Genotoxic Effects of Cylindrospermopsin, Microcystin-LR and Their Binary Mixture in Human Hepatocellular Carcinoma (HepG2) Cell Line

 , , and
, , and 
Abstract
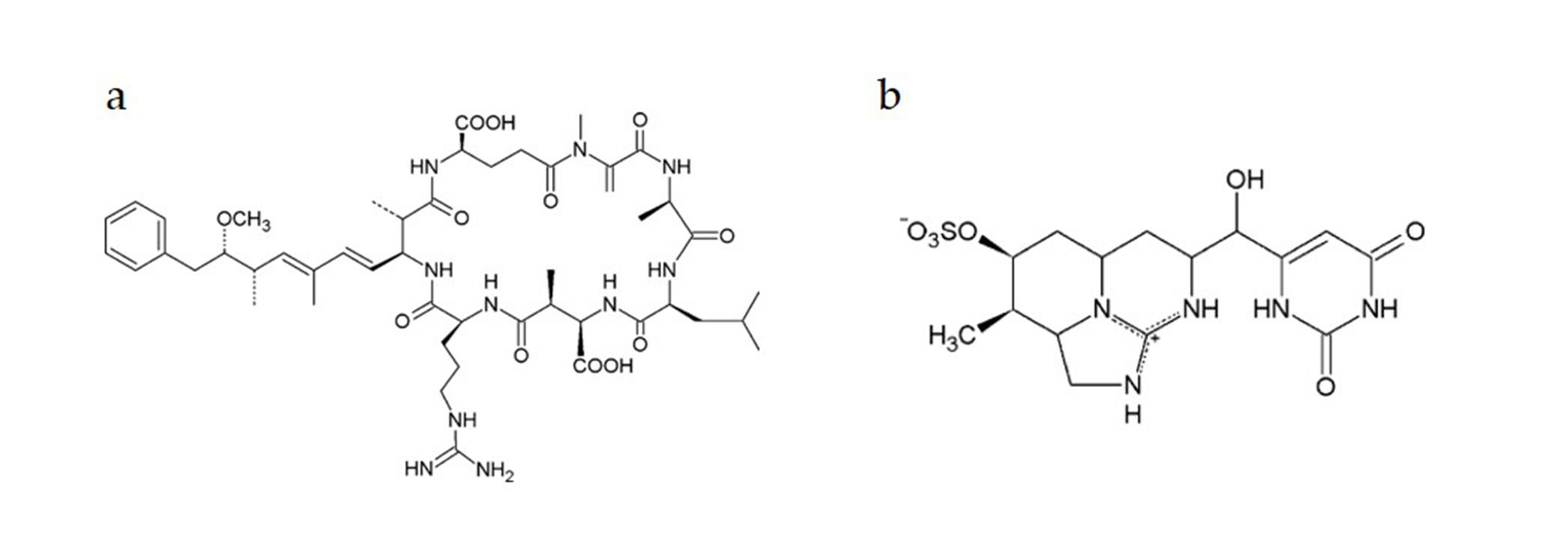
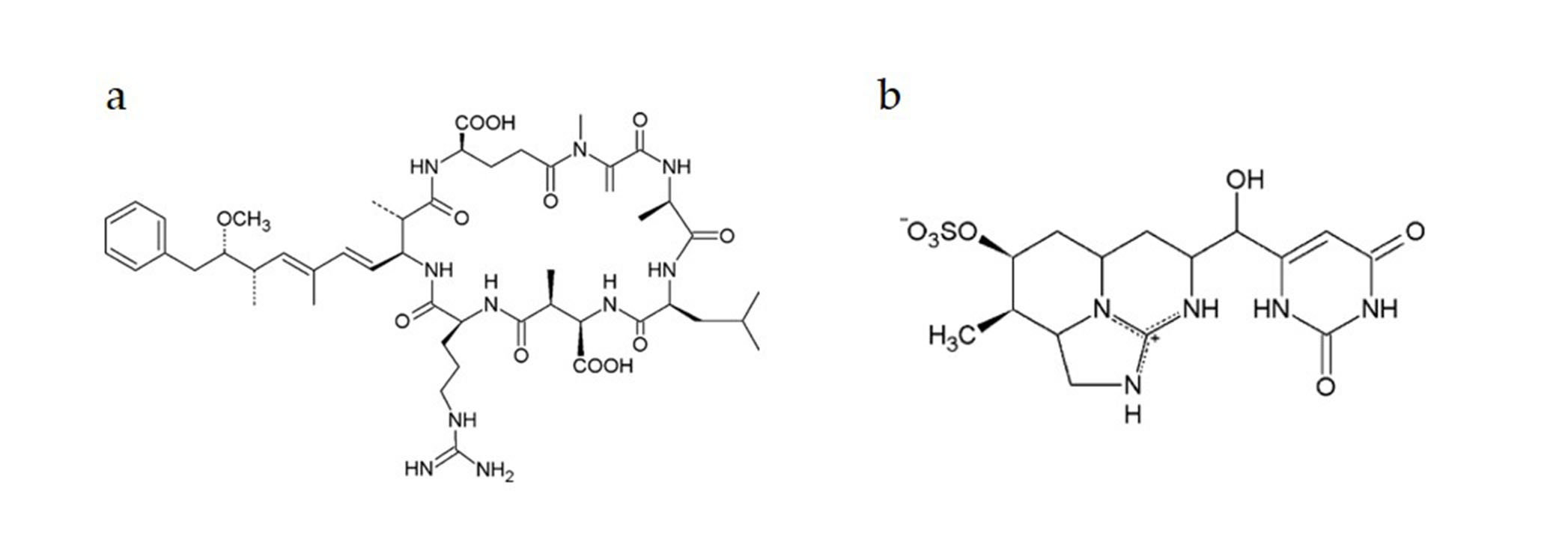
:1. Introduction
2. Results
2.1. Viability of HepG2 Cells Exposed to CYN, MCLR and Their Mixtures
2.2. The Formation of DNA Double Strand Breaks (DSBs) Induced by CYN, MCLR and Their Binary Mixture
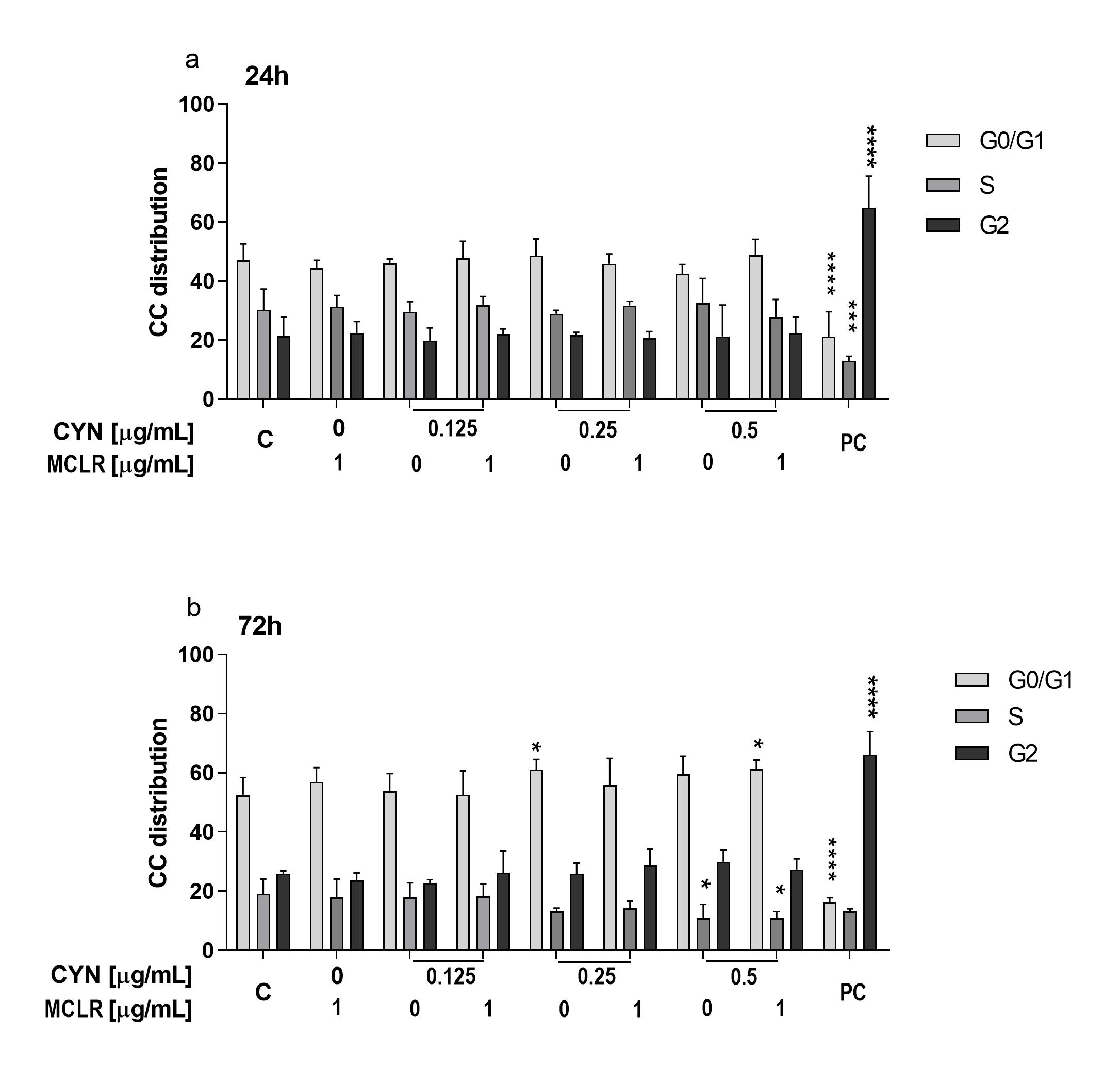
2.3. The Influence of CYN, MCLR and Their Binary Mixture on the Cell-Cycle Progression
2.4. The Effects of CYN, MCLR and Their Mixtures on the mRNA Expression
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Chemicals
5.2. Cell Culture
5.3. Concentrations of CYN, MCLR and Their Combinations for Determining Cytotoxicity, Genotoxicity and mRNA Expression
5.4. Cytotoxicity Assay-MTS Test
5.5. Influence of Cyanotoxins on DNA Double-Strand Break Induction and Cell Cycle Determined by Flow Cytometry
5.6. Gene Expression Analysis by Quantitative Real-Time PCR (qPCR)
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Testai, E.; Buratti, F.M.; Funari, E.; Manganelli, M.; Vichi, S.; Arnich, N.; Biré, R.; Fessard, V.; Sialehaamoa, A. Review and analysis of occurrence, exposure and toxicity of cyanobacteria toxins in food. EFSA Support. Publ. 2016, 13, 1–309. [Google Scholar] [CrossRef]
- Buratti, F.M.; Manganelli, M.; Vichi, S.; Stefanelli, M.; Scardala, S.; Testai, E.; Funari, E. Cyanotoxins: Producing organisms, occurrence, toxicity, mechanism of action and human health toxicological risk evaluation. Arch. Toxicol. 2017, 91, 1049–1130. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, C.; Pflugmacher, S. Ecotoxicological effects of selected cyanobacterial secondary metabolites a short review. Toxicol. Appl. Pharmacol. 2005, 203, 201–218. [Google Scholar] [CrossRef] [PubMed]
- Žegura, B.; Štraser, A.; Filipič, M. Genotoxicity and potential carcinogenicity of cyanobacterial toxins—A review. Mutat. Res. Rev. Mutat. Res. 2011, 727, 16–41. [Google Scholar] [CrossRef] [PubMed]
- Zervou, S.K.; Christophoridis, C.; Kaloudis, T.; Triantis, T.M.; Hiskia, A. New SPE-LC-MS/MS method for simultaneous determination of multi-class cyanobacterial and algal toxins. J. Hazard. Mater. 2017, 323, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Catherine, A.; Bernard, C.; Spoof, L.; Bruno, M. Microcystins and Nodularins. In Handbook of Cyanobacterial Monitoring and Cyanotoxin Analysis, 1st ed.; Meriluoto, J., Spoof, L., Cood, G.A., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2017; pp. 109–126. [Google Scholar] [CrossRef]
- Bouaïcha, N.; Miles, C.O.; Beach, D.G.; Labidi, Z.; Djabri, A.; Benayache, N.Y.; Nguyen-Quang, T. Structural Diversity, Characterization and Toxicology of Microcystins. Toxins 2019, 11, 714. [Google Scholar] [CrossRef] [Green Version]
- Díez-Quijada, L.; Puerto, M.; Gutiérrez-Praena, D.; Llana-Ruiz-Cabello, M.; Jos, A.; Cameán, A.M. Microcystin-RR: Occurrence, content in water and food and toxicological studies. A review. Environ. Res. 2019, 168, 467–489. [Google Scholar] [CrossRef]
- Diez-Quijada, L.; Prieto, A.I.; Guzmán-Guillen, R.; Jos, Á.; Cameán, A.M. Occurrence and toxicity of microcystin congeners other than MC-LR and MC-RR: A review. Food Chem. Toxicol. 2019, 125, 106–132. [Google Scholar] [CrossRef]
- Fischer, W.J.; Altheimer, S.; Cattori, V.; Meier, P.J.; Dietrich, D.R.; Hagenbuch, B. Organic anion transporting polypeptides expressed in liver and brain mediate uptake of microcystin. Toxicol. Appl. Pharmacol. 2005, 203, 257–263. [Google Scholar] [CrossRef] [Green Version]
- Žegura, B. An overview of the mechanisms of microcystin-LR genotoxicity and potential carcinogenicity. Mini Rev. Med. Chem. 2016, 16, 1042–1062. [Google Scholar] [CrossRef]
- Žegura, B.; Lah, T.T.; Filipič, M. The role of reactive oxygen species in microcystin-LR-induced DNA damage. Toxicology 2004, 200, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Puerto, M.; Pichardo, S.; Jos, Á.; Prieto, A.I.; Sevilla, E.; Frías, J.E.; Cameán, A.M. Differential oxidative stress response to pure Microcystin-LR and Microcystin-containing and non-containing cyanobacterial crude extracts on Caco-2 cells. Toxicon 2010, 55, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Lankoff, A.; Krzowski, L.; Glab, J.; Banasik, A.; Lisowska, H.; Kuszewski, T.; Góźdź, S.; Wójcik, A. DNA damage and repair in human peripheral blood lymphocytes following treatment with microcystin-LR. Mutat. Res.Genet. Toxicol. Environ. Mutagen. 2004, 559, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Ito, E.; Kondo, F.; Terao, K.; Harada, K.I. Neoplastic nodular formation in mouse liver induced by repeated intraperitoneal injections of microcystin-LR. Toxicon 1997, 35, 1453–1457. [Google Scholar] [CrossRef]
- Nishiwaki-Matsushima, R.; Ohta, T.; Nishiwaki, S.; Suganuma, M.; Kohyama, K.; Ishikawa, T.; Carmichael, W.W.; Fujiki, H. Liver tumor promotion by the cyanobacterial cyclic peptide toxin microcystin-LR. J. Cancer Res. Clin. Oncol. 1992, 118, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Svirčev, Z.; Drobac, D.; Tokodi, N.; Mijović, B.; Codd, G.A.; Meriluoto, J. Toxicology of microcystins with reference to cases of human intoxications and epidemiological investigations of exposures to cyanobacteria and cyanotoxins. Arch. Toxicol. 2017, 91, 621–650. [Google Scholar] [CrossRef]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Ingested Nitrate and Nitrite, and Cyanobacterial Peptide Toxins. 2010; World Health Organization, International Agency for Research on Cancer. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/books/NBK326544/pdf/Bookshelf_NBK326544.pdf (accessed on 28 September 2020).
- Kokocinski, M.; Cameán, A.M.; Carmeli, S.; Guzmán-Guillén, R.; Jos, Á.; Mankiewicz-Boczek, J.; Metcalf, J.S.; Moreno, I.; Prieto, A.I.; Sukenik, A. Cylindrospermopsin and congeners. In Handbook of Cyanobacterial Monitoring and Cyanotoxin Analysis, 1st ed.; Meriluoto, J., Spoof, L., Codd, G.A., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2017; pp. 127–137. [Google Scholar] [CrossRef]
- Ohtani, I.; Moore, R.E.; Runnegar, M.T.C. Cylindrospermopsin: A potent hepatotoxin from the blue-green alga Cylindrospermopsis raciborskii. J. Am. Chem. Soc. 1992, 114, 7941–7942. [Google Scholar] [CrossRef]
- Chiswell, R.K.; Shaw, G.R.; Eaglesham, G.; Smith, M.J.; Norris, R.L.; Seawright, A.A.; Moore, M.R. Stability of cylindrospermopsin, the toxin from the cyanobacterium, Cylindrospermopsis raciborskii: Effect of pH, temperature, and sunlight on decomposition. Environ. Toxicol. 1999, 14, 155–161. [Google Scholar] [CrossRef]
- Hawkins, P.R.; Runnegar, M.T.C.; Jackson, A.R.B.; Falconer, I.R. Severe hepatotoxicity caused by the tropical cyanobacterium (blue-green alga) Cylindrospermopsis raciborskii (Woloszynska) Seenaya and Subba Raju isolated from a domestic water supply reservoir. Appl. Environ. Microbiol. 1985, 50, 1292–1295. [Google Scholar] [CrossRef] [Green Version]
- Falconer, I.R.; Humpage, A.R. Cyanobacterial (blue-green algal) toxins in water supplies: Cylindrospermopsins. Environ. Toxicol. 2006, 21, 299–304. [Google Scholar] [CrossRef]
- Guzmán-Guillén, R.; Prieto, A.I.; Moyano, R.; Blanco, A.; Vasconcelos, V.; Cameán, A.M. Dietary L-carnitine prevents histopathological changes in tilapia (Oreochromis niloticus) exposed to cylindrospermopsin. Environ. Toxicol. 2017, 32, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Terao, K.; Ohmori, S.; Igarashi, K.; Ohtani, I.; Watanabe, M.F.; Harada, K.I.; Ito, E.; Watanabe, M. Electron microscopic studies on experimental poisoning in mice induced by cylindrospermopsin isolated from blue-green alga Umezakia natans. Toxicon 1994, 32, 833–843. [Google Scholar] [CrossRef]
- Froscio, S.M.; Humpage, A.R.; Burcham, P.C.; Falconer, I.R. Cylindrospermopsin-induced protein synthesis inhibition and its dissociation from acute toxicity in mouse hepatocytes. Environ. Toxicol. 2003, 18, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Runnegar, M.T.; Kong, S.-M.; Zhong, Y.-Z.; Lu, S.C. Inhibition of reduced glutathione synthesis by cyanobacterial alkaloid cylindrospermopsin in cultured rat hepatocytes. Biochem. Pharmacol. 1995, 49, 219–225. [Google Scholar] [CrossRef]
- Norris, R.L.G.; Seawright, A.A.; Shaw, G.R.; Senogles, P.; Eaglesham, G.K.; Smith, M.J.; Chiswell, R.K.; Moore, M.R. Hepatic xenobiotic metabolism of cylindrospermopsin in vivo in the mouse. Toxicon 2002, 40, 471–476. [Google Scholar] [CrossRef]
- Humpage, A.R.; Fontaine, F.; Froscio, S.; Burcham, P.; Falconer, I.R. Cylindrospermopsin genotoxicity and cytotoxicity: Role of cytochrome P-450 and oxidative stress. J. Toxicol. Environ. Health Part A 2005, 68, 739–753. [Google Scholar] [CrossRef] [PubMed]
- Pichardo, S.; Cameán, A.M.; Jos, Á. In vitro toxicological assessment of Cylindrospermopsin: A review. Toxins 2017, 9, 402. [Google Scholar] [CrossRef] [Green Version]
- Bazin, E.; Mourot, A.; Humpage, A.R.; Fessard, V. Genototoxicity of a freshwater cyanotoxin, cylindrospermopsin, in two human cell lines: Caco-2 and HepaRG. Environ. Mol. Mutat. 2010, 51, 251–259. [Google Scholar] [CrossRef]
- Štraser, A.; Filipič, M.; Žegura, B. Genotoxic effects of the cyanobacterial hepatotoxin cylindrospermopsin in the HepG2 cell line. Arch. Toxicol. 2011, 85, 1617–1626. [Google Scholar] [CrossRef]
- Štraser, A.; Filipič, M.; Žegura, B. Cylindrospermopsin induced transcriptional responses in human hepatoma HepG2 cells. Toxicol. In Vitro 2013, 27, 1809–1819. [Google Scholar] [CrossRef]
- Hercog, K.; Maisanaba, S.; Filipič, M.; Jos, Á.; Cameán, A.M.; Žegura, B. Genotoxic potential of the binary mixture of cyanotoxins microcystin-LR and cylindrospermopsin. Chemosphere 2017, 189, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Puerto, M.; Prieto, A.I.; Maisanaba, S.; Gutiérrez-Praena, D.; Mellado-García, P.; Jos, Á.; Cameán, A.M. Mutagenic and genotoxic potential of pure Cylindrospermopsin by a battery of in vitro tests. Food Chem. Toxicol. 2018, 121, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Hercog, K.; Štampar, M.; Štern, A.; Filipič, M.; Žegura, B. Application of advanced HepG2 3D cell model for studying genotoxic activity of cyanobacterial toxin cylindrospermopsin. Environ. Pollut. 2020, 265, 114965. [Google Scholar] [CrossRef] [PubMed]
- Humpage, A.R.; Fenech, M.; Thomas, P.; Falconer, I.R. Micronucleus induction and chromosome loss in transformed human white cells indicate clastogenic and aneugenic action of the cyanobacterial toxin, cylindrospermopsin. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2000, 472, 155–161. [Google Scholar] [CrossRef]
- Žegura, B.; Gajski, G.; Štraser, A.; Garaj-Vrhovac, V. Cylindrospermopsin induced DNA damage and alteration in the expression of genes involved in the response to DNA damage, apoptosis and oxidative stress. Toxicon 2011, 58, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Štraser, A.; Filipič, M.; Novak, M.; Žegura, B. Double strand breaks and cell-cycle arrest induced by the cyanobacterial toxin cylindrospermopsin in HepG2 cells. Mar. Drugs 2013, 11, 3077–3090. [Google Scholar] [CrossRef]
- Sieroslawska, A.; Rymuszka, A. Cylindrospermopsin induces oxidative stress and genotoxic effects in the fish CLC cell line. J. Appl. Toxicol. 2015, 35, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Hercog, K.; Štern, A.; Maisanaba, S.; Filipič, M.; Žegura, B. Plastics in Cyanobacterial Blooms—Genotoxic Effects of Binary Mixtures of Cylindrospermopsin and Bisphenols in HepG2 Cells. Toxins 2020, 12, 219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fastner, J.; Rücker, J.; Stüken, A.; Preußel, K.; Nixdorf, B.; Chorus, I.; Köhler, A.; Wiedner, C. Occurrence of the cyanobacterial toxin Cylindrospermopsin in northeast Germany. Environ. Toxicol. 2007, 22, 26–32. [Google Scholar] [CrossRef]
- Dantas, Ê.W.; Moura, A.N.; Bittencourt-Oliveira, M.D.C. Cyanobacterial blooms in stratified and destratified eutrophic reservoirs in semi-arid region of Brazil. An. Acad. Bras. Cienc. 2011, 83, 1327–1338. [Google Scholar] [CrossRef] [Green Version]
- Bittencourt-Oliveira, M.; Carmo, D.; Piccin-Santos, V.; Moura, A.N.; Aragão-Tavares, N.K.; Cordeiro-Araújo, M.K. Cyanobacteria, microcystins and cylindrospermopsin in public drinking supply reservoirs of Brazil. An. Acad. Bras. Cienc. 2014, 86, 297–310. [Google Scholar] [CrossRef] [Green Version]
- Jančula, D.; Straková, L.; Sadílek, J.; Maršálek, B.; Babica, P. Survey of cyanobacterial toxins in Czech water reservoirs—The first observation of neurotoxic saxitoxins. Environ. Sci. Pollut. Res. Int. 2014, 21, 8006–8015. [Google Scholar] [CrossRef] [PubMed]
- León, C.; Peñuela, G.A. Detected cyanotoxins by UHPLC MS/MS technique in tropical reservoirs of northeastern Colombia. Toxicon 2019, 167, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Lukas, J. Mammalian G1- and S-phase checkpoints in response to DNA damage. Curr. Opin. Cell Biol. 2001, 13, 738–747. [Google Scholar] [CrossRef]
- Huisman, J.; Codd, G.A.; Paerl, H.W.; Ibelings, B.W.; Verspagen, J.M.H.; Visser, P.M. Cyanobacterial blooms. Nat. Rev. Microbiol. 2018, 16, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Praena, D.; Guzmán-Guillén, R.; Pichardo, S.; Moreno, F.J.; Vasconcelos, V.; Jos, Á.; Cameán, A.M. Cytotoxic and morphological effects of microcystin-LR, cylindrospermopsin, and their combinations on the human hepatic cell line HepG2. Environ. Toxicol. 2018, 34, 240–251. [Google Scholar] [CrossRef]
- Hinojosa, M.G.; Prieto, A.I.; Gutiérrez-Praena, D.; Moreno, F.J.; Cameán, A.M.; Jos, Á. Neurotoxic assessment of Microcystin-LR, cylindrospermopsin and their combination on the human neuroblastoma SH-SY5Y cell line. Chemosphere 2019, 224, 751–764. [Google Scholar] [CrossRef]
- Clouaire, T.; Rocher, V.; Lashgari, A.; Arnould, C.; Aguirrebengoa, M.; Biernacka, A.; Skrzypczak, M.; Aymard, F.; Fongang, B.; Dojer, N.; et al. Comprehensive mapping of histone modifications at DNA double-strand breaks deciphers repair pathway chromatin signatures. Mol. Cell 2018, 72, 250–262. [Google Scholar] [CrossRef] [Green Version]
- Kuo, L.J.; Yang, L.-X. γ-H2AX—A Novel Biomarker for DNA Double-strand Breaks. In Vivo 2008, 22, 305–309. [Google Scholar]
- Watters, G.P.; Smart, D.J.; Harvey, J.S.; Austin, C.A. H2AX phosphorylation as a genotoxicity endpoint. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2009, 679, 50–58. [Google Scholar] [CrossRef]
- Fenech, M.; Kirsch-Volders, M.; Natarajan, A.T.; Suralles, J.; Crott, J.W.; Parry, J.; Norppa, H.; Eastmond, D.A.; Tucker, J.D.; Thomas, P. Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis 2011, 26, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diez-Quijada, L.; Prieto, A.I.; Jos, Á.; Cameán, A.M. In vitro mutagenic and genotoxic assessment of a mixture of the cyanotoxins Microcystin-LR and Cylindrospermopsin. Toxins 2019, 11, 318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartek, J.; Lukas, C.; Lukas, J. Checking on DNA damage in S phase. Nat. Rev. Mol. Cell Biol. 2004, 5, 792–804. [Google Scholar] [CrossRef]
- Patil, M.; Pabla, N.; Dong, Z. Checkpoint kinase 1 in DNA damage response and cell cycle regulation. Cell. Mol. Life Sci. 2013, 70, 4009–4021. [Google Scholar] [CrossRef] [Green Version]
- Poniedzialek, B.; Rzymski, P.; Wiktorowicz, K. Toxicity of cylindrospermopsin in human lymphocytes: Proliferation, viability and cell cycle studies. Toxicol. In Vitro 2014, 28, 968–974. [Google Scholar] [CrossRef] [PubMed]
- Kastan, M.B.; Lim, D.S. The many substrates and functions of ATM. Nat. Rev. Mol. Cell Biol. 2000, 1, 179–186. [Google Scholar] [CrossRef]
- Lankoff, A.; Banasik, A.; Obe, G.; Deperas, M.; Kuzminski, K.; Tarczynska, M.; Jurczak, T.; Wojcik, A. Effect of microcystin-LR and cyanobacterial extract from Polish reservoir of drinking water on cell cycle progression, mitotic spindle, and apoptosis in CHO-K1 cells. Toxicol. Appl. Pharmacol. 2003, 189, 204–213. [Google Scholar] [CrossRef]
- Liu, J.; Wang, H.; Wang, B.; Chen, T.; Wang, X.; Huang, P.; Xu, L.; Guo, Z. Microcystin-LR promotes proliferation by activating Akt/S6K1 pathway and disordering apoptosis and cell cycle associated proteins phosphorylation in HL7702 cells. Toxicol. Lett. 2016, 240, 214–225. [Google Scholar] [CrossRef]
- Manikandan, P.; Nagini, S. Cytochrome P450 Structure, Function and Clinical Significance: A Review. Curr. Drug Targets 2018, 19, 38–54. [Google Scholar] [CrossRef]
- Kittler, K.; Hurtaud-Pessel, D.; Maul, R.; Kolrep, F.; Fessard, V. In vitro metabolism of the cyanotoxin cylindrospermopsin in HepaRG cells and liver tissue fractions. Toxicon 2016, 110, 47–50. [Google Scholar] [CrossRef]
- Štraser, A.; Filipič, M.; Gorenc, I.; Žegura, B. The influence of cylindrospermopsin on oxidative DNA damage and apoptosis induction in HepG2 cells. Chemosphere 2013, 92, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Gajjar, K.; Martin-Hirsch, P.L.; Martin, F.L. CYP1B1 and hormone-induced cancer. Cancer Lett. 2012, 324, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Oren, M. Decision making by p53: Life, death and cancer. Cell Death Differ. 2003, 10, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Lakin, N.D.; Jackson, S.P. Regulation of p53 in response to DNA damage. Oncogene 1999, 18, 7644–7655. [Google Scholar] [CrossRef] [Green Version]
- Ellinger-Ziegelbauer, H.; Stuart, B.; Wahle, B.; Bomann, W.; Ahr, H.J. Comparison of the expression profiles induced by genotoxic and nongenotoxic carcinogens in rat liver. Mutat. Res. Mol. Mech. Mutagen. 2005, 575, 61–84. [Google Scholar] [CrossRef]
- Petković, J.; Žegura, B.; Stevanović, M.; Drnovšek, N.; Uskoković, D.; Novak, S.; Filipič, M. DNA damage and alterations in expression of DNA damage responsive genes induced by TiO2 nanoparticles in human hepatoma HepG2 cells. Nanotoxicology 2011, 5, 341–353. [Google Scholar] [CrossRef] [Green Version]
- Pezdirc, M.; Žegura, B.; Filipič, M. Genotoxicity and induction of DNA damage responsive genes by food-borne heterocyclic aromatic amines in human hepatoma HepG2 cells. Food Chem. Toxicol. 2013, 59, 386–394. [Google Scholar] [CrossRef]
- Wade, M.; Li, Y.-C.; Wahl, G.M. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 2013, 13, 83–96. [Google Scholar] [CrossRef] [Green Version]
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef]
- Cazzalini, O.; Scovassi, A.I.; Savio, M.; Stivala, L.A.; Prosperi, E. Multiple roles of the cell cycle inhibitor p21CDKN1A in the DNA damage response. Mutat. Res. Rev. Mutat. 2010, 704, 12–20. [Google Scholar] [CrossRef]
- Hoffman, B.; Liebermann, D.A. Gadd45 modulation of intrinsic and extrinsic stress responses in myeloid cells. J. Cell. Physiol. 2009, 218, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Fornace, A.J., Jr.; Jackman, J.; Hollander, M.C.; Hoffman-Liebermann, B.; Liebermann, D.A. Genotoxic-stress-response genes and growth-arrest genes. gadd, MyD, and other genes induced by treatments eliciting growth arrest. Ann. N. Y. Acad. Sci. 1992, 663, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Piechaczyk, M.; Farràs, R. Regulation and function of JunB in cell proliferation. Biochem. Soc. Trans. 2008, 36, 864–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knasmüller, S.; Mersch-Sundermann, V.; Kevekordes, S.; Darroudi, F.; Huber, W.W.; Hoelzl, C.; Bichler, J.; Majer, B.J. Use of human-derived liver cell lines for the detection of environmental and dietary genotoxicants; current state of knowledge. Toxicology 2004, 198, 315–328. [Google Scholar] [CrossRef]
- Bressac, B.; Galvin, K.M.; Liang, T.J.; Isselbacher, K.J.; Wands, J.R.; Ozturk, M. Abnormal structure and expression of p53 gene in human hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 1990, 87, 1973–1977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Delft, J.H.M.; van Agen, E.; van Breda, S.G.J.; Herwijnen, M.H.; Staal, Y.C.M.; Kleinjans, J.C.S. Discrimination of genotoxic from non-genotoxic carcinogens by gene expression profiling. Carcinogenesis 2004, 25, 1265–1276. [Google Scholar] [CrossRef] [Green Version]
- Donato, M.T.; Tolosa, L.; Gómez-Lechón, M.J. Culture and Functional Characterization of Human Hepatoma HepG2 Cells. Methods Mol Biol. 2015, 1250, 77–93. [Google Scholar] [CrossRef]
- Pinheiro, J.; Bates, D.; DebRoy, S.; Sarkar, D.; R Core Team. nlme: Linear and Nonlinear Mixed Effects Models. R Packag Version 3.1-150. 2020. Available online: https://www.R-project.org/ (accessed on 6 December 2020).
- Baebler, Š.; Svalina, M.; Petek, M.; Stare, K.; Rotter, A.; Pompe-Novak, M.; Gruden, K. quantGenius: Implementation of a decision support system for qPCR-based gene quantification. BMC Bioinform. 2017, 18, 1–11. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Involved Pathway | Gene Symbol | Treatment (µg/mL) | 72 h Mean ± SD | Entrez Gene Name |
|---|---|---|---|---|
| Metabolism (activation/detoxification) | CYP1A1 | 0.5 CYN 1 MCLR 0.5 CYN + 1 MCLR B(a)P 30 µM | 2.59 ± 0.89 1.03 ± 0.13 2.53 ± 1.53 48.87 ± 0.81 **** | Cytochrome P450 family 1 Subfamily A polypeptide 1 |
| UGT1A1 | 0.5 CYN 1 MCLR 0.5 CYN + 1 MCLR B(a)P 30 µM | 0.96 ± 0.45 0.97 ± 0.25 1.20 ± 0.69 2.94 ± 1.15 * | UDP glucuronosyltransferase 1 family polypeptide A1 | |
| Immediate-early response/signaling | JUNB | 0.5 CYN 1 MCLR 0.5 CYN + 1 MCLR B(a)P 30 µM | 1.22 ± 0.26 1.10 ± 0.24 1.16 ± 0.35 2.60 ± 0.45 *** | Jun B proto-oncogene |
| DNA damage responsive | TP53 | 0.5 CYN 1 MCLR 0.5 CYN + 1 MCLR B(a)P 30 µM | 0.76 ± 0.02 **** 0.96 ± 0.02 0.75 ± 0.06 **** 1.16 ± 0.04 ** | Tumor protein P53 |
| MDM2 | 0.5 CYN 1 MCLR 0.5 CYN + 1 MCLR B(a)P 30 µM | 1.25 ± 0.29 1.08 ± 0.08 1.11 ± 0.21 1.39 ± 0.08 | MDM2 oncogene, E3 ubiquitin-protein ligase | |
| CDKN1A | 0.5 CYN 1 MCLR 0.5 CYN + 1 MCLR B(a)P 30 µM | 2.44 ± 0.30 *** 1.12 ± 0.24 2.47 ± 0.43 *** 8.61 ± 0.16 **** | Cyclin-Dependent Kinase Inhibitor 1A | |
| GADD45A | 0.5 CYN 1 MCLR 0.5 CYN + 1 MCLR B(a)P 30 µM | 3.16 ± 0.54 ** 1.11 ± 0.10 3.06 ± 0.89 ** 3.76 ± 0.75 *** | Growth arrest and DNA damage-inducible gene, alpha |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Díez-Quijada, L.; Hercog, K.; Štampar, M.; Filipič, M.; Cameán, A.M.; Jos, Á.; Žegura, B. Genotoxic Effects of Cylindrospermopsin, Microcystin-LR and Their Binary Mixture in Human Hepatocellular Carcinoma (HepG2) Cell Line. Toxins 2020, 12, 778. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12120778
Díez-Quijada L, Hercog K, Štampar M, Filipič M, Cameán AM, Jos Á, Žegura B. Genotoxic Effects of Cylindrospermopsin, Microcystin-LR and Their Binary Mixture in Human Hepatocellular Carcinoma (HepG2) Cell Line. Toxins. 2020; 12(12):778. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12120778
Chicago/Turabian StyleDíez-Quijada, Leticia, Klara Hercog, Martina Štampar, Metka Filipič, Ana M. Cameán, Ángeles Jos, and Bojana Žegura. 2020. "Genotoxic Effects of Cylindrospermopsin, Microcystin-LR and Their Binary Mixture in Human Hepatocellular Carcinoma (HepG2) Cell Line" Toxins 12, no. 12: 778. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12120778