
Fetomaternal Expression of Glucose Transporters (GLUTs)—Biochemical, Cellular and Clinical Aspects

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
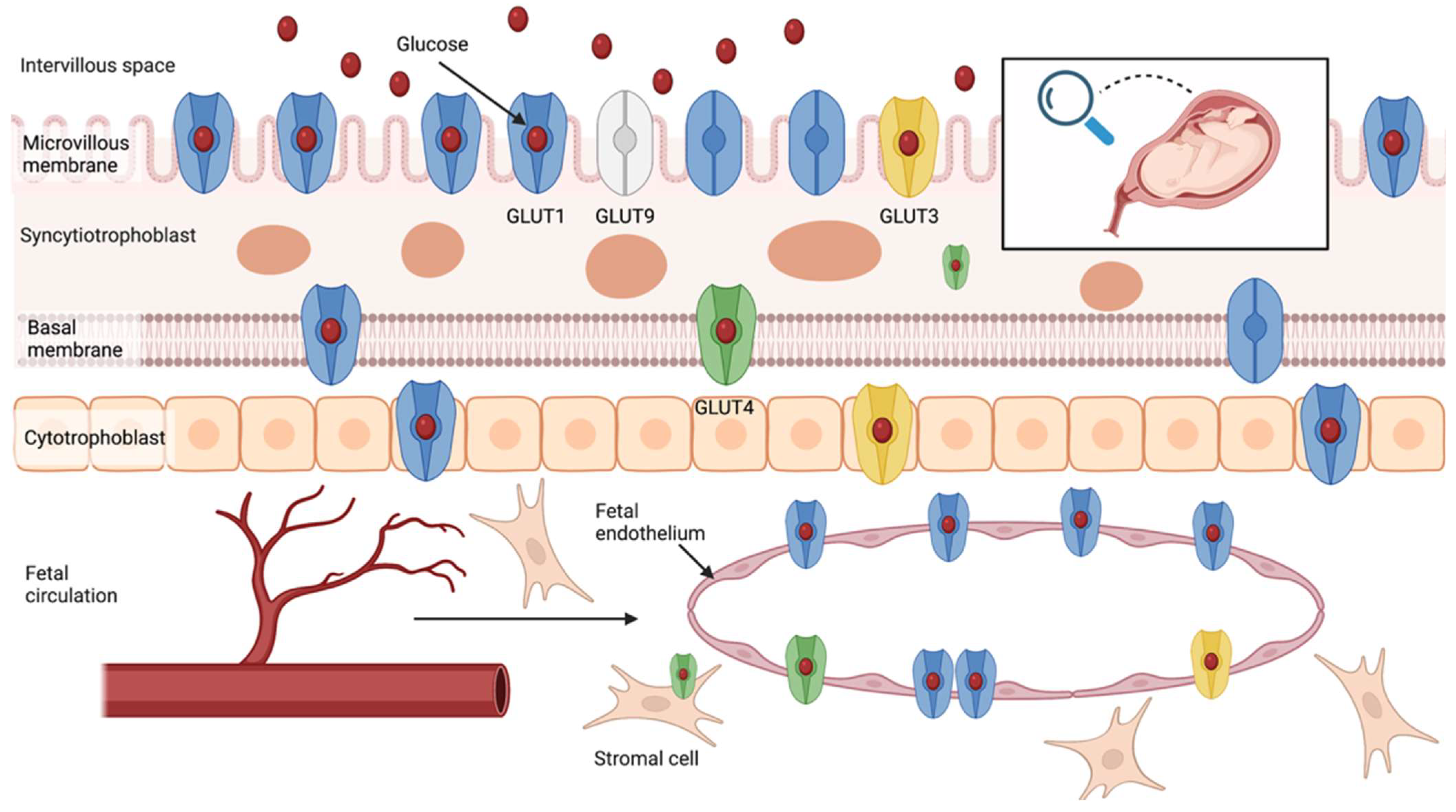
3. Glucose Transport in the Human Placenta
3.1. Glucose Transporters Family
3.1.1. GLUT1
3.1.2. GLUT3
3.1.3. GLUT4
3.1.4. GLUT9 and Other Isoforms
3.2. Potential Regulators of Glucose Transport in the Human Placenta
4. Placental Glucose Transport in Patients with Diabetes
4.1. Pregestational Diabetes Mellitus (PGDM)
4.2. Gestational Diabetes Mellitus (GDM)
5. Correlation with Fetal Growth and Other Gestational Outcomes
5.1. Fetal Macrosomia and Large-for-Gestational-Age (LGA) Newborns

{kind=link}
{kind=link}
| Clinical Characteristics, Number of Study Participants | Analyzed Parameter | Main Findings | First Author; Year; [Reference] |
|---|---|---|---|
| Growth abnormalities, 6 IUGR vs. 6 macrosomia vs. 4 AGA from IDDM mothers vs. 8 controls | GLUT3 protein GLUT4 protein | No differences No differences | Kainulainen; 1997; [131] |
| Growth abnormalities, 8 PGDM and GDM with macrosomia vs. 17 diabetes with AGA | BM GLUT1 protein | No difference | Gaither; 1999; [56] |
| Growth abnormalities, 6 GDM with LGA vs. 32 controls | MVM GLUT1 protein BM GLUT1 protein | No difference No difference | Jansson; 1999; [122] |
| Growth abnormalities, 13 GDM with macrosomia vs. 13 GDM with AGA and 13 controls | GLUT1 mRNA GLUT1 protein | Increased Increased | Yao; 2017; [128] |
| Growth abnormalities, 5 obesity with macrosomia vs. 12 obesity with AGA and 11 controls (normal BMI) | MVM GLUT1 protein BM GLUT1 protein BM GLUT4 protein | Decreased Increased Decreased | James-Allan; 2019; [37] |
| Growth abnormalities; 26 with macrosomia vs. 20 controls | GLUT1 protein GLUT3 protein GLUT8 protein GLUT12 protein | No difference No difference No difference No difference | Stanirowski; 2021; [83] |
5.2. Expression of Glucose Transporters in Fetal Growth Restriction (FGR) and Other Pregnancy-Related Pathologies
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Buch, I.; Hornnes, P.J.; Kuhl, C. Glucose tolerance in early pregnancy. Acta Endocrinol. 1986, 112, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Lesser, K.B.; Carpenter, M.W. Metabolic changes associated with normal pregnancy and pregnancy complicated by diabetes mellitus. Semin. Perinatol. 1994, 18, 399–406. [Google Scholar] [PubMed]
- Catalano, P.M.; Tyzbir, E.D.; Roman, N.M.; Amini, S.B.; Sims, E.A.H. Longitudinal changes in insulin release and insulin resistance in nonobese pregnant women. Am. J. Obstet. Gynecol. 1991, 165, 1667–1672. [Google Scholar] [CrossRef]
- Catalano, P.M.; Tyzbir, E.D.; Wolfe, R.R.; Roman, N.M.; Amini, S.B.; Sims, E.A.H. Longitudinal changes in basal hepatic glucose production and suppression during insulin infusion in normal pregnant women. Am. J. Obstet. Gynecol. 1992, 167, 913–919. [Google Scholar] [CrossRef]
- Di Cianni, G.; Miccoli, R.; Volpe, L.; Lencioni, C.; Del Prato, S. Intermediate metabolism in normal pregnancy and in gestational diabetes. Diabetes Metab. Res. Rev. 2003, 19, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Hadden, D.R.; McLaughlin, C. Normal and abnormal maternal metabolism during pregnancy. Semin. Fetal Neonatal Med. 2009, 14, 66–71. [Google Scholar] [CrossRef]
- Newbern, D.; Freemark, M. Placental hormones and the control of maternal metabolism and fetal growth. Curr. Opin. Endocrinol. Diabetes Obes. 2011, 18, 409–416. [Google Scholar] [CrossRef]
- Butte, N.F.; Hopkinson, J.M.; Mehta, N.; Moon, J.K.; Smith, E.O. Adjustments in energy expenditure and substrate utilization during late pregnancy and lactation. Am. J. Clin. Nutr. 1999, 69, 299–307. [Google Scholar] [CrossRef] [Green Version]
- Mazaki-Tovi, S.; Kanety, H.; Pariente, C.; Hemi, R.; Yissachar, E.; Schiff, E.; Cohen, O.; Sivan, E. Insulin sensitivity in late gestation and early postpartum period: The role of circulating maternal adipokines. Gynecol. Endocrinol. 2011, 27, 725–731. [Google Scholar] [CrossRef]
- Lain, K.Y.; Catalano, P.M. Metabolic Changes in Pregnancy. Clin. Obstet. Gynecol. 2007, 50, 938–948. [Google Scholar] [CrossRef]
- Desoye, G.; Hauguel-de Mouzon, S. The Human Placenta in Gestational Diabetes Mellitus. Diabetes Care 2007, 30, S120–S126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butte, N.F. Carbohydrate and lipid metabolism in pregnancy: Normal compared with gestational diabetes mellitus. Am. J. Clin. Nutr. 2000, 71, 1256S–1261S. [Google Scholar] [CrossRef] [PubMed]
- Sorenson, R.L.; Brelje, T.C. Prolactin Receptors Are Critical to the Adaptation of Islets to Pregnancy. Endocrinology 2009, 150, 1566–1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, A.E.; Cao-Minh, L.; Galasso, R.; Rizza, R.A.; Corradin, A.; Cobelli, C.; Butler, P.C. Adaptive changes in pancreatic beta cell fractional area and beta cell turnover in human pregnancy. Diabetologia 2010, 53, 2167–2176. [Google Scholar] [CrossRef] [Green Version]
- Sibiak, R.; Jankowski, M.; Gutaj, P.; Mozdziak, P.; Kempisty, B.; Wender-Ożegowska, E. Placental Lactogen as a Marker of Maternal Obesity, Diabetes, and Fetal Growth Abnormalities: Current Knowledge and Clinical Perspectives. J. Clin. Med. 2020, 9, 1142. [Google Scholar] [CrossRef]
- Negrato, C.A.; Gomes, M.B. Historical facts of screening and diagnosing diabetes in pregnancy. Diabetol. Metab. Syndr. 2013, 5, 22. [Google Scholar] [CrossRef] [Green Version]
- Hod, M.; Kapur, A.; Sacks, D.A.; Hadar, E.; Agarwal, M.; Di Renzo, G.C.; Roura, L.C.; McIntyre, H.D.; Morris, J.L.; Divakar, H. The International Federation of Gynecology and Obstetrics (FIGO) Initiative on gestational diabetes mellitus: A pragmatic guide for diagnosis, management, and care. Int. J. Gynecol. Obstet. 2015, 131, S173–S211. [Google Scholar]
- American Diabetes Association. Economic costs of diabetes in the U.S. in 2012. Diabetes Care 2013, 36, 1033–1046. [Google Scholar] [CrossRef] [Green Version]
- Hyperglycemia and Adverse Pregnancy Outcomes. N. Engl. J. Med. 2008, 358, 1991–2002. [CrossRef] [Green Version]
- Zimmer, D.M.; Golichowski, A.M.; Karn, C.A.; Brechtel, G.; Baron, A.D.; Denne, S.C. Glucose and Amino Acid Turnover in Untreated Gestational Diabetes. Diabetes Care 1996, 19, 591–596. [Google Scholar] [CrossRef]
- Atlas, D. International Diabetes Federation. IDF Diabetes Atlas, 6th ed.; International Diabetes Federation: Brussels, Belgium, 2013. [Google Scholar]
- Wendland, E.M.; Torloni, M.R.; Falavigna, M.; Trujillo, J.; Dode, M.A.; Campos, M.A.; Duncan, B.B.; Schmidt, M.I. Gestational diabetes and pregnancy outcomes-A systematic review of the World Health Organization (WHO) and the International Association of Diabetes in Pregnancy Study Groups (IADPSG) diagnostic criteria. BMC Pregnancy Childbirth 2012, 12, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Msollo, S.S.; Martin, H.D.; Mwanri, A.W.; Petrucka, P. Prevalence of hyperglycemia in pregnancy and influence of body fat on development of hyperglycemia in pregnancy among pregnant women in urban areas of Arusha region, Tanzania. BMC Pregnancy Childbirth 2019, 19, 315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larijani, B.; Hossein-nezhad, A.; Rizvi, S.W.H.; Munir, S.; Vassigh, A.-R. Cost Analysis of Different Screening Strategies for Gestational Diabetes Mellitus. Endocr. Pract. 2003, 9, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Jensen, D.M.; Ovesen, P.; Beck-Nielsen, H.; Mølsted-Pedersen, L.; Sørensen, B.; Vinter, C.; Damm, P. Gestational Weight Gain and Pregnancy Outcomes in 481 Obese Glucose-Tolerant Women. Diabetes Care 2005, 28, 2118–2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ay, L.; Kruithof, C.; Bakker, R.; Steegers, E.; Witteman, J.; Moll, H.; Hofman, A.; Mackenbach, J.; Hokken-Koelega, A.; Jaddoe, V. Maternal anthropometrics are associated with fetal size in different periods of pregnancy and at birth. The Generation R Study. BJOG Int. J. Obstet. Gynaecol. 2009, 116, 953–963. [Google Scholar] [CrossRef] [PubMed]
- Carr, D.B.; Utzschneider, K.M.; Hull, R.L.; Tong, J.; Wallace, T.M.; Kodama, K.; Shofer, J.B.; Heckbert, S.R.; Boyko, E.J.; Fujimoto, W.Y.; et al. Gestational Diabetes Mellitus Increases the Risk of Cardiovascular Disease in Women With a Family History of Type 2 Diabetes. Diabetes Care 2006, 29, 2078–2083. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Song, C.; Li, C.; Liu, P.; Sun, Z.; Yang, X. Increased risk of cardiovascular disease in women with prior gestational diabetes: A systematic review and meta-analysis. Diabetes Res. Clin. Pract. 2018, 140, 324–338. [Google Scholar] [CrossRef]
- Mosca, L.; Benjamin, E.J.; Berra, K.; Bezanson, J.L.; Dolor, R.J.; Lloyd-Jones, D.M.; Newby, L.K.; Piña, I.L.; Roger, V.L.; Shaw, L.J.; et al. Effectiveness-Based Guidelines for the Prevention of Cardiovascular Disease in Women—2011 Update. Circulation 2011, 123, 1243–1262. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; England, J.L.; Sharma, J.A.; Njoroge, T. Gestational Diabetes Mellitus and Risk of Childhood Overweight and Obesity in Offspring: A Systematic Review. Exp. Diabetes Res. 2011, 2011, 541308. [Google Scholar] [CrossRef] [Green Version]
- Philipps, L.H.; Santhakumaran, S.; Gale, C.; Prior, E.; Logan, K.M.; Hyde, M.J.; Modi, N. The diabetic pregnancy and offspring BMI in childhood: A systematic review and meta-analysis. Diabetologia 2011, 54, 1957–1966. [Google Scholar] [CrossRef] [Green Version]
- Lowe, W.L.; Scholtens, D.M.; Kuang, A.; Linder, B.; Lawrence, J.M.; Lebenthal, Y.; McCance, D.; Hamilton, J.; Nodzenski, M.; Talbot, O.; et al. Hyperglycemia and Adverse Pregnancy Outcome Follow-up Study (HAPO FUS): Maternal Gestational Diabetes Mellitus and Childhood Glucose Metabolism. Diabetes Care 2019, 42, 372–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Li, X.; Liu, G.; Han, B.; Wang, J.; Jiang, X. The association of maternal diabetes with attention deficit and hyperactivity disorder in offspring: A meta-analysis. Neuropsychiatr. Dis. Treat. 2019, 15, 675–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sędzikowska, A.; Szablewski, L. Human Glucose Transporters in Renal Glucose Homeostasis. Int. J. Mol. Sci. 2021, 22, 13522. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.M. Glucose transport families SLC5 and SLC50. Mol. Aspects Med. 2013, 34, 183–196. [Google Scholar] [CrossRef]
- Illsley, N.P. CURRENT TOPIC: Glucose Transporters in the Human Placenta. Placenta 2000, 21, 14–22. [Google Scholar] [CrossRef]
- James-Allan, L.B.; Arbet, J.; Teal, S.B.; Powell, T.L.; Jansson, T. Insulin Stimulates GLUT4 Trafficking to the Syncytiotrophoblast Basal Plasma Membrane in the Human Placenta. J. Clin. Endocrinol. Metab. 2019, 104, 4225–4238. [Google Scholar] [CrossRef]
- Ingermann, R.L.; Bissonnette, J.M.; Koch, P.L. d-glucose-sensitive and -insensitive cytochalasin B binding proteins from microvillous plasma membranes of human placenta. Identification of the d-glucose transporter. Biochim. Biophys. Acta Biomembr. 1983, 730, 57–63. [Google Scholar] [CrossRef]
- Reid, N.A.; Boyd, C.A.R. Further evidence for the presence of two facilitative glucose transporter isoforms in the brush border membrane of the syncytiotrophobast of the human full term placenta. Biochem. Soc. Trans. 1994, 22, 267S. [Google Scholar] [CrossRef] [Green Version]
- Barta, E.; Drugan, A. Glucose transport from mother to fetus—A theoretical study. J. Theor. Biol. 2010, 263, 295–302. [Google Scholar] [CrossRef]
- Vardhana, P.A.; Illsley, N.P. Transepithelial Glucose Transport and Metabolism in BeWo Choriocarcinoma Cells. Placenta 2002, 23, 653–660. [Google Scholar] [CrossRef]
- Schneider, H.; Reiber, W.; Sager, R.; Malek, A. Asymmetrical Transport of Glucose Across the In Vitro Perfused Human Placenta. Placenta 2003, 24, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Jansson, T.; Wennergren, M.; Illsley, N.P. Glucose transporter protein expression in human placenta throughout gestation and in intrauterine growth retardation. J. Clin. Endocrinol. Metab. 1993, 77, 1554–1562. [Google Scholar] [PubMed]
- Wolf, H.J.; Desoye, G. Immunohistochemical localization of glucose transporters and insulin receptors in human fetal membranes at term. Histochemistry 1993, 100, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Tadokoro, C.; Yoshimoto, Y.; Sakata, M.; Fujimiya, M.; Kurachi, H.; Adachi, E.; Maeda, T.; Miyake, A. Localization of human placental glucose transporter 1 during pregnancy. An immunohistochemical study. Histol. Histopathol. 1996, 11, 673–681. [Google Scholar] [PubMed]
- Ericsson, A.; Hamark, B.; Powell, T.L.; Jansson, T. Glucose transporter isoform 4 is expressed in the syncytiotrophoblast of first trimester human placenta. Hum. Reprod. 2005, 20, 521–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balachandiran, M.; Bobby, Z.; Dorairajan, G.; Gladwin, V.; Vinayagam, V.; Packirisamy, R.M. Decreased maternal serum adiponectin and increased insulin-like growth factor-1 levels along with increased placental glucose transporter-1 expression in gestational diabetes mellitus: Possible role in fetal overgrowth. Placenta 2021, 104, 71–80. [Google Scholar] [CrossRef]
- Hahn, T.; Hartmann, M.; Blaschitz, A.; Skofitsch, G.; Graf, R.; Dohr, G.; Desoye, G. Localisation of the high affinity factilitative glucose transporter protein GLUT 1 in the placenta of human, marmoset monkey (Callithrix jacchus) and rat at different developmental stages. Cell Tissue Res. 1995, 280, 49–57. [Google Scholar] [CrossRef]
- Takata, K.; Kasahara, T.; Kasahara, M.; Ezaki, O.; Hirano, H. Localization of erythrocyte/HepG2-type glucose transporter (GLUT1) in human placental villi. Cell Tissue Res. 1992, 267, 407–412. [Google Scholar] [CrossRef]
- Farrell, C.L.; Yang, J.; Pardridge, W.M. GLUT-1 glucose transporter is present within apical and basolateral membranes of brain epithelial interfaces and in microvascular endothelia with and without tight junctions. J. Histochem. Cytochem. 1992, 40, 193–199. [Google Scholar] [CrossRef] [Green Version]
- Jones, C.J.P.; Choudhury, R.H.; Aplin, J.D. Functional changes in Hofbauer cell glycobiology during human pregnancy. Placenta 2015, 36, 1130–1137. [Google Scholar] [CrossRef]
- Hutchinson, K.A.; Vuong, N.H.; Mohammad, S.; Everest, C.; Leung, M.L.; Bhattacharjee, J.; Adamo, K.B. Physical Activity During Pregnancy Is Associated with Increased Placental FATP4 Protein Expression. Reprod. Sci. 2020, 27, 1909–1919. [Google Scholar] [CrossRef] [PubMed]
- Hauguel-De Mouzon, S.; Leturque, A.; Alsat, E.; Loizeau, M.; Evain-Brion, D.; Girard, J. Developmental expression of Glut1 glucose transporter and c-fos genes in human placental cells. Placenta 1994, 15, 35–46. [Google Scholar] [CrossRef]
- Sakata, M.; Kurachi, H.; Imai, T.; Tadokoro, C.; Yamaguchi, M.; Yoshimoto, Y.; Oka, Y.; Miyake, A. Increase in human placental glucose transporter-1 during pregnancy. Eur. J. Endocrinol. 1995, 132, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Jansson, T.; Cowley, E.; Illsley, N. Cellular localization of glucose transporter messenger RNA in human placenta. Reprod. Fertil. Dev. 1995, 7, 1425. [Google Scholar] [CrossRef] [PubMed]
- Gaither, K.; Quraishi, A.N.; Illsley, N.P. Diabetes alters the expression and activity of the human placental GLUT1 glucose transporter. J. Clin. Endocrinol. Metab. 1999, 84, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Acosta, O.; Ramirez, V.I.; Lager, S.; Gaccioli, F.; Dudley, D.J.; Powell, T.L.; Jansson, T. Increased glucose and placental GLUT-1 in large infants of obese nondiabetic mothers. Am. J. Obstet. Gynecol. 2015, 212, e1–e227. [Google Scholar] [CrossRef] [PubMed]
- Michelsen, T.M.; Holme, A.M.; Holm, M.B.; Roland, M.C.; Haugen, G.; Powell, T.L.; Jansson, T.; Henriksen, T. Uteroplacental Glucose Uptake and Fetal Glucose Consumption: A Quantitative Study in Human Pregnancies. J. Clin. Endocrinol. Metab. 2019, 104, 873–882. [Google Scholar] [CrossRef] [Green Version]
- James-Allan, L.B.; Teal, S.; Powell, T.L.; Jansson, T. Changes in Placental Nutrient Transporter Protein Expression and Activity Across Gestation in Normal and Obese Women. Reprod. Sci. 2020, 27, 1758–1769. [Google Scholar] [CrossRef]
- Clarson, L.H.; Glazier, J.D.; Sides, M.K.; Sibley, C.P. Expression of the facilitated glucose transporters (GLUT1 and GLUT3) by a choriocarcinoma cell line (JAr) and cytotrophoblast cells in culture. Placenta 1997, 18, 333–339. [Google Scholar] [CrossRef]
- Baumann, M.U.; Schneider, H.; Malek, A.; Palta, V.; Surbek, D.V.; Sager, R.; Zamudio, S.; Illsley, N.P. Regulation of Human Trophoblast GLUT1 Glucose Transporter by Insulin-Like Growth Factor I (IGF-I). PLoS ONE 2014, 9, e106037. [Google Scholar] [CrossRef]
- Sciullo, E.; Cardellini, G.; Baroni, M.G.; Torresi, P.; Buongiorno, A.; Pozzilli, P.; Fallucca, F. Glucose transporter (Glut1, Glut3) mRNA in human placenta of diabetic and non-diabetic pregnancies. Early Pregnancy 1997, 3, 172–182. [Google Scholar] [PubMed]
- Stanirowski, P.J.; Szukiewicz, D.; Pyzlak, M.; Abdalla, N.; Sawicki, W.; Cendrowski, K. Analysis of correlations between the placental expression of glucose transporters GLUT-1, GLUT-4 and GLUT-9 and selected maternal and fetal parameters in pregnancies complicated by diabetes mellitus. J. Matern. Neonatal Med. 2019, 32, 650–659. [Google Scholar] [CrossRef] [PubMed]
- Haber, R.S.; Weinstein, S.P.; O’Boyle, E.; Morgello, S. Tissue distribution of the human GLUT3 glucose transporter. Endocrinology 1993, 132, 2538–2543. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.; Heller, D.S.; Zamudio, S.; Illsley, N.P. Glucose transporter 3 (GLUT3) protein expression in human placenta across gestation. Placenta 2011, 32, 1041–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janzen, C.; Lei, M.Y.Y.; Cho, J.; Sullivan, P.; Shin, B.-C.; Devaskar, S.U. Placental glucose transporter 3 (GLUT3) is up-regulated in human pregnancies complicated by late-onset intrauterine growth restriction. Placenta 2013, 34, 1072–1078. [Google Scholar] [CrossRef] [Green Version]
- Ogura, K.; Sakata, M.; Okamoto, Y.; Yasui, Y.; Tadokoro, C.; Yoshimoto, Y.; Yamaguchi, M.; Kurachi, H.; Maeda, T.; Murata, Y. 8-bromo-cyclicAMP stimulates glucose transporter-1 expression in a human choriocarcinoma cell line. J. Endocrinol. 2000, 164, 171–178. [Google Scholar] [CrossRef] [Green Version]
- Younes, M.; Lechago, L.V.; Somoano, J.R.; Mosharaf, M.; Lechago, J. Immunohistochemical detection of Glut3 in human tumors and normal tissues. Anticancer Res. 1997, 17, 2747–2750. [Google Scholar]
- Arnott, G.; Coghill, G.; McArdle, H.J.; Hundal, H.S. Immunolocalization of GLUT1 and GLUT3 glucose transporters in human placenta. Biochem. Soc. Trans. 1994, 22, 272S. [Google Scholar] [CrossRef] [Green Version]
- Hauguel-de Mouzon, S.; Challier, J.C.; Kacemi, A.; Caüzac, M.; Malek, A.; Girard, J. The GLUT3 Glucose Transporter Isoform Is Differentially Expressed within Human Placental Cell Types. J. Clin. Endocrinol. Metab. 1997, 82, 2689–2694. [Google Scholar] [CrossRef]
- Korgun, E.T.; Celik-Ozenci, C.; Seval, Y.; Desoye, G.; Demir, R. Do glucose transporters have other roles in addition to placental glucose transport during early pregnancy? Histochem. Cell Biol. 2005, 123, 621–629. [Google Scholar] [CrossRef]
- Hahn, D.; Blaschitz, A.; Korgun, E.T.; Lang, I.; Desoye, G.; Skofitsch, G.; Dohr, G. From maternal glucose to fetal glycogen: Expression of key regulators in the human placenta. Mol. Hum. Reprod. 2001, 7, 1173–1178. [Google Scholar] [CrossRef] [Green Version]
- Barros, L.F.; Yudilevich, D.L.; Jarvis, S.M.; Beaumont, N.; Baldwin, S.A. Quantitation and immunolocalization of glucose transporters in the human placenta. Placenta 1995, 16, 623–633. [Google Scholar] [CrossRef]
- Shepherd, P.R.; Gould, G.W.; Colville, C.A.; McCoid, S.C.; Gibbs, E.M.; Kahn, B.B. Distribution of GLUT3 glucose transporter protein in human tissues. Biochem. Biophys. Res. Commun. 1992, 188, 149–154. [Google Scholar] [CrossRef]
- Xing, A.Y.; Challier, J.C.; Lepercq, J.; Caüzac, M.; Charron, M.J.; Girard, J.; Hauguel-de Mouzon, S. Unexpected Expression of Glucose Transporter 4 in Villous Stromal Cells of Human Placenta. J. Clin. Endocrinol. Metab. 1998, 83, 4097–4101. [Google Scholar] [CrossRef]
- Zhang, W.; Su, R.; Lin, L.; Yang, H. ARHGEF11 affecting the placental insulin signaling pathway in fetal macrosomia of normal glucose tolerance pregnant women. Placenta 2018, 63, 7–14. [Google Scholar] [CrossRef]
- Ermini, L.; Nuzzo, A.M.; Ietta, F.; Romagnoli, R.; Moretti, L.; Masturzo, B.; Paulesu, L.; Rolfo, A. Placental Glucose Transporters and Response to Bisphenol A in Pregnancies from of Normal and Overweight Mothers. Int. J. Mol. Sci. 2021, 22, 6625. [Google Scholar] [CrossRef]
- Kuzmicki, M.; Telejko, B.; Wawrusiewicz-kurylonek, N.; Nikolajuk, A.; Zwierz-gugala, D.; Jelski, W.; Kolodziejczak, M.; Zonenberg, A.; Wilczynski, J.; Kretowski, A.; et al. Retinol-binding protein 4 in adipose and placental tissue of women with gestational diabetes. Gynecol. Endocrinol. 2011, 27, 1065–1069. [Google Scholar] [CrossRef]
- Augustin, R.; Carayannopoulos, M.O.; Dowd, L.O.; Phay, J.E.; Moley, J.F.; Moley, K.H. Identification and Characterization of Human Glucose Transporter-like Protein-9 (GLUT9). J. Biol. Chem. 2004, 279, 16229–16236. [Google Scholar] [CrossRef] [Green Version]
- Gude, N.M.; Stevenson, J.L.; Rogers, S.; Best, J.D.; Kalionis, B.; Huisman, M.A.; Erwich, J.J.H.M.; Timmer, A.; King, R.G. GLUT12 Expression in Human Placenta in First Trimester and Term. Placenta 2003, 24, 566–570. [Google Scholar] [CrossRef]
- Gude, N.M.; Stevenson, J.L.; Murthi, P.; Rogers, S.; Best, J.D.; Kalionis, B.; King, R.G. Expression of GLUT12 in the fetal membranes of the human placenta. Placenta 2005, 26, 67–72. [Google Scholar] [CrossRef]
- Stanirowski, P.J.; Szukiewicz, D.; Majewska, A.; Wątroba, M.; Pyzlak, M.; Bomba-Opoń, D.; Wielgoś, M. Placental expression of glucose transporters GLUT-1, GLUT-3, GLUT-8 and GLUT-12 in pregnancies complicated by gestational and type 1 diabetes mellitus. J. Diabetes Investig. 2021, 13, 560–570. [Google Scholar] [CrossRef]
- Stanirowski, P.J.; Szukiewicz, D.; Majewska, A.; Wątroba, M.; Pyzlak, M.; Bomba-Opoń, D.; Wielgoś, M. Differential Expression of Glucose Transporter Proteins GLUT-1, GLUT-3, GLUT-8 and GLUT-12 in the Placenta of Macrosomic, Small-for-Gestational-Age and Growth-Restricted Foetuses. J. Clin. Med. 2021, 10, 5833. [Google Scholar] [CrossRef]
- Acevedo, C.G.; Márquez, J.L.; Rojas, S.; Bravo, I. Insulin and nitric oxide stimulates glucose transport in human placenta. Life Sci. 2005, 76, 2643–2653. [Google Scholar] [CrossRef]
- Gordon, M.C.; Zimmerman, P.D.; Landon, M.B.; Gabbe, S.G.; Kniss, D.A. Insulin and glucose modulate glucose transporter messenger ribonucleic acid expression and glucose uptake in trophoblasts isolated from first-trimester chorionic villi. Am. J. Obstet. Gynecol. 1995, 173, 1089–1097. [Google Scholar] [CrossRef]
- Illsley, N.P.; Sellers, M.C.; Wright, R.L. Glycaemic regulation of glucose transporter expression and activity in the human placenta. Placenta 1998, 19, 517–524. [Google Scholar] [CrossRef]
- Shah, S.W.; Zhao, H.; Low, S.Y.; Mcardle, H.J.; Hundal, H.S. Characterization of Glucose Transport and Glucose Transporters in the Human Choriocarcinoma Cell Line, BeWo. Placenta 1999, 20, 651–659. [Google Scholar] [CrossRef]
- Hahn, T.; Barth, S.; Weiss, U.; Mosgoeller, W.; Desoye, G. Sustained hyperglycemia in vitro down-regulates the GLUT1 glucose transport system of cultured human term placental trophoblast: A mechanism to protect fetal development? FASEB J. 1998, 12, 1221–1231. [Google Scholar] [CrossRef] [Green Version]
- Hahn, T.; Hahn, D.; Blaschitz, A.; Korgun, E.T.; Desoye, G.; Dohr, G. Hyperglycaemia-induced subcellular redistribution of GLUT1 glucose transporters in cultured human term placental trophoblast cells. Diabetologia 2000, 43, 173–180. [Google Scholar] [CrossRef] [Green Version]
- Novakovic, B.; Gordon, L.; Robinson, W.P.; Desoye, G.; Saffery, R. Glucose as a fetal nutrient: Dynamic regulation of several glucose transporter genes by DNA methylation in the human placenta across gestation. J. Nutr. Biochem. 2013, 24, 282–288. [Google Scholar] [CrossRef]
- Song, T.-R.; Su, G.-D.; Chi, Y.-L.; Wu, T.; Xu, Y.; Chen, C.-C. Dysregulated miRNAs contribute to altered placental glucose metabolism in patients with gestational diabetes via targeting GLUT1 and HK2. Placenta 2021, 105, 14–22. [Google Scholar] [CrossRef]
- Borges, M.H.; Pullockaran, J.; Catalano, P.M.; Baumann, M.U.; Zamudio, S.; Illsley, N.P. Human placental GLUT1 glucose transporter expression and the fetal insulin-like growth factor axis in pregnancies complicated by diabetes. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2411–2419. [Google Scholar] [CrossRef] [PubMed]
- Visiedo, F.; Bugatto, F.; Carrasco-Fernández, C.; Sáez-Benito, A.; Mateos, R.M.; Cózar-Castellano, I.; Bartha, J.L.; Perdomo, G. Hepatocyte growth factor is elevated in amniotic fluid from obese women and regulates placental glucose and fatty acid metabolism. Placenta 2015, 36, 381–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumann, M.U.; Zamudio, S.; Illsley, N.P. Hypoxic upregulation of glucose transporters in BeWo choriocarcinoma cells is mediated by hypoxia-inducible factor-1. Am. J. Physiol. Physiol. 2007, 293, C477–C485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, M.; Sakata, M.; Takeda, T.; Tahara, M.; Yamamoto, T.; Minekawa, R.; Isobe, A.; Tasaka, K.; Murata, Y. Hypoxia Up-Regulates Hypoxia-Inducible Factor-1α Expression through RhoA Activation in Trophoblast Cells. J. Clin. Endocrinol. Metab. 2005, 90, 1712–1719. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, M.; Sakata, M.; Takeda, T.; Yamamoto, T.; Okamoto, Y.; Sawada, K.; Kimura, A.; Minekawa, R.; Tahara, M.; Tasaka, K.; et al. Induction of glucose transporter 1 expression through hypoxia-inducible factor 1α under hypoxic conditions in trophoblast-derived cells. J. Endocrinol. 2004, 183, 145–154. [Google Scholar] [CrossRef] [Green Version]
- Rajakumar, A.; Conrad, K.P. Expression, Ontogeny, and Regulation of Hypoxia-Inducible Transcription Factors in the Human Placenta1. Biol. Reprod. 2000, 63, 559–569. [Google Scholar] [CrossRef] [Green Version]
- Cindrova-Davies, T.; van Patot, M.T.; Gardner, L.; Jauniaux, E.; Burton, G.J.; Charnock-Jones, D.S. Energy status and HIF signalling in chorionic villi show no evidence of hypoxic stress during human early placental development. MHR Basic Sci. Reprod. Med. 2015, 21, 296–308. [Google Scholar] [CrossRef] [Green Version]
- Vaughan, O.R.; Thompson, F.; Lorca, R.A.; Julian, C.G.; Powell, T.L.; Moore, L.G.; Jansson, T. Effect of high altitude on human placental amino acid transport. J. Appl. Physiol. 2020, 128, 127–133. [Google Scholar] [CrossRef]
- Zamudio, S.; Torricos, T.; Fik, E.; Oyala, M.; Echalar, L.; Pullockaran, J.; Tutino, E.; Martin, B.; Belliappa, S.; Balanza, E.; et al. Hypoglycemia and the Origin of Hypoxia-Induced Reduction in Human Fetal Growth. PLoS ONE 2010, 5, e8551. [Google Scholar] [CrossRef] [Green Version]
- Zamudio, S.; Baumann, M.U.; Illsley, N.P. Effects of chronic hypoxia in vivo on the expression of human placental glucose transporters. Placenta 2006, 27, 49–55. [Google Scholar] [CrossRef] [Green Version]
- Francois, L.N.; Gorczyca, L.; Du, J.; Bircsak, K.M.; Yen, E.; Wen, X.; Tu, M.-J.; Yu, A.-M.; Illsley, N.P.; Zamudio, S.; et al. Down-regulation of the placental BCRP/ABCG2 transporter in response to hypoxia signaling. Placenta 2017, 51, 57–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandrasiri, U.P.; Chua, C.L.L.; Umbers, A.J.; Chaluluka, E.; Glazier, J.D.; Rogerson, S.J.; Boeuf, P. Insight Into the Pathogenesis of Fetal Growth Restriction in Placental Malaria: Decreased Placental Glucose Transporter Isoform 1 Expression. J. Infect. Dis. 2014, 209, 1663–1667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vota, D.; Torti, M.; Paparini, D.; Giovannoni, F.; Merech, F.; Hauk, V.; Calo, G.; Ramhorst, R.; Garcia, C.; Pérez Leirós, C. Zika virus infection of first trimester trophoblast cells affects cell migration, metabolism and immune homeostasis control. J. Cell. Physiol. 2021, 236, 4913–4925. [Google Scholar] [CrossRef]
- Cowell, W.; Deyssenroth, M.; Chen, J.; Wright, R.J. Maternal stress in relation to sex-specific expression of placental genes involved in nutrient transport, oxygen tension, immune response, and the glucocorticoid barrier. Placenta 2020, 96, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Di Simone, N.; Di Nicuolo, F.; Marzioni, D.; Castellucci, M.; Sanguinetti, M.; D’lppolito, S.; Caruso, A. Resistin modulates glucose uptake and glucose transporter-1 (GLUT-1) expression in trophoblast cells. J. Cell. Mol. Med. 2009, 13, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Merech, F.; Soczewski, E.; Hauk, V.; Paparini, D.; Ramhorst, R.; Vota, D.; Pérez Leirós, C. Vasoactive Intestinal Peptide induces glucose and neutral amino acid uptake through mTOR signalling in human cytotrophoblast cells. Sci. Rep. 2019, 9, 17152. [Google Scholar] [CrossRef]
- Xu, J.; Lu, C.; Wang, J.; Zhang, R.; Qian, X.; Zhu, H. Regulation of Human Trophoblast GLUT3 Glucose Transporter by Mammalian Target of Rapamycin Signaling. Int. J. Mol. Sci. 2015, 16, 13815–13828. [Google Scholar] [CrossRef] [Green Version]
- Hahn, T.; Barth, S.; Graf, R.; Engelmann, M.; Beslagic, D.; Reul, J.M.H.M.; Holsboer, F.; Dohr, G.; Desoye, G. Placental Glucose Transporter Expression Is Regulated by Glucocorticoids. J. Clin. Endocrinol. Metab. 1999, 84, 1445–1452. [Google Scholar] [CrossRef]
- Kipmen-Korgun, D.; Ozmen, A.; Unek, G.; Simsek, M.; Demir, R.; Korgun, E.T. Triamcinolone up-regulates GLUT 1 and GLUT 3 expression in cultured human placental endothelial cells. Cell Biochem. Funct. 2012, 30, 47–53. [Google Scholar] [CrossRef]
- Mateos, R.M.; Jiménez, G.; Álvarez-Gil, C.; Visiedo, F.; Rivera-Rodríguez, F.; Santos-Rosendo, C.; Rodriguez-Pareja, A.; Perdomo, G.; Lechuga-Sancho, A.M. Excess Hydrocortisone Hampers Placental Nutrient Uptake Disrupting Cellular Metabolism. Biomed Res. Int. 2018, 2018, 5106174. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Lv, C.; Xu, C.; Li, Y.; Cui, X.; Gu, H.; Ni, X. Differential Regulation of Glucose Transporters Mediated by CRH Receptor Type 1 and Type 2 in Human Placental Trophoblasts. Endocrinology 2012, 153, 1464–1471. [Google Scholar] [CrossRef] [PubMed]
- Ericsson, A.; Hamark, B.; Jansson, N.; Johansson, B.R.; Powell, T.L.; Jansson, T. Hormonal regulation of glucose and system A amino acid transport in first trimester placental villous fragments. Am. J. Physiol. Integr. Comp. Physiol. 2005, 288, R656–R662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benincasa, L.; Mandalà, M.; Paulesu, L.; Barberio, L.; Ietta, F. Prenatal Nutrition Containing Bisphenol A Affects Placenta Glucose Transfer: Evidence in Rats and Human Trophoblast. Nutrients 2020, 12, 1375. [Google Scholar] [CrossRef] [PubMed]
- Rajakumar, C.; Guan, H.; Langlois, D.; Cernea, M.; Yang, K. Bisphenol A disrupts gene expression in human placental trophoblast cells. Reprod. Toxicol. 2015, 53, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Díaz, P.; Dimasuay, K.G.; Koele-Schmidt, L.; Jang, B.; Barbour, L.A.; Jansson, T.; Powell, T.L. Glyburide treatment in gestational diabetes is associated with increased placental glucose transporter 1 expression and higher birth weight. Placenta 2017, 57, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Lappas, M.; Andrikopoulos, S.; Permezel, M. Hypoxanthine–xanthine oxidase down-regulates GLUT1 transcription via SIRT1 resulting in decreased glucose uptake in human placenta. J. Endocrinol. 2012, 213, 49–57. [Google Scholar] [CrossRef] [Green Version]
- Barta, E.; Drugan, A. A clinical study which relates to a theoretical simulation of the glucose transport in the human placenta under various diabetic conditions. J. Perinat. Med. 2016, 44, 405–410. [Google Scholar] [CrossRef]
- Jansson, T.; Wennergren, M.; Powell, T.L. Placental glucose transport and GLUT 1 expression in insulin-dependent diabetes. Am. J. Obstet. Gynecol. 1999, 180, 163–168. [Google Scholar] [CrossRef]
- Stanirowski, P.J.; Szukiewicz, D.; Pyzlak, M.; Abdalla, N.; Sawicki, W.; Cendrowski, K. Impact of pre-gestational and gestational diabetes mellitus on the expression of glucose transporters GLUT-1, GLUT-4 and GLUT-9 in human term placenta. Endocrine 2017, 55, 799–808. [Google Scholar] [CrossRef]
- Castillo-Castrejon, M.; Yamaguchi, K.; Rodel, R.L.; Erickson, K.; Kramer, A.; Hirsch, N.M.; Rolloff, K.; Jansson, T.; Barbour, L.A.; Powell, T.L. Effect of type 2 diabetes mellitus on placental expression and activity of nutrient transporters and their association with birth weight and neonatal adiposity. Mol. Cell. Endocrinol. 2021, 532, 111319. [Google Scholar] [CrossRef]
- Jansson, T.; Ekstrand, Y.; Wennergren, M.; Powell, T.L. Placental glucose transport in gestational diabetes mellitus. Am. J. Obstet. Gynecol. 2001, 184, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Colomiere, M.; Permezel, M.; Riley, C.; Desoye, G.; Lappas, M. Defective insulin signaling in placenta from pregnancies complicated by gestational diabetes mellitus. Eur. J. Endocrinol. 2009, 160, 567–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dekker Nitert, M.; Barrett, H.L.; Kubala, M.H.; Scholz Romero, K.; Denny, K.J.; Woodruff, T.M.; McIntyre, H.D.; Callaway, L.K. Increased Placental Expression of Fibroblast Growth Factor 21 in Gestational Diabetes Mellitus. J. Clin. Endocrinol. Metab. 2014, 99, E591–E598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rong, C.; Cui, X.; Chen, J.; Qian, Y.; Jia, R.; Hu, Y. DNA Methylation Profiles in Placenta and Its Association with Gestational Diabetes Mellitus. Exp. Clin. Endocrinol. Diabetes 2015, 123, 282–288. [Google Scholar] [CrossRef]
- Zhang, B.; Jin, Z.; Sun, L.; Zheng, Y.; Jiang, J.; Feng, C.; Wang, Y. Expression and correlation of sex hormone-binding globulin and insulin signal transduction and glucose transporter proteins in gestational diabetes mellitus placental tissue. Diabetes Res. Clin. Pract. 2016, 119, 106–117. [Google Scholar] [CrossRef]
- Muralimanoharan, S.; Maloyan, A.; Myatt, L. Mitochondrial function and glucose metabolism in the placenta with gestational diabetes mellitus: Role of miR-143. Clin. Sci. 2016, 130, 931–941. [Google Scholar] [CrossRef] [Green Version]
- Yao, G.; Zhang, Y.; Wang, D.; Yang, R.; Sang, H.; Han, L.; Zhu, Y.; Lu, Y.; Tan, Y.; Shang, Z. GDM-Induced Macrosomia Is Reversed by Cav-1 via AMPK-Mediated Fatty Acid Transport and GLUT1-Mediated Glucose Transport in Placenta. PLoS ONE 2017, 12, e0170490. [Google Scholar] [CrossRef]
- Osmond, D.T.D.; King, R.G.; Brennecke, S.P.; Gude, N.M. Placental glucose transport and utilisation is altered at term in insulin-treated, gestational-diabetic patients. Diabetologia 2001, 44, 1133–1139. [Google Scholar] [CrossRef] [Green Version]
- Desoye, G.; Korgun, E.T.; Ghaffari-Tabrizi, N.; Hahn, T. Is fetal macrosomia in adequately controlled diabetic women the result of a placental defect?–A hypothesis. J. Matern. Neonatal Med. 2002, 11, 258–261. [Google Scholar]
- Kainulainen, H.; Järvinen, T.; Heinonen, P. Placental Glucose Transporters in Fetal Intrauterine Growth Retardation and Macrosomia. Gynecol. Obstet. Investig. 1997, 44, 89–92. [Google Scholar] [CrossRef]
- Jansson, T.; Ylvén, K.; Wennergren, M.; Powell, T.L. Glucose Transport and System A Activity in Syncytiotrophoblast Microvillous and Basal Plasma Membranes in Intrauterine Growth Restriction. Placenta 2002, 23, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Janzen, C.; Lei, M.Y.Y.; Jeong, I.S.D.; Ganguly, A.; Sullivan, P.; Paharkova, V.; Capodanno, G.; Nakamura, H.; Perry, A.; Shin, B.-C.; et al. Humanin (HN) and glucose transporter 8 (GLUT8) in pregnancies complicated by intrauterine growth restriction. PLoS ONE 2018, 13, e0193583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lüscher, B.P.; Marini, C.; Joerger-Messerli, M.S.; Huang, X.; Hediger, M.A.; Albrecht, C.; Baumann, M.U.; Surbek, D.V. Placental glucose transporter (GLUT)-1 is down-regulated in preeclampsia. Placenta 2017, 55, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Choi, S.-Y.; Kang, B.-H.; Yoo, H.-J.; Song, S.-Y.; Seong, I.-O.; Suh, K.-S.; Kim, K.-H. Differential microscopic finding and glucose transporter 3 expression in terminal chorionic villi among birth weight-discordant twin placentas. Histol. Histopathol. 2015, 30, 955–962. [Google Scholar]
- Chang, Y.-L.; Chao, A.-S.; Chang, S.-D.; Cheng, P.-J. Placental glucose transporter 1 and 3 gene expression in Monochorionic twin pregnancies with selective fetal growth restriction. BMC Pregnancy Childbirth 2021, 21, 260. [Google Scholar] [CrossRef]
- Wei, W.; Hu, Y.Y. Expression of hypoxia-regulated genes and glycometabolic genes in placenta from patients with intrahepatic cholestasis of pregnancy. Placenta 2014, 35, 732–736. [Google Scholar] [CrossRef]
- Dong, J.; Wen, L.; Guo, X.; Xiao, X.; Jiang, F.; Li, B.; Jin, N.; Wang, J.; Wang, X.; Chen, S.; et al. The increased expression of glucose transporters in human full-term placentas from assisted reproductive technology without changes of mTOR signaling. Placenta 2019, 86, 4–10. [Google Scholar] [CrossRef]
- Bloise, E.; Braga, J.R.S.; Andrade, C.B.V.; Imperio, G.E.; Martinelli, L.M.; Antunes, R.A.; Silva, K.R.; Nunes, C.B.; Cobellis, L.; Bloise, F.F.; et al. Altered Umbilical Cord Blood Nutrient Levels, Placental Cell Turnover and Transporter Expression in Human Term Pregnancies Conceived by Intracytoplasmic Sperm Injection (ICSI). Nutrients 2021, 13, 2587. [Google Scholar] [CrossRef]
- Ozmen, A.; Kipmen-Korgun, D.; Isenlik, B.S.; Erman, M.; Sakinci, M.; Berkkanoglu, M.; Coetzee, K.; Ozgur, K.; Cetindag, E.; Yanar, K.; et al. Does fresh or frozen embryo transfer affect imprinted gene expressions in human term placenta? Acta Histochem. 2021, 123, 151694. [Google Scholar] [CrossRef]
- Imudia, A.N.; Suzuki, Y.; Kilburn, B.A.; Yelian, F.D.; Diamond, M.P.; Romero, R.; Armant, D.R. Retrieval of trophoblast cells from the cervical canal for prediction of abnormal pregnancy: A pilot study. Hum. Reprod. 2009, 24, 2086–2092. [Google Scholar] [CrossRef] [Green Version]


| Clinical Characteristics, Number of Study Participants | Analyzed Parameter | Main Findings | First Author; Year; [Reference] |
|---|---|---|---|
| PGDM + GDM, 10 patients with diabetes (5 IDDM, 2 NIDDM, 3 GDM) vs. 9 controls | GLUT1 mRNA GLUT3 mRNA | No difference No difference | Sciullo; 1997; [62] |
| Pregestational diabetes, 6 PGDM vs. 7 controls | MVM GLUT1 protein BM GLUT1 protein | No difference Increased | Gaither; 1999; [56] |
| Pregestational diabetes, 7 White’s class D vs. 21 controls | MVM GLUT1 protein BM GLUT1 protein | No difference Increased | Jansson; 1999; [119] |
| PGDM + GDM, 12 IDDM (6 PGDM, 6 GDMA2) vs. 25 controls | GLUT4 protein GLUT9 protein | Increased Increased | Stanirowski; 2017; [120] |
| Pregestational diabetes, 6 PGDM vs. 25 controls | GLUT1 protein | Increased | Stanirowski; 2017; [120] |
| Pregestational diabetes, 14 T1D, class B vs. 19 controls | MVM GLUT1 protein BM GLUT1 protein | No difference Increased | Borges; 2019; [92] |
| Pregestational diabetes, 20 PGDM vs. 23 controls and 60 GDM | GLUT1 protein GLUT3 protein GLUT8 protein GLUT12 protein | Increased No difference No difference No difference | Stanirowski; 2021; [82] |
| Pregestational diabetes, 9 T2D vs. 9 controls | MVM GLUT1 protein BM GLUT1 protein BM GLUT4 protein | No difference Increased Decreased | Castillo-Castrejon; 2021; [121] |
| Gestational diabetes, 7 GDMA2 vs. 12 controls | GLUT4 mRNA GLUT4 protein | No difference No difference | Xing; 1998; [75] |
| Gestational diabetes, 16 GDM vs. 7 controls | MVM GLUT1 protein BM GLUT1 protein | No difference Increased | Gaither; 1999; [56] |
| Gestational diabetes, 18 GDM vs. 32 controls | MVM GLUT1 protein BM GLUT1 protein | No difference No difference | Jansson; 1999; [122] |
| Gestational diabetes, 6 GDMA2 vs. 7 controls (non-obese) | GLUT1 mRNA GLUT1 protein GLUT4 mRNA GLUT4 protein | No difference Increased Decreased Decreased | Colomiere; 2009; [123] |
| Gestational diabetes, 6 GDMA2 vs. 7 controls (obese) | GLUT1 mRNA GLUT1 protein GLUT4 mRNA GLUT4 protein | No difference No difference Decreased No difference | Colomiere; 2009; [123] |
| Gestational diabetes, 20 GDM vs. 18 controls | GLUT4 mRNA | No difference | Kuzmicki; 2011; [78] |
| Gestational diabetes, 19 GDM vs. 19 controls | GLUT1 mRNA GLUT3 mRNA GLUT4 mRNA | No difference Increased Increased | Dekker; 2014; [124] |
| Gestational diabetes, 36 GDM vs. 40 controls | GLUT3 mRNA | Increased | Rong; 2015; [125] |
| Gestational diabetes, 10 GDM vs. 10 controls | GLUT1 mRNA GLUT1 protein GLUT3 mRNA GLUT3 protein GLUT4 mRNA GLUT4 protein | No difference No difference No difference Decreased Decreased Decreased | Zhang; 2016; [126] |
| Gestational diabetes, 6 GDMA2 vs. 6 GDMA1 vs. 6 controls | GLUT1 mRNA GLUT1 protein | Increased in GDMA2 Increased in GDMA2 | Muralimanoharan; 2016; [127] |
| Gestational diabetes, 26 GDM vs. 13 controls | GLUT1 mRNA GLUT1 protein | Increased Increased | Yao; 2017; [128] |
| Gestational diabetes, 24 GDMA2 vs. 19 controls | MVM GLUT1 protein BM GLUT1 protein | No difference Increased | Borges; 2019; [92] |
| Gestational diabetes, 31 GDM vs. 20 controls | GLUT1 mRNA GLUT1 protein GLUT4 mRNA GLUT4 protein GLUT9 mRNA GLUT9 protein | No difference Increased No difference No difference No difference No difference | Song; 2021; [91] |
| Gestational diabetes, 20 GDM vs. 20 controls | GLUT1 mRNA GLUT1 protein GLUT4 protein | Increased Increased No difference | Balachandiran; 2021; [47] |
| Gestational diabetes, 60 GDM vs. 23 controls | GLUT1 protein GLUT3 protein GLUT8 protein GLUT12 protein | No difference No difference No difference No difference | Stanirowski; 2021; [82] |
| Medical Condition | GLUTs Expression; Main Findings | Association with the Fetal Growth |
|---|---|---|
| Pregestational diabetes mellitus | Multiple conflicting results— most likely increased GLUTs expression | No evidence of association with macrosomia. Positive correlation between GLUT1, GLUT3, and GLUT4 expression and fetal birth weight. |
| Gestational diabetes mellitus | Multiple conflicting results— less pronounced increase in GLUTs expression, more likely in insulin-dependent patients | Not enough evidence supporting association with macrosomia. Potential positive correlation between GLUT1, and GLUT4 expression and fetal birth weight. |
| Abnormal Doppler examination results suggesting the placental insufficiency | Compensatory increased GLUT3 protein expression stimulated by hypoxia | Association with the fetal growth restriction |
| Preeclampsia | Decreased MVM GLUT1 and increased GLUT3 expression | - |
| Maternal obesity | No differences in GLUT1, GLUT4 and GLUT3 placental protein expression in obese and control individuals | Positive correlation between BM GLUT1 expression and fetal birth weight in obese mothers |
| Intrahepatic cholestasis of pregnancy | Increased GLUT1 protein expression | - |
| ART pregnancies | Markedly altered GLUTs mRNA expression | No association |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sibiak, R.; Ozegowska, K.; Wender-Ozegowska, E.; Gutaj, P.; Mozdziak, P.; Kempisty, B. Fetomaternal Expression of Glucose Transporters (GLUTs)—Biochemical, Cellular and Clinical Aspects. Nutrients 2022, 14, 2025. https://0-doi-org.brum.beds.ac.uk/10.3390/nu14102025
Sibiak R, Ozegowska K, Wender-Ozegowska E, Gutaj P, Mozdziak P, Kempisty B. Fetomaternal Expression of Glucose Transporters (GLUTs)—Biochemical, Cellular and Clinical Aspects. Nutrients. 2022; 14(10):2025. https://0-doi-org.brum.beds.ac.uk/10.3390/nu14102025
Chicago/Turabian StyleSibiak, Rafal, Katarzyna Ozegowska, Ewa Wender-Ozegowska, Pawel Gutaj, Paul Mozdziak, and Bartosz Kempisty. 2022. "Fetomaternal Expression of Glucose Transporters (GLUTs)—Biochemical, Cellular and Clinical Aspects" Nutrients 14, no. 10: 2025. https://0-doi-org.brum.beds.ac.uk/10.3390/nu14102025