
Development and Comparison of Seminested PCR, qPCR, and LAMP for the Rapid Detection of Arthrinium phaeospermum, the Causal Agent of Bamboo Blight

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains
2.2. Culture of the Tested Strains and DNA Extraction
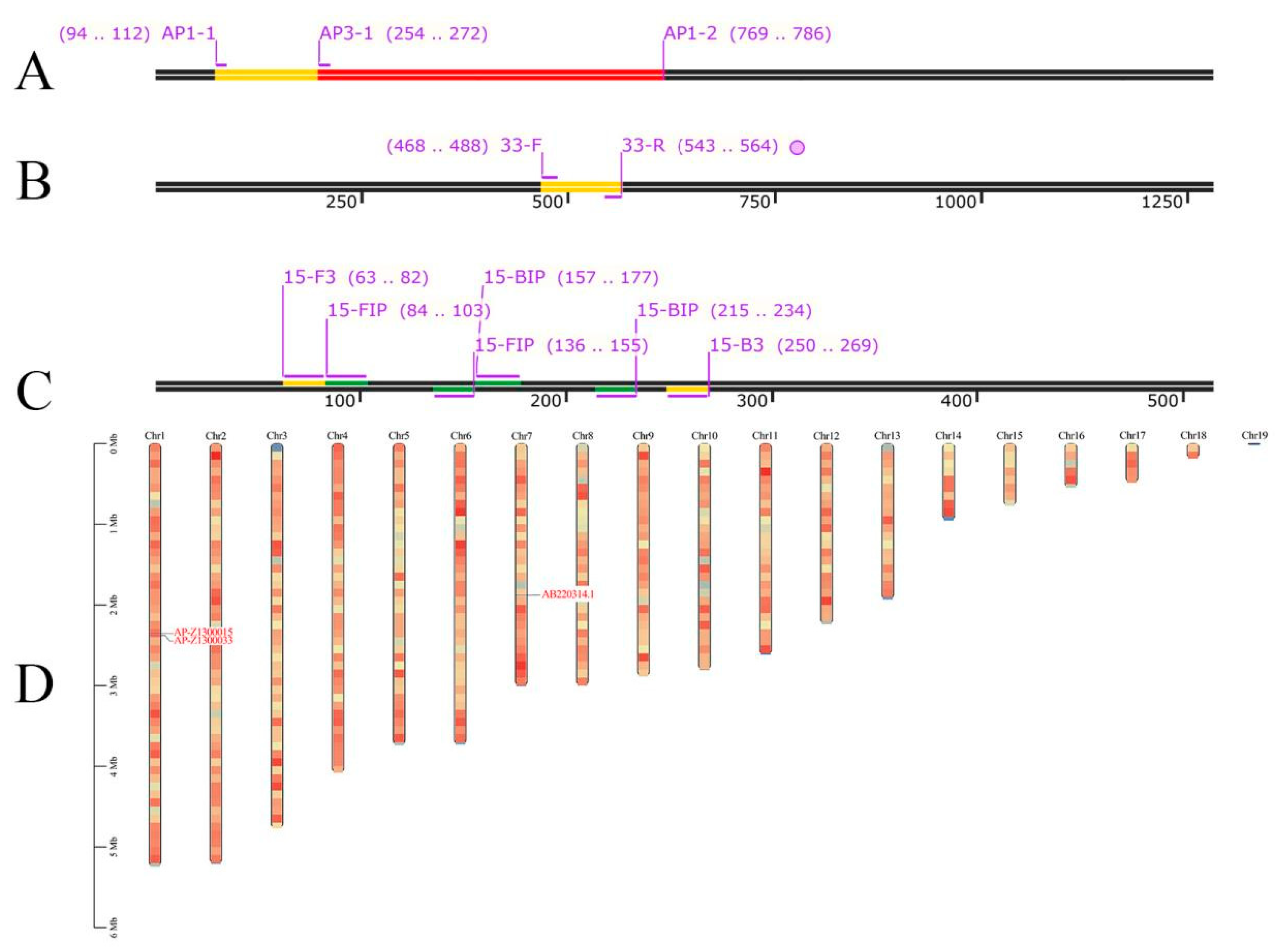
2.3. Primer Design
2.4. Seminested PCR, qPCR, and LAMP System Assays
2.4.1. Seminested PCR Conditions
2.4.2. qPCR Conditions
2.4.3. LAMP Conditions
2.5. Specificity of the Assays
2.6. Sensitivity of the Assays
2.7. Detection of Artificially Inoculated and Field-Collected Samples
2.7.1. Artificially Inoculated Sample Preparation
2.7.2. Field-Collected Sample Preparation
3. Results
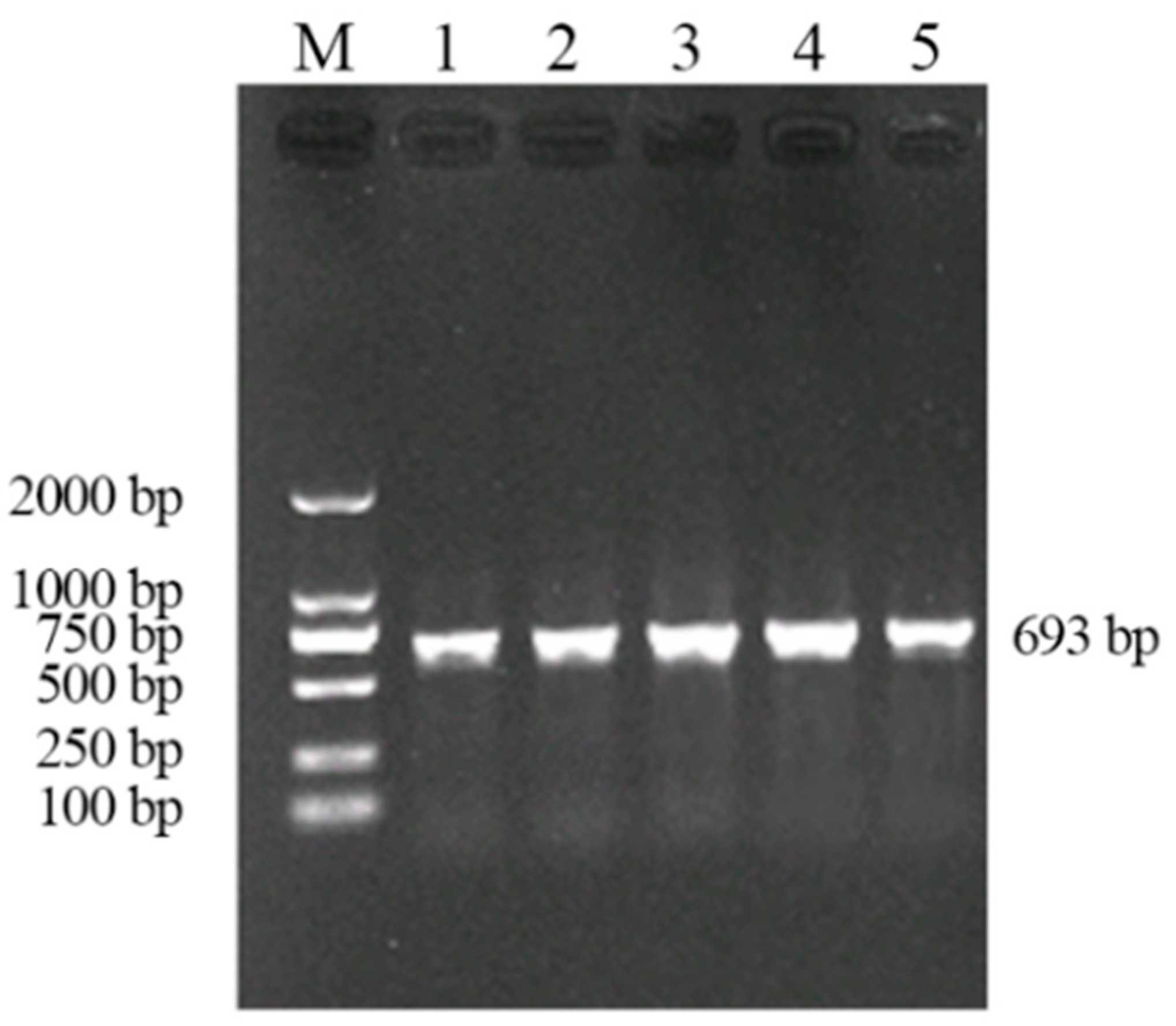
3.1. Establishment of Seminested PCR
3.1.1. Optimization of the Reaction Conditions of Seminested PCR
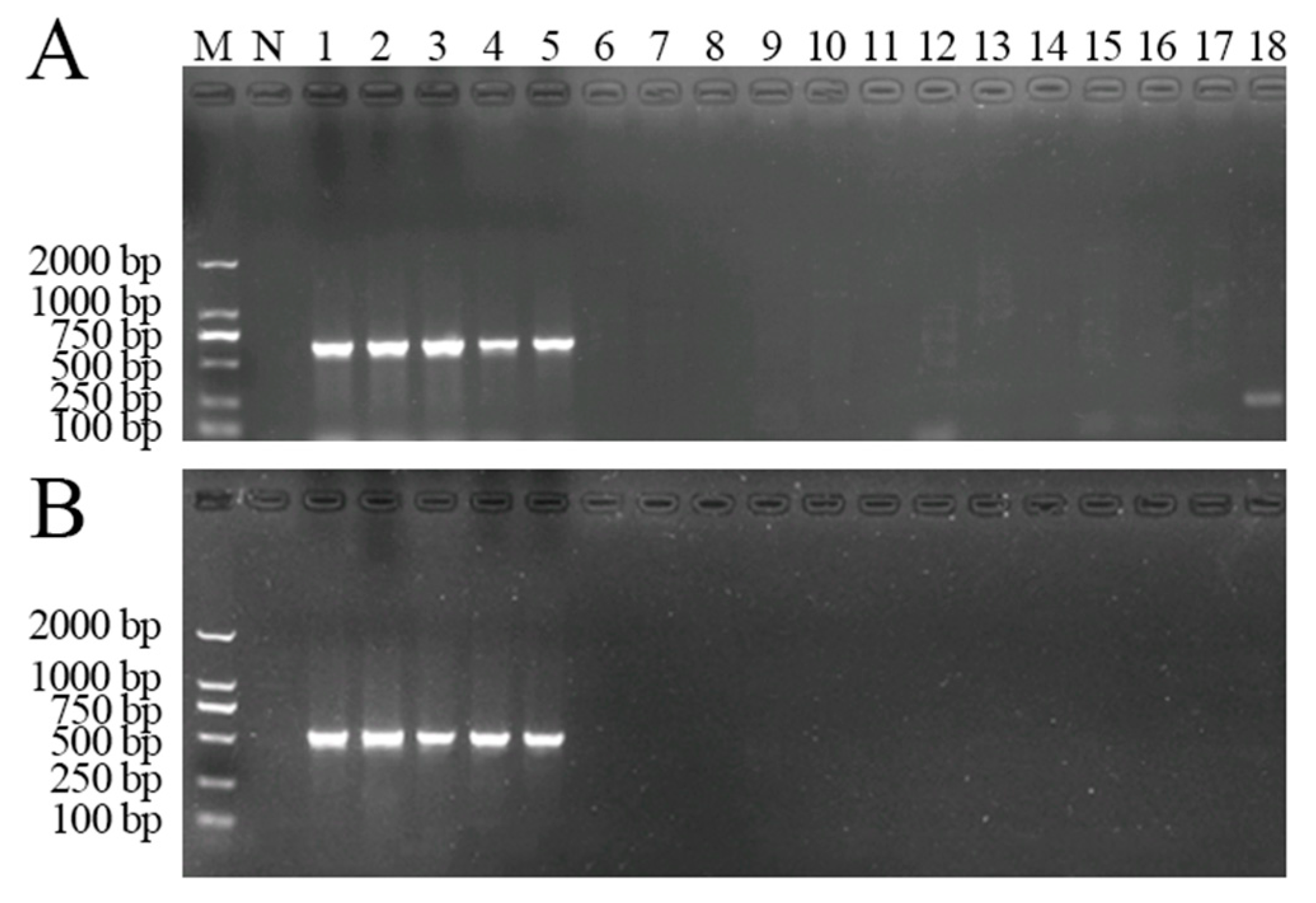
3.1.2. Specificity of the Seminested PCR Assay
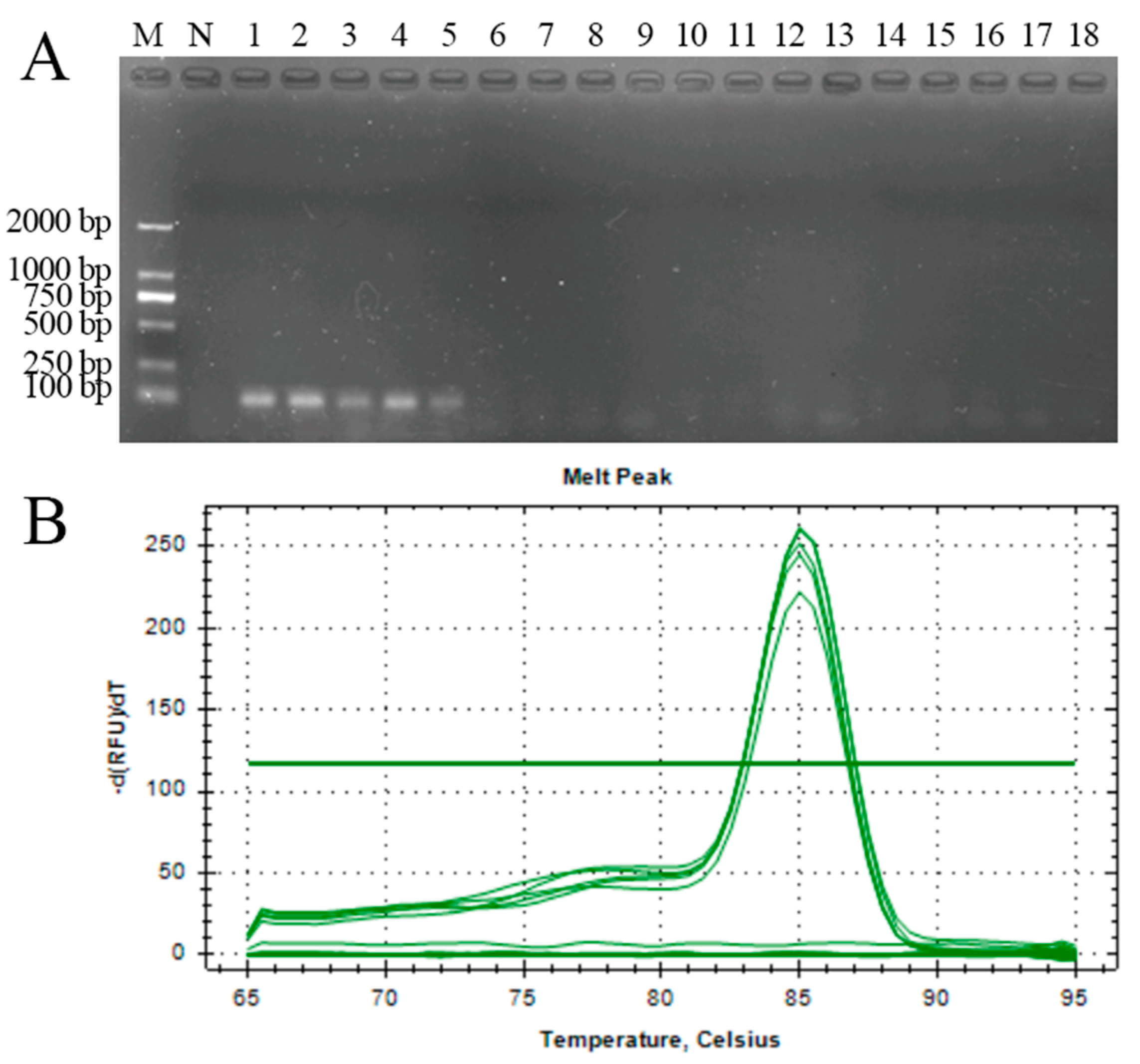
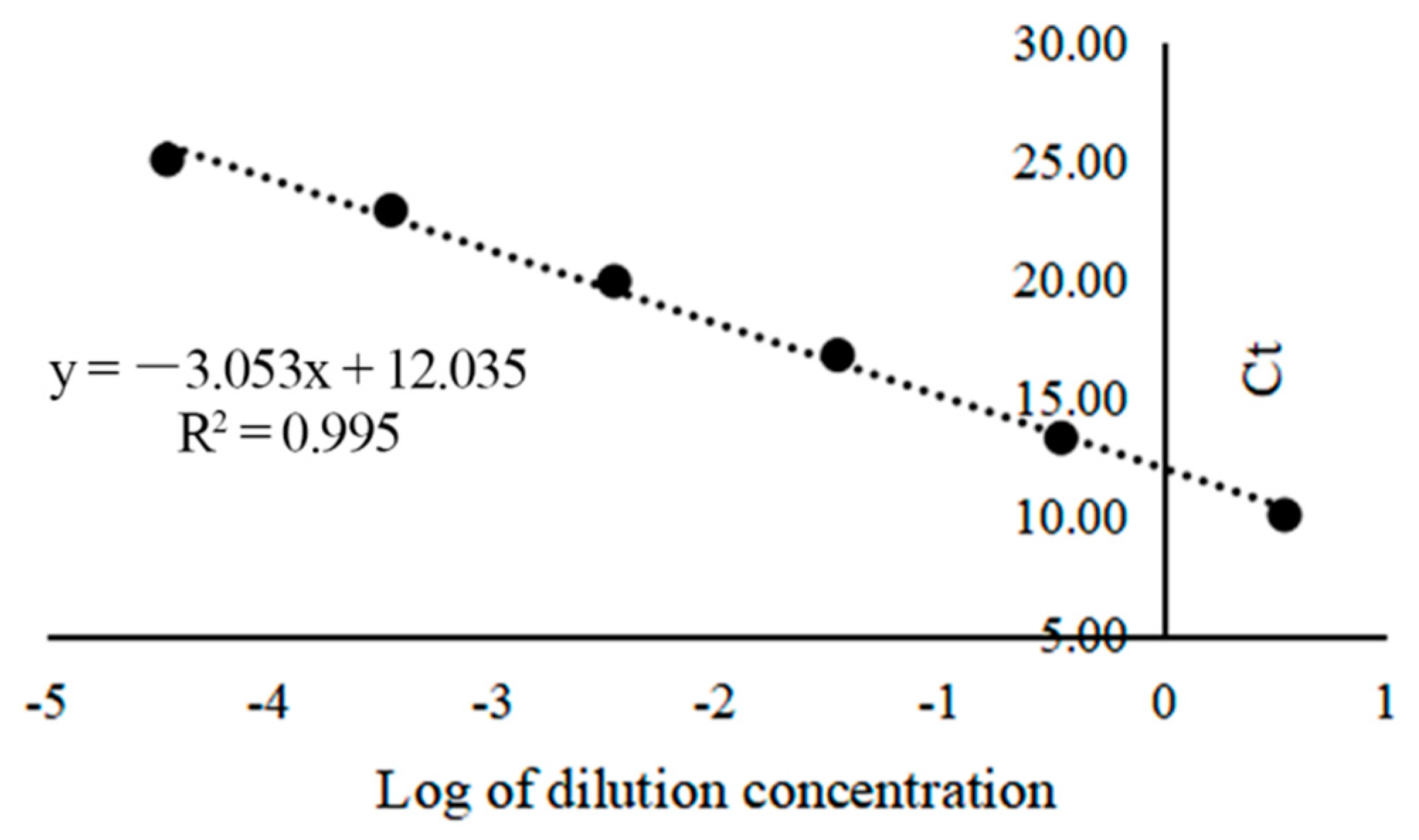
3.2. Establishment of Real-Time Quantitative PCR
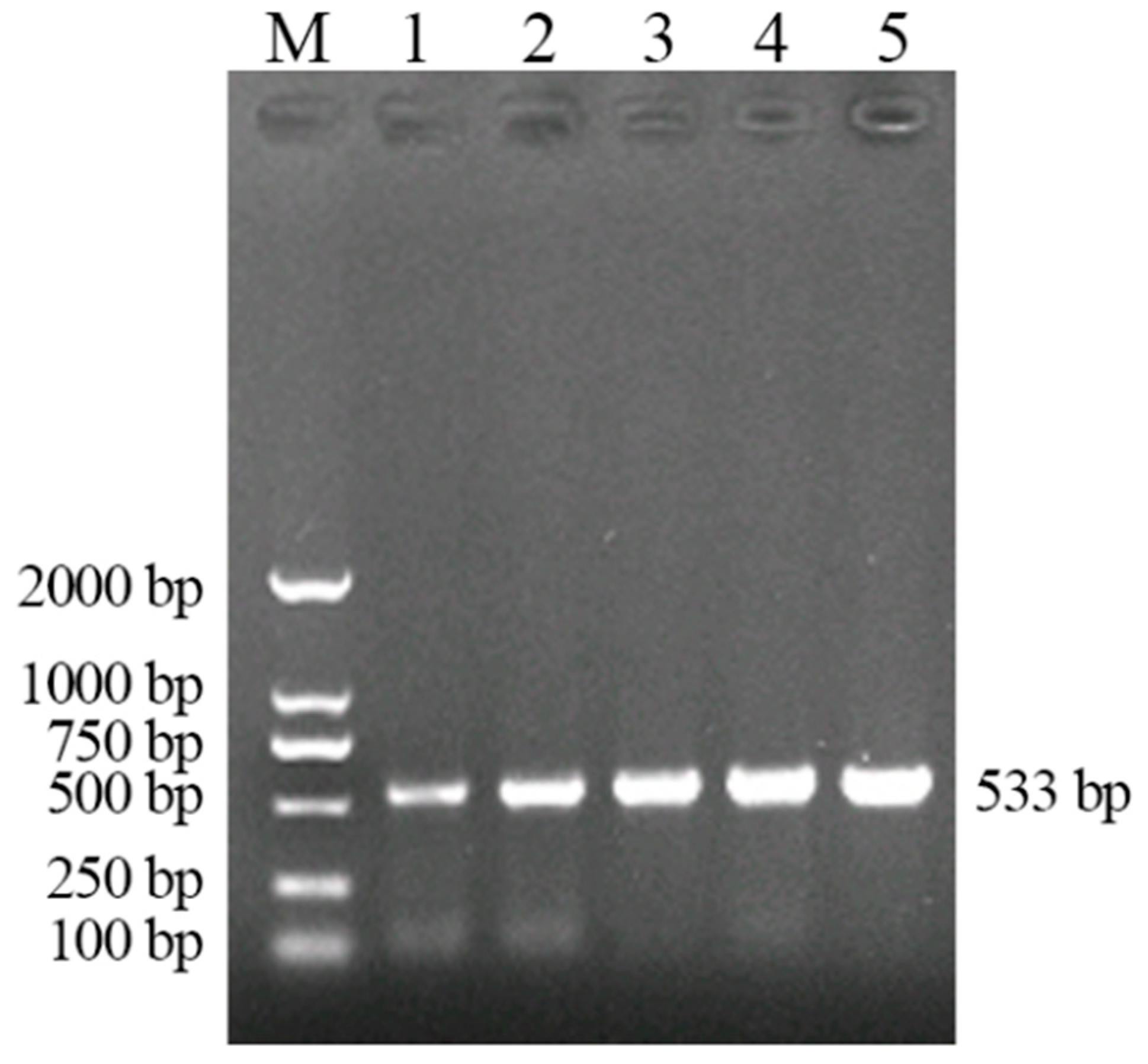
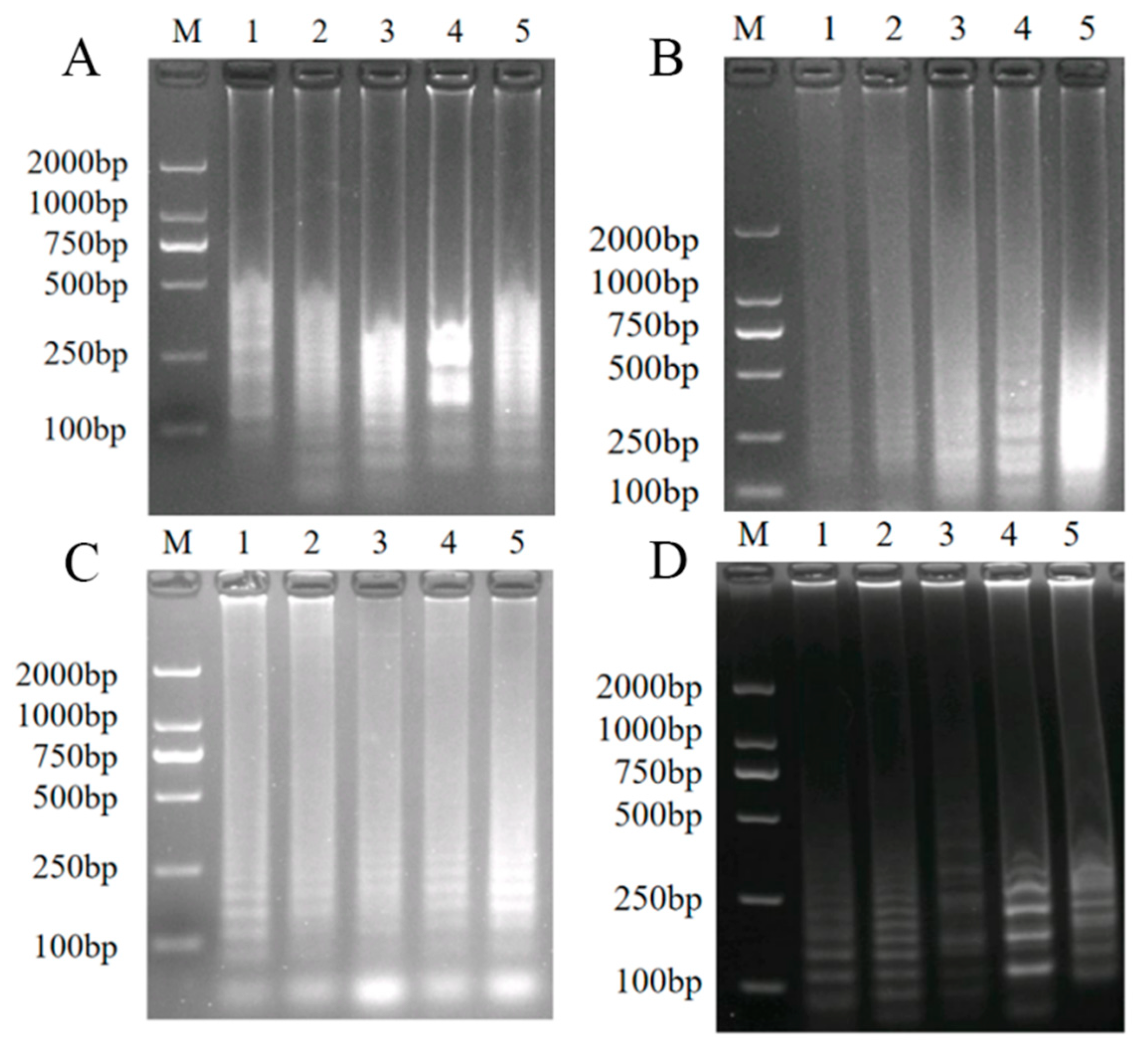
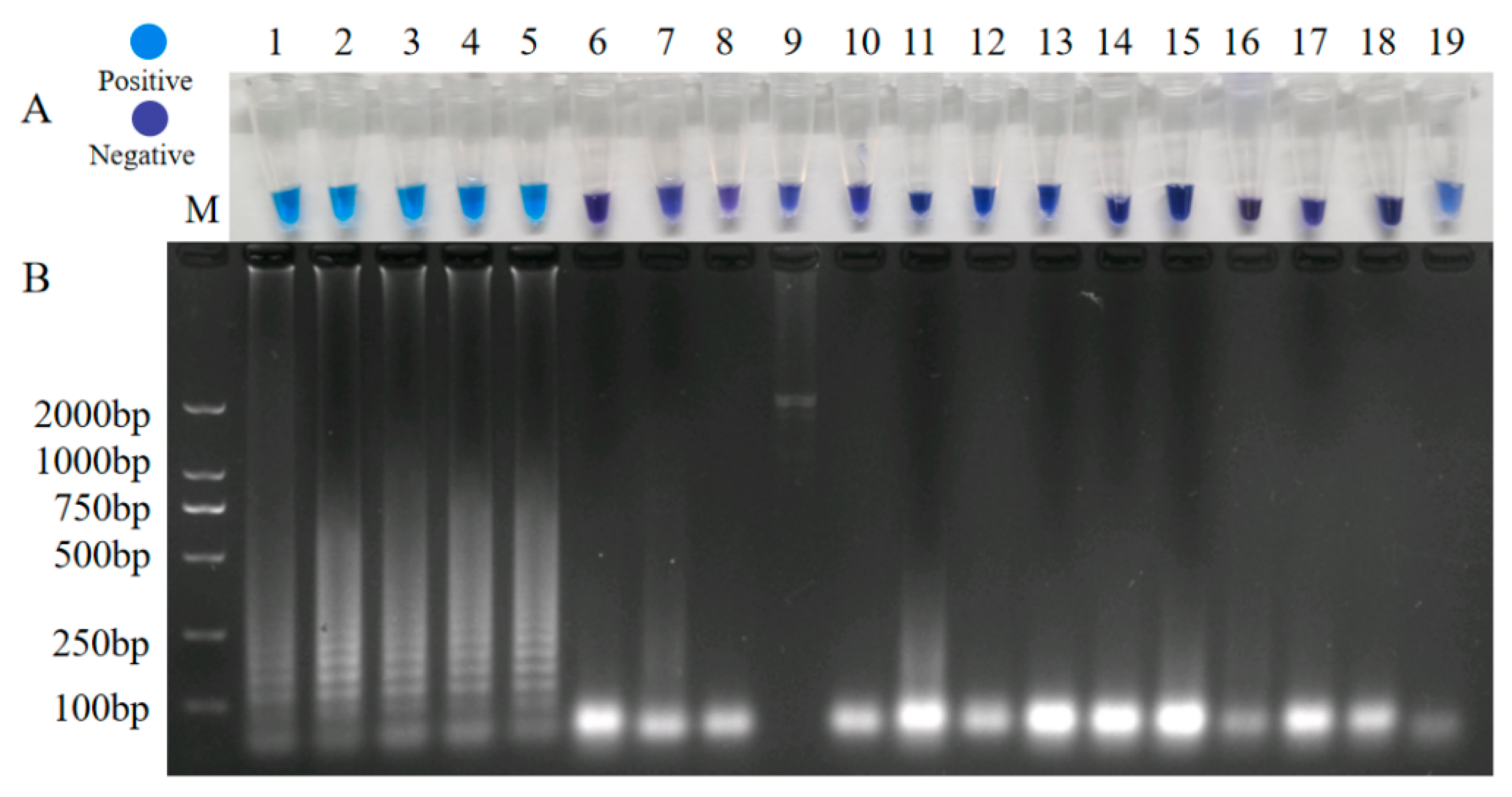
3.3. Establishment of LAMP
3.3.1. Optimization of the Reaction Conditions of LAMP
3.3.2. Specificity Test of LAMP for the Detection of A. phaeospermum
3.4. Sensitivity Test of the Seminested PCR, Real-Time Quantitative PCR, and LAMP for the Detection of A. phaeospermum
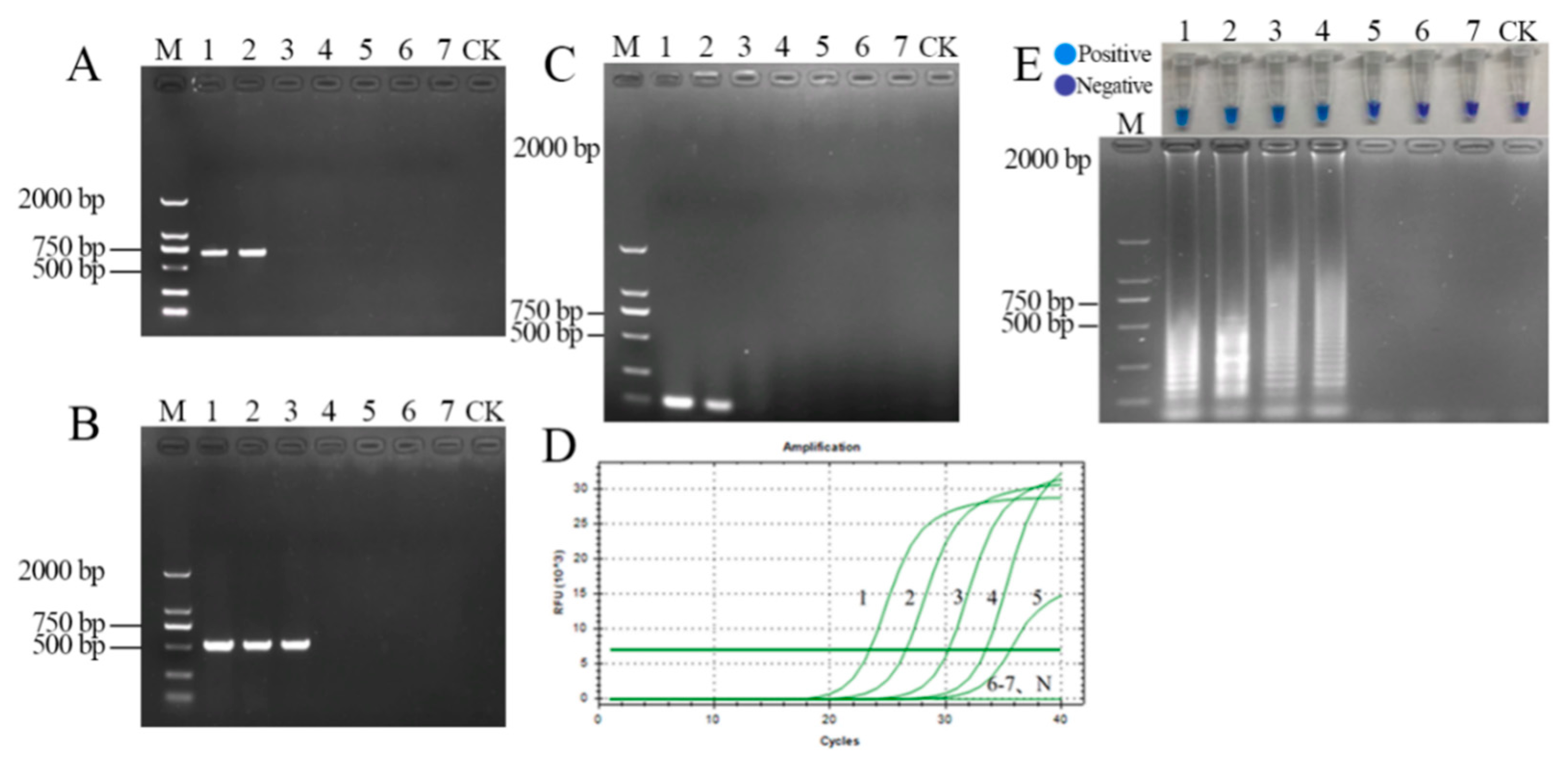
3.5. Detection of Artificially Inoculated and Field-Collected Samples
4. Discussion
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, Q. Research Status of B. pervariabilis × D. Daii and Cost-benefifit analysis. J. Shandong For. Sci. Technol. 2007, 3, 101–103. [Google Scholar]
- Yang, Z.Z.; Ye, J.R. Identifification on pathogen of hybrid bamboo blight. J. Sichuan Agric. Univ. 2004, 22, 225–227. [Google Scholar]
- Li, S.; He, Q.; Peng, Q.; Fang, X.; Zhu, T.; Qiao, T.; Han, S. Metabolomics responses of Bambusa pervariabilis × Dendrocalamopsis grandis varieties to Biotic (pathogenic fungus) stress. Phytochemistry 2019, 167, 112087. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.H.; Huang, Z.C.; Gao, Q.Z. Pathogen and occurrence regularity of Bambusa ervariabilis × Dendrocalamopsis daii blight. For. Pest Dis. 2009, 28, 10–12. [Google Scholar]
- Zhao, Y.M.; Deng, C.R.; Chen, X. Arthrinium phaeospermum causing dermatomycosis, a new record of China. Mycosystema 1990, 3, 232–235. [Google Scholar]
- Rai, M.K. Mycosis in man due to Arthrinium phaeospermum var. indicum. First Case Report: Mykose durch Arthrinium phaeospermum var. indicum beim Menschen. Erstbericht. Mycoses 2010, 32, 472–475. [Google Scholar] [CrossRef]
- Khan, K.R.; Sullia, S.B. Arthrinium phaeospermium var. indicum var. nov., a new market pathogen of cowpea, garden pea and french bean. Acta Bot. Indica 1980, 8, 103–104. [Google Scholar]
- Liu, X.J.; Luo, X.Y.; Li, X.F.; Chen, Q.T. Taxonomic determination of toxigenic Arthrinium strains isolated from poisoning sugarcane. Mycosystema 1988, 7, 221–225. [Google Scholar]
- Xia, L.M.; Zhang, S.X.; Huang, J.H.; Zhang, W.M. Studies on Arthrinium phaeospermum causing moso bamboo foot rot. J. Nanjing Fores. Univ. 1995, 19, 1–7. [Google Scholar]
- Ma, G.L.; Hu, G.L.; Yu, C.Z.; Wu, J.L.; Xu, B.C. Phyllostachys prominens plum shoot wilt pathogenic fungoid and its biological characteristics. Zhejiang For. Coll. 2003, 20, 44–48. [Google Scholar]
- Piccolo, S.L.; Mondello, V.; Giambra, S.; Conigliaro, G.; Torta, L.; Burruano, S. Arthrinium phaeospermum, Phoma cladoniicola and Ulocladium consortiale, New Olive Pathogens in Italy. J. Phytopathol. 2014, 162, 258–263. [Google Scholar] [CrossRef]
- Li, B.J.; Liu, P.Q.; Jiang, Y.; Weng, Q.Y.; Chen, Q.H. First Report of Culm Rot Caused by Arthrinium phaeospermum on Phyllostachys viridis in China. Plant Dis. 2016, 100, 10–13. [Google Scholar] [CrossRef]
- Gong, H.Y.; Li, Q.Y.; Zhang, H.; Ye, L.; Shi, L.; Feng, Y.H. Development and comparison of qPCR and qLAMP for rapid detection of the decapod iridescent virus 1 (DIV1). J. Invertebr. Pathol. 2021, 182, 107567. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y.; Gong, J.Z.; Yang, B.; Fan, Y.M.; Yao, N.; Wang, C.R. Development of a nest-PCR for detection of Fasciola hepatica DNA in the intermediate snail host, Radix cucunorica, and the prevalence in northwestern China. Infect. Genet. Evol. 2019, 75, 103984. [Google Scholar] [CrossRef] [PubMed]
- Matthew, M.A.; Christie, J.; Yang, N.; Yao, C. A Loop-Mediated Isothermal Amplification (LAMP) Assay Specific to Trichomonas tenax Is Suitable for Use at Point-of-Care. Microorganisms 2022, 10, 594. [Google Scholar] [CrossRef] [PubMed]
- Smolejová, M.; Cihová, I.; Sulo, P. Reliable and Sensitive Nested PCR for the Detection of Chlamydia in Sputum. Microorganisms 2021, 9, 935. [Google Scholar] [CrossRef]
- Kamber, T.; Malpica-López, N.; Messmer, M.M.; Oberhänsli, T.; Arncken, C.; Alkemade, J.A.; Hohmann, P. A qPCR Assay for the Fast Detection and Quantification of Colletotrichum lupini. Plants 2021, 10, 1548. [Google Scholar] [CrossRef]
- Coats, K.; DeBauw, A.; Lakshman, D.K.; Roberts, D.P.; Ismaiel, A.; Chastagner, G. Detection and Molecular Phylogenetic-Morphometric Characterization of Rhizoctonia tuliparum, Causal Agent of Gray Bulb Rot of Tulips and Bulbous Iris. J. Fungi 2022, 8, 163. [Google Scholar] [CrossRef]
- Wang, J.; Jacobs, J.L.; Byrne, J.M.; Chilvers, M.I. Improved diagnoses and quantification of Fusarium virguliforme, causal agent of soybean sudden death syndrome. Phytopathology 2015, 105, 378–387. [Google Scholar] [CrossRef] [Green Version]
- Notomi, T.; Mori, Y.; Tomita, N.; Kanda, H. Loop-mediated isothermal amplification (LAMP): Principle, features, and future prospects. J. Microbiol. 2015, 53, 1–5. [Google Scholar] [CrossRef]
- Li, S.; Tang, Y.; Fang, X.; Qiao, T.; Han, S.; Zhu, T. Whole-genome sequence of Arthrinium phaeospermum, a globally distributed pathogenic fungus. Genomics 2020, 112, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Maestu, A.; Azinheiro, S.; Carvalho, J.; Prado, M. Rapid and sensitive detection of viable Listeria monocytogenes in food products by a filtration-based protocol and qPCR. Food Microbiol. 2018, 73, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Schneider, L.; Blakely, H.; Tripathi, A. Mathematical model to reduce loop mediated isothermal amplification (LAMP) false-positive diagnosis. Electrophoresis 2019, 40, 2706–2717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Sun, Z.; Yan, J.; Luo, Y.; Wang, H.; Ma, Z. Rapid and Precise Detection of Latent Infections of Wheat Stripe Rust in Wheat Leaves using Loop-Mediated Isothermal Amplification. J. Phytopathol. 2011, 159, 582–584. [Google Scholar] [CrossRef]
- Yan, H.; Zhang, J.; Ma, D.; Yin, J. qPCR and loop mediated isothermal amplification for rapid detection of Ustilago tritici. PeerJ 2019, 7, e7766. [Google Scholar] [CrossRef] [Green Version]
- Almasi, M.A.; Dehabadi, S.H.; Moradi, A.; Eftekhari, Z.; Ojaghkandi, M.A.; Aghaei, S. Development and application of loop-mediated isothermal amplifification assay for rapid detection of Fusarium oxysporum f. sp. lycopersici. J. Plant Pathol. Microbiol. 2013, 4, 177. [Google Scholar]
- Sagcan, H.; Turgut Kara, N. Detection of Potato ring rot Pathogen Clavibacter michiganensis subsp. sepedonicus by Loop-mediated isothermal amplification (LAMP) assay. Sci. Rep. 2019, 9, 20393. [Google Scholar] [CrossRef] [Green Version]
- Durand, L.; La Carbona, S.; Geffard, A.; Possenti, A.; Dubey, J.P.; Lalle, M. Comparative evaluation of loop-mediated isothermal amplification (LAMP) vs qPCR for detection of Toxoplasma gondii oocysts DNA in mussels. Exp. Parasitol. 2020, 208, 107809. [Google Scholar] [CrossRef]
- Gill, P.; Ghaemi, A. Nucleic acid isothermal amplification technologies—A review. Nucleosides Nucleotides Nucleic Acids 2008, 27, 224–243. [Google Scholar] [CrossRef]
- Zhao, B.; Yang, D.; Zhang, Y.; Xu, Y.; Zhao, X.; Liang, J.; Fan, X.; Du, Y.; Zhu, Z.; Shi, B.; et al. Rapid visual detection of lily mottle virus using a loop-mediated isothermal amplification method. Arch. Virol. 2018, 163, 545–548. [Google Scholar] [CrossRef]
- Hopkins, H.; González, I.J.; Polley, S.D.; Angutoko, P.; Ategeka, J.; Asiimwe, C.; Bell, D. Highly sensitive detection of malaria parasitemia in a malaria-endemic setting: Performance of a new loop-mediated isothermal amplification kit in a remote clinic in Uganda. J. Infect. Dis. 2013, 208, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Cesbron, S.; Dupas, E.; Beaurepère, Q.; Briand, M.; Montes-Borrego, M.; Velasco-Amo, M.D.P.; Jacques, M.A. Development of a Nested-MultiLocus Sequence Typing approach for a highly sensitive and specific identification of Xylella fastidiosa subspecies directly from plant samples. Agronomy 2020, 10, 1099. [Google Scholar] [CrossRef]
- Puig, A.S. Detection of Cacao Mild Mosaic Virus (CaMMV) Using Nested PCR and Evidence of Uneven Distribution in Leaf Tissue. Agronomy 2021, 11, 1842. [Google Scholar] [CrossRef]
- Coelho, C.; Vieira-Pinto, M.; Vilares, A.; Gargaté, M.J.; Rodrigues, M.; Cardoso, L.; Lopes, A.P. PCR detection of Toxoplasma gondii in European wild rabbit (Oryctolagus cuniculus) from Portugal. Microorganisms 2020, 8, 1926. [Google Scholar] [CrossRef]
- Yuan, J.; Mi, T.; Zhen, Y.; Yu, Z. Development of a rapid detection and quantification method of Karenia mikimotoi by real-time quantitative PCR. Harmful Algae 2012, 17, 83–91. [Google Scholar] [CrossRef]
- Foulds, I.V.; Granacki, A.; Xiao, C.; Krull, U.J.; Castle, A.; Horgen, P.A. Quantification of microcystin-producing cyanobacteria and E. coli in water by 5′-nuclease PCR. J. Appl. Microbiol. 2002, 93, 825–834. [Google Scholar] [CrossRef] [Green Version]
- Abd-Elsalam, K.; Bahkali, A.; Moslem, M.; Amin, O.E.; Niessen, L. An optimized protocol for DNA extraction from wheat seeds and loop-mediated isothermal amplification (LAMP) to detect Fusarium graminearum contamination of wheat grain. Int. J. Mol. Sci. 2011, 12, 3459–3472. [Google Scholar] [CrossRef] [Green Version]
- Bustin, S.; Huggett, J. qPCR primer design revisited. Biomol. Detect. Quantif. 2017, 14, 19–28. [Google Scholar] [CrossRef]
- Garrido-Maestu, A.; Azinheiro, S.; Fuciños, P.; Carvalho, J.; Prado, M. Highly sensitive detection of gluten-containing cereals in food samples by real-time Loop-mediated isothermal AMPlification (qLAMP) and real-time polymerase chain reaction (qPCR). Food Chem. 2018, 246, 156–163. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Species | Host | Source |
|---|---|---|---|
| 1 | Arthrinium phaeospermum (Corda) M.B. Ellis | B. pervariabilis × D. grandis | Tianquan, Sichuan, China |
| 2 | Arthrinium phaeospermum (Corda) M.B. Ellis | B. pervariabilis × D. grandis | Ya’an, Sichuan, China |
| 3 | Arthrinium phaeospermum (Corda) M.B. Ellis | B. pervariabilis × D. grandis | Changning, Sichuan, China |
| 4 | Arthrinium phaeospermum (Corda) M.B. Ellis | Phyllostachys edulis | Baoxing, Sichuan, China |
| 5 | Arthrinium phaeospermum (Corda) M.B. Ellis | Phyllostachys edulis | Xinyang, Henan, China |
| 6 | Arthrinium kogelbergense | Dendrocalamus farinosus | Changning, Sichuan, China |
| 7 | Arthrinium marii Larrondo & Calvo | Phyllostachys edulis | Qingdao, Shandong, China |
| 8 | Arthrinium arundinis (Corda) Dyko & B. Sutton | Arundo donax | Xinyang, Henan, China |
| 9 | Arthrinium haricotospora (Sacc.) Y.L. Zhang & T.Y. Zhang, comb. nov. | Phyllostachys edulis | Qingdao, Shandong, China |
| 10 | Diaporthe guangxinsis | Dendrocalamus latifloru | Yibin, Sichuan, China |
| 11 | Neofusicoccum parvum | Juglans regia | Mianyang, Sichuan, China |
| 12 | Fusarium tricinctum (Corda) Sacc. | Zanthoxylum bungeanum | Hanyuan, Sichuan, China |
| 13 | Nigrospora sphaerica (Sacc.) E.W. Mason | Arundo donax | Xinyang, Henan, China |
| 14 | Apiospora montagnei Sacc. | Phyllostachys praecox | Tianshui, Gansu, China |
| 15 | Arthrinium rasikravindrii Shiv M. Singh, L.S. Yadav, P.N. Sin | B. pervariabilis × D. grandis | Jiangan, Sichuan, China |
| 16 | Fusarium proliferatum (Matsush.) Nirenberg | B. pervariabilis × D. grandis | Changning, Sichuan, China |
| 17 | Nigrospora guilinensis Mei Wang & L. Cai | B. pervariabilis × D. grandis | Changning, Sichuan, China |
| 18 | Nigrospora oryzae (Berk. & Broome) Petch | B. pervariabilis × D. grandis | Jiangan, Sichuan, China |
| Assay | Primer Name | Sequence (5′–3′) | Length |
|---|---|---|---|
| qPCR | 33-F | CACTCCGAGCCATGCTACTAC | 21 |
| 33-R | GCATAGCGATCCAACAGGTAGA | 22 | |
| Seminested PCR | AP1-1 | GGCACCGACCCCTTGATTG | 19 |
| AP1-2 | CGAAGTTGTCGGGGCGGA | 18 | |
| AP3-1 | CTCCAGACCGGTCAATGCG | 19 | |
| LAMP | 15-F3 | GCATCGAACAAGACGGAAAC | 20 |
| 15-B3 | GTTGACCCTGCTCTCGTTTC | 20 | |
| 15-FIP | CTCTTCGCCGGGGCGTATGCCCTGGACCTCGGCATCAC | 38 | |
| 15-BIP | TTGTGTGGACCGTCTTCGTCGCCACCAGGGCCAGTTTC | 38 |
| No. | The Time of Inoculation | Seminested PCR | LAMP | qPCR |
|---|---|---|---|---|
| 1 | 4 d | − | − | + |
| 2 | − | − | − | |
| 3 | − | + | + | |
| 4 | 8 d | − | + | + |
| 5 | − | + | + | |
| 6 | − | + | + | |
| 7 | 12 d | + | + | + |
| 8 | + | + | + | |
| 9 | + | + | + | |
| 10 | 16 d | + | + | + |
| 11 | + | + | + | |
| 12 | + | + | + |
| No. | Symptom Rank | Seminested PCR | LAMP | qPCR |
|---|---|---|---|---|
| 1 | asymptomatic | − | + | + |
| 2 | − | + | + | |
| 3 | − | − | − | |
| 4 | Mild | + | + | + |
| 5 | + | + | + | |
| 6 | + | + | + | |
| 7 | severe | + | + | + |
| 8 | + | + | + | |
| 9 | + | + | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Yang, W.; Xie, L.; Zhu, T.; Li, S.; Han, S.; Lin, T.; Li, S. Development and Comparison of Seminested PCR, qPCR, and LAMP for the Rapid Detection of Arthrinium phaeospermum, the Causal Agent of Bamboo Blight. Forests 2022, 13, 850. https://0-doi-org.brum.beds.ac.uk/10.3390/f13060850
Zhang H, Yang W, Xie L, Zhu T, Li S, Han S, Lin T, Li S. Development and Comparison of Seminested PCR, qPCR, and LAMP for the Rapid Detection of Arthrinium phaeospermum, the Causal Agent of Bamboo Blight. Forests. 2022; 13(6):850. https://0-doi-org.brum.beds.ac.uk/10.3390/f13060850
Chicago/Turabian StyleZhang, Han, Weiyi Yang, Liling Xie, Tianhui Zhu, Shuying Li, Shan Han, Tiantian Lin, and Shujiang Li. 2022. "Development and Comparison of Seminested PCR, qPCR, and LAMP for the Rapid Detection of Arthrinium phaeospermum, the Causal Agent of Bamboo Blight" Forests 13, no. 6: 850. https://0-doi-org.brum.beds.ac.uk/10.3390/f13060850