
Stability of FeVO4-II under Pressure: A First-Principles Study


1
División de Materiales Avanzados, IPICYT, Camino a la presa de San José 2055 Col. Lomas 4a sección, San Luis Potosí 78126, Mexico
2
CONACYT—División de Materiales Avanzados, IPICYT, Camino a la presa de San José 2055 Col. Lomas 4a sección, San Luis Potosí 78126, Mexico
3
Centro Nacional de Supercómputo, IPICYT, Camino a la presa de San José 2055 Col. Lomas 4a sección, San Luis Potosí 78126, Mexico
4
Departamento de Física Aplicada, Instituto de Ciencias de Materiales, MALTA Consolider Team, Universidad de Valencia, 46100 Valencia, Spain
*
Author to whom correspondence should be addressed.
Crystals 2022, 12(12), 1835; https://0-doi-org.brum.beds.ac.uk/10.3390/cryst12121835
Submission received: 18 November 2022
/
Revised: 5 December 2022
/
Accepted: 13 December 2022
/
Published: 15 December 2022
(This article belongs to the Special Issue Pressure-Induced Phase Transformations (Volume II))
Abstract
:In this work, we report first-principles calculations to study FeVO in the CrVO-type (phase II) structure under pressure. Total-energy calculations were performed in order to analyze the structural parameters, the electronic, elastic, mechanical, and vibrational properties of FeVO-II up to 9.6 GPa for the first time. We found a good agreement in the structural parameters with the experimental results available in the literature. The electronic structure analysis was complemented with results obtained from the Laplacian of the charge density at the bond critical points within the Quantum Theory of Atoms in Molecules methodology. Our findings from the elastic, mechanic, and vibrational properties were correlated to determine the elastic and dynamic stability of FeVO-II under pressure. Calculations suggest that beyond the maximum pressure covered by our study, this phase could undergo a phase transition to a wolframite-type structure, such as in CrVO and InVO.
1. Introduction
High-pressure studies on O compounds have increased in the last two decades due to the evolution of the diamond-anvil cell (DAC) and its impact on other relevant areas, such as materials sciences [1,2]. Among O compounds, those with a CrVO-type structure (Space Group, SG, , No. 63, Z = 4, with [6 − 4] coordination for the [A-B cations]) have received less attention. CrVO-type compounds are a family formed mainly by phosphates, vanadates, chromates, and sulfates [3]. Given their position in the Bastide diagram [4], it has been reported that they could have a structural phase transition driven by pressure to the wolframite [5,6] or zircon [7,8] structure. This implies a change in the coordination to [6 − 4 + 2] and [8 − 4] in the high-pressure phases, respectively.
According to the literature, AVO vanadates with CrVO-type structures have been used in applications as photocatalyst materials [1,9,10], cathodoluminescent materials [11,12], and scintillators [13,14], among others. The properties of these compounds are reported to be highly dependent on the occupation of the d valence electrons of the transition metal cations [9,10,11,12,13,14]. Until now, only four vanadates with a CrVO-type structure have been reported: CrVO [6], FeVO [15], InVO [16], and the less studied TlVO [3]. Unlike the other vanadates, FeVO presents a great polymorphism, crystallizing at ambient conditions in the triclinic AlVO-type [17] structure (SG: , No. 2, Z = 6) [15], while the CrVO-type phase was obtained from high-pressure-high-temperature studies [18] and synthesized at ambient conditions by hydrothermal method [19]. Further, the antiferromagnetic order in FeVO-II was observed after the characterization of its magnetic properties [19]. However, until now, no more experimental or theoretical studies have been reported to characterize this phase. It is worth mentioning that most of the studies carried out at high pressure on this compound have focused on characterizing the crystal structure of the different polymorphs [15,18,20,21,22].
In this work, we performed a first-principles characterization of FeVO-II up to 9.6 GPa. The main goal is to determine the elastic and dynamic stability of the studied polymorph at ambient conditions and under pressure. In this sense, a complete report of the elastic constants, mechanical properties, elastic anisotropy, and vibrational properties is presented. Furthermore, given the relevance of the electronic structure of this compound in materials science, we computed the density of states and the band structure together with an analysis of the charge density with the QTAIM methodology [23,24].
2. Computational Details
Total energy calculations were performed within the framework of the density functional theory (DFT) [25] and the projector-augmented wave (PAW) [26,27] method as implemented in the Vienna Ab initio Simulation Package (VASP) [28,29,30,31]. We used a plane-wave energy cutoff of 520 eV to ensure high precision in our calculations. The exchange-correlation energy was described within the generalized gradient approximation (GGA) in the Perdew–Burke–Ernzerhof for solids (PBEsol) formulation [32]. The GGA+U was used to account for the strong correlation between the electrons in the d shell, on the basis of Dudarev’s method [33]. In this method, the Coulomb interaction U and the onsite exchange interaction are treated together as = U - . For our GGA+U calculations, we choose U = 6 eV and = 0.95 eV. Similar values were previously used with relative success in the study of other iron and vanadate compounds [15,20,34,35,36,37].
The Monkhorst-Pack scheme was employed for the Brillouin-zone (BZ) integrations [38] with a mesh 4 × 3 × 3, which corresponds to a set of 8 special k-points in the irreducible BZ. In the relaxed equilibrium configuration, the forces are less than 0.3 meV/Å per atom in each Cartesian direction. The highly converged results on forces are required for the calculations of the dynamical matrix using the direct force constant approach [39], which allows us to identify the irreducible representation and the character of the phonon modes at the zone center ( point). The phonon density of states (PDOS) were obtained from the calculation of the phonons in the whole BZ with a supercell 2 × 2 × 2 times the conventional unit cell by using the PHONON software. The calculations of elastic constants were made following the methodology validated in Ref. [40] with a k-points mesh of 8 × 6 × 6, a plane-wave energy cutoff of 570 eV, and a POTIM parameter of 0.016. The elastic tensor is determined by performing six finite distortions of the lattice and deriving the elastic constants from the strain–stress relationship [41]. The electron density was computed with a refined grid to be analyzed within the fundaments of Quantum Theory of Atoms in Molecules (QTAIM) [42] with CRITIC [43,44], and AIM [45] codes.
3. Results and Discussion
3.1. Crystal Structure
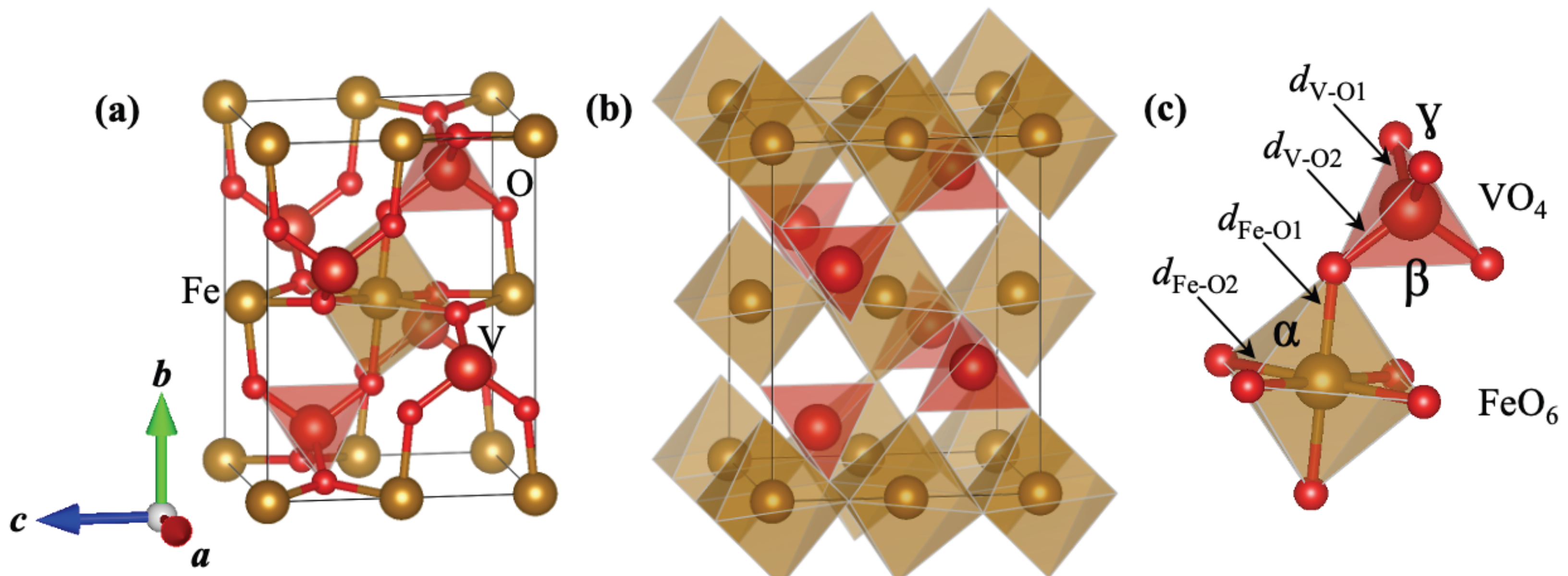
According to the literature, FeVO has been successfully synthesized as a metastable polymorph at ambient conditions in the orthorhombic CrVO-type structure (SG: , No. 63, Z = 4) [19], also called the FeVO-II phase, see Figure 1a. In this structure, there are four non-equivalent Wyckoff positions (WP): Fe occupies the 4a (0, 0, 0), V the 4c (0, y, 1/4), and O the 8f (0, y, z), and 8g (x, y, 1/4). Internally, the structure consists of edge-sharing FeO distorted octahedra along the c direction. The –FeO–FeO– chains are linked together by means of the VO distorted tetrahedra, while the VO tetrahedra are not linked to each other, as can be seen in Figure 1b. As shown in Figure 1c, there are two non-equivalent interatomic distances in the FeO octahedra, the apical Fe-O distance () and the equatorial Fe-O distance (). In the VO tetrahedra, there are two V–O interatomic distances ( and ). In both polyhedra, the respective interatomic distance is larger than (X = Fe, V). Inside the polyhedrons, there are important angles to mention that are relevant for the structural changes in the polyhedra under pressure. The angle is formed between the equatorial plane of the FeO octahedra and the polar bond, while the () angle is between the two () interatomic distances of the VO tetrahedra.
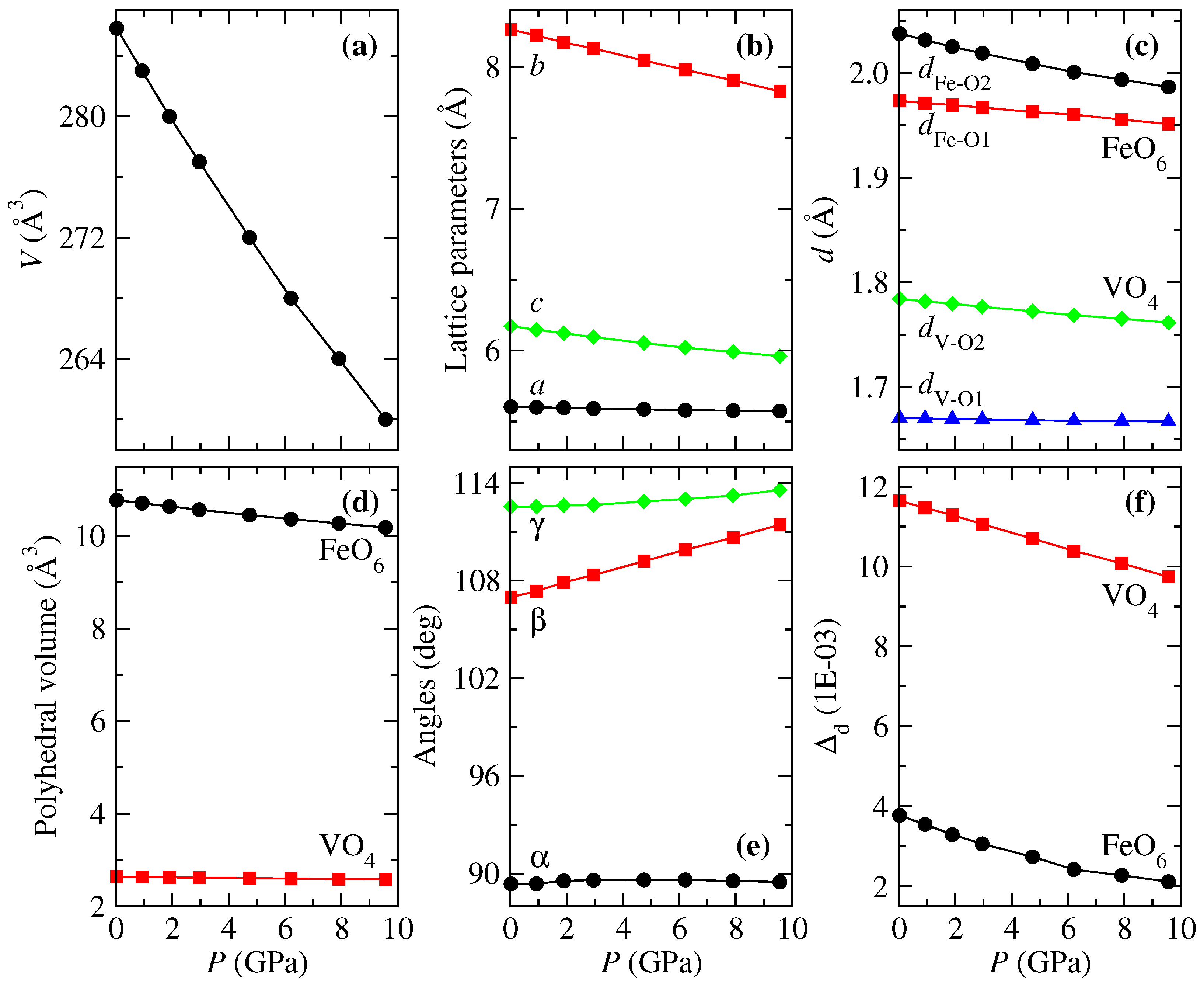
The equilibrium volume and unit-cell parameters at GPa were computed by minimizing the total crystal energy for different volumes ranging from −4 to 10 GPa while allowing the internal atomic positions and the lattice parameters to relax. We used the third-order Birch–Murnaghan [47] equation-of-state (EOS) to fit the volume–energy curve. Our results showed that the lowest energy configuration is obtained when an antiferromagnetic coupling between Fe ions is obtained, in accordance with the experimental results from Mössbauer effect [19]. The optimized lattice parameters (a, b, c), volume (V), interatomic distances (, , , ), internal angles (, , ), bulk modulus (B) and pressure derivative of B (B’) appear in Table 1 together with experimental results from the literature [19]. Furthermore, we have included the reported parameters for other CrVO-type structure vanadates for comparison. According to Table 1, the equilibrium volume at ambient pressure of AVO (A = Cr, Fe, In, Tl) compounds increases with the atomic mass of cation A, while the opposite occurs for bulk modulus. The obtained results for FeVO–II are in excellent agreement with experiments, with a difference of less than 1% in the lattice parameters. For this volume, the WPs are 4a (0, 0, 0) for Fe, 4c (0, 0.35788, 1/4) for V, 8f (0, 0.76239, 0.96753), and 8g (0.26481, 0.47778, 1/4) for O. Following Table 1, the equilibrium volume at ambient pressure of AVO (A = Cr, Fe, In, Tl) compounds increases with the atomic mass of cation A. In contrast, the opposite occurs for bulk modulus.
According to the most recent high-pressure experiments carried out on FeVO [15,20], this compound undergoes the following phase transition sequence, -MnMoO-type, driven by pressure. However, previous high-pressure studies reported the next phase transition sequence CrVO-type → wolframite in a small range of pressure [49]. Therefore, it is unknown if the CrVO-type structure of FeVO would transition to wolframite when high-pressure experiments are carried out starting from the CrVO-type structure, as occurs with CrVO [6] and InVO [5,16], and was predicted through first-principles calculations [15]. Therefore, from now on, we will only deal with studying the behavior of FeVO-II under pressure without considering whether the CrVO wolframite transition occurs. Figure 2 shows the pressure evolution of the structural parameters for the FeVO-II phase. According to Figure 2a, a volume reduction of = −9% is observed for a range of pressure of ≈10 GPa. Considering the anisotropy of the orthorhombic crystal structure, is more reflected in the [010] and [001] directions, while the change in [100] is practically negligible, see Figure 2b. Indeed the compressibility of the [001] direction is comparable to the linear compressibility of diamond, making the studied material a near zero linear compressibility material along [001]. The anisotropy can also be understood with the help of the other internal parameters from Figure 2c–f. As seen in Figure 2c, the interatomic distances of FeO undergo more changes than the distances of VO, which is reflected in the larger compressibility of octahedra against the minimal change in , see Figure 2d. Hence, the large compressibility of the y-axis is due primarily to the compressibility of octahedra due to the reduction in . Whereas, despite the reduction in , the and angles adjust in such a way that remains almost constant with pressure. On the other hand, the distortion parameter presents an important change at 6.2 GPa, which will be discussed in detail in Section 3.3.
3.2. Electronic Structure
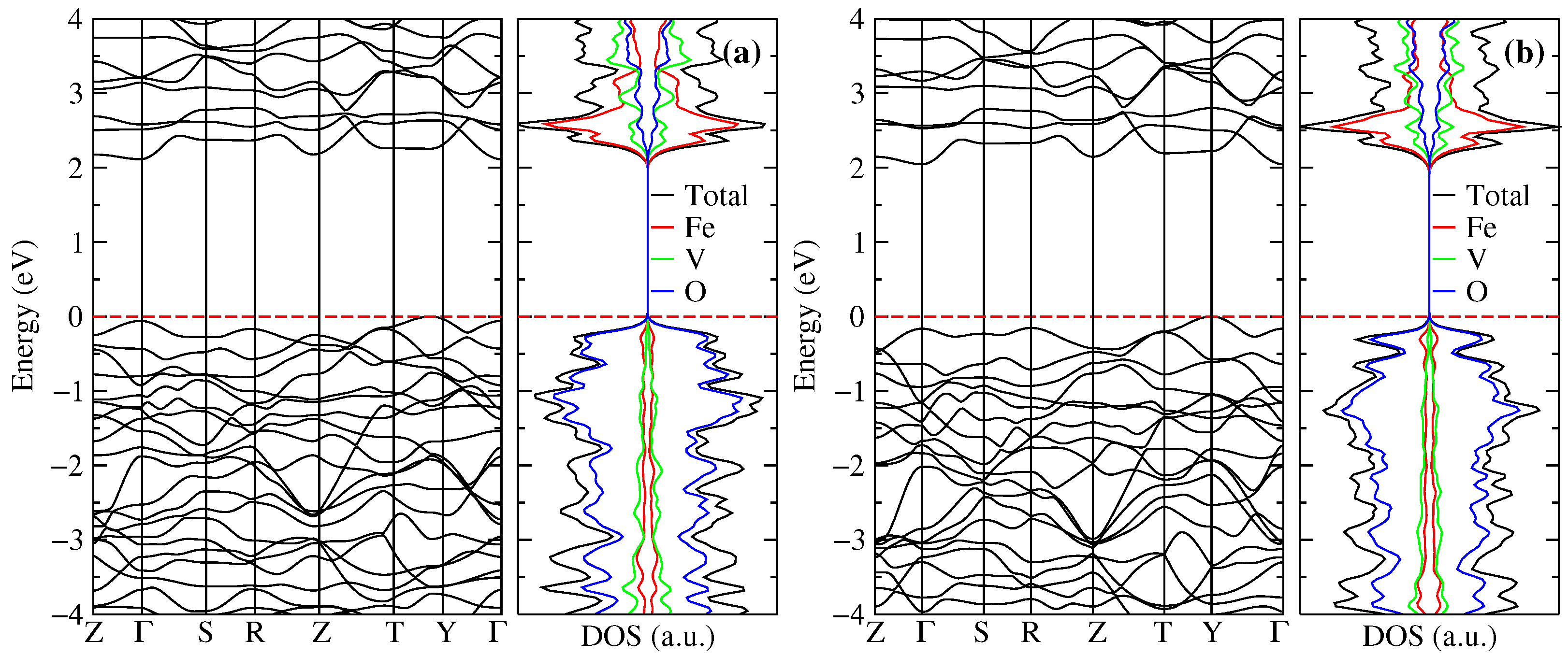
Figure 3a,b displays our calculated density of states (DOS) and band structures along the high-symmetry path Z––S–R–Z–T–Y– of the FeVO-II phase at ambient pressure and 9.6 GPa, respectively. Red dashed lines at 0 eV indicate the Fermi level. At ambient pressure, the valence band maximum (VBM) is located at an H (, , ) point (between T and Y), mainly occupied by 2p () states from oxygen. In contrast, the conduction band minima (CBM) is located at the point with contributions of the 3d states from Fe, mostly from sub-orbital. Therefore, FeVO-II phase presents an indirect energy band-gap = 2.11 eV. In comparison, the direct – energy band-gap is 2.17 eV. In this case, the maximum at below the Fermi level is occupied by 2p ( and ) states from oxygen, as happens in other O compounds [40]. It has been reported that CBM of many rare-earth AVO vanadates is dominated by vanadium 3d states, which makes these compounds present energy band-gaps close to 3.8 eV [50,51]. However, in the case of vanadates InVO (phase II), CrVO (phase II), and FeVO (phase I), the energy band-gap lowers to 3.2, 2.6, and 2.1 eV, respectively, due to contribution from cation A to the valence and conduction band [5,6,50,52]. Hence, our value follows the observed results for those vanadates. According to partial electronic DOS of Figure 3a, the states below the Fermi level mainly belong to the O atoms. In contrast, the states above the conduction bands are due to Fe, followed by V and O.
As pressure increases up to 9.6 GPa, the VBM shifts to the high-symmetry point Y while the CBM is still at the point with the same band occupation observed at ambient pressure, see Figure 3b. Hence, at this pressure, the band-gap is still indirect with = 2.05 eV, which gives a pressure coefficient of = −7.1 meV/GPa. In contrast, the pressure coefficients for InVO and CrVO are positive, with values of 13.5 [52] (8.9 [5]) meV for InVO and 1.9 (4.1) meV for CrVO [6], obtained by optical absorption measurements (DFT calculations). On the other hand, the forbidden direct – band-gap increases with pressure up to 2.21 eV at 9.6 GPa, which gives a value of = 4.2 meV, similar to the obtained values for the other AVO vanadates.
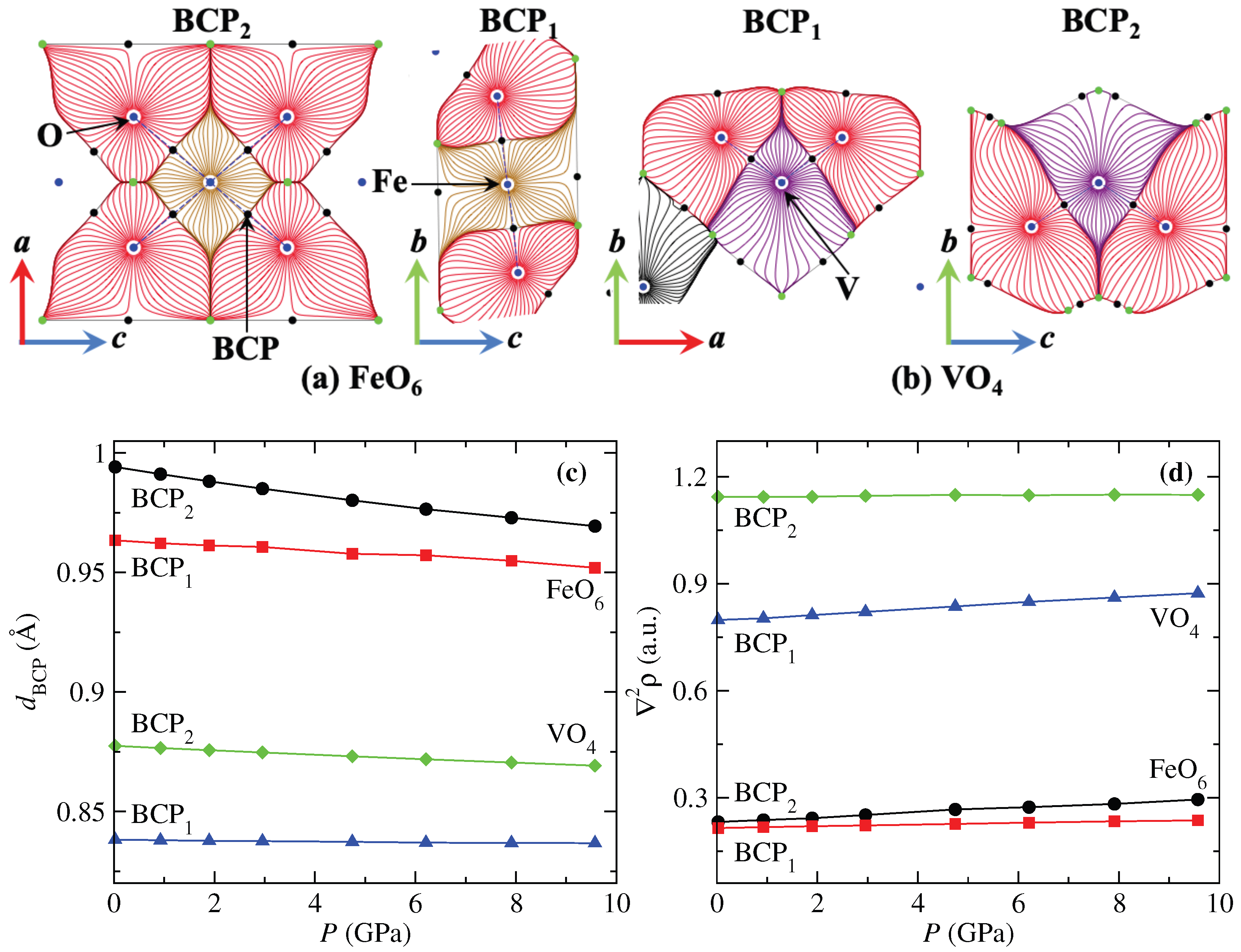
We performed a topological analysis of the charge density () with the QTAIM methodology to complement the study of the electronic structure of FeVO-II. Figure 4a,b show the gradient vector field of the electron density () of FeO and VO polyhedra, respectively (gold for Fe, purple for V, and red for O). Blue points indicate the atomic nucleus, corresponding to the maximum critical points (MCP). The minimal CP (mCP) are in green, and the bond CPs (BCPs) are in black and are located in the bond path (blue dashed lines) between Fe (V) and O. The distance between Fe (V) and the BCP () is plotted as a function of pressure in Figure 4c. As seen, the distances follow the same trend observed in the interatomic distances of Figure 2c. Therefore, the crystal structure can be interpreted in terms of charge density. In this sense, the Laplacian of the charge density () at the BCP gives an idea of the ionicity () or covalency () of a system [53]. According to Figure 4d, the Fe–O and V–O bonds have an ionic nature. The obtained values of (0.214–0.295 a.u.) for Fe–O bonds in the distorted octahedra are larger than those reported for A–O bonds in AWO (0.086–0.147 a.u.) [54], AMoO (0.085–0.22 a.u.) [53], and ATcO (0.045–0.153 a.u.) [40] compounds. In the case of the VO distorted tetrahedra, there is a significative difference in the values of and , which is related to the interatomic distances (see Table 1) being significantly larger than those obtained for the FeO distorted octahedra. This is the reason for the low compressibility of the VO polyhedra. As the pressure grows, the Laplacian values increase, strengthening the Fe–O and V–O bonds and the resistance of the polyhedrons to compression.
3.3. Elastic Constants and Mechanical Properties
The elastic constants () determine the resistance to deformation in the directions and planes of a crystal, making them highly dependent on the system’s symmetry. For the orthorhombic crystalline system, there are nine independent elastic constants: , and for longitudinal compression, , and for transverse expansion, , , and for pure shear [55]. Table 2 shows the elastic constants of FeVO-II at 0 GPa and the respective pressure coefficients . The necessary and sufficient Born stability criteria for orthorhombic systems are [56]: , , , , , and , which are fulfilled for FeVO-II. Under hydrostatic pressure (P), the stability criteria are [57]: (i = 1 to 6), , , , and , which are also meet. According to Table 2, the elastic constant presents the higher value, followed by and , indicating that longitudinally the crystal is more resistant to deformation in the a-axis; this was also observed in the evolution of the lattice parameters under pressure in Figure 2b. As would be expected, the largest transverse expansion constant is , followed by and , being the smallest of the nine elastic constants. At the same time, the shear elastic constants are generally small against those from longitudinal compression and transverse expansion. We found that elastic constants follow a linear trend with pressure, where only presents a negative pressure coefficient related to the trends observed in the structural parameters with pressure.
The elastic constants and compliances were used to compute the bulk (B) and shear (G) modulus with the Hill procedure [58], which is the average of the Voigt (V) [59] and Reuss (R) [60] methods. The bulk modulus in the Voigt and Reuss methods is computed as follows: = ( + + + 2 + 2 + 2), and = ( + + + 2 + 2 + 2), respectively. While the shear modulus is obtained with the following equations: 15 = ( + + − − − + 3 + 3 + 3), and = [4( + + ) − 4( + + ) + 3( + + )], respectively. B and G in the Hill average are obtained by means of and , respectively. On the other hand, Young’s modulus (E) and Poisson’s ratio () are given by and , respectively. The Vickers hardness was calculated with the approximation of Tian et al. [61]: = 0.92 , .
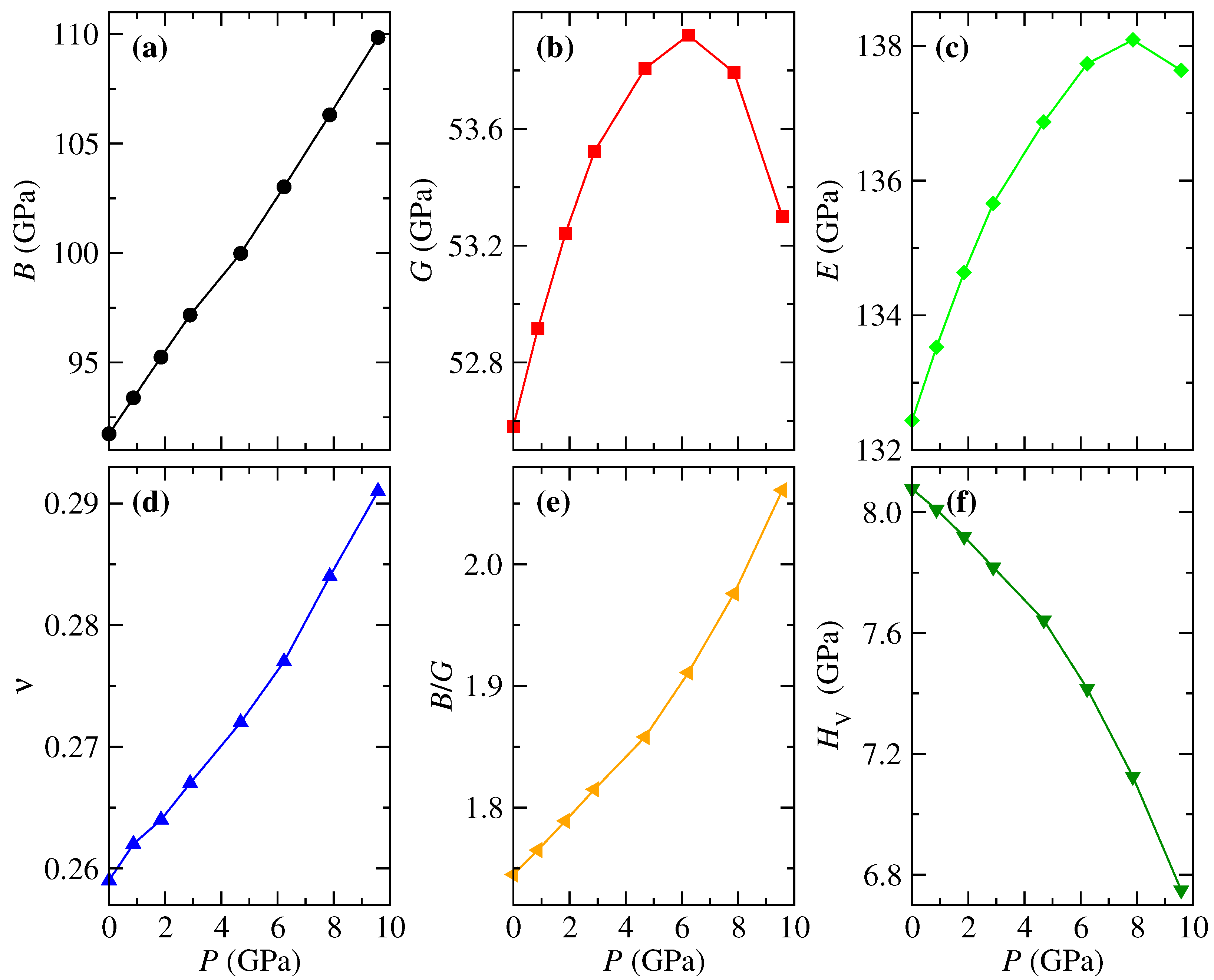
The mechanical properties and their evolution with pressure are provided in Figure 5. As seen, B increases almost linearly with pressure, which means that the resistance to deformation increases for this pressure range. Whereas G undergoes a change in the linear trend above 3 GPa, reaching a maximum value of 53.9 at 6.2 GPa and falling down to 53.3 GPa at 9.6 GPa. There is no significant change in G for this pressure range, but the change in trend will substantially affect the other properties that depend on G. Interestingly, in other materials, such change in the slope of the pressure dependence of the shear modulus G has been identified as a precursor effect of a phase transition observed at higher pressures [62]. According to the equations of Voigt and Reuss for G, the behavior of G is due to the negative pressure coefficient for and the significant values of for the transverse expansion elastic constants. On the other hand, B behaves linearly because () only sums the longitudinal and transverse elastic constants (compliances), while () decreases with pressure, so the value of affects B by a small constant factor.
The behavior of Young’s modulus is similar to G due to its relation with this mechanical property. According to Figure 5c, E increases by 4.3 % in a pressure range of 7.86 GPa; above this pressure, it decreases from 138.1 to 137.6 GPa. This behavior indicates that FeVO could undergo a phase transition from a CrVO-type phase to another one at a relatively small pressure since it has been observed that FeVO undergoes several phase transitions below 10 GPa, see Refs. [15,20] and references therein. The Poisson’s ratio follows an increasing trend, taking values between 0.26 and 0.29, which are in the range for ceramic semiconductors (0.25–0.42) [63] and are similar for other O compounds [40,54,64]. These values indicate that ionic contribution dominates in FeVO-II. The Pugh relation, Figure 5e, represents the ductility or brittleness of the system. A value below 1.75 [11] reveals a tendency to brittleness, while a value greater than this indicates that the material behaves in a ductile manner. FeVO-II presents a value of 1.745 at ambient pressure and increases to 2.06 at 9.6 GPa, which means that ductility increases with pressure. As was expected, FeVO-II, as other O compounds, presents a small Vickers hardness due to the relatively small values of B and G. Given the relation between and , it is natural that the Vickers hardness decreases with pressure, Figure 5f.
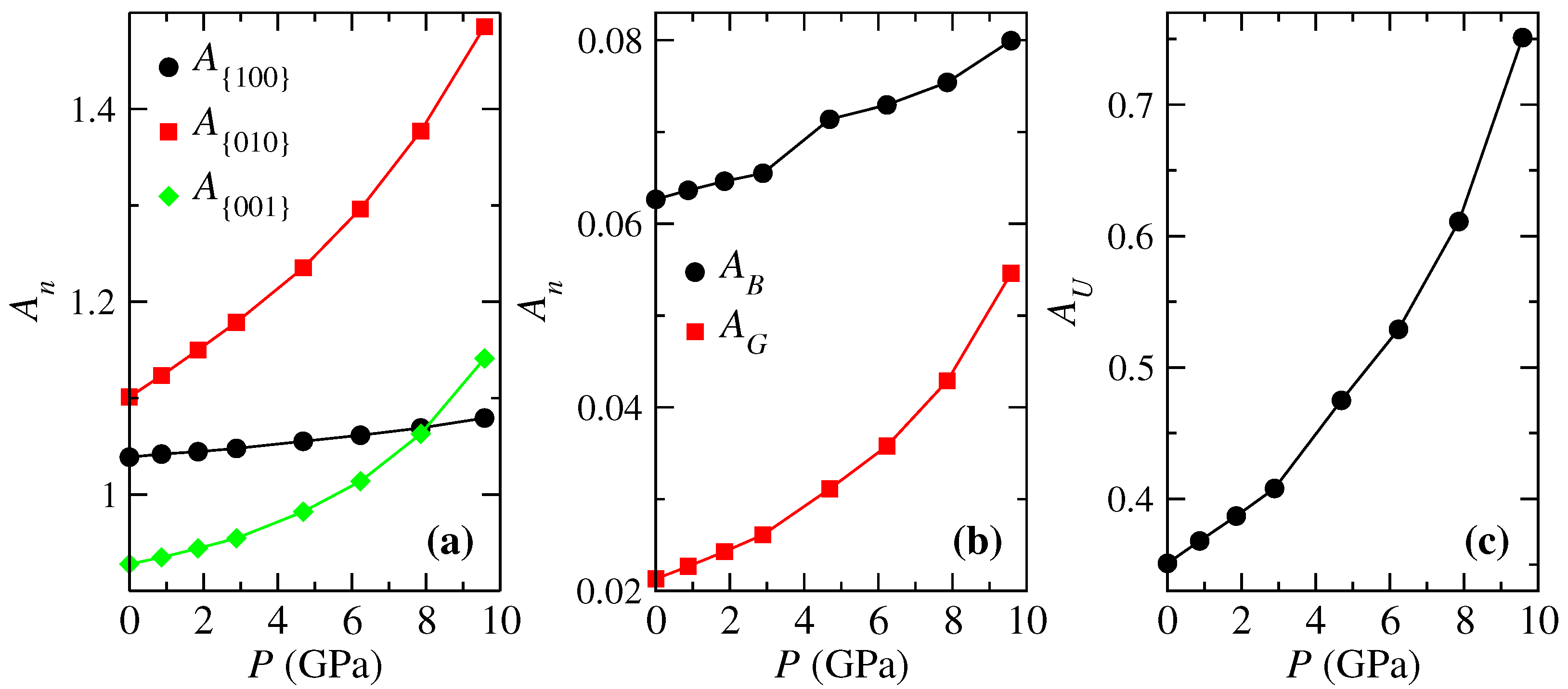
On the other hand, elastic anisotropy has important implications in engineering science and crystal physics because it is commonly responsible for the formation of microcracks in materials [65]. In this work, we analyzed several anisotropic factors discussed below. The anisotropic shear factors provide a measure of the degree of anisotropy in the bonding between atoms in different crystallographic planes. The shear anisotropic factor for the {100} shear planes between the and directions is . For the {010} shear planes between and directions it is . For the {001} shear planes between and directions, it is . The factors , , and must be one for isotropic crystals, and any value greater or smaller than one measures the degree of elastic anisotropy. According to Figure 6a, presents the smallest deviation from one, followed by and . However, once pressure is applied, is more affected than the others. Factor is related to the equatorial interatomic distances of the distorted octahedra [, see Figure 2c], which makes quite a bit of sense since it is that undergoes the greatest change with pressure. While is more related to , with this interatomic distance being the second one most affected by pressure.
Other important factors are the universal anisotropy , the percentage of elastic anisotropy in bulk [] and shear [] modulus, where a value of zero is associated with isotropic elastic constants, which means that and . For the entire range of pressure, is larger than because the difference between and is bigger than between and . However, the pressure affects more than as happens with G. This difference increases above 6.2 GPa as presents the same behavior of G, while is always increasing. is larger than and since the anisotropies of both modules are added.
3.4. Vibrational Properties
According to the group theory, the space group has the following Raman, infrared (IR), and silent phonon modes at the zone center = 5 + 4 + 2 + 4, = 6 + 7 + 5, and = 3, respectively. These modes can be further classified into internal (symmetric stretching, , symmetric bending, , asymmetric stretching, , and asymmetric bending, ) and external (translational, T, and rotational, R) modes of VO units. The calculated Raman, IR, and silent phonon frequencies, the pressure coefficients (), and the Grüneisen parameters ( = -∂ln()/∂lnV) at the point appear in Table 3.
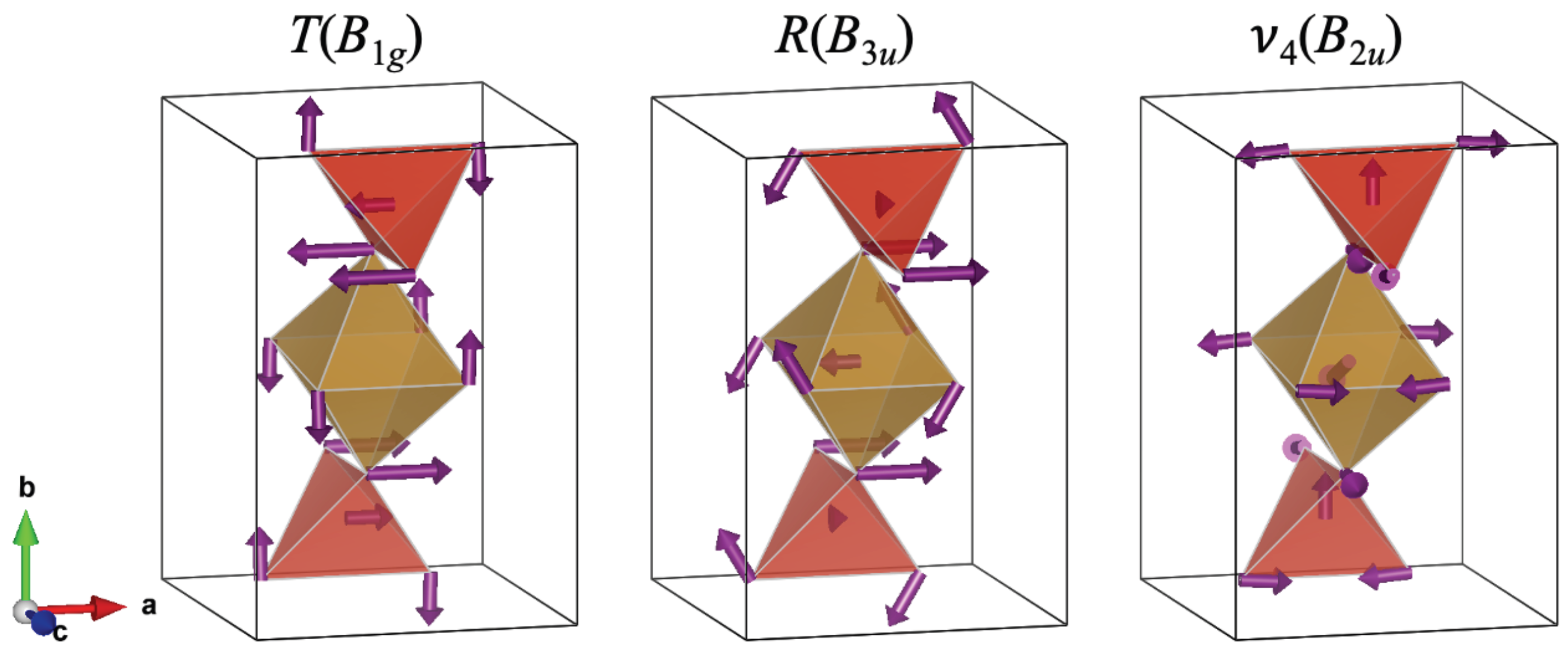
According to Table 3, one Raman, , and two IR modes, and , have negative pressure coefficients and Grüneisen parameters (soften with pressure), which are related to the instability of the CrVO-type structure under pressure [2]. Such behavior was already observed in InVO [5] and CrVO [20]. The soft translational Raman mode involves a translation of the VO polyhedra accompanied by a rotation of the FeO units around the z-axis as rigid units, see Figure 7, whose freedom of movement is affected by the decrease in the lattice parameter b, which is the most compressible under pressure. In contrast, the remaining Raman modes harden with pressure. The IR soft mode is quite similar to the , but in this case, the FeO vibrates asymmetrically. While the other soft mode implies the asymmetric bending in the VO polyhedra with a translation of the Fe cation in the c direction, which produces a stretching of the apical interatomic distances. Therefore, the softening of R and IR modes is related to the compression of the y-axis. Interestingly, phosphates with CrVO-type structure, such as InPO and TiPO, only present some IR soft modes. The main difference being that Ti(In)PO is more compressible in the c(b)-axis [7].
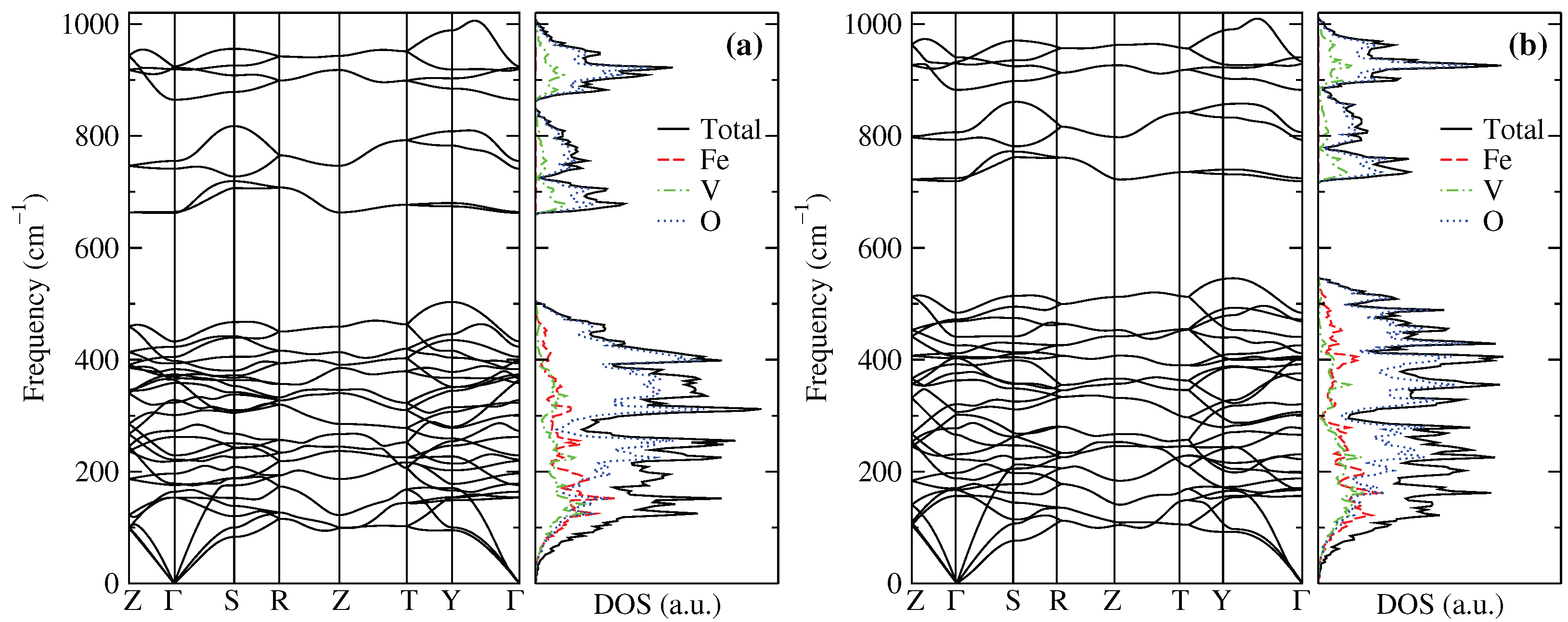
The phonon dispersion relation and the phonon density of states (DOS) are displayed in Figure 8 at (a) ambient pressure and (b) 9.6 GPa. As can be seen in Figure 8a, the CrVO-type structure of FeVO complains with the dynamic stability criteria at P = 0, which implies that (q, s)> 0 for all wave vectors q and polarization s (longitudinal and transverse modes) [66]. Therefore, FeVO-II complains with the elastic and dynamic stability criteria to be experimentally synthesized [19], such as InVO [5] and CrVO [20]. We can see no significant differences in the phonon spectrum at 9.6 GPa, which ensures that the FeVO-II phase would be stable in a pressure range where the isostructural vanadates InVO and CrVO undergo a phase transition to the wolframite-type structure [5,20].
The phonon DOS of FeVO-II is more similar to CrVO than InVO because In is more than double the mass of Cr and Fe. The partial phonon DOS of the first lower frequency zone (up to ≈ 505 cm) is due to external (R and T) and internal ( and ) phonon modes, with Fe and V similar contributions. In contrast, the two high-frequency zones are due to pure internal vibrations and , which indicates that phonon DOS is only due to O and V.
4. Conclusions
First-principles calculations were performed to study FeVO in the CrVO-type structure up to 9.6 GPa. Our results indicate that this phase is elastically and dynamically stable for this pressure range. FeVO presents an indirect band-gap = 2.11 eV, similar to the value reported for the triclinic phase of FeVO at ambient pressure. The Laplacian charge density at BCPs in both polyhedra is larger than those reported for AMoO and AWO compounds and increases with pressure. It was observed that the y-axis is the most compressible, related to the reduction in the interatomic distances of the FeO polyhedra. These changes in the structural parameters have strong effects on the phonon modes with negative pressure coefficients. We found that the shear modulus G softens with pressure, which suggests that the FeVO-II phase could undergo a phase transition below 10 GPa as was predicted in a previous study and happen in CrVO and InVO, where these compounds have a phase transition from CrVO-type to the wolframite structure. We hope this work encourages other research groups to perform high-pressure characterization studies with the diamond-anvil cell device in order to increase the knowledge of less studied orthovanadates with CrVO-type structure, such as FeVO and TlVO.
Author Contributions
Methodology, P.B.R.-V., S.L.-M.; calculations, P.B.R.-V., S.L.-M.; writing—original draft preparation, P.B.R.-V., S.L.-M.; supervision, S.L.-M.; discussion, S.L.-M., D.E.; writing—review and editing, P.B.R.-V., S.L.-M., D.E. All authors have read and agreed to the published version of the manuscript.
Funding
S.L.-M. thanks CONACYT of Mexico for financial support through the program “Programa de Investigadoras e Investigadores por México”. D.E. thanks the financial support from the Spanish Ministerio de Ciencia e Investigación (10.13039/501100011033) under Project PID2019-106383GB-41, as well as through the MALTA Consolider Team research network (RED2018-102612-T) and from Generalitat Valenciana under Grants PROMETEO CIPROM/2021/075-GREENMAT and MFA/2022/007.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All relevant data that support the findings of this study are available from the corresponding authors upon request.
Acknowledgments
The authors gratefully acknowledge the computing time granted by LANCAD and CONACYT on the supercomputer Miztli at LSVP DGTIC UNAM. Furthermore, the IPICYT Supercomputing National Center for Education and Research, grant TKII-R2022-SLM1/PBRV1.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Errandonea, D.; Manjon, F. Pressure effects on the structural and electronic properties of ABX4 scintillating crystals. Prog. Mater. Sci. 2008, 53, 711–773. [Google Scholar] [CrossRef]
- Errandonea, D.; Garg, A.B. Recent progress on the characterization of the high-pressure behaviour of AVO4 orthovanadates. Prog. Mater. Sci. 2018, 97, 123–169. [Google Scholar] [CrossRef] [Green Version]
- Baran, E. Materials belonging to the CrVO4 structure type: Preparation, crystal chemistry and physicochemical properties. J. Mater. Sci. 1998, 33, 2479–2497. [Google Scholar] [CrossRef]
- de Jesus Pereira, A.L.; Santamaría-Pérez, D.; Vilaplana, R.; Errandonea, D.; Popescu, C.; da Silva, E.L.; Sans, J.A.; Rodríguez-Carvajal, J.; Muñoz, A.; Rodríguez-Hernández, P.; et al. Experimental and Theoretical Study of SbPO4 under Compression. Inorg. Chem. 2020, 59, 287–307. [Google Scholar] [CrossRef] [PubMed]
- López-Moreno, S.; Rodríguez-Hernández, P.; Munoz, A.; Errandonea, D. First-principles study of InVO4 under pressure: Phase transitions from CrVO4-to AgMnO4-type structure. Inorg. Chem. 2017, 56, 2697–2711. [Google Scholar] [CrossRef] [Green Version]
- Botella, P.; López-Moreno, S.; Errandonea, D.; Manjón, F.J.; Sans, J.; Vie, D.; Vomiero, A. High-pressure characterization of multifunctional CrVO4. J. Phys. Condens. Matter. 2020, 32, 385403. [Google Scholar] [CrossRef]
- López-Moreno, S.; Errandonea, D. Ab initio prediction of pressure-induced structural phase transitions of CrVO4-type orthophosphates. Phys. Rev. B 2012, 86, 104112. [Google Scholar] [CrossRef]
- Dwivedi, A.; Kaiwart, R.; Varma, M.; Velaga, S.; Poswal, H. High-pressure structural investigations on InPO4. J. Solid State Chem. 2020, 282, 121065. [Google Scholar] [CrossRef]
- Butcher, D.P., Jr.; Gewirth, A.A. Photoelectrochemical response of TlVO4 and InVO4: TlVO4 composite. Chem. Mater. 2010, 22, 2555–2562. [Google Scholar] [CrossRef]
- Zhao, C.; Tan, G.; Yang, W.; Xu, C.; Liu, T.; Su, Y.; Ren, H.; Xia, A. Fast interfacial charge transfer in α-Fe2O3- δCδ/FeVO4-x+ δC x-δ bulk heterojunctions with controllable phase content. Sci. Rep. 2016, 6, 38603. [Google Scholar] [CrossRef]
- Balamurugan, M.; Yun, G.; Ahn, K.S.; Kang, S.H. Revealing the beneficial effects of FeVO4 nanoshell layer on the BiVO4 inverse opal core layer for photoelectrochemical water oxidation. J. Phys. Chem. C 2017, 121, 7625–7634. [Google Scholar] [CrossRef]
- Marberger, A.; Elsener, M.; Ferri, D.; Sagar, A.; Schermanz, K.; Krocher, O. Generation of NH3 selective catalytic reduction active catalysts from decomposition of supported FeVO4. ACS Catal. 2015, 5, 4180–4188. [Google Scholar] [CrossRef]
- Yi, X.; Li, J.; Chen, Z.; Tok, A. Single-crystalline InVO4 nanotubes by self-template-directed fabrication. J. Am. Ceram. Soc. 2010, 93, 596–600. [Google Scholar] [CrossRef]
- Yu, Y.; Ju, P.; Zhang, D.; Han, X.; Yin, X.; Zheng, L.; Sun, C. Peroxidase-like activity of FeVO4 nanobelts and its analytical application for optical detection of hydrogen peroxide. Sens. Actuators B 2016, 233, 162–172. [Google Scholar] [CrossRef] [Green Version]
- López-Moreno, S.; Errandonea, D.; Pellicer-Porres, J.; Martínez-García, D.; Patwe, S.J.; Achary, S.N.; Tyagi, A.K.; Rodríguez-Hernández, P.; Muñoz, A.; Popescu, C. Stability of FeVO4 under Pressure: An X-ray Diffraction and First-Principles Study. Inorg. Chem. 2018, 57, 7860–7876. [Google Scholar] [CrossRef]
- Errandonea, D.; Gomis, O.; García-Domene, B.; Pellicer-Porres, J.; Katari, V.; Achary, S.N.; Tyagi, A.K.; Popescu, C. New polymorph of InVO4: A high-pressure structure with six-coordinated vanadium. Inorg. Chem. 2013, 52, 12790–12798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arisi, E.; Sánchez, S.; Leccabue, F.; Watts, B.; Bocelli, G.; Calderón, F.; Calestani, G.; Righi, L. Preparation and characterization of AlVO4 compound. J. Mater. Sci. 2004, 39, 2107–2111. [Google Scholar] [CrossRef]
- Muller, J.; Joubert, J. Synthese sous haute pression d’oxygene d’une forme dense ordonne e de FeVO4 et mise en evidence d’une varie te allotropique de structure CrVO4. J. Solid State Chem. 1975, 14, 8–13. [Google Scholar] [CrossRef]
- Oka, Y.; Yao, T.; Yamamoto, N.; Ueda, Y.; Kawasaki, S.; Azuma, M.; Takano, M. Hydrothermal synthesis, crystal structure, and magnetic properties of FeVO4-II. J. Solid State Chem. 1996, 123, 54–59. [Google Scholar] [CrossRef]
- Gonzalez-Platas, J.; López-Moreno, S.; Bandiello, E.; Bettinelli, M.; Errandonea, D. Precise characterization of the rich structural landscape induced by pressure in multifunctional FeVO4. Inorg. Chem. 2020, 59, 6623–6630. [Google Scholar] [CrossRef]
- Young, A.; Schwartz, C. High pressure forms of CrVO4 and FeVO4. Acta Crystallogr. Sect. A Found. Crystallogr. 1962, 15, 1305. [Google Scholar] [CrossRef]
- Laves, F.; Young, A.P.; Schwartz, C.M. On the high-pressure form of FeVO4. Acta Cryst. 1964, 17, 1476–1477. [Google Scholar] [CrossRef]
- Ouahrani, T.; Garg, A.B.; Rao, R.; Rodríguez-Hernández, P.; Muñoz, A.; Badawi, M.; Errandonea, D. High-Pressure Properties of Wolframite-Type ScNbO4. J. Phys. Chem. C 2022, 126, 4664–4676. [Google Scholar] [CrossRef]
- Bader, R.F. Atoms in molecules. Accounts Chem. Res. 1985, 18, 9–15. [Google Scholar] [CrossRef]
- Jones, R.O. Density functional theory: Its origins, rise to prominence, and future. Rev. Mod. Phys. 2015, 87, 897–923. [Google Scholar] [CrossRef] [Green Version]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Csonka, G.I.; Perdew, J.P.; Ruzsinszky, A.; Philipsen, P.H.T.; Lebègue, S.; Paier, J.; Vydrov, O.A.; Ángyán, J.G. Assessing the performance of recent density functionals for bulk solids. Phys. Rev. B 2009, 79, 155107. [Google Scholar] [CrossRef]
- Dudarev, S.L.; Botton, G.A.; Savrasov, S.Y.; Humphreys, C.J.; Sutton, A.P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 1998, 57, 1505–1509. [Google Scholar] [CrossRef]
- Ruiz-Fuertes, J.; Errandonea, D.; López-Moreno, S.; González, J.; Gomis, O.; Vilaplana, R.; Manjón, F.J.; Muñoz, A.; Rodríguez-Hernández, P.; Friedrich, A.; et al. High-pressure Raman spectroscopy and lattice-dynamics calculations on scintillating MgWO4: Comparison with isomorphic compounds. Phys. Rev. B 2011, 83, 214112. [Google Scholar] [CrossRef] [Green Version]
- López, S.; Romero, A.H.; Mejía-López, J.; Mazo-Zuluaga, J.; Restrepo, J. Structure and electronic properties of iron oxide clusters: A first-principles study. Phys. Rev. B 2009, 80, 085107. [Google Scholar] [CrossRef] [Green Version]
- López-Moreno, S.; Romero, A.H.; Mejía-López, J.; Muñoz, A.; Roshchin, I.V. First-principles study of electronic, vibrational, elastic, and magnetic properties of FeF2 as a function of pressure. Phys. Rev. B 2012, 85, 134110. [Google Scholar] [CrossRef] [Green Version]
- Mejía-López, J.; Mazo-Zuluaga, J.; López-Moreno, S.; Muñoz, F.; Duque, L.F.; Romero, A.H. Physical properties of quasi-one-dimensional MgO and Fe3O4-based nanostructures. Phys. Rev. B 2014, 90, 035411. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Parlinski, K. Computer Code PHONON. 2008. Available online: http://wolf.ifj.edu.pl/phonon (accessed on 5 December 2022).
- Romero-Vázquez, P.B.; López-Moreno, S.; Errandonea, D. First-principles study of ATcO4 pertechnetates. J. Phys. Chem. Solids 2022, 171, 110979. [Google Scholar] [CrossRef]
- Le Page, Y.; Saxe, P. Symmetry-general least-squares extraction of elastic data for strained materials from ab initio calculations of stress. Phys. Rev. B 2002, 65, 104104. [Google Scholar] [CrossRef]
- Popelier, P.L.A. The QTAIM perspective of chemical bonding. In The Chemical Bond: Fundamental Aspects of Chemical Bonding; Chapter 8; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2014; pp. 271–308. [Google Scholar] [CrossRef]
- Otero-de-la Roza, A.; Blanco, M.; Pendás, A.M.; Luaña, V. Critic: A new program for the topological analysis of solid-state electron densities. Comput. Phys. Commun. 2009, 180, 157–166. [Google Scholar] [CrossRef]
- Otero-de-la Roza, A.; Johnson, E.R.; Luaña, V. Critic2: A program for real-space analysis of quantum chemical interactions in solids. Comput. Phys. Commun. 2014, 185, 1007–1018. [Google Scholar] [CrossRef]
- Vega, D.; Almeida, D. AIM-UC: An application for QTAIM analysis. J. Comput. Methods Sci. Eng. 2014, 14, 131–136. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallog. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Birch, F. Finite elastic strain of cubic crystals. Phys. Rev. 1947, 71, 809–824. [Google Scholar] [CrossRef]
- Touboul, M.; Ingrain, D. Syntheses et propriétés thermiques de InVO4 et TlVO4. J. Less Common Met. 1980, 71, 55–62. [Google Scholar] [CrossRef]
- Hotta, Y.; Ueda, Y.; Nakayama, N.; Kosuge, K.; Kachi, S.; Shimada, M.; Koizumi, M. Pressure-products diagram of FeVO4 system (0≤ x ≤ 0.5). J. Solid State Chem. 1984, 55, 314–319. [Google Scholar] [CrossRef]
- Errandonea, D. High pressure crystal structures of orthovanadates and their properties. J. Appl. Phys. 2020, 128, 040903. [Google Scholar] [CrossRef]
- Díaz-Anichtchenko, D.; Errandonea, D. Comparative Study of the Compressibility of M3V2O8 (M = Cd, Zn, Mg, Ni) Orthovanadates. Crystals 2022, 12, 1544. [Google Scholar] [CrossRef]
- Botella, P.; Errandonea, D.; Garg, A.; Rodriguez-Hernandez, P.; Muñoz, A.; Achary, S.; Vomiero, A. High-pressure characterization of the optical and electronic properties of InVO4, InNbO4, and InTaO4. SN Appl. Scien. 2019, 1, 389. [Google Scholar] [CrossRef] [Green Version]
- Monteseguro, V.; Ruiz-Fuertes, J.; Contreras-García, J.; Rodríguez-Hernández, P.; Muñoz, A.; Errandonea, D. High pressure theoretical and experimental analysis of the bandgap of BaMoO4, PbMoO4, and CdMoO4. Appl. Phys. Lett. 2019, 115, 012102. [Google Scholar] [CrossRef]
- Romero-Vázquez, P.B.; López-Moreno, S. Ab initio study of RaWO4: Comparison with isoelectronic tungstates. J. Solid State Chem. 2023, 317, 123709. [Google Scholar] [CrossRef]
- Nye, J.F. Physical Properties of Crystals: Their Representation by Tensors and Matrices. Oxford University Press: Oxford, UK, 1985. [Google Scholar]
- Mouhat, F.; Coudert, F.X. Necessary and sufficient elastic stability conditions in various crystal systems. Phys. Rev. B 2014, 90, 224104. [Google Scholar] [CrossRef] [Green Version]
- ping Feng, L.; tang Liu, Z.; jun Liu, Q. Structural, elastic and mechanical properties of orthorhombic SrHfO3 under pressure from first-principles calculations. Phys. B Condens. Matter 2012, 407, 2009–2013. [Google Scholar] [CrossRef]
- Najafvandzadeh, N.; López-Moreno, S.; Errandonea, D.; Pavone, P.; Draxl, C. First-principles study of elastic and thermal properties of scheelite-type molybdates and tungstates. Mater. Today Commun. 2020, 24, 101089. [Google Scholar] [CrossRef]
- Voigt, W. Lehrbuch Kristallphysik; Springer: Berlin/Heidelberg, Germany, 1928. [Google Scholar] [CrossRef]
- Reuss, A. Berechnung der Fliebgrenze von Mischkristallen auf Grund der Plastizitatsbedingung für Einkristalle. Angew Appl. Math. Mech. 1929, 9, 49–58. [Google Scholar] [CrossRef]
- Tian, Y.; Xu, B.; Zhao, Z. Microscopic theory of hardness and design of novel superhard crystals. Int. J. Refract. Met. Hard. Mater. 2012, 33, 93–106. [Google Scholar] [CrossRef]
- Singh, J.; Sahoo, S.S.; Venkatakrishnan, K.; Vaitheeswaran, G.; Errandonea, D. High-pressure study of the aurophilic topological Dirac material AuI. J. Alloys Compd. 2022, 928, 167178. [Google Scholar] [CrossRef]
- Prawoto, Y. Seeing auxetic materials from the mechanics point of view: A structural review on the negative Poisson’s ratio. Comput. Mater. Sci. 2012, 58, 140–153. [Google Scholar] [CrossRef]
- Liu, Y.; Jia, D.; Zhou, Y.; Zhou, Y.; Zhao, J.; Li, Q.; Liu, B. Discovery of ABO4 scheelites with the extra low thermal conductivity through high-throughput calculations. J. Mater. 2020, 6, 702–711. [Google Scholar] [CrossRef]
- Tvergaard, V.; Hutchinson, J.W. Microcracking in Ceramics Induced by Thermal Expansion or Elastic Anisotropy. J. Am. Ceram. Soc. 1988, 71, 157–166. [Google Scholar] [CrossRef]
- Grimvall, G.; Magyari-Köpe, B.; Ozoliņš, V.; Persson, K.A. Lattice instabilities in metallic elements. Rev. Mod. Phys. 2012, 84, 945. [Google Scholar] [CrossRef]
Figure 1.
(a) Crystal structure of FeVO-II polymporph, (b) polyhedral representation of the structure, and (c) irregular polyhedra of FeO and VO units. The different bond distances mentioned in the text are labeled as well as the bond angles. We used the VESTA software [46] to build the structures.
Figure 1.
(a) Crystal structure of FeVO-II polymporph, (b) polyhedral representation of the structure, and (c) irregular polyhedra of FeO and VO units. The different bond distances mentioned in the text are labeled as well as the bond angles. We used the VESTA software [46] to build the structures.

Figure 2.
Structural parameters for FeVO-II as a function of pressure: (a) volume, (b) lattice parameters, (c) interatomic distances, (d) polyhedral volumes, (e) angles, and (f) polyhedral distortion parameter.
Figure 2.
Structural parameters for FeVO-II as a function of pressure: (a) volume, (b) lattice parameters, (c) interatomic distances, (d) polyhedral volumes, (e) angles, and (f) polyhedral distortion parameter.

Figure 3.
Band structure and electronic density of states at (a) ambient pressure and (b) 9.6 GPa of FeVO-II phase.
Figure 3.
Band structure and electronic density of states at (a) ambient pressure and (b) 9.6 GPa of FeVO-II phase.

Figure 4.
Gradient vector field of the electron density () for (a) FeO and (b) VO polyhedra. (c) Evolution of the interatomic distances from the cations to the critical bond points and (d) Laplacian of the charge density at the critical bond point as a function of pressure for FeVO.
Figure 4.
Gradient vector field of the electron density () for (a) FeO and (b) VO polyhedra. (c) Evolution of the interatomic distances from the cations to the critical bond points and (d) Laplacian of the charge density at the critical bond point as a function of pressure for FeVO.

Figure 5.
Pressure dependence of mechanical properties obtained from elastic constants with the Hill average: (a) bulk modulus, B, (b) shear modulus, G, (c) Young’s modulus, E, (d) Poisson’s ratio, , (e) Pugh’s ratio, , and (f) Vickers hardness, , for FeVO-II.
Figure 5.
Pressure dependence of mechanical properties obtained from elastic constants with the Hill average: (a) bulk modulus, B, (b) shear modulus, G, (c) Young’s modulus, E, (d) Poisson’s ratio, , (e) Pugh’s ratio, , and (f) Vickers hardness, , for FeVO-II.

Figure 6.
(a) Pressure dependence of anisotropy factors: (a) by crystallographic direction, (b) percentage bulk and shear anisotropy, and (c) universal anisotropy for FeVO-II.
Figure 6.
(a) Pressure dependence of anisotropy factors: (a) by crystallographic direction, (b) percentage bulk and shear anisotropy, and (c) universal anisotropy for FeVO-II.

Figure 7.
Eigenvectors of the phonon frequencies with negative pressure coefficients of FeVO-II. We used the VESTA software [46] to build the structures.
Figure 7.
Eigenvectors of the phonon frequencies with negative pressure coefficients of FeVO-II. We used the VESTA software [46] to build the structures.

Figure 8.
Phonon spectrum and phonon DOS at (a) 0 GPa, and (b) 9.6 GPa of FeVO-II.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Calculated structural parameters for FeVO-II phase. a, b, and c are the lattice parameters, V is the volume, (X = Fe, V; n = 1, 2) and , , and , are the interatomic distances and angles inside the crystal structure, respectively (see Figure 1). B is the bulk modulus and is the pressure derivative of B. Results from this work are denoted with 🟉. We also include experimental results from the literature of AVO compounds for comparison.
Table 1.
Calculated structural parameters for FeVO-II phase. a, b, and c are the lattice parameters, V is the volume, (X = Fe, V; n = 1, 2) and , , and , are the interatomic distances and angles inside the crystal structure, respectively (see Figure 1). B is the bulk modulus and is the pressure derivative of B. Results from this work are denoted with 🟉. We also include experimental results from the literature of AVO compounds for comparison.
| a (Å) | b (Å) | c (Å) | V (Å) | (Å) | (Å) | (Å) | (Å) | () | () | () | B (GPa) | Ref. | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FeVO | 5.6038 | 8.2624 | 6.1726 | 285.80 | 1.9734 | 2.0377 | 1.6705 | 1.7842 | 89.271 | 106.990 | 112.547 | 88.8 | 2.8 | 🟉 |
| 5.5941 | 8.3216 | 6.2252 | 289.80 | 1.9784 | 2.0521 | 1.6544 | 1.7762 | 88.3 | 3.2 | DFT [15] | ||||
| 5.6284 | 8.2724 | 6.1118 | 284.57 | 2.0060 | 2.0290 | 1.6520 | 1.7920 | 89.600 | 107.200 | 112.500 | Exp. [19] | |||
| CrVO | 5.5785 | 8.2830 | 6.0576 | 279.90 | 93.0 | 4.0 | DFT [6] | |||||||
| 5.5680 | 8.2080 | 5.9770 | 273.16 | 63.0 | 4.0 | Exp. [6] | ||||||||
| InVO | 5.7547 | 8.6168 | 6.6751 | 331.00 | 2.1877 | 2.1392 | 1.6690 | 1.7824 | 71.0 | 4.0 | DFT [5] | |||
| 5.7380 | 8.4920 | 6.5820 | 320.72 | 69.0 | 4.0 | Exp. [16] | ||||||||
| TlVO | 5.8390 | 8.6870 | 6.8000 | 344.92 | Exp. [48] |
Table 2.
Elastic constants (in GPa) at 0 GPa and pressure coefficients.
| 234.2 | 125.5 | 144.8 | 62.98 | 52.49 | 47.04 | 78.46 | 68.21 | 39.81 | |
| 2.41 | −0.87 | 3.78 | 0.74 | 1.04 | 0.15 | 2.39 | 2.18 | 2.57 |
Table 3.
Calculated Raman, infrared, and silent phonon frequencies ( in cm), pressure coefficients (), and Grüneisen parameters () of FeVO-II.
Table 3.
Calculated Raman, infrared, and silent phonon frequencies ( in cm), pressure coefficients (), and Grüneisen parameters () of FeVO-II.
| Mode | Raman | Mode | Infrared | Mode | Silent | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 153.7 | 1.37 | 0.57 | 153.7 | 0.24 | 0.15 | 164.4 | 1.29 | 0.77 | |||
| 177.4 | −1.05 | −0.82 | 180.8 | 1.68 | 0.86 | 220.3 | 0.55 | 0.23 | |||
| 219.6 | 0.52 | 0.22 | 201.2 | −0.01 | −0.03 | 404.9 | 7.45 | 1.68 | |||
| 221.8 | 3.23 | 1.29 | 274.9 | 1.12 | 0.37 | ||||||
| 261.8 | 4.25 | 1.47 | 298.8 | 1.51 | 0.46 | ||||||
| 355.1 | 0.30 | 0.07 | 315.1 | 3.82 | 0.25 | ||||||
| 361.7 | 1.36 | 0.33 | 327.9 | −0.88 | −1.06 | ||||||
| 366.4 | 3.78 | 0.94 | 363.1 | 2.41 | 0.48 | ||||||
| 374.0 | 3.58 | 0.88 | 381.5 | 6.61 | 1.61 | ||||||
| 394.2 | 1.55 | 0.36 | 398.3 | 4.93 | 1.13 | ||||||
| 431.3 | 5.90 | 1.25 | 420.6 | 4.38 | 0.92 | ||||||
| 652.8 | 5.71 | 0.78 | 654.2 | 5.64 | 0.77 | ||||||
| 751.0 | 5.74 | 0.72 | 735.8 | 5.47 | 0.67 | ||||||
| 918.9 | 1.33 | 0.12 | 866.8 | 2.58 | 0.26 | ||||||
| 922.0 | 2.44 | 0.24 | 924.3 | 1.38 | 0.13 | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Romero-Vázquez, P.B.; López-Moreno, S.; Errandonea, D. Stability of FeVO4-II under Pressure: A First-Principles Study. Crystals 2022, 12, 1835. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst12121835
AMA Style
Romero-Vázquez PB, López-Moreno S, Errandonea D. Stability of FeVO4-II under Pressure: A First-Principles Study. Crystals. 2022; 12(12):1835. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst12121835
Chicago/Turabian StyleRomero-Vázquez, Pricila Betbirai, Sinhué López-Moreno, and Daniel Errandonea. 2022. "Stability of FeVO4-II under Pressure: A First-Principles Study" Crystals 12, no. 12: 1835. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst12121835
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.