RNAseq Analysis of Rhizomania-Infected Sugar Beet Provides the First Genome Sequence of Beet Necrotic Yellow Vein Virus from the USA and Identifies a Novel Alphanecrovirus and Putative Satellite Viruses

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Plant Growth and Virus Recovery
2.3. Virus Enrichment
2.4. RNAseq Analyses
2.5. Sequence Analysis of BNYVV Strains
2.6. Construction and Inoculation of BNYVV RNA1 and 2 Infectious Clones
2.7. Putative Alphanecrovirus and Satellite Virus Sequence Validation and Characterization
2.8. Construction and Inoculation of Novel Alphanecrovirus Infectious Clones
3. Results
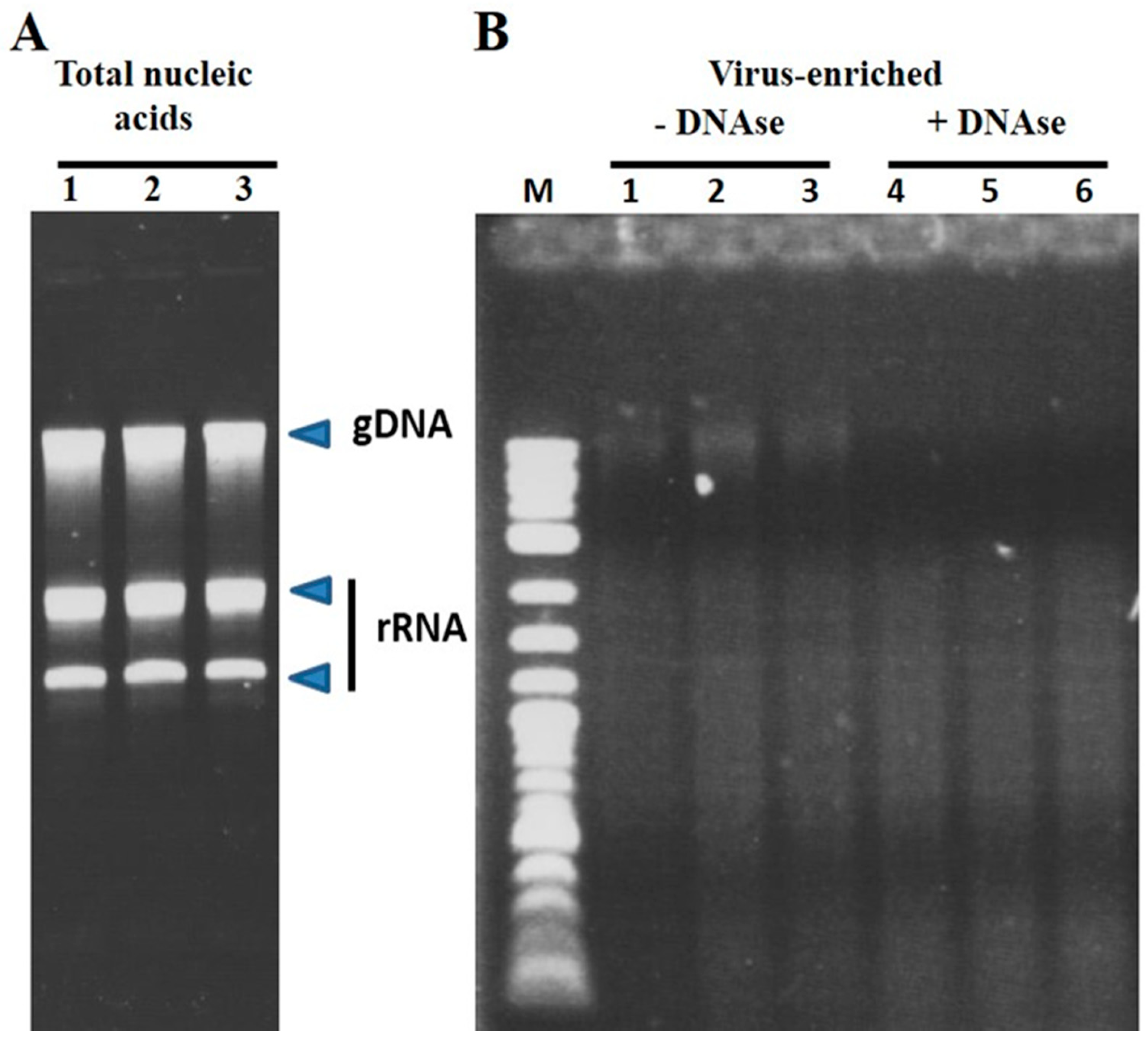
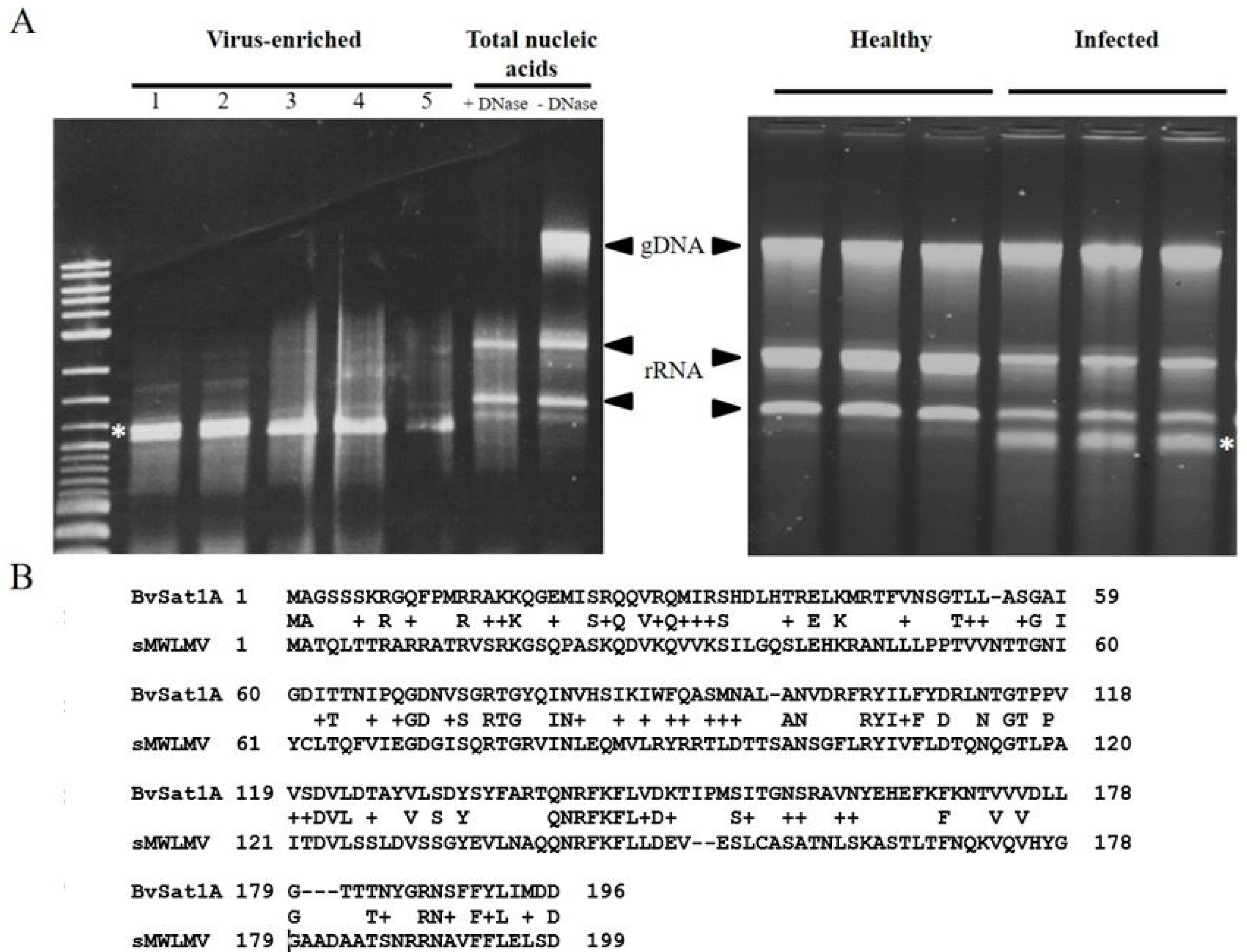
3.1. Virus Enrichment and RNAseq Analysis
3.2. Taxonomic Grouping of US BNYVV Isolates and Biological Validation of Sequences
3.3. Development of Infectious Clones of BNYVV RNAs 1 and 2
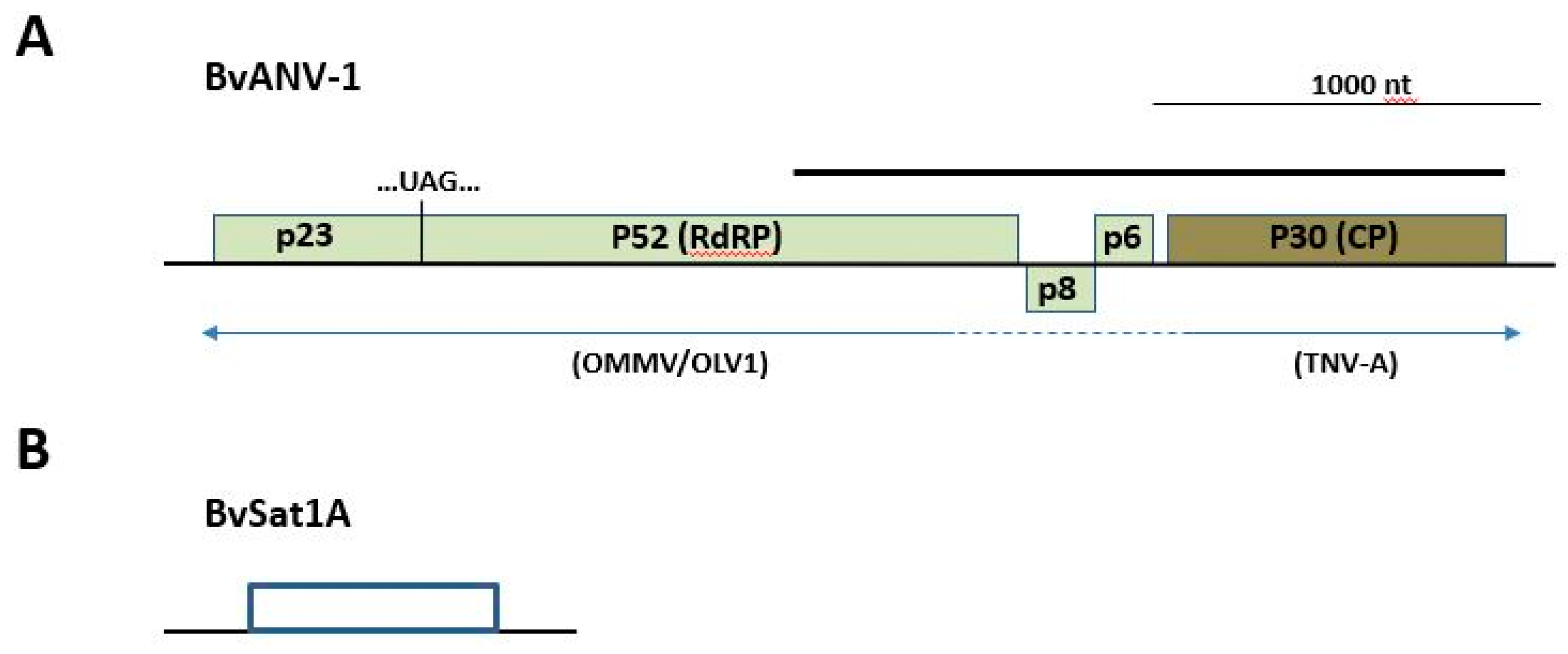
3.4. Novel Virus Discovery in Sugar Beet through RNAseq
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gilardi, G.; Garibaldi, A.; Gullino, M.L. Emerging pathogens as a consequence of globalization and climate change: Leafy vegetables as a case study. Phytopathol. Mediterr. 2018, 57, 146–152. [Google Scholar]
- Jones, R.A.C.; Naidu, R.A. Global dimensions of plant virus diseases: Current status and future perspectives. Annu. Rev. Virol. 2019, 6, 387–409. [Google Scholar] [CrossRef] [PubMed]
- Friesen, T.L.; Stukenbrock, E.H.; Liu, Z.; Meinhardt, S.; Ling, H.; Faris, J.D.; Rasmussen, J.B.; Solomon, P.S.; McDonald, B.A.; Oliver, R.P. Emergence of a new disease as a result of interspecific virulence gene transfer. Nat. Genet. 2006, 38, 953–956. [Google Scholar] [CrossRef] [PubMed]
- Kado, C.I. Horizontal gene transfer: Sustaining pathogenicity and optimizing host–pathogen interactions. Mol. Plant Pathol. 2009, 10, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Koenig, R.; Loss, S.; Specht, J.; Varrelmann, M.; Lüddecke, P.; Deml, G. A single U/C nucleotide substitution changing alanine to valine in the Beet necrotic yellow vein virus P25 protein promotes increased virus accumulation in roots of mechanically inoculated, partially resistant sugar beet seedlings. J. Gen. Virol. 2009, 90, 759–763. [Google Scholar] [CrossRef] [PubMed]
- Rush, C.M.; Liu, H.Y.; Lewellen, R.T.; Acosta-Leal, R. The continuing saga of rhizomania of sugar beets in the United States. Plant Dis. 2006, 90, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Biancardi, E.; Lewellen, R.T.; De Biaggi, M.; Erichsen, A.W.; Stevanato, P. The origin of rhizomania resistance in sugar beet. Euphytica 2002, 127, 383–397. [Google Scholar] [CrossRef]
- Canova, A.; Giunchedi, L.; Biancardi, E. History and current status. In Rhizomania; Biancardi, E., Tamada, T., Eds.; Springer International Publishing: Basel, Switzerland, 2016; pp. 29–51. [Google Scholar]
- Richards, K.; Tamada, T. Mapping functions on the multipartite genome of Beet necrotic yellow vein virus. Annu. Rev. Phytopathol. 1992, 30, 291–313. [Google Scholar] [CrossRef]
- Tamada, T. General features of Beet necrotic yellow vein virus. In Rhizomania; Biancardi, E., Tamada, T., Eds.; Springer International Publishing: Basel, Switzerland, 2016; pp. 55–83. [Google Scholar]
- Peltier, C.; Hleibieh, K.; Thiel, H.; Klein, E.; Bragard, C.; Gilmer, D. oMlecular biology of the Beet necrotic yellow vein virus. Plant Viruses 2008, 2, 14–24. [Google Scholar]
- Laufer, M.; Mohammad, H.; Maiss, E.; Richert-Pöggeler, K.; Dall’Ara, M.; Ratti, C.; Gilmer, D.; Liebe, S.; Varrelmann, M. Biological properties of Beet soil-borne mosaic virus and Beet necrotic yellow vein virus cDNA clones produced by isothermal in vitro recombination: Insights for reassortant appearance. Virology 2018, 518, 25–33. [Google Scholar] [CrossRef]
- Gilmer, D. Molecular biology and replication of Beet Necrotic Yellow Vein Virus. In Rhizomania; Biancardi, E., Tamada, T., Eds.; Springer International Publishing: Basel, Switzerland, 2016; pp. 85–107. [Google Scholar]
- Acosta-Leal, R.; Bryan, B.K.; Smith, J.T.; Rush, C.M. Breakdown of host resistance by independent evolutionary lineages of Beet necrotic yellow vein virus involves a parallel C/U mutation in its p25 gene. Phytopathology 2010, 100, 127–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bornemann, K.; Hanse, B.; Varrelmann, M.; Stevens, M. Occurrence of resistance-breaking strains of Beet necrotic yellow vein virus in sugar beet in northwestern Europe and identification of a new variant of the viral pathogenicity factor P25. Plant Pathol. 2015, 64, 25–34. [Google Scholar] [CrossRef]
- Liu, H.Y.; Lewellen, R.T. Distribution and molecular characterization of resistance-breaking isolates of Beet necrotic yellow vein virus in the United States. Plant Dis. 2007, 91, 847–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiland, J.J.; Bornemann, K.; Neubauer, J.D.; Khan, M.F.R.; Bolton, M.D. Prevalence and Distribution of Beet Necrotic Yellow Vein Virus Strains in North Dakota and Minnesota. Plant. Dis. 2019, 103, 2083–2089. [Google Scholar] [CrossRef] [PubMed]
- Lecuit, M.; Eloit, M. The potential of whole genome NGS for infectious disease diagnosis. Expert Rev. Mol. Diagn. 2015, 15, 1517–1519. [Google Scholar] [CrossRef] [PubMed]
- Villamor, D.E.V.; Ho, T.; Al Rwahnih, M.; Martin, R.R.; Tzanetakis, I.E. High Throughput Sequencing For Plant Virus Detection and Discovery. Phytopathology 2019, 109, 716–725. [Google Scholar] [CrossRef] [PubMed]
- Filloux, D.; Dallot, S.; Delaunay, A.; Galzi, S.; Jacquot, E.; Roumagnac, P. Metagenomics Approaches Based on Virion-Associated Nucleic Acids (VANA): An Innovative Tool for Assessing Without A Priori Viral Diversity of Plants. In Plant Pathology: Techniques and Protocols; Lacomme, C., Ed.; Springer: New York, NY, USA, 2015; pp. 249–257. [Google Scholar]
- Ma, Y.; Marais, A.; Lefebvre, M.; Theil, S.; Svanella-Dumas, L.; Faure, C.; Candresse, T. Phytovirome Analysis of Wild Plant Populations: Comparison of Double-Stranded RNA and Virion-Associated Nucleic Acid Metagenomic Approaches. J. Virol. 2019, 94. [Google Scholar] [CrossRef] [PubMed]
- Greninger, A.L. A decade of RNA virus metagenomics is (not) enough. Virus Res. 2018, 244, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Lane, L.C. Propagation and purification of RNA plant viruses. In Methods in Enzymology; Weissbach, A., Weissbach, H., Eds.; Elsevier: Amsterdam, The Netherlands, 2004; Volume 118, pp. 687–696. [Google Scholar]
- Weiland, J.J.; Edwards, M.C. Evidence That the αa Gene of Barley Stripe Mosaic Virus Encodes Determinants of Pathogenicity to Oat (Avena sativa). Virology 1994, 201, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Chiba, S.; Kondo, H.; Miyanishi, M.; Andika, I.B.; Han, C.; Tamada, T. The evolutionary history of Beet necrotic yellow vein virus deduced from genetic variation, geographical origin and spread, and the breaking of host resistance. Mol. Plant Microbe Interact. 2011, 24, 207–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petty, I.T.D.; Donald, R.G.K.; Jackson, A.O. Multiple Genetic Determinants of Barley Stripe Mosaic Virus Influence Lesion Phenotype on Chenopodium amaranticolor. Virology 1994, 198, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Edwards, M.C.; Weiland, J.J.; Todd, J.; Stewart, L.R. Infectious Maize rayado fino virus from Cloned cDNA. Phytopathology 2015, 105, 833–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiland, J.J.; Van Winkle, D.; Edwards, M.C.; Larson, R.L.; Shelver, W.L.; Freeman, T.P.; Liu, H.Y. Characterization of a U.S. isolate of Beet black scorch virus. Phytopathology 2007, 97, 1245–1254. [Google Scholar] [CrossRef] [PubMed]
- Heidel, G.B.; Rush, C.M.; Kendall, T.L.; Lommel, S.A.; French, R.C. Characteristics of Beet Soilborne Mosaic Virus, a Furo-like Virus Infecting Sugar Beet. Plant Dis. 1997, 81, 1070–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, C.M.; Jones, R.A.C.; Coutts, R.H.A. Occurrence of a soil-borne virus of sugar beet in England. Plant Pathol. 1986, 35, 585–591. [Google Scholar] [CrossRef]
- Koenig, R.; Pleij, C.W.; Beier, C.; Commandeur, U. Genome properties of beet virus Q, a new furo-like virus from sugarbeet, determined from unpurified virus. J. Gen. Virol. 1998, 79, 2027–2036. [Google Scholar] [CrossRef] [Green Version]
- Weiland, J.J.; Larson, R.L.; Freeman, T.P.; Edwards, M.C. First Report of Beet black scorch virus in the United States. Plant Dis. 2006, 90, 828. [Google Scholar] [CrossRef]
- Lee, L.; Telford, E.B.; Batten, J.S.; Scholthof, K.B.G.; Rush, C.M. Complete nucleotide sequence and genome organization of Beet soilborne mosaic virus, a proposed member of the genus Benyvirus. Arch. Virol. 2001, 146, 2443–2453. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, A.; Link, D.; Cognat, V.; Moury, B.; Beuve, M.; Meunier, A.; Bragard, C.; Gilmer, D.; Lemaire, O. Phylogenetic analysis of isolates of Beet necrotic yellow vein virus collected worldwide. J. Gen. Virol 2005, 86 Pt 10, 2897–2911. [Google Scholar] [CrossRef]
- Zhuo, N.; Jiang, N.; Zhang, C.; Zhang, Z.Y.; Zhang, G.Z.; Han, C.G.; Wang, Y. Genetic diversity and population structure of Beet necrotic yellow vein virus in China. Virus Res. 2015, 205, 54–62. [Google Scholar] [CrossRef]
- Zhang, L.; Zitter, T.A.; Palukaitis, P. Helper virus-dependent replication, nucleotide sequence and genome organization of the satellite virus of maize white line mosaic virus. Virology 1991, 180, 467–473. [Google Scholar] [CrossRef]
- Meulewaeter, F.; Seurinck, J.; Emmelo, J.V. Genome structure of tobacco necrosis virus strain A. Virology 1990, 177, 699–709. [Google Scholar] [CrossRef]
- Cardoso, J.M.S.; Félix, M.R.; Clara, M.I.E.; Oliveira, S.J. The complete genome sequence of a new necrovirus isolated from Olea europaea L. Arch. Virol. 2005, 150, 815–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Félix, M.R.; Cardoso, J.; Varanda, C.M.; Oliveira, S.; Clara, M.I.E. Complete nucleotide sequence of an Olive latent virus 1 isolate from olive trees. Arch. Virol. 2005, 150, 2403–2406. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C. The virome hunters. Nature 2018, 36, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Corney, D.C.; Basturea, G.N. RNA-Seq using next generation sequencing. Mater. Methods 2013, 3. [Google Scholar] [CrossRef]
- Acosta-Leal, R.; Fawley, M.W.; Rush, C.M. Changes in the intraisolate genetic structure of Beet necrotic yellow vein virus populations associated with plant resistance breakdown. Virology 2008, 376, 60–68. [Google Scholar] [CrossRef] [Green Version]
- Liebe, S.; Wibberg, D.; Maiss, E.; Varrelmann, M. Application of a Reverse Genetic System for Beet Necrotic Yellow Vein Virus to Study Rz1 Resistance Response in Sugar Beet. Front. Plant Sci. 2020, 10, 1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delbianco, A.; Lanzoni, C.; Klein, E.; Rubies Autonell, C.; Gilmer, D.; Ratti, C. Agroinoculation of Beet necrotic yellow vein virus cDNA clones results in plant systemic infection and efficient Polymyxa betae transmission. Mol. Plant Pathol. 2013, 14, 422–428. [Google Scholar] [CrossRef]
- Jiang, N.; Zhang, C.; Liu, J.Y.; Guo, Z.H.; Zhang, Z.Y.; Han, C.G.; Wang, Y. Development of Beet necrotic yellow vein virus-based vectors for multiple-gene expression and guide RNA delivery in plant genome editing. Plant Biotechnol. J. 2019, 17, 1302–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasin, F.; Menzel, W.; Daròs, J.A. Harnessed viruses in the age of metagenomics and synthetic biology: An update on infectious clone assembly and biotechnologies of plant viruses. Plant Biotechnol. J. 2019, 17, 1010–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Smith, S.A. Optimizing de novo assembly of short-read RNA-seq data for phylogenomics. BMC Genom. 2013, 14, 328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.Y.; Duffus, J.E.; Wisler, G.C. Etiology of Vascular Necrosis Syndrome of Sugarbeet; Rothamsted Research: Harpenden, UK, 1996; pp. 161–164. [Google Scholar]
- Guo, L.H.; Cao, Y.-H.; Li, D.W.; Niu, S.N.; Cai, Z.N.; Han, C.G.; Zhai, Y.F.; Yu, J.-L. Analysis of Nucleotide Sequences and Multimeric Forms of a Novel Satellite RNA Associated with Beet Black Scorch Virus. J. Virol. 2005, 79, 3664–3674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, T.; Tzanetakis, I.E. Development of a virus detection and discovery pipeline using next generation sequencing. Virology 2014, 471–473, 54–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weirather, J.L.; De Cesare, M.; Wang, Y.; Piazza, P.; Sebastiano, V.; Wang, X.J.; Buck, D.; Au, K.F. Comprehensive comparison of Pacific Biosciences and Oxford Nanopore Technologies and their applications to transcriptome analysis. F1000Res 2017, 6, 100. [Google Scholar] [CrossRef] [PubMed]
- Matranga, C.B.; Andersen, K.G.; Winnicki, S.; Busby, M.; Gladden, A.D.; Tewhey, R.; Stremlau, M.; Berlin, A.; Gire, S.K.; England, E.; et al. Enhanced methods for unbiased deep sequencing of Lassa and Ebola RNA viruses from clinical and biological samples. Genome Biol. 2014, 15, 519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metsky, H.C.; Matranga, C.B.; Wohl, S.; Schaffner, S.F.; Freije, C.A.; Winnicki, S.M.; West, K.; Qu, J.; Baniecki, M.L.; Gladden-Young, A.; et al. Zika virus evolution and spread in the Americas. Nature 2017, 546, 411–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Beet Necrotic Yellow Vein Virus (GCF_000854885.1; 15,914 nt) a | ||||||||||||||
| RNA 1 (NC_003514.1; 6746 nt) b | RNA 2 (NC_003515.1; 4609 nt) | RNA 3 (NC_003516.1; 1774 nt) | RNA 4 (NC_003517.1; 1465 nt) | |||||||||||
| # | Start | End | Reads Mapped | Start | End | Reads Mapped | Start | End | Reads Mapped | Start | End | Reads Mapped | Total Reads Mapped | Total Reads |
| S1 | 12 | 6735 | 46,701 | 14 | 4594 | 82,921 | 17 | 1753 | 36,298 | 12 | 1465 | 37,957 | 203,877 | 66,078,080 |
| S2 | 12 | 6734 | 121,686 | 16 | 4595 | 240,749 | 14 | 1760 | 71,911 | 14 | 1451 | 123,978 | 558,325 | 69,414,580 |
| S3 | 10 | 6734 | 97,617 | 11 | 4595 | 235,333 | 17 | 1758 | 52,887 | 14 | 1448 | 93,530 | 479,367 | 71,362,360 |
| S4 | 2 | 6724 | 15,646 | 18 | 4609 | 21,455 | 12 | 1766 | 12,608 | 14 | 1465 | 5373 | 55,082 | 62,246,430 |
| S5 | 6 | 6727 | 8369 | 19 | 4596 | 15,041 | 22 | 1761 | 6411 | 14 | 1465 | 2840 | 32,661 | 59,595,934 |
| S6 | 5 | 6744 | 26,060 | 16 | 4609 | 40,054 | 1 | 1744 | 25,411 | 14 | 1465 | 19,449 | 110,974 | 73,945,544 |
| Beet Soilborne Mosaic Virus (GCA_002867265.1; 14,744 nt) | ||||||||||||||
| RNA 1 (JF513082.1; 6679 nt) | RNA 2 (JF513083.1; 4615 nt) | RNA 3 (EU410955.1; 1720 nt) | RNA 4 (FJ424610.2; 1730 nt) | |||||||||||
| # | Start | End | Reads Mapped | Start | End | Reads Mapped | Start | End | Reads Mapped | Start | End | Reads Mapped | Total Reads Mapped | Total Reads |
| S1 | 12 | 6671 | 140,396 | 16 | 4597 | 424,451 | 1 | 1720 | 245,901 | 1 | 1714 | 67,780 | 878,528 | 66,078,080 |
| S2 | 12 | 6671 | 519,420 | 16 | 4595 | 1,428,890 | 15 | 1720 | 827,273 | 13 | 1721 | 131,926 | 2,902,509 | 69,414,580 |
| S3 | 12 | 6668 | 153,627 | 16 | 4599 | 677,253 | 13 | 1720 | 217,594 | 13 | 1717 | 162,226 | 1,210,700 | 71,362,360 |
| S4 | 7 | 6676 | 8696 | 16 | 4615 | 16,635 | 1 | 1720 | 16,177 | 13 | 1730 | 2210 | 43,718 | 62,246,430 |
| S5 | 6 | 6679 | 83,687 | 17 | 4615 | 157,895 | 13 | 1720 | 133,978 | 1 | 1730 | 56,128 | 431,688 | 59,595,934 |
| S6 | 6 | 6646 | 738 | 33 | 4598 | 853 | 16 | 1702 | 136 | 24 | 1714 | 24 | 1751 | 73,945,544 |
| Country of Origin | BNYVV RNA 1 | BNYVV RNA 2 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Accession # a | Identity (%) | Aligned | Mismatch | Gaps | Accession # | Identity (%) | Aligned | Mismatch | Gaps | Type b | |
| Brazil | MH106726.1 | 99.85 | 6616 | 10 | 0 | MH106727.1 | 99.43 | 4560 | 26 | 0 | A |
| Spain | EU330453.1 | 99.83 | 6616 | 11 | 0 | EU330452.1 | 99.52 | 4543 | 22 | 0 | A |
| Sweden | EU330450.1 | 99.80 | 6616 | 13 | 0 | EU330451.1 | 99.44 | 4533 | 25 | 0 | A |
| Yugoslavia | KX665536.1 | 99.82 | 6616 | 12 | 0 | KX665537.1 | 98.84 | 4560 | 32 | 1 | A |
| Japan | D84410.1 | 99.37 | 6616 | 42 | 0 | D84411.1 | 98.47 | 4560 | 70 | 0 | A |
| France (Pithiviers) | HM126464.1 | 99.41 | 6616 | 39 | 0 | HM117903.1 | 98.42 | 4560 | 72 | 0 | A(P) |
| China | KM434313.1 | 99.43 | 6616 | 38 | 0 | KM434314.1 | 95.66 | 4563 | 195 | 1 | B |
| France | X05147.1 | 98.46 | 6616 | 102 | 0 | X04197.1 | 95.35 | 4563 | 209 | 1 | B |
| Virus a | Accession # b | 5′ UTR c | P23 | P52 | P8 | P6 | CP (P30) | 3′ UTR |
|---|---|---|---|---|---|---|---|---|
| TNV-A | GCA_000857065.1 | 93.22 | 87.13 | 94.24 | 89.04 | 98.21 | 93.04 | 78.04% |
| OMMV | GCF_000858865.1 | 86.44 | 96.04 | 95.01 | 93.15 | 98.21 | 54.13 | 76.74% |
| OLV-1 | GCF_000855965.1 | 83.05 | 90.10 | 95.59 | 95.89 | 100.00 | 49.07 | 75.80% |
| CSNV | MF125267.1 | 89.83 | 86.63 | 93.67 | 86.30 | 98.21 | 51.65 | 75.46% |
| PoNV | NC_029900.1 | 70.00 | 86.63 | 89.83 | 73.97 | 96.43 | 50.84 | 76.05% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weiland, J.J.; Sharma Poudel, R.; Flobinus, A.; Cook, D.E.; Secor, G.A.; Bolton, M.D. RNAseq Analysis of Rhizomania-Infected Sugar Beet Provides the First Genome Sequence of Beet Necrotic Yellow Vein Virus from the USA and Identifies a Novel Alphanecrovirus and Putative Satellite Viruses. Viruses 2020, 12, 626. https://0-doi-org.brum.beds.ac.uk/10.3390/v12060626
Weiland JJ, Sharma Poudel R, Flobinus A, Cook DE, Secor GA, Bolton MD. RNAseq Analysis of Rhizomania-Infected Sugar Beet Provides the First Genome Sequence of Beet Necrotic Yellow Vein Virus from the USA and Identifies a Novel Alphanecrovirus and Putative Satellite Viruses. Viruses. 2020; 12(6):626. https://0-doi-org.brum.beds.ac.uk/10.3390/v12060626
Chicago/Turabian StyleWeiland, John J., Roshan Sharma Poudel, Alyssa Flobinus, David E. Cook, Gary A. Secor, and Melvin D. Bolton. 2020. "RNAseq Analysis of Rhizomania-Infected Sugar Beet Provides the First Genome Sequence of Beet Necrotic Yellow Vein Virus from the USA and Identifies a Novel Alphanecrovirus and Putative Satellite Viruses" Viruses 12, no. 6: 626. https://0-doi-org.brum.beds.ac.uk/10.3390/v12060626