
The Microstructure of γ-Alumina


1
Department of Chemistry, Drexel University, Philadelphia, PA 19104, USA
2
Department of Materials Science & Engineering, Drexel University, Philadelphia, PA 19104, USA
*
Author to whom correspondence should be addressed.
Energies 2021, 14(20), 6472; https://0-doi-org.brum.beds.ac.uk/10.3390/en14206472
Submission received: 19 August 2021
/
Revised: 30 September 2021
/
Accepted: 4 October 2021
/
Published: 10 October 2021
Abstract
:Though γ-Al2O3 has played a central role in heterogeneous catalysis for more than two centuries, its microstructure continues to be debated. Specifically, the positions of Al3+ cations within the crystal lattice have been discussed extensively in the literature. Many authors uphold that the cations primarily occupy spinel sites, while others endorse the occupation of non-spinel sites. The other main point of dispute is whether the structure contains interstitial hydrogen, with some authors supporting a partially hydrated model and others claiming that the structure must be completely dehydrated. The use of different structural models directly affects the predicted geometry of γ-Al2O3 at the surface, which in turn has significant implications for its catalytic utility. A comparison of theoretical data to experimental infrared (IR), X-ray diffraction (XRD), and selected area electron diffraction (SAED) evidence suggests that γ-Al2O3 features cations primarily in spinel positions, while IR and nuclear magnetic resonance (NMR) data indicate that interstitial hydrogen is present within the bulk structure.
1. Introduction
Alumina (Al2O3) is an abundant material that is under continuous study due to the vast number of industrial and technological applications in which it finds use and its role in geological processes. It can exist either as the thermodynamically stable polymorph α-alumina (corundum) or as any one of several metastable/transitional phases (η, γ, χ, δ, κ, and θ) [1]. Transition aluminas are most often utilized as adsorbents, catalysts, and catalytic supports, though they progressively degrade into α-alumina upon heating [1]. Of the metastable phases, γ-alumina has the most commercial/industrial utility due to its high specific surface area and defect crystal structure. It is regularly employed as a catalyst support and washcoat in automotive catalytic converter systems [2] because of its exemplary electrical insulating properties (favorable for exothermic catalytic reactions) [3] and proclivity for dispersing non-noble metal-based catalysts [4], respectively. Despite the material’s usefulness, the surface and bulk structures of γ-alumina have been the subject of sustained debate. The main points of controversy have been the positions of Al cations and the possible presence of hydrogen in the bulk unit cell.
Though the discrepancy over cation positions and the presence of a few hydrogen atoms in the bulk unit cell may seem trivial, these properties have an outsized influence on surface chemistry. Surface structure is of central importance in many of γ-alumina’s applications, especially heterogeneous catalysis. Detailed models of the bulk and surface would be of enormous benefit to understanding the material and especially in catalytic design. Here, ‘bulk’ refers to an approximation of the arrangements of atoms interior to the solid material, as described by a three-dimensional unit cell which repeats infinitely through space. The ‘surface’ is a boundary separating two phases (i.e., the interface between a solid material and air). Because the bulk unit cell is too miniscule for visual observation, information about it is gleaned from experimental diffraction techniques (neutron/electron/x-ray) and computational modeling strategies. The predicted catalytic properties of γ-alumina are highly dependent on the arrangement of atoms at its surface, which is conditional upon the atomic arrangement specified by the bulk unit cell [5]. Since γ-alumina is an important catalytic material, it would be highly desirable to reach a consensus on which bulk unit cell most accurately represents its interior structure so that consistent predictions can be made about its surface structure and properties.
2. Methods of Literature Search
This review highlights the progress that has been made toward resolving the controversies over cation positions and hydrogen content in alumina since a review by Sohlberg and colleagues in 2000 [6]. Each reference is obtained by a literature search, most with a filter for publication after the year 2000, with appropriate citations listed below. Data were collected after careful analysis of each reference and organized based upon the crystal structure of γ-alumina supported by each reference. Experimental results were organized based on method of analysis, i.e., infrared (IR), X-ray diffraction (XRD), nuclear magnetic resonance (NMR), transmission electron microscopy (TEM), and selected area electron diffraction (SAED) techniques. Theoretical results were organized based on computational methods, i.e., first principles methods such as density functional theory (DFT) and Newtonian-based methods such as molecular dynamics (MD). Studies were determined to be eligible for inclusion in this review if their results were deemed compatible with the topics of boehmite, corundum, and transition alumina microstructure, alumina surface studies. Studies of the atomic-level structure of metal oxides analogous to alumina are also reviewed where they support the above topics.
3. Boehmite: Transition Aluminas
Before delving into the controversies, we begin with alumina characteristics that are generally agreed upon. The precursor of γ-alumina is boehmite (AlOOH), a hydroxide which contains a sublattice of cubic close-packed (ccp) O2- anions, with Al3+ cations interstitially situated in octahedral positions. Upon thermal treatment, boehmite loses water and progressively degrades via the sequence shown in Figure 1.
This transition sequence is idealized because the structural characteristics of transition aluminas differ based on the surface properties/morphology of the starting material and its thermal calcination conditions [7]. Alumina phase transformations are also accompanied by changes in symmetry that can lead to a number of variants for both δ and θ polymorphs [1,8]. The transition aluminas are especially difficult to characterize due to their similar diffraction patterns [7,8,9,10], low crystallinity [1], and small particle size [11,12]. Metastable aluminas all inherit a ccp oxygen sublattice from their hydroxide precursors [7,13], but differ in terms of the arrangement of ions in their cation sublattices. Boehmite has a very ordered structure (see Figure 2 [14]) of cation and anion sublattices with interstitial hydrogen, but as it is heated, some (but probably not all) of the hydrogen atoms migrate out of the structure in the form of water.
This dehydration leaves holes in the crystal lattice that either stay vacant or become occupied by aluminum cations that shift their positions and coordination number. This phase transition creates structurally disordered metastable aluminas, with the γ form having the least ordered cation sublattice of octahedrally (oct) and tetrahedrally (tet) coordinated aluminum cations. This disorder is likely the result of the ‘new’ tet positions that are formed upon boehmite dehydration. As a result, γ is the least thermodynamically stable alumina form, which is troublesome for those who desire to utilize the material at elevated temperatures. The disordered tet lattice can be experimentally observed via XRD analysis by the wide (220) reflection [10], whereas the sharp (222) reflection provides evidence of a well-ordered oxygen anion sublattice [7] (see Figure 3).
When γ-alumina is heated, its cation sublattice becomes more ordered as the material gradually transitions into the δ and θ phases, which have more ordered structures [7,9] and are thus increasingly stable. This increase in thermodynamic stability, however, is paired with a decrease in surface area (~200 m2/g for γ-alumina versus ~30 m2/g for θ-alumina) [11].
There is currently no consensus on the structure of δ-alumina, which is considered an intermediate between the γ and θ forms [15]. XRD evidence supports the δ form exhibiting a spinel superstructure [16], built by aggregating three spinel units with oct vacancies [10,13]. The δ microstructure has also been described as an intergrowth of two crystallographic variants, δ1- and δ2-alumina [17]. This proposed structure has a P212121 space group, consistent with earlier studies of δ-alumina structure based on electron diffraction [8], and contains 37.5% tet cations. In opposition to these characterizations, some studies have reported that the δ and γ forms are indistinguishable [18], but more evidence is required to support this conclusion and in general, the microstructure of δ-alumina merits further investigation.
The structure of θ-alumina is generally understood to be monoclinic with a C2/m space group (a = 11.854, b = 2.904, c = 5.622 Å, β = 103.83°) and Al3+ cations evenly split between oct and tet sites [7,13]. The thermodynamically stable α phase differs from transition aluminas by containing a hexagonally close-packed (hcp) anion sublattice (ABAB). It is the only polymorph present at 1573 K [13] and is also completely dehydrated. Therefore, the main question regarding the degradation sequence is: at what point does alumina lose all its water? The majority of experimental evidence supports the description of transition aluminas as incompletely dehydrated aluminum hydroxides [7,11,19]. Therefore, the γ structure contains some “water,” which is more correctly viewed as interstitial hydrogen. Others assert that boehmite loses all its water in the transition to γ-alumina [20], thus rendering the structure completely dehydrated. The question as to the completeness of dehydration is important because the presence/absence of hydrogen directly affects the shape of the unit cell (cubic/monoclinic/triclinic/tetragonal), which positions cations occupy, and the surface structure of γ-alumina (factors which govern catalytic activity).
4. Spinel-like Structure
Until the early 2000s, γ-alumina was consistently reported as having a cubic spinel-like lattice structure [7,21], similar to that of γ-Fe2O3 (ferric oxide) [22]. Spinels are formulaically represented as AB2X4, in which X anions form a ccp lattice with A and B cations occupying interstitial oct and tet lattice sites, respectively [23]. Spinels generally have an Fdm space group and are known to exhibit remarkable magnetic and electrical properties (as conductors or insulators depending on composition) [23,24]. Magnesium-spinel, namesake of the mineral group, is formulaically represented as (Mg2+)tet(Al3+)2octO4 [6]. Its cubic unit cell contains 96 interstices (32 oct and 64 smaller tet sites) between the oxygen atoms with cations located at 8a (tet) and 16d (oct) Wyckoff positions, known as spinel sites. Because γ-alumina does not contain any Mg ions, it possesses vacant spinel sites (VSS) at cation positions to retain its Al2O3 stoichiometry [25]. The distribution and mobility of VSS between the bulk and surface layers of γ-alumina have been matters of considerable disagreement, with published results suggesting that they primarily occupy oct positions [26,27,28,29], tet positions [8,30], or a mix of both [12,31]. As will be discussed below, there is also controversial evidence that Al may occupy non-spinel positions in the crystal lattice [20,30,32].
From early XRD patterns of γ-alumina and γ-ferric oxide structures, Verwey estimated that vacancies are primarily situated at octahedral sites [22]. This theory has since been supported by several first-principles studies which provide evidence of Al’s energetic preference (ranging from 5.0*10−3–3.0 eV) towards the occupation of tet sites [28,33,34]. Gutiérrez et al. [21] performed an extensive set of DFT total energy calculations to demonstrate that the lowest-energy cubic spinel unit cell (a = 7.887 Å) features two maximally separated oct VSS. Their primitive structure contains only 40 atoms and has fully occupied lattice positions (rather than fractional), which is advantageous for computational modeling. The model has thus been used for theoretical studies [35,36]. However, as the authors note, it may suffer from being oversimplified in its description of γ-alumina’s interior geometry. The structurally similar bulk unit cell of Pinto et al. [37] is twice as large, featuring 80 atom positions with two oct VSS separated by ~7.45 Å. Rather than cubic, the unit cell is monoclinic (a = b = 5.663 Å, c = 13.71 Å, α = β = 90.6°, γ = 60.401°) with a C2/m space group, like θ-alumina. All cations are in spinel positions with a ratio of 38% tet and 62% oct. The Pinto et al. monoclinic spinel-like unit cell has also been used as the bulk structure for several subsequent theoretical studies [38,39,40].
It has been shown that oct VSS are repelled from surface layers and from each other. Thus, they tend to occupy interior layers of γ-alumina [34] as far away from each other as possible [21,37]. It is also unlikely for VSS to move from their original positions in the absence of significant thermal activation, due to a large energy barrier (0.55 eV/Al2O3) for vacancy migration [41]. The positions of VSS are significant because they affect the surface structure and properties of γ-alumina [38], potentially altering its catalytic properties and capability for surface chemistry processes [42,43]. While there is continuous discourse on vacancy positions within the lattice, spinel-like aluminas must feature defects in order to reach valency balance. These defects can take the form of VSS, hydrogen atoms, or a combination of both.
5. Hydrogenated Spinel Structure
The presence of interstitial hydrogen in the γ-alumina structure is supported by numerous experimental [7,11,12,13,19,44,45,46] and theoretical [35,47] studies. The γ form was first described as a perfect hydrogenated spinel in 1952 by de Boer and Houben [44], formulaically represented as HAl5O8, in which cation vacancies are occupied by hydrogen atoms. The computed equilibrium volume and bandgap of this model are in agreement with experimental evidence [35]. The dehydration experiments of Soled [11] and Zhou and Snyder [7] both provide evidence that γ-alumina releases water (0.84%) as it transitions into corundum, with Soled’s results corresponding to the structure containing one OH group per primitive unit cell (consistent with de Boer and Houben’s early predictions). Zhou and Snyder’s dehydration experiment [7] also shows that the η, γ, and θ forms are only partially dehydrated after calcination, with each successive polymorph containing a decreased fraction of water (η = Al2O3∙(0.057)H2O, γ = Al2O3∙(0.048)H2O, θ = Al2O3∙(0.006)H2O). This progressive loss of water could explain the thermodynamic instability of the transition aluminas. However, the studies of Soled and Zhou and Snyder describe γ-alumina as a crystalline hydrate with Al2O3∙nH2O notation, which inaccurately conveys that the material’s dehydration produces stoichiometric alumina. Hydrogen atoms are mobile [19] and can hop between positions within a metal oxide lattice [47,48]. After hydrogens migrate out of the γ-alumina structure, Al cations can take their place, resulting in increasingly ordered and thermodynamically stable structures. Li et al. [35] (p. 9) demonstrated that the diffusion rate of hydrogen is extremely slow due to vacancies trapping H atoms. Therefore, hydrogen should be considered ‘a locked-in impurity in γ-alumina’ under conditions of normal temperature and pressure.
In contrast to the crystalline hydrate (Al2O3∙nH2O) representation, Sohlberg et al. [47] employed DFT modeling to propose a H3mAl2-mO3 notation for hydrogenated spinel γ-alumina, in which m = 2n/(n + 3) to provide a notation for tabulating the material’s degree of dehydration. They suggest that the structure exists with a range of hydrogen contents and compared it to a sponge, with the ability to reactively store and release water. When a water molecule adsorbs onto the surface of γ-alumina, it breaks apart into its atomic constituents, with H entering the bulk structure while O stays at the surface. Aluminum cations then migrate from the bulk to the surface, where they recombine with oxygen atoms and extend the crystal matrix. In the reverse process, for every three hydrogens that leave the alumina structure, one Al cation must go into the bulk from the surface to satisfy valence requirements [47]. The H3mAl2-mO3 notation tabulates the interchange between vacancies and hydrogen atoms in the spinel structure while maintaining a balanced valence, with n < 0.2 corresponding with a hydrogen-poor regime exhibiting VSS, while n > 0.2 coincides with a lattice of fully occupied cation sites exhibiting additional interstitial hydrogen atoms. The hydrogenated spinel notation is also consistent with previous descriptions of transition aluminas, with n = 1/5 (m = 1/8) corresponding to the ideal hydrogen spinel model of de Boer and Houben [44]. Indeed, the n = m = 0 limit corresponds to the fully dehydrated defect spinel structure, consistent with the description of δ-alumina as a triple block spinel [10,13].
Sohlberg et al. [47] also provide vibrational frequency evidence of hydroxyl group stretching within the γ lattice at oct and tet positions. Previous infrared spectroscopy data of Tsyganenko et al. [45] exhibited two absorbance bands attributed to OH ions with H at a nominal tet/oct cation sites in the spinel lattice. The more intense band at 3500 cm−1 was assigned by Tsyganenko et al. to OH groups in oct sites because the anion sublattice exhibits a majority of oct interstices, while the band at 3300 cm−1 was assigned to OH groups in tet vacancies [45]. Sohlberg et al. reversed this band assignment, deducing from DFT calculations that H-oct absorbs at 3306 cm−1 and H-tet absorbs at 3449 cm−1. This allocation has since been supported by the theoretical study of Dyan et al. [49], who observed oct-coordinated OH absorbing at a lower frequency than OH in tet positions. Because tet H atoms exhibit higher thermodynamic stability than H atoms at oct sites, the band at ~3500 cm−1 associated with the lower-energy structure should be more intense at low temperatures. Only at high temperatures where thermal energy is sufficient to statistically populate the sites should the ~3300 cm−1 band become stronger, which is completely consistent with Tsyganenko’s temperature-dependent IR spectra. Though the frequency assignments are reversed, the works of Sohlberg et al. [47] and Tsyganenko et al. [45] provide matching theoretical and experimental evidence that the bulk unit cell of γ-alumina contains interstitial hydrogen.
Some criticism of the hydrogenated spinel γ-alumina structure seems to be strictly semantic, i.e., the structure of Al2O3 cannot contain hydrogen due to its generally accepted aluminum oxide nomenclature. This is generally a myopic argument since the material was named ‘alumina’ many years before it was postulated that the structure could contain hydrogen. Multiple metal oxides are known to exist with a range of hydrogen contents because defects can act as hydrogen trapping sites [50]. For example, silica (SiO2) [51] and zirconia (ZrO2) [52] are two technologically important metal oxides that have also been shown to contain interstitial hydrogen. As previously mentioned, γ-Fe2O3 features the same cubic spinel lattice as γ-alumina, and its thermodynamic stability depends on partial-hydration for ‘the oxide cannot be freed from H2O without the formation of α-Fe2O3′ [22] (p. 2). Indeed, David and Welch [53] provide XRD data supporting that the presence of water is ‘essential’ to the characteristic spinel structure of γ-ferric oxide. Additionally, hydrogen content is highly dependent on the thermal history of the sample [6], so the complete dismissal of interstitial hydrogen in γ-alumina seems spurious. Furthermore, the structure of γ-alumina has been characterized via proton NMR measurements [54], which would not be possible for a structure completely devoid of hydrogen.
6. Non-Spinel Structure
In 2001, a ‘non-spinel’ model was suggested by Krokidis et al. [20], which has subsequently become quite controversial. They employed a combination of results from XRD, DFT, and MD methods to inform their proposal of a stepwise mechanism of boehmite dehydration that would occur in the industrial production of γ-alumina. The non-spinel structural model features the familiar ccp oxygen anion sublattice but is unique in its assignment of cation positions. Rather than restricting Al to the 8a (tet) and 16d (oct) spinel positions, cations also occupy 8b, 48f (tet), and 16c (oct) sites [55]. Thus, the equilibrium structure is tetragonally deformed and devoid of interstitial hydrogen. Krokidis et al. [20] (p. 5) mention that incomplete dehydration may result in a ‘number of H atoms and OH groups [remaining] within the structure’ but assert that the equilibrium structure of γ-alumina is formed after ‘100% of water molecules have been extracted from boehmite’. The dehydrated non-spinel model’s simulated XRD patterns are comparable to experimental findings [20]. Though the ratio of tet:oct cations (25:75) for the non-spinel model is supported by 27Al NMR experiments [56,57], the recent work of Khivantsev et al. [58] criticizes the relatively low percentage of tet cations, using XRD to advocate that ~30-35% of cations are in tet positions. The non-spinel structural model is also at odds with a vast amount of previous research supporting the presence of hydrogen in γ-alumina [7,11,12,13,44,45,47].
Digne et al. [32,59] became strong proponents of the non-spinel model, criticizing spinel-like structures for ‘arbitrarily [imposing] constraints on the type and number of interstices occupied by aluminum atoms’ [32] (p. 3). They used DFT calculations to further describe this dehydrated monoclinic (P21/m space group) non-spinel bulk unit cell. It benefits from being relatively small (40 atoms) and having fully occupied lattice sites, making it relatively easy to use for materials modeling simulations. Like the unit cell of Gutiérrez et al. [21], Digne et al.’s model may suffer from being ‘too simplified’ to accurately describe the material’s complex geometry. Nevertheless, the dehydrated non-spinel unit cell model has gained popularity in the 21st century, (quite possibly due to the simplicity and small size of its unit cell) and has been used as the basis for several computational studies [60,61,62,63].
The atypical non-spinel model has also paved the way for consideration of other alternative structures that do not conform to spinel cation positions. In 2003, Paglia et al. [64] proposed a fully tetragonal model for γ-alumina with I41/amd symmetry (a = 5.652, c = 7.871 Å) based on neutron diffraction and TEM data. The unit cell is relatively small (36 atoms) and allows fractionally occupied lattice sites to better capture the complexity of γ-alumina’s geometry. The Paglia model features cations in non-spinel 4a, 8d, and 8c Wyckoff positions with 31% of cations in tet sites and the remaining 69% in oct sites. The authors utilized loss on ignition analysis to determine that γ-alumina contains 2.26% wt of hydrogen species within the bulk, however, this was ‘assumed to be in the form of water’ [64] (p. 7). Thus, Paglia and colleagues concluded that ‘hydrogen is not interstitially present’ within the bulk of their tetragonal γ-alumina model, ‘but rather is in the form of water, within the amorphous content’ [64] (p. 9). A second publication by Paglia et al. [65] also utilized loss on ignition analysis, this time to show that their tetragonal sample of γ-alumina contained 3.9% wt hydrogen at 500 °C, a value which decreased to 0.4% wt at 800 °C. Again, the authors proclaimed that ‘the presence of protons in the structure…is limited to amorphous regions’ [65] (p. 7). These results are largely a reconfirmation of earlier meticulous loss-on-ignition analysis performed by Zhou and Snyder, [7] which demonstrated that a small but non-trivial amount of hydrogen remains even after samples have been calcined for an hour at 873K. It should be noted that because loss-on-ignition analysis does not provide further structural information, such results in isolation could be equally well interpreted as supporting the hydrogen-containing spinel structure of γ-alumina. A third publication by Paglia et al. [66] (p. 14–15) reaffirmed their earlier finding that the calcination of boehmite forms tetragonal γ-alumina containing 31% tet cations and 69% oct cations. They stated that while γ-alumina was formed at temperatures between 450 and 750 °C, ‘δ-Al2O3 was not observed’ above 750 °C, but rather a ‘new phase’ which they label γ’-alumina. It should be noted, however, that they characterize γ’-alumina as a transition state between the γ and θ forms with the structure of a ‘triple cell of γ-Al2O3′, two descriptions that had previously been [10,15] and continue to be [13] used to describe δ-alumina. It is interesting, therefore, that Paglia and colleagues make the distinction that their ‘new’ polymorph is not δ-alumina when the exact microstructure of δ-alumina remains a topic of debate.
Also inspired by the non-spinel model, Smrčok et al. [67] proposed a majority spinel-like unit cell for γ-alumina, in which most of the cations are in spinel sites, except for 6%, which are allowed to occupy 16c (oct) and 48f (tet) non-spinel positions. This unit cell does not deviate extensively from the spinel-like structure of Gutiérrez et al. [21] with its cubic Fdm space group (a = 7.9382 Å) and 37% of the cations in tet positions. Rather than being ‘too simplified’, the Smrčok et al. unit cell has the deficit of being cumbersome to model computationally due to its large size (120 atoms) and fractionally occupied cation sites, which impedes the creation of supercell models for surface studies. The bulk model aligns with Zhou and Snyder’s work [7], which indicates that γ-alumina cations can populate both spinel and non-spinel (8b and 48f tet, 16c oct) lattice sites, but the latter are poorly occupied if at all.
Digne et al. [32] proposed that their non-spinel model is 0.05 eV/Al2O3 unit more stable than the spinel-like model of Gutiérrez et al. [21], though the true morphology of γ-alumina is not necessarily described by the lowest energy structure as it is a metastable polymorph. Theoretical studies by Cai et al. [41] have shown that γ-alumina exhibits a thermodynamically spontaneous relaxation in which Al ions migrate from spinel to non-spinel sites in the bulk structure, a process that leads to an approximate unit cell of θ- rather than γ-alumina. An elegant experimental study of non-spinel site occupancy was reported by Zhou and Snyder [7] who demonstrated by doping samples with sodium chloride that the tetragonal deformation of η and γ signifies the onset of their degradation into θ-alumina. Upon heating, the pure samples exhibited more pronounced tetragonal characteristics until completing their degradation into θ-alumina, while the doped samples did not exhibit further tetragonal deformation up to 1373 K. The diffusion of Al to tet sites was hindered by large Na ions, thus preventing the γ-θ phase transition. Occupancy of non-spinel sites therefore appears more an indication of the incipient transition to the θ phase than a characteristic of the γ phase.
7. Why Does Structure Matter? Surfaces
The basic structures present on γ-alumina surfaces were characterized by Knözinger and Ratnasamy in 1978 [68]. With the increased emphasis on computational research in recent years, the choice of structural model and its atomic-scale details have taken on greater significance. The optimization of a bulk unit cell is generally the first step when performing a theoretical study on a solid material. The use of an inaccurate bulk model has significant implications for the general understanding of γ-alumina as a catalyst. As will be described below, the use of different unit cells has already led to inconsistent descriptions of γ-alumina’s surface structure.
Slab models of spinel-like γ-alumina can either be terminated by low-density (LD) layers, which contain only tet cations, or high-density (HD) layers that contain oxygen and oct aluminum [34]. The exposure of HD terminations result in more stable surfaces with VSS located in interior atomic layers rather than at the surface [34], while LD terminations result in extensive atomic rearrangement at the surface [69], with low-coordinated surface cations sometimes collapsing into sub-surface sites [41,43,70]. The (100) and (110) surfaces of γ-alumina are the most thermodynamically stable, with the (100) face exhibiting penta-coordinated AlV ions and the (110) face containing lower-coordinated cations [70].
Several theoretical studies employing the dehydrated non-spinel unit cell have reported finding AlIII centers on the (110) [62,63] and (111)n [61] surfaces. Tri-coordinated cations will be exposed on γ-alumina surfaces for certain cleavages of the bulk structure, but the results of Sohlberg et al. [70] using the spinel-like model show that tri-coordinated AlIII ions are unstable and drop down from the surface into oct VSS and become AlVI upon reconstruction. This conclusion is supported by other theoretical studies [71] and experimental 27Al NMR [72] results.
There are a number of subsequent NMR studies on the surface topology of γ-alumina with varying results. The work of Taoufik et al. [73] utilized a combination of 1H and 27Al NMR experimental techniques to explore the topology of hydroxyl groups bonding to surface of γ-alumina. Their hydroxyl group assignments reflected the presence of only AlIV, AlV, and AlVI centers, consistent with the earlier theoretical [70] and experimental [72] studies. The recent work of Khivantsev et al. [58] utilizes a combination of IR and high-field 27Al NMR spectroscopy to also provide evidence against the presence of tri-coordinated Al cations on the surface of γ-alumina. Both of these studies, however, are in conflict with the 1H and 27Al NMR results of Delago et al. [74] which support the presence of AlIII ions on γ-alumina. From these inconsistencies, it is clear that the use of different bulk models can result in decidedly different slab models and therefore divergent descriptions of γ-alumina surface structure, which is of practical importance since its catalytic properties are directly affected by adsorption site geometry and the presence (or lack) of low-coordinated cations [43].
8. Comparison of Theoretical and Experimental Results: Which Structure Fits Best?
Several studies have sought to resolve the inconsistent use of different bulk models by directly comparing predictions based on the theoretical non-spinel and spinel-like structures to experimental XRD, IR, and SAED data [5,36,75]. Sun et al. [75] used DFT simulations and Rietveld refinement to compare the dehydrated non-spinel model [20,32] and three nonequivalent spinel-like structures (with varying VSS distributions and hydrogen content) to powder γ-alumina spectra. All the spinel-like models fit the experimental synchrotron XRD patterns better than the non-spinel model (see Figure 4a). Additionally, their spinel-like structure accurately reproduced the lattice parameters and other structural features of γ-alumina, while the non-spinel model did not.
The above XRD patterns would appear to be convincing evidence that the spinel-like structure is a better model of γ-alumina than the dehydrated non-spinel models, but these results were strongly opposed by Digne et al. [55] and Paglia et al. [76], who criticized the commercially obtained sample of γ-alumina used for XRD spectra and claimed that it had degraded into δ/θ-alumina. Digne et al. [55] (p. 1) claimed that the distinction between ‘γ-, δ-, and θ-alumina is mainly based on significant differences of their diffraction patterns’, which conflicts with previous studies highlighting the similarities between transition alumina’s XRD patterns [7,9,10].
Specifically, Digne et al. and Paglia et al. cite the (220), (400), and (440) reflections of Sun et al.’s XRD data being a better match to δ/θ-alumina than the γ phase [55,76]. Using the XRD spectra obtained by Krokidis et al. [20] (Figure 4b) as a reference, it is true that the (220) reflection in Sun et al.’s data is sharper and more split than that seen for the γ form at 600 °C and that the (400) reflection features a shoulder. According to Zhou and Snyder [7], however, the (220) reflection of γ-alumina results from the presence of the tet cation sublattice. A sharper reflection would coincide with more tet Al ions, which is more consistent with spinel-like models rather than non-spinel models. Zhou and Snyder also stated that ‘a slight splitting, or obvious shoulder, of the (400) reflection’ is characteristic of the γ structure [7] (p. 6). Nevertheless, the criticisms of Digne et al. and Paglia et al. do not provide sufficient evidence that the sample used by Sun et al. is anything other than γ-alumina because XRD analysis does not allow the unambiguous characterization of alumina phases present in the film, as described by the work of Boumaza et al. [13].
It has been consistently shown that the γ-to-θ phase transition is initiated by the Al migration to reduce strong Al-Al interactions while oxygen atoms remain fixed [13,20,41,77,78]. Starting at the surface, degradation is signified by the near-surface region exhibiting θ characteristics while the core structure remains γ-alumina [77]. As a bulk technique, XRD is ineffective for detecting structural properties of surfaces at an atomic level [1,77]. Therefore, the γ-to-θ transition would be undetectable until the structure shows a majority of θ-alumina characteristics. XRD methods suffer from serious disadvantages when applied in isolation to transition aluminas because the materials have complicated structures (all with very similar d-spacings) that continuously transform upon heating [1]. It is also notoriously difficult to produce γ-alumina crystals of sufficient size and purity for accurate XRD analysis [5,35]. Furthermore, Digne et al. and Paglia et al. did not provide an explanation for Sun et al.’s simulated spinel-like structures qualitatively matching the patterns of the ‘degraded’ sample. The criticism would be more convincing if Digne et al. and Paglia et al. had demonstrated that dehydrated non-spinel γ-alumina degrades into spinel-like δ and θ forms, but such a non-spinel to spinel progression seems unlikely. Despite criticism, the work of Sun et al. provides good correlation between experimental data and theoretical defect spinel structures.
An extensive study by Ferreira et al. [36] compared Digne’s dehydrated non-spinel unit cell [32] and the spinel-like model of Gutiérrez [21,79] to experimental data [80]. The authors used density-functional perturbation theory calculations to simulate IR vibrational modes for both models and compared them to the frequencies provided by Saniger in 1995 [80]. Their results, presented in Figure 5, show that the simulated spectra for the spinel-like model contain peaks that qualitatively match the previously reported vibrational frequencies of powder γ-alumina, while the simulated spectra for the non-spinel model do not [36].
Despite the 2011 Ferreira et al. work promoting ‘the spinel-like model as more adequate to describe γ-alumina’ [36] (p. 7), the main author proceeds to use the non-spinel model in a 2013 surface study, claiming it is ‘the most advanced structural models for the (100) and (110) surfaces of γ-alumina available today’ [63] (p. 2). The above XRD and IR patterns would appear to be unequivocal evidence that the spinel-like structure is a better model of γ-alumina than the dehydrated non-spinel models. Thus, the authors question why Ferreira et al. decided to publish results based on a less adequate bulk model.
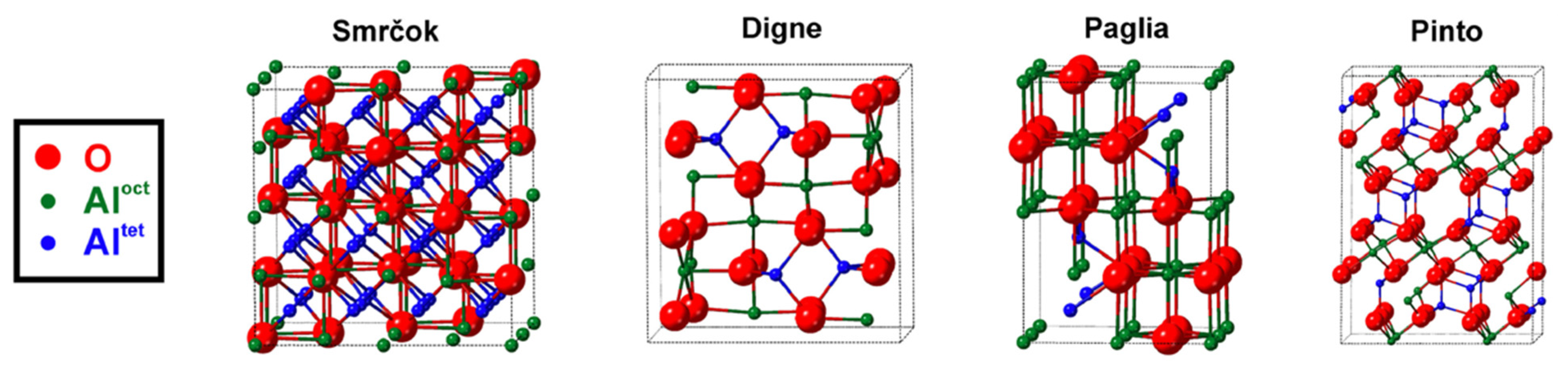
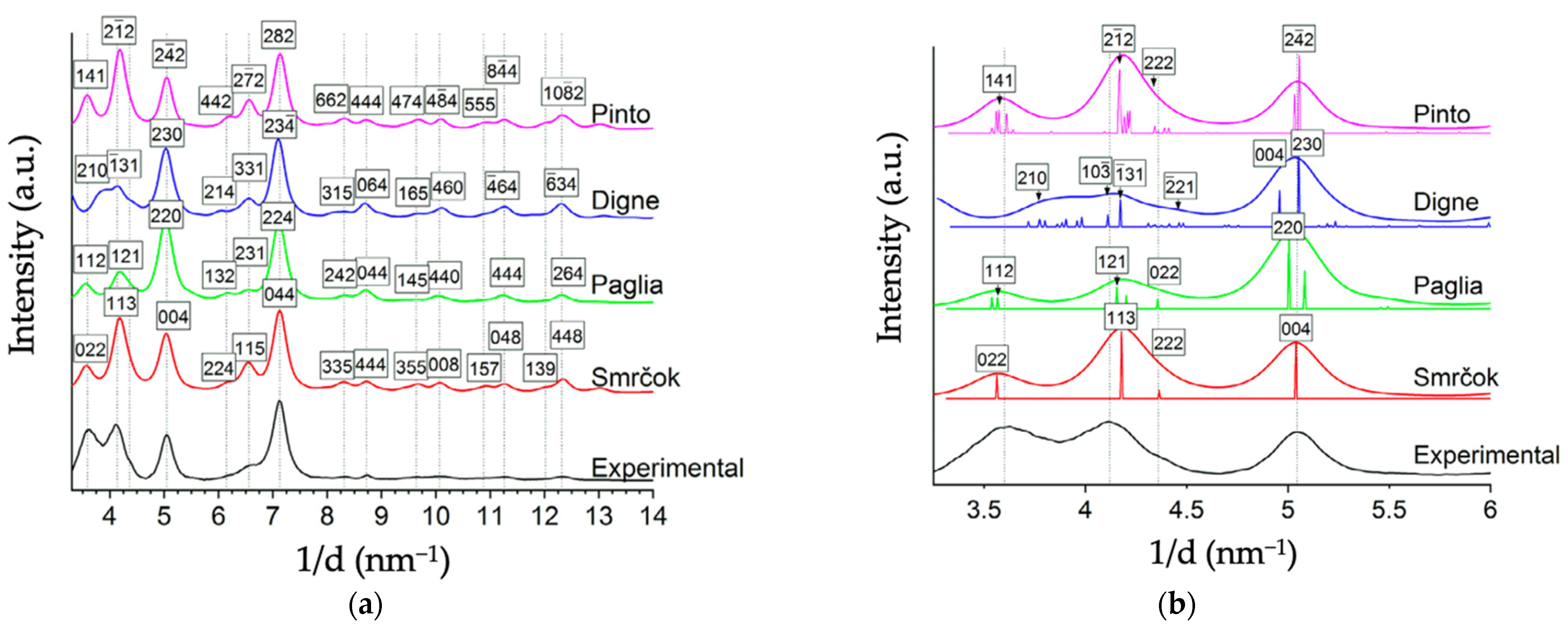
More recently, Ayoola et al. [5] provided notable SAED and TEM spectra comparing the bulk unit cells of Smrčok [67], Pinto [37], Digne [32], and Paglia [64] to commercial γ-alumina nano-powder. Relative to XRD, SAED with TEM is a superior experimental method of analysis for transitional aluminas because its two-dimensional high spatial resolution can distinguish structures of different symmetry [5], despite the models’ nearly identical fcc anion sublattices. Below in Figure 6 are reproductions of Ayoola et al.’s visual representations of each model’s unit cell, their instructive table of structural details for each model in Table 1, and their SAED spectra of each model in Figure 7.
Generally, the spinel-like structures (Smrčok, Pinto) fit the experimental patterns better than the non-spinel (Digne, Paglia). Smrčok’s cubic spinel-like model exhibits the best match in terms of peak shapes, positions, and relative intensities (which correspond to a good match for Al cation distribution). The monoclinic non-spinel model of Digne et al. exhibits the worst fit to experimental data (and the other models) based on peak shapes and positions, especially in the range of 3–4 nm−1 (see Figure 7). Thus far, Digne has not responded to these results.
9. Recommendations for Future Research
As previously mentioned, the structure of γ-alumina has been compared to γ-ferric oxide [22]. It would be reasonable for these metal oxides to have similar lattice structures, resulting from their similar hydroxide precursors (γ-AlOOH → γ-Al2O3 as γ-FeOOH → γ-Fe2O3). Unlike γ-alumina, γ-ferric oxide has a fully elucidated geometry, containing a cubic spinel structure (absent of cations in non-spinel positions) with oct VSS. It is also considered to be a hydrate complex containing a small percentage of water [53], depending on its initial hydrogen concentration [81]. A recent review by Prins [82] includes detailed comparisons between the (spinel-like and non-spinel) structures of γ-alumina and (spinel-like) γ-ferric oxide, which the authors recommend for further reading. This description is similar to the hydrogenated spinel model for γ-alumina (H3mAl2-mO3) proposed by Sohlberg et al. [47] More comparison studies between alumina and ferric oxide could be a valuable addition to the literature.
A recent publication by Wang et al. [30] proposes a ‘robust and reliable’ (p. 7) method of determining the oxygen structure in γ-alumina via two-dimensional (2D) solid-state NMR spectroscopy at high field. Using 17O NMR analysis, they report finding a majority of cation vacancies located at tet positions for 17O-enriched γ-alumina, which conflicts with studies endorsing primary vacancy occupation of oct sites [26,27,28,29]. The results of Wang et al. suggest that their prepared γ-alumina sample features cations in non-spinel positions, though the authors do not relate their experimental results to any existing theoretical structural models. A reviewer of the present manuscript has suggested performing theoretical 2D NMR spectra simulations of varying structural models to see which, if any, can recreate the NMR spectra of Wang et al., which the present authors endorse as a potentially valuable exercise
Returning to the discrepancy mentioned in Section 7, regarding cleaved surfaces of γ-alumina exhibiting/not exhibiting tri-coordinated Al cations, the authors offer the following possible explanation. It is conceivable that a perfectly clean cleaved surface does not exhibit any AlIII because the cations will drop into the bulk, as seen in theoretical work [70,71], but in the presence of certain adsorbates, the cations “pop up” again to bond to the adsorbate, if that adsorbate does a better job of saturating the Al valance than the subsurface environment. Since no experimental surface will be rigorously adsorbate free, different surface preparations of the same material could lead to different results. This speculation could beneficially be subjected to experimental testing.
10. Conclusions
Though γ-alumina is an essential catalytic material whose utility depends on its bulk and surface structure, these properties have been the subject of spirited debate. The material is most commonly described as a defective cubic spinel with Al cations in 8a, 16d Wyckoff positions. The defects may take the form of VSS or H atoms. It may therefore be considered a range of compounds based on the concentration of hydrogen in the bulk material [6,47] and can be described via H3mAl2-mO3 notation [47]. When the structure is fully dehydrated, vacant octahedral cation positions are required to maintain proper Al2O3 stoichiometry. The presence of hydrogen cations within the structure is compensated by Al deficiency to satisfy the rules of valence. In opposition to this spinel-like structure, there is (limited) evidence that γ-alumina is consistently fully dehydrated and features cations in non-spinel positions. The distinction is important because adequate knowledge of microscopic structures is required for optimizing its application [83]. Though the concentration of hydrogen in all γ-alumina structures varies, there is more than sufficient evidence to conclude that some samples do indeed contain interstitial hydrogen [7,11,35,44,45,47]. Though the dehydrated non-spinel bulk unit cells benefit from being easier to use via computational modeling, these models almost universally lead to predictions of physical observables that are in poorer agreement with experimental results than analogous predictions based on spinel-like models. The authors strongly suggest skepticism towards surface studies based on these models.
Author Contributions
Conceptualization, N.M.S. and K.S.; writing—original draft preparation, N.M.S.; writing—review and editing, K.S. and N.M.S.; supervision, K.S.; project administration, K.S.; funding acquisition, K.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded in-part by ACS PRF #58323-ND10, for which the KS group thanks the donors of the American Chemical Society Petroleum Research Fund.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors thank all members of the KS group for manuscript feedback and thoughtful discussion.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Levin, I.; Brandon, D. Metastable Alumina Polymorphs: Crystal Structures and Transition Sequences. J. Am. Ceram. Soc. 1998, 81, 1995–2012. [Google Scholar] [CrossRef]
- Kašpar, J.; Fornasiero, P.; Hickey, N. Automotive catalytic converters: Current status and some perspectives. Catal. Today 2003, 77, 419–449. [Google Scholar] [CrossRef]
- Kitzner, E.; Rhodes, A.; Shachter, M. Catalytic Converter with Electrically Resistive Catalyst Support. U.S. Patent 3,770,389, 6 November 1973. [Google Scholar]
- Labhsetwar, N.K.; Watanabe, A.; Biniwale, R.; Kumar, R.; Mitsuhashi, T. Alumina supported, perovskite oxide based catalytic materials and their auto-exhaust application. Appl. Catal. B Environ. 2001, 33, 165–173. [Google Scholar] [CrossRef]
- Ayoola, H.O.; House, S.D.; Bonifacio, C.S.; Kisslinger, K.; Saidi, W.A.; Yang, J.C. Evaluating the accuracy of common γ-Al2O3 structure models by selected area electron diffraction from high-quality crystalline γ-Al2O3. Acta Mater. 2020, 182, 257–266. [Google Scholar] [CrossRef]
- Sohlberg, K.; Pennycook, S.J.; Pantelides, S.T. The bulk and surface structure of γ-alumina. Chem. Eng. Commun. 2000, 181, 107–135. [Google Scholar] [CrossRef]
- Zhou, R.-S.; Snyder, R.L. Structures and transformation mechanisms of the [eta], [gamma] and [theta] transition aluminas. Acta Crystallogr. Sect. B 1991, 47, 617–630. [Google Scholar] [CrossRef]
- Jayaram, V.; Levi, C. The structure of δ-alumina evolved from the melt and the γ→ δ transformation. Acta Metall. 1989, 37, 569–578. [Google Scholar] [CrossRef]
- Lippens, B.; De Boer, J. Study of phase transformations during calcination of aluminum hydroxides by selected area electron diffraction. Acta Crystallogr. 1964, 17, 1312–1321. [Google Scholar] [CrossRef]
- Lippens, B.C. Structure and Texture of Aluminas; Waltman: Delft, The Netherlands, 1961. [Google Scholar]
- Soled, S. γ-Al2O3 viewed as a defect oxyhydroxide. J. Catal. 1983, 81, 252–257. [Google Scholar] [CrossRef]
- Wang, J.; Bokhimi, X.; Morales, A.; Novaro, O.; Lopez, T.; Gomez, R. Aluminum local environment and defects in the crystalline structure of Sol− Gel alumina catalyst. J. Phys. Chem. B 1999, 103, 299–303. [Google Scholar] [CrossRef]
- Boumaza, A.; Favaro, L.; Lédion, J.; Sattonnay, G.; Brubach, J.; Berthet, P.; Huntz, A.; Roy, P.; Tétot, R. Transition alumina phases induced by heat treatment of boehmite: An X-ray diffraction and infrared spectroscopy study. J. Solid State Chem. 2009, 182, 1171–1176. [Google Scholar] [CrossRef]
- Peintinger, M.F.; Kratz, M.J.; Bredow, T. Quantum-chemical study of stable, meta-stable and high-pressure alumina polymorphs and aluminum hydroxides. J. Mater. Chem. A 2014, 2, 13143–13158. [Google Scholar] [CrossRef] [Green Version]
- Wefers, K.; Misra, C. Oxides and Hydroxides of Aluminum; Alcoa Laboratories: Pittsburgh, PA, USA, 1987; Volume 19. [Google Scholar]
- Narayanan, C.; Srinivasan, S.; Datye, A.; Gorte, R.; Biaglow, A. The effect of alumina structure on surface sites for alcohol dehydration. J. Catal. 1992, 138, 659–674. [Google Scholar] [CrossRef]
- Kovarik, L.; Bowden, M.; Genc, A.; Szanyi, J.; Peden, C.H.; Kwak, J.H. Structure of δ-alumina: Toward the atomic level understanding of transition alumina phases. J. Phys. Chem. C 2014, 118, 18051–18058. [Google Scholar] [CrossRef]
- French, R.H.; Müllejans, H.; Jones, D.J. Optical properties of aluminum oxide: Determined from vacuum ultraviolet and electron energy-loss spectroscopies. J. Am. Ceram. Soc. 1998, 81, 2549–2557. [Google Scholar] [CrossRef]
- Caldararu, M.; Postole, G.; Hornoiu, C.; Bratan, V.; Dragan, M.; Ionescu, N. Electrical conductivity of γ-Al2O3 at atmospheric pressure under dehydrating/hydrating conditions. Appl. Surf. Sci. 2001, 181, 255–264. [Google Scholar] [CrossRef]
- Krokidis, X.; Raybaud, P.; Gobichon, A.-E.; Rebours, B.; Euzen, P.; Toulhoat, H. Theoretical study of the dehydration process of boehmite to γ-alumina. J. Phys. Chem. B 2001, 105, 5121–5130. [Google Scholar] [CrossRef]
- Gutiérrez, G.; Taga, A.; Johansson, B. Theoretical structure determination of γ− Al 2 O 3. Phys. Rev. B 2001, 65, 012101. [Google Scholar] [CrossRef]
- Verwey, E. The crystal structure of γ-Fe2O3 and γ-Al2O3. Z. Für Krist. Cryst. Mater. 1935, 91, 65–69. [Google Scholar] [CrossRef]
- Sickafus, K.E.; Wills, J.M.; Grimes, N.W. Structure of Spinel. J. Am. Ceram. Soc. 1999, 82, 3279–3292. [Google Scholar] [CrossRef]
- Grimes, N. The spinels: Versatile materials. Phys. Technol. 1975, 6, 22. [Google Scholar] [CrossRef]
- Verwey, E. Electrolytic conduction of a solid insulator at high fields The formation of the anodic oxide film on aluminium. Physica 1935, 2, 1059–1063. [Google Scholar] [CrossRef]
- Sinha, K.; Sinha, A. Vacancy distribution and bonding in some oxides of spine1 structure. J. Phys. Chem. 1957, 61, 758–761. [Google Scholar] [CrossRef]
- Kryukova, G.; Klenov, D.; Ivanova, A.; Tsybulya, S. Vacancy ordering in the structure of γ-Al2O3. J. Eur. Ceram. Soc. 2000, 20, 1187–1189. [Google Scholar] [CrossRef]
- Taniike, T.; Tada, M.; Morikawa, Y.; Sasaki, T.; Iwasawa, Y. Density functional theoretical calculations for a Co2/γ-Al2O3 model catalyst: Structures of the γ-Al2O3 bulk and surface and attachment sites for Co2+ ions. J. Phys. Chem. B 2006, 110, 4929–4936. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.G.; Bronsveld, P.M.; DeHosson, J.T.M.; Djuričić, B.; McGarry, D.; Pickering, S. Ordering of octahedral vacancies in transition aluminas. J. Am. Ceram. Soc. 1998, 81, 1655–1660. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Li, W.; Hung, I.; Mentink-Vigier, F.; Wang, X.; Qi, G.; Wang, X.; Gan, Z.; Xu, J.; Deng, F. Mapping the oxygen structure of γ-Al 2 O 3 by high-field solid-state NMR spectroscopy. Nat. Commun. 2020, 11, 1–9. [Google Scholar]
- Streitz, F.; Mintmire, J. Energetics of aluminum vacancies in gamma alumina. Phys. Rev. B 1999, 60, 773. [Google Scholar] [CrossRef]
- Digne, M.; Sautet, P.; Raybaud, P.; Euzen, P.; Toulhoat, H. Use of DFT to achieve a rational understanding of acid–basic properties of γ-alumina surfaces. J. Catal. 2004, 226, 54–68. [Google Scholar] [CrossRef]
- Wolverton, C.; Hass, K.C. Phase stability and structure of spinel-based transition aluminas. Phys. Rev. B 2000, 63, 024102. [Google Scholar] [CrossRef]
- Vijay, A.; Mills, G.; Metiu, H. Structure of the (001) surface of γ alumina. J. Chem. Phys. 2002, 117, 4509–4516. [Google Scholar] [CrossRef]
- Li, Y.; Lousada, C.M.; Korzhavyi, P.A. The nature of hydrogen in γ-alumina. J. Appl. Phys. 2014, 115, 203514. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, A.R.; Martins, M.J.; Konstantinova, E.; Capaz, R.B.; Souza, W.F.; Chiaro, S.S.X.; Leitão, A.A. Direct comparison between two γ-alumina structural models by DFT calculations. J. Solid State Chem. 2011, 184, 1105–1111. [Google Scholar] [CrossRef]
- Pinto, H.P.; Nieminen, R.M.; Elliott, S.D. Ab initio study of γ− Al 2 O 3 surfaces. Phys. Rev. B 2004, 70, 125402. [Google Scholar] [CrossRef] [Green Version]
- Bermudez, V.M. Quantum-Chemical Study of the Adsorption of DMMP and Sarin on γ-Al2O3. J. Phys. Chem. C 2007, 111, 3719–3728. [Google Scholar] [CrossRef]
- Bermudez, V.M. Computational Study of Environmental Effects in the Adsorption of DMMP, Sarin, and VX on γ-Al2O3: Photolysis and Surface Hydroxylation. J. Phys. Chem. C 2009, 113, 1917–1930. [Google Scholar] [CrossRef]
- Chen, Y.; Ouyang, C.; Shi, S.; Sun, Z.; Song, L. Density functional theory study of Ir atom deposited on γ-Al2O3 (001) surface. Phys. Lett. A 2009, 373, 277–281. [Google Scholar] [CrossRef]
- Cai, S.; Sohlberg, K.; Rashkeev, S.; Pantelides, S.T. Phase transformation mechanism between gamma- and theta-alumina. Phys. Rev. B 2003, 67. [Google Scholar] [CrossRef] [Green Version]
- Védrine, J.C. Heterogeneous catalysis on metal oxides. Catalysts 2017, 7, 341. [Google Scholar] [CrossRef] [Green Version]
- Sohlberg, K.; Pantelides, S.T.; Pennycook, S.J. Surface Reconstruction and the Difference in Surface Acidity between γ- and η-Alumina. J. Am. Chem. Soc. 2001, 123, 26–29. [Google Scholar] [CrossRef]
- De Boer, J.H.; Houben, G.; Terpstra, R. Proceedings of the International Symposium on the Reactivity of Solids; Royal Swedish Academy of Engineering Sciences: Göteburg, Sweden, 1952.
- Tsyganenko, A.; Smirnov, K.; Rzhevskij, A.; Mardilovich, P. Infrared spectroscopic evidence for the structural OH groups of spinel alumina modifications. Mater. Chem. Phys. 1990, 26, 35–46. [Google Scholar] [CrossRef]
- Hall, W.K.; Lutinski, F.; Gerberich, H. Studies of the hydrogen held by solids: VI. The hydroxyl groups of alumina and silica-alumina as catalytic sites. J. Catal. 1964, 3, 512–527. [Google Scholar] [CrossRef]
- Sohlberg, K.; Pennycook, S.J.; Pantelides, S.T. Hydrogen and the Structure of the Transition Aluminas. J. Am. Chem. Soc. 1999, 121, 7493–7499. [Google Scholar] [CrossRef]
- Wexler, R.B.; Sohlberg, K. Role of Proton Hopping in Surface Charge Transport on Tin Dioxide as Revealed by the Thermal Dependence of Conductance. J. Phys. Chem. A 2014, 118, 12031–12040. [Google Scholar] [CrossRef]
- Dyan, A.; Cenedese, P.; Dubot, P. Physical properties of γ alumina surface hydroxyls revisited through a large scale periodic quantum-chemistry approach. J. Phys. Chem. B 2006, 110, 10041–10050. [Google Scholar] [CrossRef]
- Lee, H.G.; Lee, J.-Y. Hydrogen trapping by TiC particles in iron. Acta Metall. 1984, 32, 131–136. [Google Scholar] [CrossRef]
- Van de Walle, C.G.; Tuttle, B.R. Microscopic theory of hydrogen in silicon devices. IEEE Trans. Electron Devices 2000, 47, 1779–1786. [Google Scholar] [CrossRef]
- Bokhimi, X.; Morales, A.; Novaro, O.; Portilla, M.; Lopez, T.; Tzompantzi, F.; Gomez, R. Tetragonal nanophase stabilization in nondoped sol–gel zirconia prepared with different hydrolysis catalysts. J. Solid State Chem. 1998, 135, 28–35. [Google Scholar] [CrossRef]
- David, I.; Welch, A. The oxidation of magnetite and related spinels. Constitution of gamma ferric oxide. Trans. Faraday Soc. 1956, 52, 1642–1650. [Google Scholar] [CrossRef]
- Pearson, R. Wide line nuclear magnetic resonance studies on transition aluminas—Distribution of protons between surface and bulk phases. J. Catal. 1971, 23, 388–394. [Google Scholar] [CrossRef]
- Digne, M.; Raybaud, P.; Sautet, P.; Rebours, B.; Toulhoat, H. Comment on “Examination of Spinel and Nonspinel Structural Models for γ-Al2O3 by DFT and Rietveld Refinement Simulations”. J. Phys. Chem. B 2006, 110, 20719–20720. [Google Scholar] [CrossRef]
- John, C.; Alma, N.; Hays, G. Characterization of transitional alumina by solid-state magic angle spinning aluminium NMR. Appl. Catal. 1983, 6, 341–346. [Google Scholar] [CrossRef]
- Lee, M.-H.; Cheng, C.-F.; Heine, V.; Klinowski, J. Distribution of tetrahedral and octahedral A1 sites in gamma alumina. Chem. Phys. Lett. 1997, 265, 673–676. [Google Scholar] [CrossRef]
- Khivantsev, K.; Jaegers, N.R.; Kwak, J.H.; Szanyi, J.; Kovarik, L. Precise identification and characterization of catalytically active sites on the surface of γ-alumina. Angew. Chem. 2021, 133, 17663–17671. [Google Scholar] [CrossRef]
- Digne, M.; Sautet, P.; Raybaud, P.; Euzen, P.; Toulhoat, H. Hydroxyl groups on γ-alumina surfaces: A DFT study. J. Catal. 2002, 211, 1–5. [Google Scholar] [CrossRef]
- Valero, M.C.; Digne, M.; Sautet, P.; Raybaud, P. DFT Study of the Interaction of a single Palladium Atom with-Alumina Surfaces: The Role of Hydroxylation. Oil Gas Sci. Technol.-Rev. De L’ifp 2006, 61, 535–545. [Google Scholar] [CrossRef]
- Gu, J.; Wang, J.; Leszczynski, J. Structure and Energetics of (111) Surface of γ-Al2O3: Insights from DFT Including Periodic Boundary Approach. ACS Omega 2018, 3, 1881–1888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wischert, R.; Copéret, C.; Delbecq, F.; Sautet, P. Dinitrogen: A selective probe for tri-coordinate Al “defect” sites on alumina. Chem. Commun. 2011, 47, 4890–4892. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.R.; Küçükbenli, E.; De Gironcoli, S.; Souza, W.F.; Chiaro, S.S.X.; Konstantinova, E.; Leitão, A.A. Structural models of activated γ-alumina surfaces revisited: Thermodynamics, NMR and IR spectroscopies from ab initio calculations. Chem. Phys. 2013, 423, 62–72. [Google Scholar] [CrossRef]
- Paglia, G.; Buckley, C.; Rohl, A.; Hunter, B.; Hart, R.; Hanna, J.; Byrne, L. Tetragonal structure model for boehmite-derived γ-alumina. Phys. Rev. B 2003, 68, 144110. [Google Scholar] [CrossRef]
- Paglia, G.; Buckley, C.E.; Udovic, T.J.; Rohl, A.L.; Jones, F.; Maitland, C.F.; Connolly, J. Boehmite-derived γ-alumina system. 2. consideration of hydrogen and surface effects. Chem. Mater. 2004, 16, 1914–1923. [Google Scholar] [CrossRef]
- Paglia, G.; Buckley, C.E.; Rohl, A.L.; Hart, R.D.; Winter, K.; Studer, A.J.; Hunter, B.A.; Hanna, J.V. Boehmite derived γ-alumina system. 1. Structural evolution with temperature, with the identification and structural determination of a new transition phase, γ ‘-alumina. Chem. Mater. 2004, 16, 220–236. [Google Scholar] [CrossRef]
- Smrčok, Ľ.; Langer, V.; Křesťan, J. γ-Alumina: A single-crystal X-ray diffraction study. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2006, 62, i83–i84. [Google Scholar] [CrossRef] [PubMed]
- Knözinger, H.; Ratnasamy, P. Catalytic aluminas: Surface models and characterization of surface sites. Catal. Rev. Sci. Eng. 1978, 17, 31–70. [Google Scholar] [CrossRef]
- Ouyang, C.; Šljivančanin, Ž.; Baldereschi, A. First-principles study of γ-Al 2 O 3 (100) surface. Phys. Rev. B 2009, 79, 235410. [Google Scholar] [CrossRef]
- Sohlberg, K.; Pennycook, S.J.; Pantelides, S.T. Explanation of the Observed Dearth of Three-Coordinated Al on γ-Alumina Surfaces. J. Am. Chem. Soc. 1999, 121, 10999–11001. [Google Scholar] [CrossRef]
- Ionescu, A.; Allouche, A.; Aycard, J.-P.; Rajzmann, M.; Hutschka, F. Study of γ-alumina surface reactivity: Adsorption of water and hydrogen sulfide on octahedral aluminum sites. J. Phys. Chem. B 2002, 106, 9359–9366. [Google Scholar] [CrossRef]
- Coster, D.; Blumenfeld, A.; Fripiat, J. Lewis acid sites and surface aluminum in aluminas and zeolites: A high-resolution NMR study. J. Phys. Chem. 1994, 98, 6201–6211. [Google Scholar] [CrossRef]
- Taoufik, M.; Szeto, K.C.; Merle, N.; Rosal, I.D.; Maron, L.; Trébosc, J.; Tricot, G.; Gauvin, R.M.; Delevoye, L. Heteronuclear NMR Spectroscopy as a Surface-Selective Technique: A Unique Look at the Hydroxyl Groups of γ-Alumina. Chem. Eur. J. 2014, 20, 4038–4046. [Google Scholar] [CrossRef]
- Delgado, M.; Delbecq, F.O.; Santini, C.C.; Lefebvre, F.; Norsic, S.; Putaj, P.; Sautet, P.; Basset, J.-M. Evolution of structure and of grafting properties of γ-alumina with pretreatment temperature. J. Phys. Chem. C 2012, 116, 834–843. [Google Scholar] [CrossRef]
- Sun, M.; Nelson, A.E.; Adjaye, J. Examination of spinel and nonspinel structural models for γ-Al2O3 by DFT and Rietveld refinement simulations. J. Phys. Chem. B 2006, 110, 2310–2317. [Google Scholar] [CrossRef] [PubMed]
- Paglia, G.; Buckley, C.E.; Rohl, A.L. Comment on “Examination of Spinel and Nonspinel Structural Models for γ-Al2O3 by DFT and Rietveld Refinement Simulations”. J. Phys. Chem. B 2006, 110, 20721–20723. [Google Scholar] [CrossRef]
- Kwak, J.H.; Peden, C.H.F.; Szanyi, J. Using a Surface-Sensitive Chemical Probe and a Bulk Structure Technique to Monitor the γ- to θ-Al2O3 Phase Transformation. J. Phys. Chem. C 2011, 115, 12575–12579. [Google Scholar] [CrossRef]
- Cai, S.-H.; Rashkeev, S.N.; Pantelides, S.T.; Sohlberg, K. Atomic scale mechanism of the transformation of γ-alumina to θ-alumina. Phys. Rev. Lett. 2002, 89, 235501. [Google Scholar] [CrossRef] [PubMed]
- Menéndez-Proupin, E.; Gutiérrez, G. Electronic properties of bulk γ-Al2O3. Phys. Rev. B 2005, 72, 035116. [Google Scholar] [CrossRef]
- Saniger, J. Al-O infrared vibrational frequencies of γ-alumina. Mater. Lett. 1995, 22, 109–113. [Google Scholar] [CrossRef]
- Baudisch, O.; Albrecht, W.H. Gamma-ferric oxide hydrate. J. Am. Chem. Soc. 1932, 54, 943–947. [Google Scholar] [CrossRef]
- Prins, R. On the structure of γ-Al2O3. J. Catal. 2020, 392, 336–346. [Google Scholar] [CrossRef]
- Trueba, M.; Trasatti, S.P. γ-Alumina as a support for catalysts: A review of fundamental aspects. Eur. J. Inorg. Chem. 2005, 2005, 3393–3403. [Google Scholar] [CrossRef]
Figure 1.
Thermal diagram of transition aluminas derived from calcination of boehmite.

Figure 2.
Structure of boehmite. Grey, red, and white spheres represent Al, O, and H atoms, respectively. Atoms enclosed inside rectangle represent the bulk unit cell. Reproduced from Ref. [14] with permission from the Royal Society of Chemistry.
Figure 2.
Structure of boehmite. Grey, red, and white spheres represent Al, O, and H atoms, respectively. Atoms enclosed inside rectangle represent the bulk unit cell. Reproduced from Ref. [14] with permission from the Royal Society of Chemistry.

Figure 3.
XRD powder pattern of γ-alumina. Values given above the peaks are hkl’s, FWHM and relative intensity. Figure reproduced from Zhou & Snyder (1991). Acta Cryst. B 47, 617–630, https://0-doi-org.brum.beds.ac.uk/10.1107/S0108768191002719 with permission of the International Union of Crystallography.
Figure 3.
XRD powder pattern of γ-alumina. Values given above the peaks are hkl’s, FWHM and relative intensity. Figure reproduced from Zhou & Snyder (1991). Acta Cryst. B 47, 617–630, https://0-doi-org.brum.beds.ac.uk/10.1107/S0108768191002719 with permission of the International Union of Crystallography.

Figure 4.
(a) Simulated (blue) XRD patterns for γ-alumina models compared with experimental (red) *; (b) Experimental XRD patterns for boehmite at different temperatures **. * Reprinted (adapted) with permission from Sun, M.; Nelson, A.E.; Adjaye, J. Examination of spinel and nonspinel structural models for γ-Al2O3 by DFT and Rietveld refinement simulations. J. Phys. Chem. B 2006, 110, 2310–2317. Copyright 2006 American Chemical Society. ** Reprinted (adapted) with permission from Krokidis, X.; Raybaud, P.; Gobichon, A.-E.; Rebours, B.; Euzen, P.; Toulhoat, H. Theoretical study of the dehydration process of boehmite to γ-alumina. J. Phys. Chem. B 2001, 105, 5121–5130. Copyright 2001 American Chemical Society.
Figure 4.
(a) Simulated (blue) XRD patterns for γ-alumina models compared with experimental (red) *; (b) Experimental XRD patterns for boehmite at different temperatures **. * Reprinted (adapted) with permission from Sun, M.; Nelson, A.E.; Adjaye, J. Examination of spinel and nonspinel structural models for γ-Al2O3 by DFT and Rietveld refinement simulations. J. Phys. Chem. B 2006, 110, 2310–2317. Copyright 2006 American Chemical Society. ** Reprinted (adapted) with permission from Krokidis, X.; Raybaud, P.; Gobichon, A.-E.; Rebours, B.; Euzen, P.; Toulhoat, H. Theoretical study of the dehydration process of boehmite to γ-alumina. J. Phys. Chem. B 2001, 105, 5121–5130. Copyright 2001 American Chemical Society.

Figure 5.
Simulated IR spectra for (a) non-spinel and (b) defect spinel structures, dashed black lines are experimental frequencies. Reprinted from the Journal of Solid State Chemistry, Vol 184 Issue 5, Ary R. Ferreira, Mateus J.F. Martins, Elena Konstantinova, Rodrigo B. Capaz, Wladmir F. Souza, Sandra Shirley X. Chiaro, Alexandre A. Leitão, Direct comparison between two γ—alumina structural models by DFT calculations, pp 1105–1111, Copyriht 2011, with permission from Elsevier.
Figure 5.
Simulated IR spectra for (a) non-spinel and (b) defect spinel structures, dashed black lines are experimental frequencies. Reprinted from the Journal of Solid State Chemistry, Vol 184 Issue 5, Ary R. Ferreira, Mateus J.F. Martins, Elena Konstantinova, Rodrigo B. Capaz, Wladmir F. Souza, Sandra Shirley X. Chiaro, Alexandre A. Leitão, Direct comparison between two γ—alumina structural models by DFT calculations, pp 1105–1111, Copyriht 2011, with permission from Elsevier.

Figure 6.
Schematic representations of γ-alumina bulk models. Unit cell boundaries are indicated by dotted black lines. Reprinted from Acta Materialia, Vol 182, Henry O. Ayoola, Stephen D. House, Cecile S. Bonifacio, Kim Kisslinger, Wissam A. Saidi, Judith C. Yang, Evaluating the accuracy of common γ-Al2O3 structure models by selected area electron diffraction from high-quality crystalline γAl2O3, pp 257–266, Copyright 2020, with permission from Elsevier.
Figure 6.
Schematic representations of γ-alumina bulk models. Unit cell boundaries are indicated by dotted black lines. Reprinted from Acta Materialia, Vol 182, Henry O. Ayoola, Stephen D. House, Cecile S. Bonifacio, Kim Kisslinger, Wissam A. Saidi, Judith C. Yang, Evaluating the accuracy of common γ-Al2O3 structure models by selected area electron diffraction from high-quality crystalline γAl2O3, pp 257–266, Copyright 2020, with permission from Elsevier.

Figure 7.
(a) Azimuthally averaged line profile of experimental SAED pattern obtained by Ayoola et al., plotted with the simulated diffraction data for each of the theoretical models considered. Dashed lines indicate the peaks in the experimental diffraction profile. (b) A closeup image of the region from 3.0–6.0 nm−1 from (a) showing the contributing peaks to the profile. The diffraction profiles of the models agree well overall with the experimental profile; however, the monoclinic non-spinel (Digne) model lacks the first peak present in the experimental pattern. Reprinted (adapted) from Acta Materialia, Vol 182, Henry O. Ayoola, Stephen D. House, Cecile S. Bonifacio, Kim Kisslinger, Wissam A. Saidi, Judith C. Yang, Evaluating the accuracy of common γ-Al2O3 structure models by selected area electron diffraction from high-quality crystalline γAl2O3, pp 257–266, Copyright 2020, with permission from Elsevier.
Figure 7.
(a) Azimuthally averaged line profile of experimental SAED pattern obtained by Ayoola et al., plotted with the simulated diffraction data for each of the theoretical models considered. Dashed lines indicate the peaks in the experimental diffraction profile. (b) A closeup image of the region from 3.0–6.0 nm−1 from (a) showing the contributing peaks to the profile. The diffraction profiles of the models agree well overall with the experimental profile; however, the monoclinic non-spinel (Digne) model lacks the first peak present in the experimental pattern. Reprinted (adapted) from Acta Materialia, Vol 182, Henry O. Ayoola, Stephen D. House, Cecile S. Bonifacio, Kim Kisslinger, Wissam A. Saidi, Judith C. Yang, Evaluating the accuracy of common γ-Al2O3 structure models by selected area electron diffraction from high-quality crystalline γAl2O3, pp 257–266, Copyright 2020, with permission from Elsevier.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Structural details of four γ-alumina bulk models. Reprinted from Acta Materialia, Vol 182, Henry O. Ayoola, Stephen D. House, Cecile S. Bonifacio, Kim Kisslinger, Wissam A. Saidi, Judith C. Yang, Evaluating the accuracy of common γ-Al2O3 structure models by selected area electron diffraction from high-quality crystalline γAl2O3, pp 257–266, Copyright 2020, with permission from Elsevier.
Table 1.
Structural details of four γ-alumina bulk models. Reprinted from Acta Materialia, Vol 182, Henry O. Ayoola, Stephen D. House, Cecile S. Bonifacio, Kim Kisslinger, Wissam A. Saidi, Judith C. Yang, Evaluating the accuracy of common γ-Al2O3 structure models by selected area electron diffraction from high-quality crystalline γAl2O3, pp 257–266, Copyright 2020, with permission from Elsevier.
| Model | Symmetry (Space Group) | Dimensions | Number of Sites per Unit Cell | Partially Occupied Al Sites? | Al Site Positions | Al Ratio in Tetrahedral vs. Octahedral | Al Ratio in Spinel vs. Non-Spinel Sites | Refs |
|---|---|---|---|---|---|---|---|---|
| Smrčok | Cubic (Fdm) | a = 7.9382 Å | 120 | Yes | 8a, 16d, 16c, 48f | 37:63 | 94:6 | [67] |
| Paglia | Tetragonal (I41/amd) | a = 5.652 Å | 36 | Yes | 4a, 8d, 8c | 31:69 | 73:27 | [64] |
| Pinto | Monoclinic (C2/m) | a = b = 5.663 Å c = 13.71 Å α = β = 90.6° γ = 60.401° | 80 | No | n/a | 38:62 | 100:0 | [37] |
| Digne | Monoclinic (P21/m) | a = 5.587 Å b = 8.413 Å c = 8.068 Å α = γ = 90° β = 90.59° | 40 | No | n/a | 25:75 | 32:68 | [32] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Stuart, N.M.; Sohlberg, K. The Microstructure of γ-Alumina. Energies 2021, 14, 6472. https://0-doi-org.brum.beds.ac.uk/10.3390/en14206472
AMA Style
Stuart NM, Sohlberg K. The Microstructure of γ-Alumina. Energies. 2021; 14(20):6472. https://0-doi-org.brum.beds.ac.uk/10.3390/en14206472
Chicago/Turabian StyleStuart, Natalie M., and Karl Sohlberg. 2021. "The Microstructure of γ-Alumina" Energies 14, no. 20: 6472. https://0-doi-org.brum.beds.ac.uk/10.3390/en14206472
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.