In Vitro and In Vivo Antiviral Activity of Nylidrin by Targeting the Hemagglutinin 2-Mediated Membrane Fusion of Influenza A Virus


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Compounds and Cytopathic Effect (CPE) Reduction Assay
2.3. Field-Based Pharmacophore Modeling
2.4. Immunoassays
2.5. Plaque Assay
2.6. Confocal Microscopy
2.7. Polykaryon Assay
2.8. Trypsin Protection Assay
2.9. In Vivo Study
2.10. Statistical Analysis
3. Results
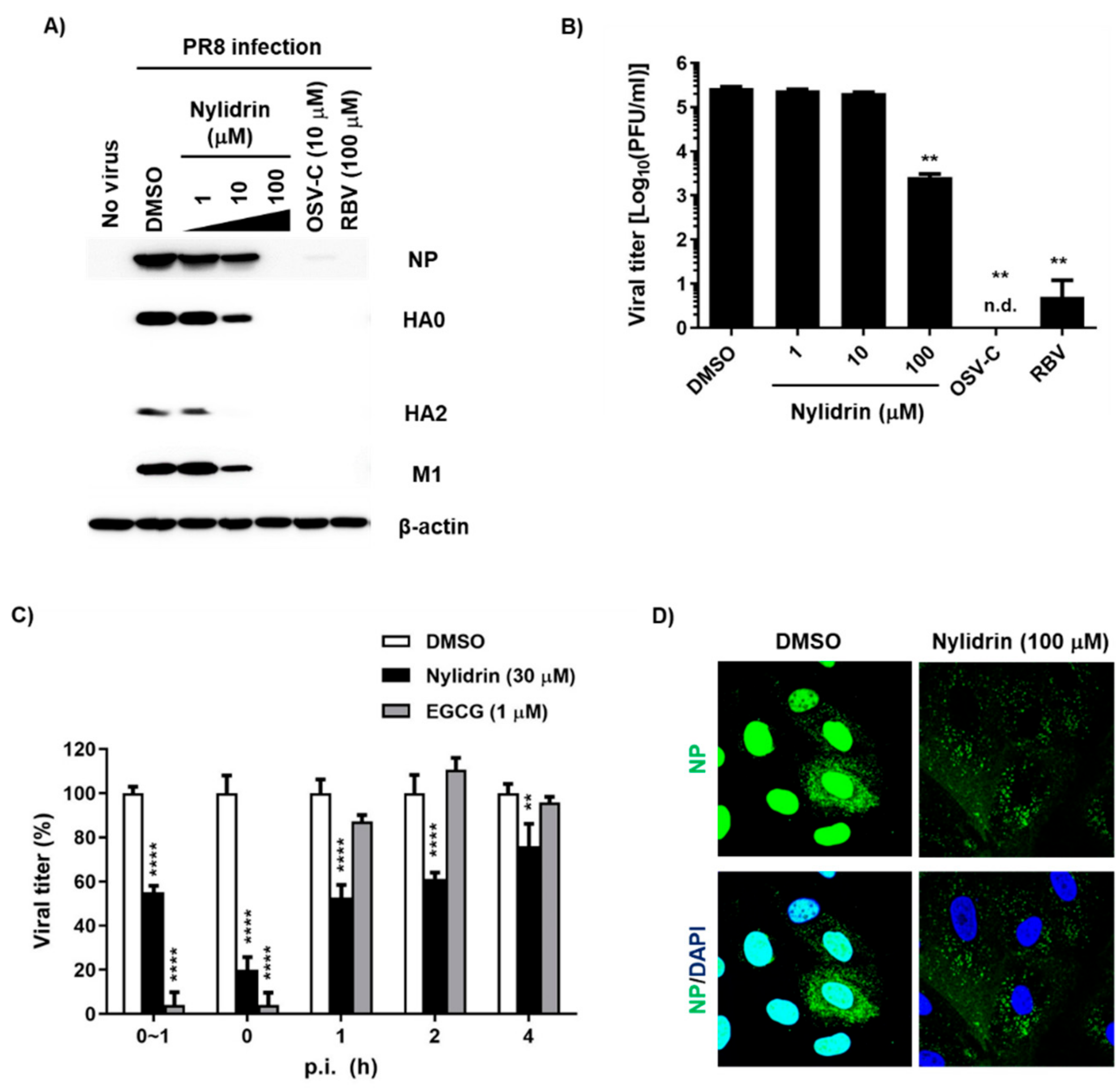
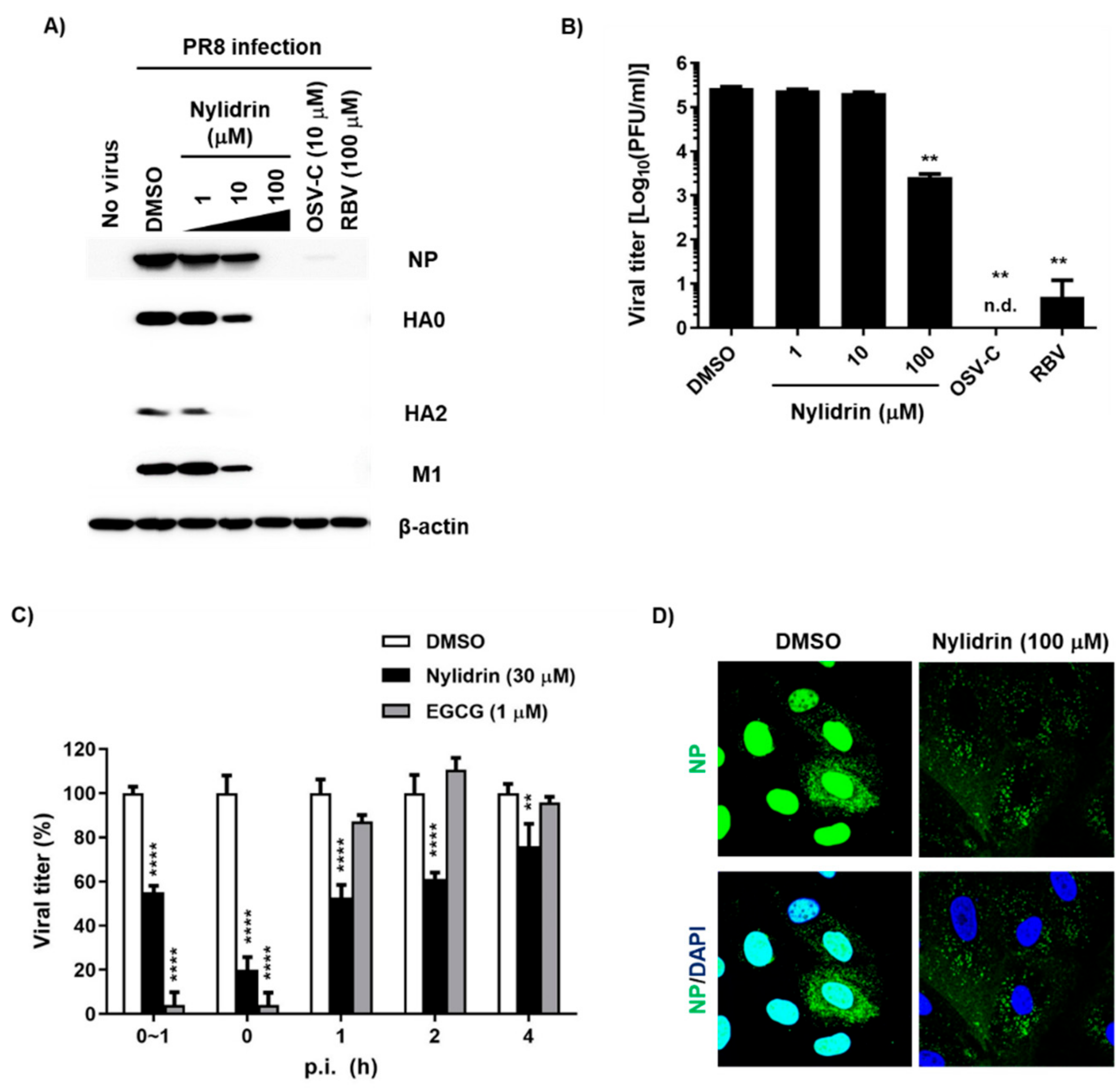
3.1. Anti-Influenza Viral Activity of Nylidrin and Its Analogues
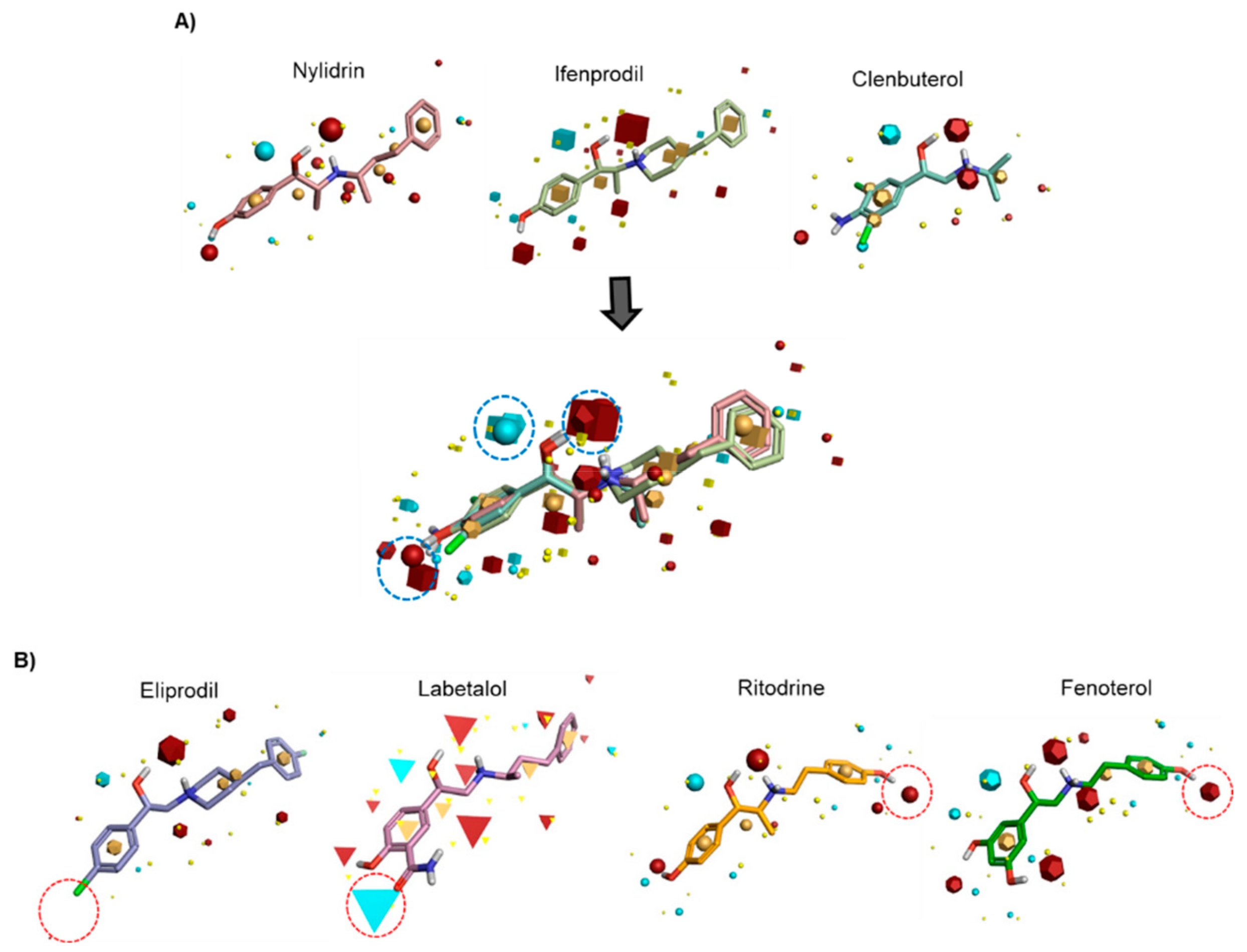
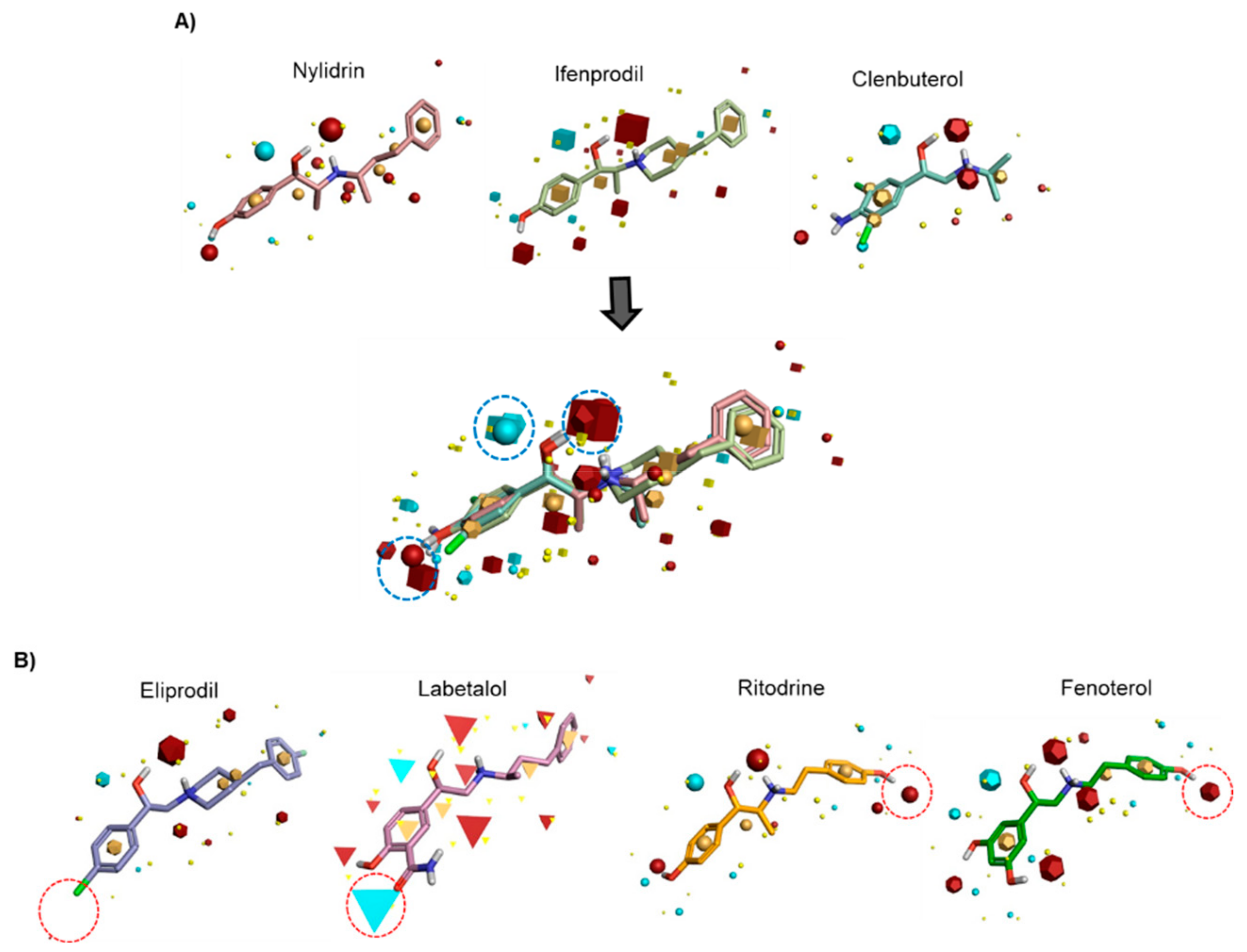
3.2. Field-Based Pharmacophore Analysis of Phenyl Aminoethanol Compounds
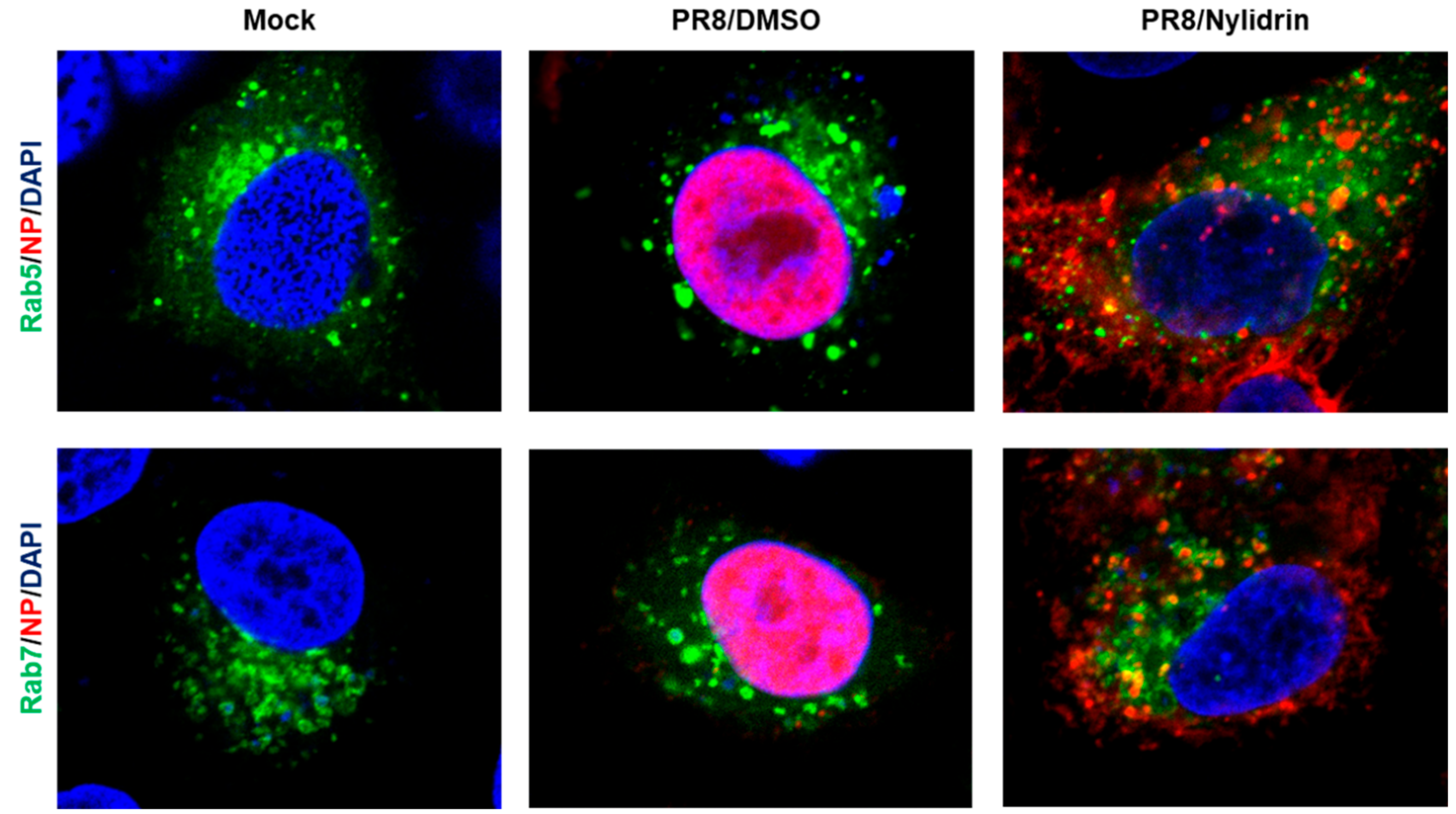
3.3. Abnormal Accumulation of Nucleoprotein (NP) in the Cytoplasm by Nylidrin
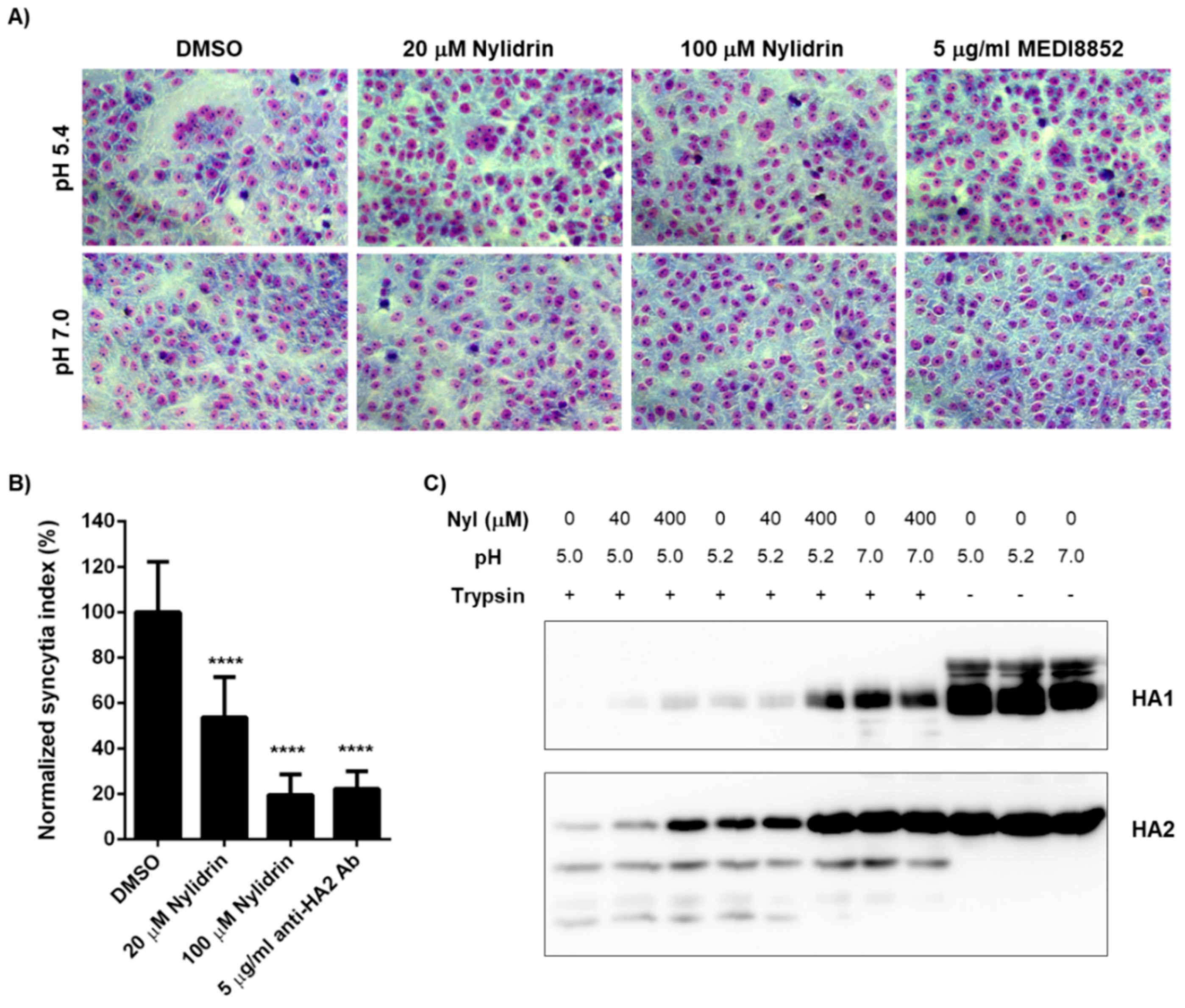
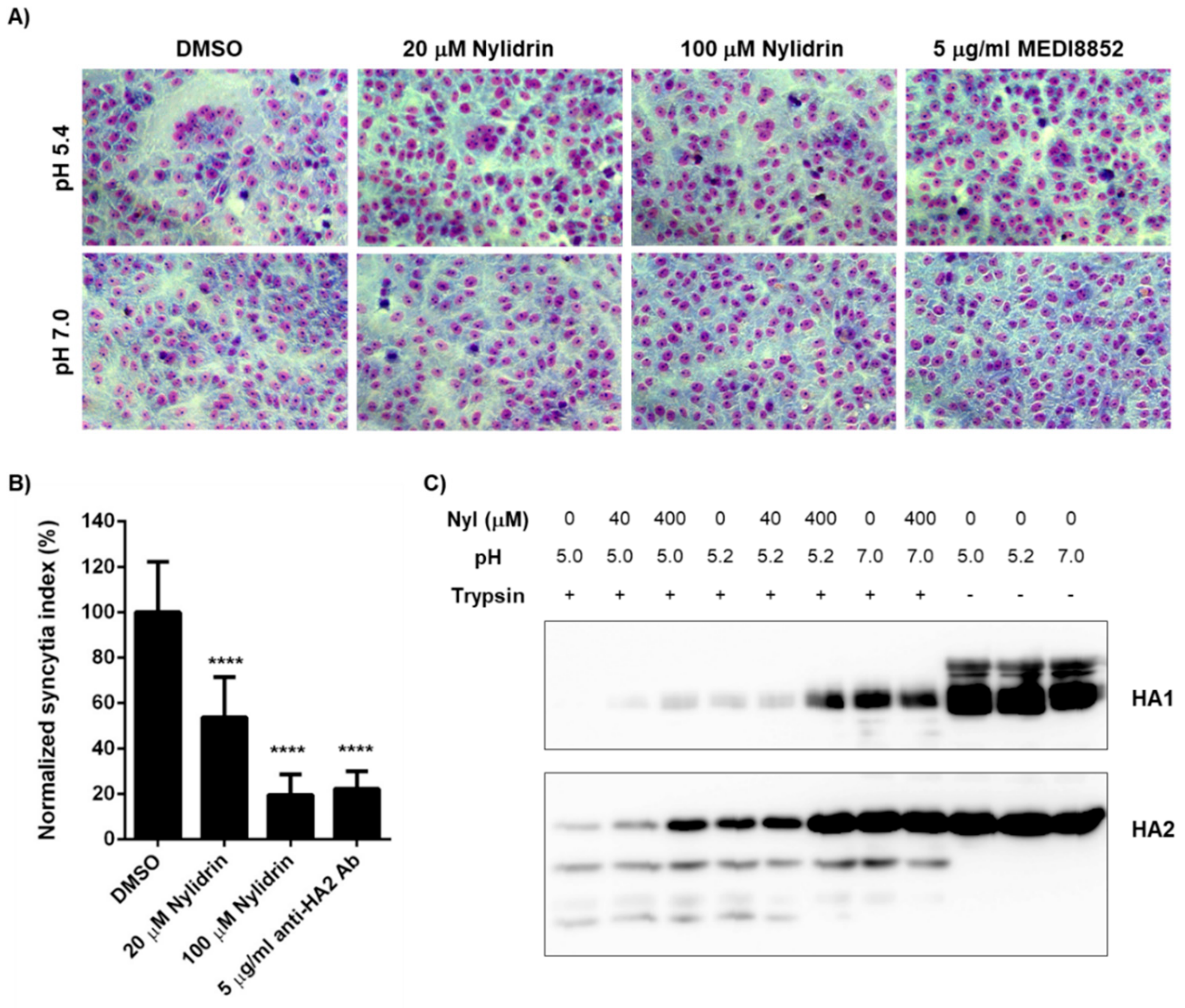
3.4. Inhibition of HA2 Fusion Activity by Nylidrin
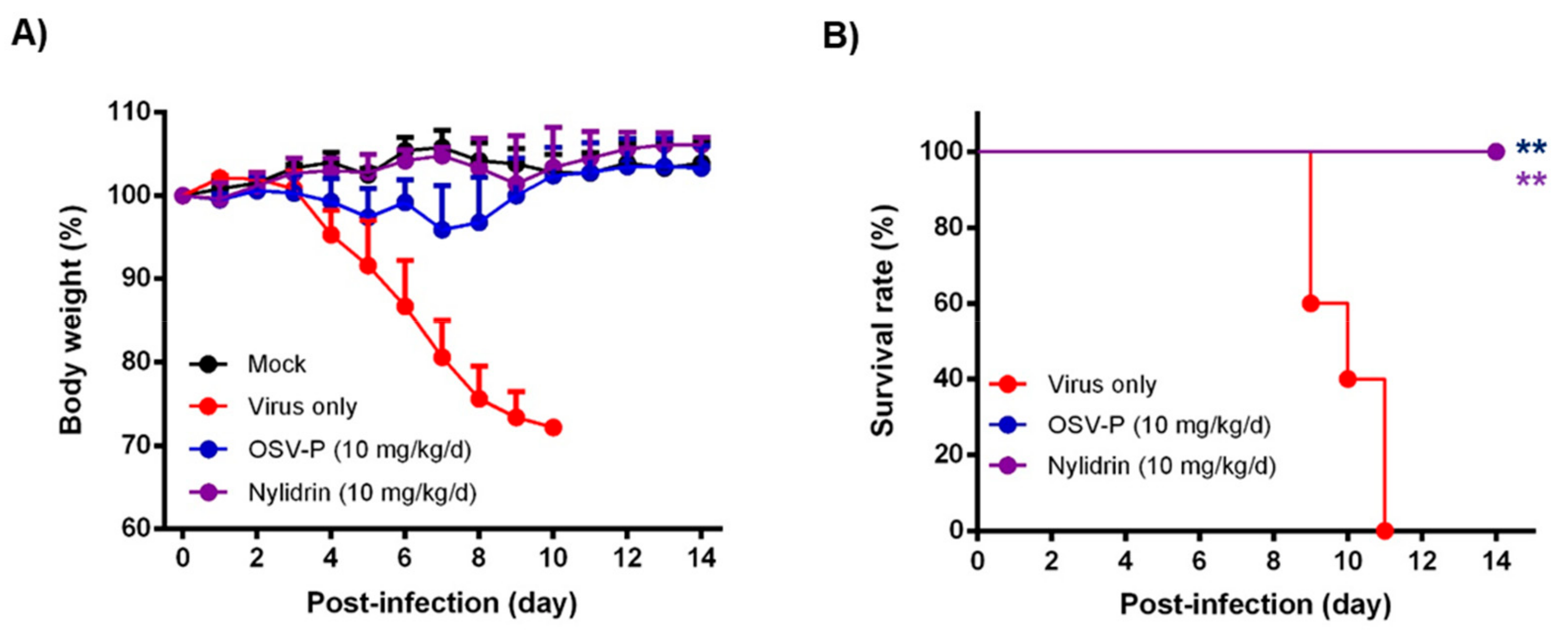
3.5. In Vivo Antiviral Efficacy of Nylidrin
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Arranz, R.; Coloma, R.; Chichon, F.J.; Conesa, J.J.; Carrascosa, J.L.; Valpuesta, J.M.; Ortin, J.; Martin-Benito, J. The structure of native influenza virion ribonucleoproteins. Science 2012, 338, 1634–1637. [Google Scholar] [CrossRef] [PubMed]
- Pflug, A.; Guilligay, D.; Reich, S.; Cusack, S. Structure of influenza A polymerase bound to the viral RNA promoter. Nature 2014, 516, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Nakatsu, S.; Sagara, H.; Sakai-Tagawa, Y.; Sugaya, N.; Noda, T.; Kawaoka, Y. Complete and Incomplete Genome Packaging of Influenza A and B Viruses. mBio 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weis, W.; Brown, J.H.; Cusack, S.; Paulson, J.C.; Skehel, J.J.; Wiley, D.C. Structure of the influenza virus haemagglutinin complexed with its receptor, sialic acid. Nature 1988, 333, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Gamblin, S.J.; Skehel, J.J. Influenza hemagglutinin and neuraminidase membrane glycoproteins. J. Biol. Chem. 2010, 285, 28403–28409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bullough, P.A.; Hughson, F.M.; Skehel, J.J.; Wiley, D.C. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature 1994, 371, 37–43. [Google Scholar] [CrossRef]
- Pinto, L.H.; Lamb, R.A. The M2 proton channels of influenza A and B viruses. J. Biol. Chem. 2006, 281, 8997–9000. [Google Scholar] [CrossRef] [Green Version]
- Neumann, G.; Castrucci, M.R.; Kawaoka, Y. Nuclear import and export of influenza virus nucleoprotein. J. Virol. 1997, 71, 9690–9700. [Google Scholar] [CrossRef] [Green Version]
- Abraham, G.M.; Morton, J.B.; Saravolatz, L.D. Baloxavir: A Novel Antiviral Agent in the Treatment of Influenza. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- De Clercq, E. Antiviral agents active against influenza A viruses. Nat. Rev. Drug Discov. 2006, 5, 1015–1025. [Google Scholar] [CrossRef]
- Bragstad, K.; Hungnes, O.; Litleskare, I.; Nyrerod, H.C.; Dorenberg, D.H.; Hauge, S.H. Community spread and late season increased incidence of oseltamivir-resistant influenza A(H1N1) viruses in Norway 2016. Influenza Other Respir. Viruses 2019, 13, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Pielak, R.M.; Schnell, J.R.; Chou, J.J. Mechanism of drug inhibition and drug resistance of influenza A M2 channel. Proc. Natl. Acad. Sci. USA 2009, 106, 7379–7384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurt, A.C.; Besselaar, T.G.; Daniels, R.S.; Ermetal, B.; Fry, A.; Gubareva, L.; Huang, W.; Lackenby, A.; Lee, R.T.; Lo, J.; et al. Global update on the susceptibility of human influenza viruses to neuraminidase inhibitors, 2014-2015. Antiviral Res. 2016, 132, 178–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurt, A.C.; Chotpitayasunondh, T.; Cox, N.J.; Daniels, R.; Fry, A.M.; Gubareva, L.V.; Hayden, F.G.; Hui, D.S.; Hungnes, O.; Lackenby, A.; et al. Antiviral resistance during the 2009 influenza A H1N1 pandemic: Public health, laboratory, and clinical perspectives. Lancet Infect. Dis. 2012, 12, 240–248. [Google Scholar] [CrossRef]
- Checkmahomed, L.; M’Hamdi, Z.; Carbonneau, J.; Venable, M.C.; Baz, M.; Abed, Y.; Boivin, G. Impact of the Baloxavir-Resistant Polymerase Acid I38T Substitution on the Fitness of Contemporary Influenza A(H1N1) pdm09 and A(H3N2) Strains. J. Infect. Dis. 2020, 221, 63–70. [Google Scholar] [CrossRef]
- Jang, Y.; Shin, J.S.; Yoon, Y.S.; Go, Y.Y.; Lee, H.W.; Kwon, O.S.; Park, S.; Park, M.S.; Kim, M. Salinomycin Inhibits Influenza Virus Infection by Disrupting Endosomal Acidification and Viral Matrix Protein 2 Function. J. Virol. 2018, 92, e01441-18. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Kim, S.Y.; Lee, H.W.; Shin, J.S.; Kim, P.; Jung, Y.S.; Jeong, H.S.; Hyun, J.K.; Lee, C.K. Inhibition of influenza virus internalization by (-)-epigallocatechin-3-gallate. Antiviral Res. 2013, 100, 460–472. [Google Scholar] [CrossRef]
- Niemeyer, G.; Cottier, D.; Resch, H. Effects of buphenine (nylidrin) on the perfused mammalian eye. Graefes Arch. Clin. Exp. Ophthalmol. 1987, 225, 33–38. [Google Scholar] [CrossRef]
- Shin, J.S.; Ku, K.B.; Jang, Y.; Yoon, Y.S.; Shin, D.; Kwon, O.S.; Go, Y.Y.; Kim, S.S.; Bae, M.A.; Kim, M. Comparison of anti-influenza virus activity and pharmacokinetics of oseltamivir free base and oseltamivir phosphate. J. Microbiol. 2017, 55, 979–983. [Google Scholar] [CrossRef]
- Jang, Y.; Lee, H.W.; Shin, J.S.; Go, Y.Y.; Kim, C.; Shin, D.; Malpani, Y.; Han, S.B.; Jung, Y.S.; Kim, M. Antiviral activity of KR-23502 targeting nuclear export of influenza B virus ribonucleoproteins. Antiviral Res. 2016, 134, 77–88. [Google Scholar] [CrossRef]
- Vinter, J.G. Extended electron distributions applied to the molecular mechanics of some intermolecular interactions. J. Comput Aided Mol Des. 1994, 8, 653–668. [Google Scholar] [CrossRef] [PubMed]
- Vanderlinden, E.; Goktas, F.; Cesur, Z.; Froeyen, M.; Reed, M.L.; Russell, C.J.; Cesur, N.; Naesens, L. Novel inhibitors of influenza virus fusion: Structure-activity relationship and interaction with the viral hemagglutinin. J. Virol. 2010, 84, 4277–4288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kallewaard, N.L.; Corti, D.; Collins, P.J.; Neu, U.; McAuliffe, J.M.; Benjamin, E.; Wachter-Rosati, L.; Palmer-Hill, F.J.; Yuan, A.Q.; Walker, P.A.; et al. Structure and Function Analysis of an Antibody Recognizing All Influenza A Subtypes. Cell 2016, 166, 596–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, H.; Song, J.M.; Ryu, C.J.; Kim, Y.G.; Lee, E.K.; Kang, S.; Kim, S.J. An efficient strategy for cell-based antibody library selection using an integrated vector system. BMC Biotechnol. 2012, 12, 62. [Google Scholar] [CrossRef] [Green Version]
- Mittag, T.W.; Tormay, A.; Messenger, M.; Podos, S.M. Ocular hypotension in the rabbit. Receptor mechanisms of pirbuterol and nylidrin. Invest. Ophthalmol. Vis. Sci. 1985, 26, 163–169. [Google Scholar]
- Liggett, S.B. Update on current concepts of the molecular basis of beta2-adrenergic receptor signaling. J. Allergy Clin. Immunol. 2002, 110, S223–S227. [Google Scholar] [CrossRef]
- Johnson, M. Molecular mechanisms of beta (2)-adrenergic receptor function, response, and regulation. J. Allergy Clin. Immunol. 2006, 117, 18–24. [Google Scholar] [CrossRef]
- Wieduwild, E.; Girard-Madoux, M.J.; Quatrini, L.; Laprie, C.; Chasson, L.; Rossignol, R.; Bernat, C.; Guia, S.; Ugolini, S. beta2-adrenergic signals downregulate the innate immune response and reduce host resistance to viral infection. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Matsui, K.; Ozawa, M.; Kiso, M.; Yamashita, M.; Maekawa, T.; Kubota, M.; Sugano, S.; Kawaoka, Y. Stimulation of alpha2-adrenergic receptors impairs influenza virus infection. Sci. Rep. 2018, 8, 4631. [Google Scholar] [CrossRef] [Green Version]
- Li, T.W.; Cheng, S.F.; Tseng, Y.T.; Yang, Y.C.; Liu, W.C.; Wang, S.C.; Chou, M.J.; Lin, Y.J.; Wang, Y.; Hsiao, P.W.; et al. Development of single-chain variable fragments (scFv) against influenza virus targeting hemagglutinin subunit 2 (HA2). Arch. Virol. 2016, 161, 19–31. [Google Scholar] [CrossRef]
- Gocnik, M.; Fislova, T.; Sladkova, T.; Mucha, V.; Kostolansky, F.; Vareckova, E. Antibodies specific to the HA2 glycopolypeptide of influenza A virus haemagglutinin with fusion-inhibition activity contribute to the protection of mice against lethal infection. J. Gen. Virol. 2007, 88, 951–955. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, N.; Prabakaran, M.; Ho, H.T.; Velumani, S.; Qiang, J.; Goutama, M.; Kwang, J. Monoclonal antibodies against the fusion peptide of hemagglutinin protect mice from lethal influenza A virus H5N1 infection. J. Virol. 2009, 83, 2553–2562. [Google Scholar] [CrossRef] [Green Version]
- Basu, A.; Antanasijevic, A.; Wang, M.; Li, B.; Mills, D.M.; Ames, J.A.; Nash, P.J.; Williams, J.D.; Peet, N.P.; Moir, D.T.; et al. New small molecule entry inhibitors targeting hemagglutinin-mediated influenza a virus fusion. J. Virol. 2014, 88, 1447–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, K.M.; De Jesus, P.; Chen, Z.; Abreu, P., Jr.; Barile, E.; Mak, P.A.; Anderson, P.; Nguyen, Q.T.; Inoue, A.; Stertz, S.; et al. A Potent Anti-influenza Compound Blocks Fusion through Stabilization of the Prefusion Conformation of the Hemagglutinin Protein. ACS Infect. Dis. 2015, 1, 98–109. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | CC50 (μM) a to MDCK Cells | EC50 (μM) b against Influenza Viruses (S.I. c) | Function | ||
|---|---|---|---|---|---|
| A/ Puerto Rico/8/1934 (H1N1) | A/Hong Kong/8/1968 (H3N2) | B/Lee/40 | |||
| Nylidrin | 549.2 ± 0.5 | 7.2 ± 0.2 | 12.1 ± 1.6 | >100.0 | β2-Adrenergic receptor agonist |
| (76.3) | (45.4) | (N.D.) | |||
| Ifenprodil | 488.2 ± 0.5 | 6.6 ± 0.4 | 16.9 ± 3.0 | >100.0 | N-methyl-D-aspartate receptor 2B antagonist |
| (74.0) | (29.0) | (N.D. d) | |||
| Labetalol | 498.1 ± 0.5 | >100.0 | 44.0 ± 9.7 | >100.0 | α1- and β1/β2-Adrenergic receptor antagonist |
| (N.D.) | (>11.3) | (N.D.) | |||
| Ritodrine | >900.0 | >100.0 | >100.0 | >100.0 | β2-Adrenergic receptor agonist |
| (N.D.) | (N.D.) | (N.D.) | |||
| Fenoterol | >900.0 | >100.0 | >100.0 | >100.0 | β2-Adrenergic receptor agonist |
| (N.D.) | (N.D.) | (N.D.) | |||
| Eliprodil | >900.0 | >100.0 | 58.1 ± 6.4 | >100.0 | N-methyl-D-aspartate antagonist |
| (N.D.) | (>15.5) | (N.D.) | |||
| Clenbuterol | >900.0 | 9.4 ± 4.6 | >100.0 | >100.0 | β2-Adrenergic receptor agonist |
| (>96.3) | (N.D.) | (N.D.) | |||
| Bambuterol | >900.0 | >100.0 | >100.0 | >100.0 | β2-Adrenergic receptor agonist |
| (N.D.) | (N.D.) | (N.D.) | |||
| AMTe | >900.0 | >100.0 | 0.8 ± 0.1 | >100.0 | - f |
| (N.D.) | (>1200) | (N.D.) | |||
| RBV g | >900.0 | 18.4 ± 3.4 | 15.7 ± 2.7 | 13.8 ± 0.2 | - |
| (>48.9) | (>57.5) | (>65.5) | |||
| OSV-C h | >900.0 | 0.04 ± 0.01 | <0.005 | 0.8 ± 0.03 | - |
| (>25,714) | (>180,000) | (>1125) | |||
| Compound | CC50 (μM) a to MDCK Cells | EC50 (μM) b against Influenza Viruses (S.I. c) | ||||||
|---|---|---|---|---|---|---|---|---|
| A/California /7/2009 (H1N1) | A/Brisbane /59/2007 (H1N1) | A/Seoul /11/1988 (H3N2) | A/Perth /16/2009 (H3N2) | A/Brisbane /10/2007 (H3N2) | A/Victoria /361/2011-like (H3N2) | B/Brisbane /60/2008 | ||
| Nylidrin | 549.2 ± 0.5 | 1.7 ± 0.5 | 3.5 ± 1.6 | 38.8 ± 19.4 | >100.0 | >100.0 | >100.0 | >100.0 |
| (332.8) | (156.9) | (14.3) | (N.D. d) | (N.D.) | (N.D.) | (N.D.) | ||
| Ifenprodil | 488.2 ± 0.5 | 1.1 ± 0.1 | 5.1 ± 0.3 | 37.9 ± 7.7 | >100.0 | >100.0 | >100.0 | >100.0 |
| (465.0) | (96.7) | (14.5) | (N.D.) | (N.D.) | (N.D.) | (N.D.) | ||
| Labetalol | 498.1 ± 0.5 | >100.0 | >100.0 | >100.0 | >100.0 | >100.0 | >100.0 | >100.0 |
| (N.D.) | (N.D.) | (N.D.) | (N.D.) | (N.D.) | (N.D.) | (N.D.) | ||
| Eliprodil | >900.0 | >100.0 | 53.1 ± 0.3 | >100.0 | >100.0 | >100.0 | >100.0 | >100.0 |
| (N.D.) | (>16.9) | (N.D.) | (N.D.) | (N.D.) | (N.D.) | (N.D.) | ||
| Clenbuterol | >900.0 | 13.7 ± 3.6 | 12.9 ± 0.3 | >100.0 | >100.0 | >100.0 | >100.0 | >100.0 |
| (>65.9) | (>69.8) | (N.D.) | (N.D.) | (N.D.) | (N.D.) | (N.D.) | ||
| AMT e | >900.0 | >100.0 | 1.2 ± 0.4 | 0.3 ± 0.1 | >100.0 | >100.0 | >100.0 | >100.0 |
| (N.D.) | (>750) | (>1831) | (N.D.) | (N.D.) | (N.D.) | (N.D.) | ||
| RBV f | >900.0 | 52.5 ± 1.5 | 24.1 ± 0.5 | 10.5 ± 4.2 | 33.8 ± 11.6 | 12.6 ± 6.4 | 33.4 ± 8.8 | 25.6 ± 0.3 |
| (>17.2) | (>37.3) | (>52.3) | (>26.6) | (>71.4) | (>16.4) | (>35.2) | ||
| OSV-C g | >900.0 | 0.02 ± 0.0 | 0.02 ± 0.0 | <0.005 | 0.03 ± 0.02 | 0.53 ± 0.16 | <0.005 | 53.1 ± 25.25 |
| (>58,065) | (>58,065) | (>180,000) | (>30,000) | (>1714) | (>180,000) | (>17.0) | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jang, Y.; Shin, J.S.; Lee, J.-Y.; Shin, H.; Kim, S.J.; Kim, M. In Vitro and In Vivo Antiviral Activity of Nylidrin by Targeting the Hemagglutinin 2-Mediated Membrane Fusion of Influenza A Virus. Viruses 2020, 12, 581. https://0-doi-org.brum.beds.ac.uk/10.3390/v12050581
Jang Y, Shin JS, Lee J-Y, Shin H, Kim SJ, Kim M. In Vitro and In Vivo Antiviral Activity of Nylidrin by Targeting the Hemagglutinin 2-Mediated Membrane Fusion of Influenza A Virus. Viruses. 2020; 12(5):581. https://0-doi-org.brum.beds.ac.uk/10.3390/v12050581
Chicago/Turabian StyleJang, Yejin, Jin Soo Shin, Joo-Youn Lee, Heegwon Shin, Sang Jick Kim, and Meehyein Kim. 2020. "In Vitro and In Vivo Antiviral Activity of Nylidrin by Targeting the Hemagglutinin 2-Mediated Membrane Fusion of Influenza A Virus" Viruses 12, no. 5: 581. https://0-doi-org.brum.beds.ac.uk/10.3390/v12050581