriction through ameliorating placental inflammation and vascular development in late pregnancy in rats. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [Green Version]
- Tsoyi, K.; Jang, H.J.; Kim, J.W.; Chang, H.K.; Lee, Y.S.; Pae, H.O.; Kim, H.J.; Seo, H.G.; Lee, J.H.; Chung, H.T.; et al. Stimulation of alpha7 nicotinic acetylcholine receptor by nicotine attenuates inflammatory response in macrophages and improves survival in experimental model of sepsis through heme oxygenase-1 induction. Antioxid. Redox. Signal 2011, 14, 2057–2070. [Google Scholar] [CrossRef]
- Nizri, E.; Irony-Tur-Sinai, M.; Lory, O.; Orr-Urtreger, A.; Lavi, E.; Brenner, T. Activation of the cholinergic anti-inflammatory system by nicotine attenuates neuroinflammation via suppression of Th1 and Th17 responses. J. Immunol. 2009, 183, 6681–6688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Liao, H.; Ochani, M.; Justiniani, M.; Lin, X.; Yang, L.; Al-Abed, Y.; Wang, H.; Metz, C.; Miller, E.J.; et al. Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat. Med. 2004, 10, 1216–1221. [Google Scholar] [CrossRef] [PubMed]
- Sugano, N.; Shimada, K.; Ito, K.; Murai, S. Nicotine inhibits the production of inflammatory mediators in U937 cells through modulation of nuclear factor-kappaB activation. Biochem. Biophys. Res. Commun. 1998, 252, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Bondy-Carey, J.L.; Galicia, J.; Bagaitkar, J.; Potempa, J.S.; Potempa, B.; Kinane, D.F.; Veillard, F.; Scott, D.A. Neutrophils alter epithelial response to Porphyromonas gingivalis in a gingival crevice model. Mol. Oral Microbiol. 2013, 28, 102–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishioka, T.; Tada, H.; Ibaragi, S.; Chen, C.; Sasano, T. Nicotine exposure induces the proliferation of oral cancer cells through the α7 subunit of the nicotinic acetylcholine receptor. Biochem. Biophys. Res. Commun. 2019, 509, 514–520. [Google Scholar] [CrossRef] [PubMed]
- Gursoy, U.K.; Kononen, E. Understanding the roles of gingival beta-defensins. J. Oral. Microbiol. 2012, 4. [Google Scholar] [CrossRef]
- Hatipoglu, O.; Saydam, F. Association between rs11362 polymorphism in the beta-defensin 1 (DEFB1) gene and dental caries: A meta-analysis. J. Oral Biosci. 2020. [Google Scholar] [CrossRef]
- Wang, W.M.; Ye, P.; Qian, Y.J.; Gao, Y.F.; Li, J.J.; Sun, F.F.; Zhang, W.Y.; Wang, X. Effects of whole cigarette smoke on human beta defensins expression and secretion by oral mucosal epithelial cells. Tob. Induc. Dis. 2015, 13, 3. [Google Scholar] [CrossRef] [Green Version]
- Offenbacher, S.; Barros, S.P.; Paquette, D.W.; Winston, J.L.; Biesbrock, A.R.; Thomason, R.G.; Gibb, R.D.; Fulmer, A.W.; Tiesman, J.P.; Juhlin, K.D.; et al. Gingival transcriptome patterns during induction and resolution of experimental gingivitis in humans. J. Periodontol. 2009, 80, 1963–1982. [Google Scholar] [CrossRef]
- Bayirli, B.A.; Ozturk, A.; Avci, B. Serum vitamin D concentration is associated with antimicrobial peptide level in periodontal diseases. Arch. Oral Biol. 2020, 117, 104827. [Google Scholar] [CrossRef]
- Pereira, A.G.; Costa, L.C.M.; Soldati, K.R.; Guimaraes de Abreu, M.H.N.; Costa, F.O.; Zandim-Barcelos, D.L.; Cota, L.O.M. Gingival crevicular fluid levels of human beta-defensin 2 and 3 in healthy and diseased sites of individuals with and without periodontitis. J. Int. Acad. Periodontol. 2020, 22, 90–99. [Google Scholar] [PubMed]
- Niyonsaba, F.; Ushio, H.; Nakano, N.; Ng, W.; Sayama, K.; Hashimoto, K.; Nagaoka, I.; Okumura, K.; Ogawa, H. Antimicrobial peptides human beta-defensins stimulate epidermal keratinocyte migration, proliferation and production of proinflammatory cytokines and chemokines. J. Invest. Dermatol. 2007, 127, 594–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowdish, D.M.E.; Davidson, D.J.; Hancock, R.E.W. Immunomodulatory properties of defensins and cathelicidins. Curr. Top. Microbiol. 2006, 306, 27–66. [Google Scholar]
- Wolgin, M.; Liodakis, S.; Pries, A.R.; Zakrzewicz, A.; Kielbassa, A.M. HBD-1 and hBD-2 expression in HaCaT keratinocytes stimulated with nicotine. Arch. Oral Biol. 2012, 57, 814–819. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Saitoh, M.; Yamazaki, M.; Nishimura, M.; Kurashige, Y.; Arakawa, T.; Takuma, T.; Kaku, T.; Abiko, Y. Nicotine induces upregulated expression of beta defensin-2 via the p38MAPK pathway in the HaCaT human keratinocyte cell line. Med. Mol. Morphol. 2010, 43, 204–210. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
An, N.; Holl, J.; Wang, X.; Rausch, M.A.; Andrukhov, O.; Rausch-Fan, X. Potential Suppressive Effect of Nicotine on the Inflammatory Response in Oral Epithelial Cells: An In Vitro Study. Int. J. Environ. Res. Public Health 2021, 18, 483. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph18020483
An N, Holl J, Wang X, Rausch MA, Andrukhov O, Rausch-Fan X. Potential Suppressive Effect of Nicotine on the Inflammatory Response in Oral Epithelial Cells: An In Vitro Study. International Journal of Environmental Research and Public Health. 2021; 18(2):483. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph18020483
Chicago/Turabian StyleAn, Na, Jasmin Holl, Xuekui Wang, Marco Aoqi Rausch, Oleh Andrukhov, and Xiaohui Rausch-Fan. 2021. "Potential Suppressive Effect of Nicotine on the Inflammatory Response in Oral Epithelial Cells: An In Vitro Study" International Journal of Environmental Research and Public Health 18, no. 2: 483. https://0-doi-org.brum.beds.ac.uk/10.3390/ijerph18020483