
Precise Tuning of Polymeric Fiber Dimensions to Enhance the Mechanical Properties of Alginate Hydrogel Matrices


Abstract
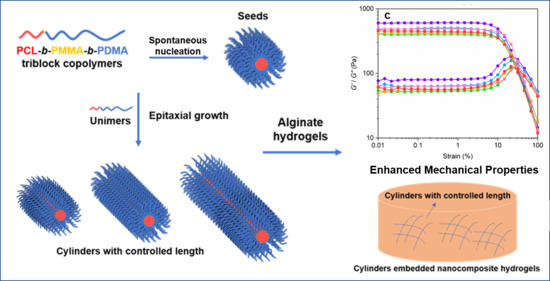
:
1. Introduction
2. Materials and Methods
2.1. Materials
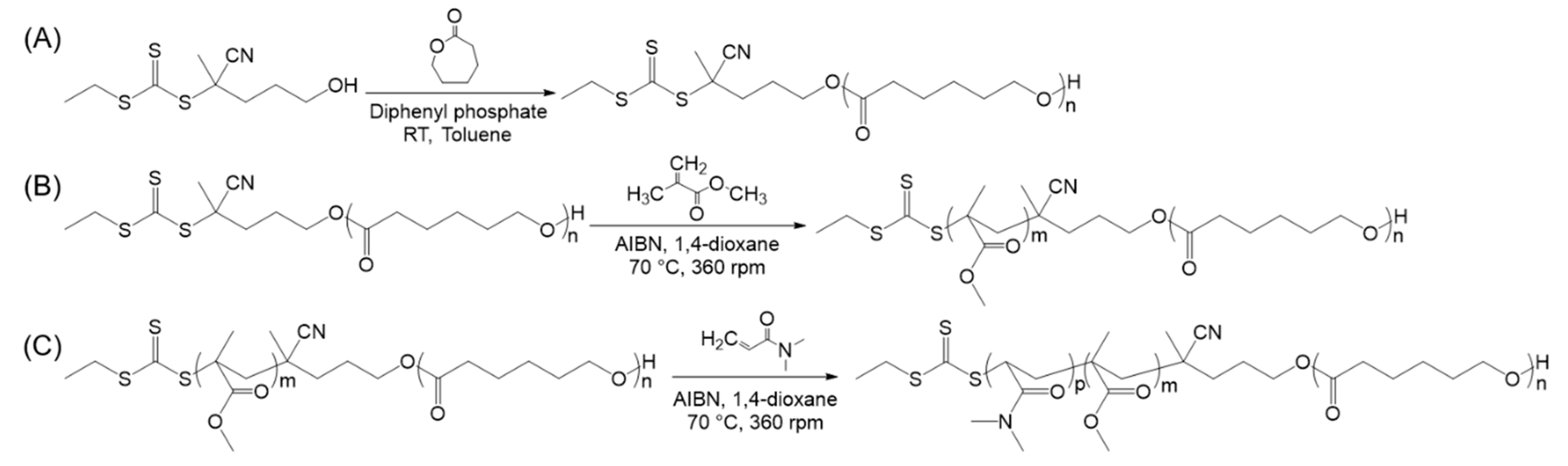
2.2. Typical Procedure for the ROP of PCL Homopolymers
2.3. Typical Procedure for the Synthesis of PCL-b-PMMA Diblock Copolymers
2.4. Typical Procedure for the Synthesis of PCL-b-PMMA-b-PDMA Triblock Copolymers
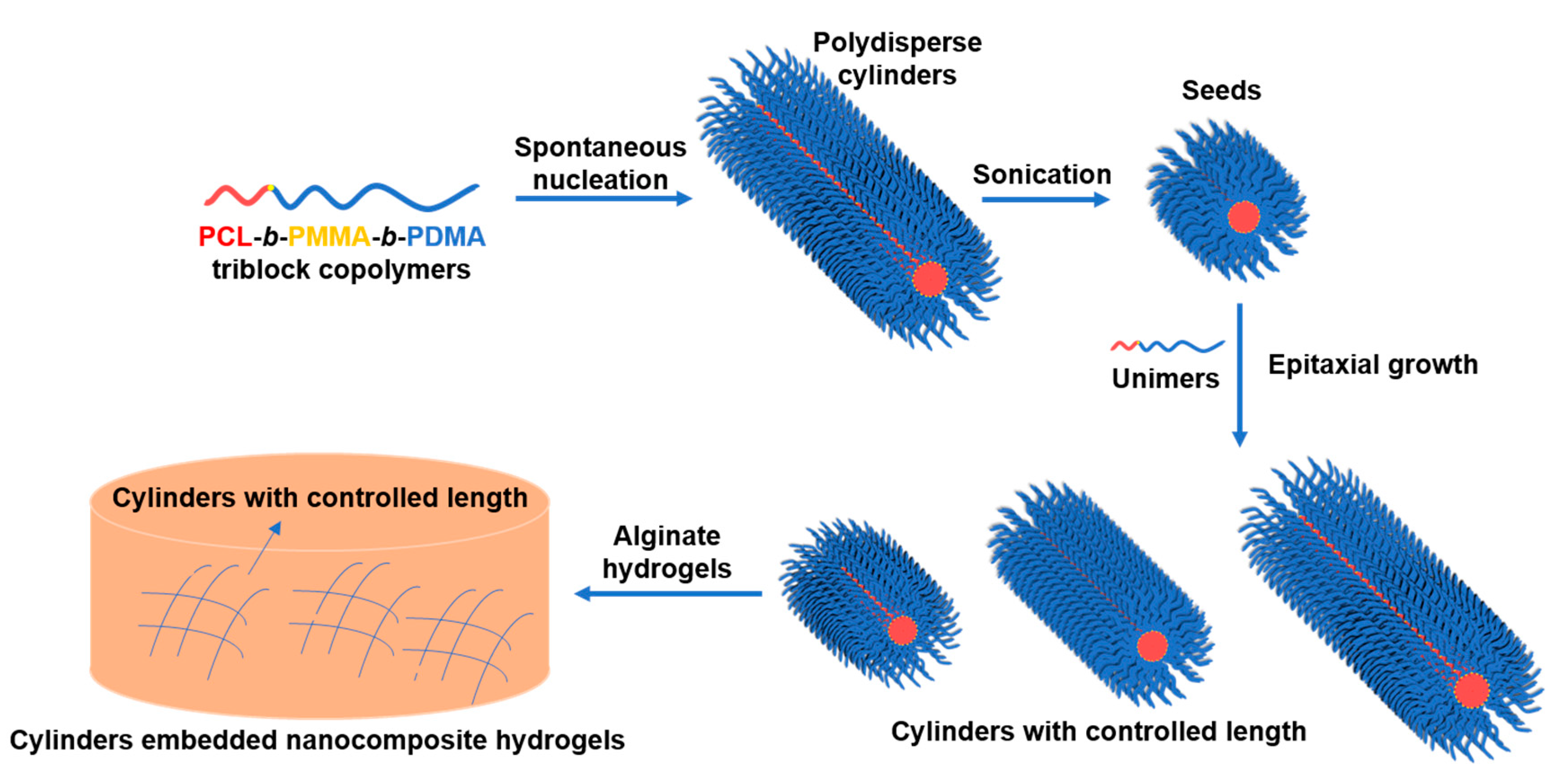
2.5. Typical Crystallization-Driven Self-Assembly Method for the Self-Nucleation of PCL Block Copolymers
2.6. Typical Gel Formation of Nanocomposite Calcium Alginates
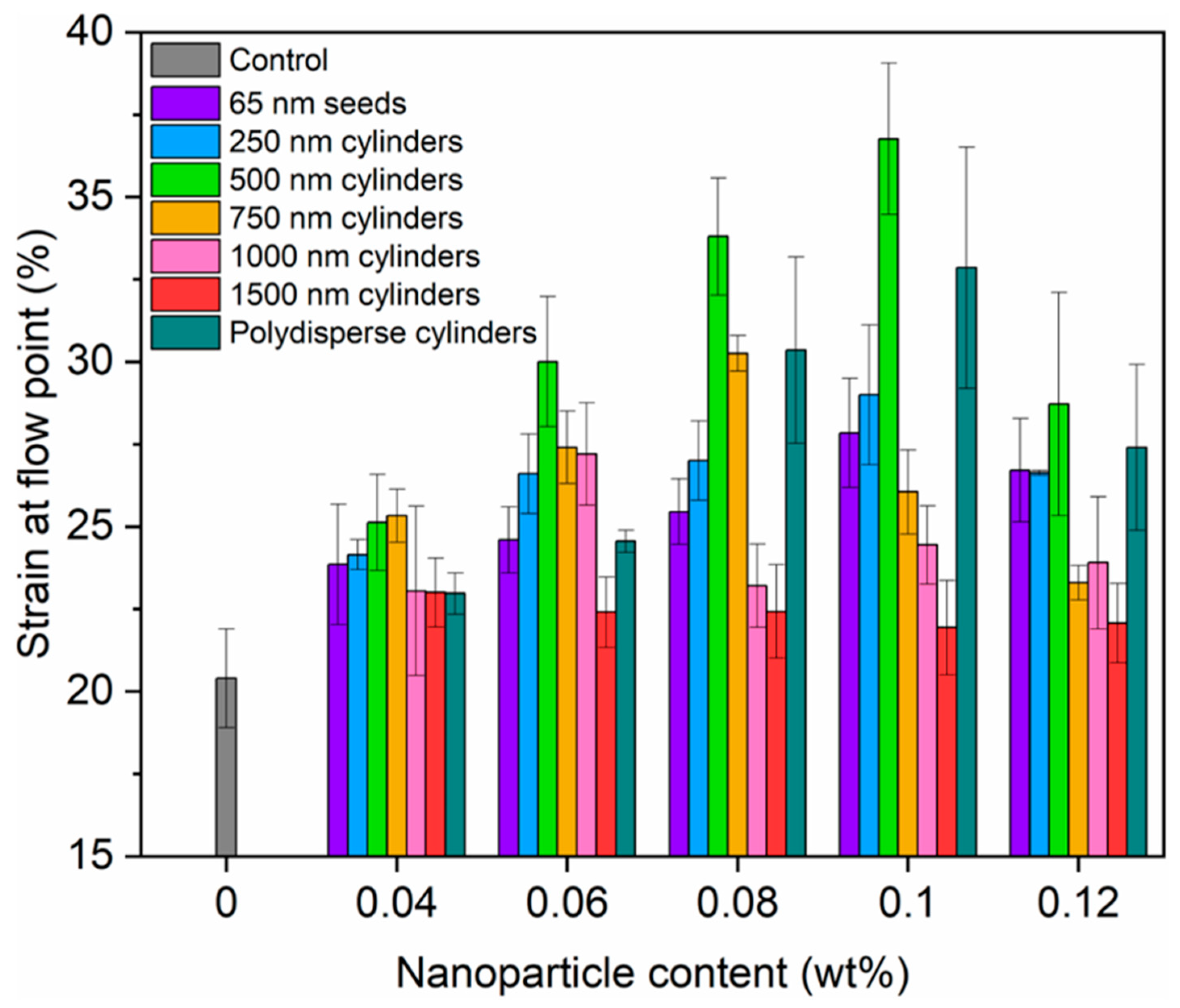
3. Results and Discussion
3.1. Triblock Copolymer Synthesis and Characterization
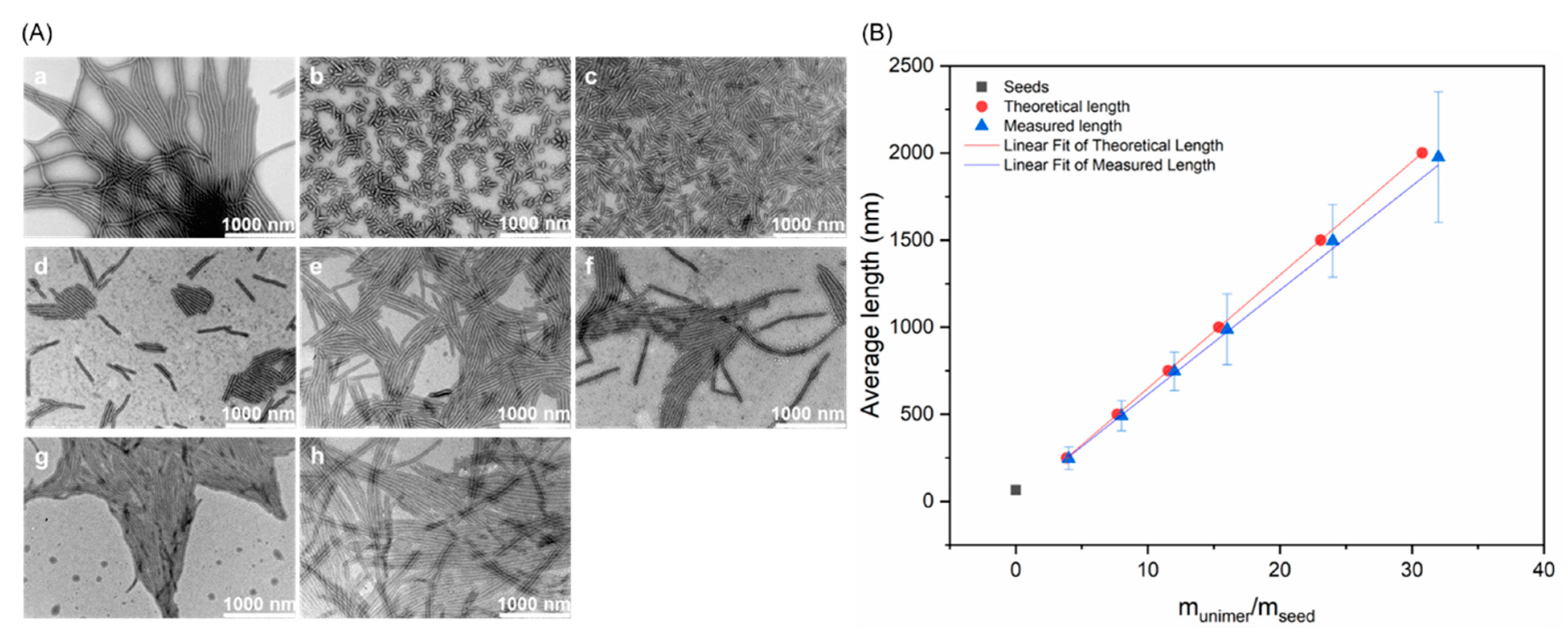
3.2. Crystallization-Driven Self-Assembly for the Production of Cylindrical Micelles
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gelse, K.; Pöschl, E.; Aigner, T. Collagens—structure, function, and biosynthesis. Adv. Drug Deliv. Rev. 2003, 55, 1531–1546. [Google Scholar] [CrossRef] [Green Version]
- Bierbaum, S.; Hintze, V.; Scharnweber, D. 2.8 Artificial extracellular matrices to functionalize biomaterial surfaces. In Comprehensive Biomaterials II; Ducheyne, P., Ed.; Elsevier: Oxford, UK, 2017; pp. 147–178. [Google Scholar]
- Theocharis, A.D.; Skandalis, S.S.; Gialeli, C.; Karamanos, N.K. Extracellular matrix structure. Adv. Drug Deliv. Rev. 2016, 97, 4–27. [Google Scholar] [CrossRef]
- Bosman, F.T.; Stamenkovic, I. Functional structure and composition of the extracellular matrix. J. Pathol. 2003, 200, 423–428. [Google Scholar] [CrossRef]
- Wozniak, M.A.; Chen, C.S. Mechanotransduction in development: A growing role for contractility. Nat. Rev. Mol. Cell Biol. 2009, 10, 34–43. [Google Scholar] [CrossRef] [Green Version]
- DuFort, C.C.; Paszek, M.J.; Weaver, V.M. Balancing forces: Architectural control of mechanotransduction. Nat. Rev. Mol. Cell Biol. 2011, 12, 308–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dworatzek, E.; Baczko, I.; Kararigas, G. Effects of aging on cardiac extracellular matrix in men and women. Proteom. Clin. Appl. 2016, 10, 84–91. [Google Scholar] [CrossRef]
- Swynghedauw, B. Molecular mechanisms of myocardial remodeling. Physiol. Rev. 1999, 79, 215–262. [Google Scholar] [CrossRef]
- Baldwin, A.K.; Simpson, A.; Steer, R.; Cain, S.A.; Kielty, C.M. Elastic fibres in health and disease. Expert Rev. Mol. Med. 2013, 15, e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prince, E.; Kumacheva, E. Design and applications of man-made biomimetic fibrillar hydrogels. Nat. Rev. Mater. 2019, 4, 99–115. [Google Scholar] [CrossRef]
- Zhan, H.; Löwik, D.W.P.M. A Hybrid Peptide Amphiphile Fiber PEG Hydrogel Matrix for 3D Cell Culture. Adv. Funct. Mater. 2019, 29, 1808505. [Google Scholar] [CrossRef]
- Nowak, A.P.; Breedveld, V.; Pakstis, L.; Ozbas, B.; Pine, D.J.; Pochan, D.; Deming, T.J. Rapidly recovering hydrogel scaffolds from self-assembling diblock copolypeptide amphiphiles. Nature 2002, 417, 424–428. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, J.; Magli, S.; Rabbachin, L.; Sampaolesi, S.; Nicotra, F.; Russo, L. 3D Extracellular Matrix Mimics: Fundamental Concepts and Role of Materials Chemistry to Influence Stem Cell Fate. Biomacromolecules 2020, 21, 1968–1994. [Google Scholar] [CrossRef]
- Cohen, D.L.; Malone, E.; Lipson, H.O.D.; Bonassar, L.J. Direct freeform fabrication of seeded hydrogels in arbitrary geometries. Tissue Eng. 2006, 12, 1325–1335. [Google Scholar] [CrossRef] [PubMed]
- Hinderer, S.; Layland, S.L.; Schenke-Layland, K. ECM and ECM-like materials—Biomaterials for applications in regenerative medicine and cancer therapy. Adv. Drug Deliv. Rev. 2016, 97, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Mooney, D.J. Hydrogels for tissue engineering. Chem. Rev. 2001, 101, 1869–1880. [Google Scholar] [CrossRef]
- Kim, M.S.; Kim, G. Three-dimensional electrospun polycaprolactone (PCL)/alginate hybrid composite scaffolds. Carbohydr. Polym. 2014, 114, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Zhu, J.; Londono, J.D.; Pochan, D.J.; Jia, X. Mechano-responsive hydrogels crosslinked by block copolymer micelles. Soft Matter 2012, 8, 10233–10237. [Google Scholar] [CrossRef] [Green Version]
- Xiao, L.; Liu, C.; Zhu, J.; Pochan, D.J.; Jia, X. Hybrid, elastomeric hydrogels crosslinked by multifunctional block copolymer micelles. Soft Matter 2010, 6, 5293–5297. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Jiang, R.; Chen, J.; Wang, M.; Ge, X. In situ synthesis and self-reinforcement of polymeric composite hydrogel based on particulate macro-RAFT agents. RSC Adv. 2017, 7, 1513–1519. [Google Scholar] [CrossRef] [Green Version]
- Tan, M.; Zhao, T.; Huang, H.; Guo, M. Highly stretchable and resilient hydrogels from the copolymerization of acrylamide and a polymerizable macromolecular surfactant. Polym. Chem. 2013, 4, 5570–5576. [Google Scholar] [CrossRef]
- Pek, Y.S.; Wan, A.C.; Shekaran, A.; Zhuo, L.; Ying, J.Y. A thixotropic nanocomposite gel for three-dimensional cell culture. Nat. Nanotechnol. 2008, 3, 671–675. [Google Scholar] [CrossRef]
- Merino, S.; Martin, C.; Kostarelos, K.; Prato, M.; Vazquez, E. Nanocomposite Hydrogels: 3D Polymer–Nanoparticle Synergies for On-Demand Drug Delivery. ACS Nano 2015, 9, 4686–4697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arno, M.C.; Inam, M.; Weems, A.C.; Li, Z.; Binch, A.L.; Platt, C.I.; Richardson, S.M.; Hoyland, J.A.; Dove, A.P.; O’Reilly, R.K. Exploiting the role of nanoparticle shape in enhancing hydrogel adhesive and mechanical properties. Nat. Commun. 2020, 11, 1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tibbitt, M.W.; Rodell, C.B.; Burdick, J.A.; Anseth, K.S. Progress in material design for biomedical applications. Proc. Natl. Acad. Sci. USA 2015, 112, 14444–14451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kesireddy, V.; Kasper, F.K. Approaches for building bioactive elements into synthetic scaffolds for bone tissue engineering. J. Mater. Chem. B 2016, 4, 6773–6786. [Google Scholar] [CrossRef]
- Kharkar, P.M.; Kiick, K.L.; Kloxin, A.M. Designing degradable hydrogels for orthogonal control of cell microenvironments. Chem. Soc. Rev. 2013, 42, 7335–7372. [Google Scholar] [CrossRef] [Green Version]
- Vedadghavami, A.; Minooei, F.; Mohammadi, M.H.; Khetani, S.; Kolahchi, A.R.; Mashayekhan, S.; Sanati-Nezhad, A. Manufacturing of hydrogel biomaterials with controlled mechanical properties for tissue engineering applications. Acta Biomater. 2017, 62, 42–63. [Google Scholar] [CrossRef] [PubMed]
- Caló, E.; Khutoryanskiy, V.V. Biomedical applications of hydrogels: A review of patents and commercial products. Eur. Polym. J. 2015, 65, 252–267. [Google Scholar] [CrossRef] [Green Version]
- Sano, K.; Ishida, Y.; Aida, T. Synthesis of anisotropic hydrogels and their applications. Angew. Chem. Int. Ed. 2018, 57, 2532–2543. [Google Scholar] [CrossRef]
- Rose, J.C.; Gehlen, D.B.; Haraszti, T.; Köhler, J.; Licht, C.J.; De Laporte, L. Biofunctionalized aligned microgels provide 3D cell guidance to mimic complex tissue matrices. Biomaterials 2018, 163, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Lutolf, M.P.; Hubbell, J.A. Synthetic biomaterials as instructive extracellular microenvironments for morphogenesis in tissue engineering. Nat. Biotechnol. 2005, 23, 47–55. [Google Scholar] [CrossRef]
- Münster, S.; Jawerth, L.M.; Leslie, B.A.; Weitz, J.I.; Fabry, B.; Weitz, D.A. Strain history dependence of the nonlinear stress response of fibrin and collagen networks. Proc. Natl. Acad. Sci. USA 2013, 110, 12197–12202. [Google Scholar] [CrossRef] [Green Version]
- Brown, A.E.X.; Litvinov, R.I.; Discher, D.E.; Purohit, P.K.; Weisel, J.W. Multiscale Mechanics of Fibrin Polymer: Gel Stretching with Protein Unfolding and Loss of Water. Science 2009, 325, 741–744. [Google Scholar] [CrossRef] [Green Version]
- Guvendiren, M.; Lu, H.D.; Burdick, J.A. Shear-thinning hydrogels for biomedical applications. Soft Matter 2012, 8, 260–272. [Google Scholar] [CrossRef]
- Black, L.D.; Allen, P.G.; Morris, S.M.; Stone, P.J.; Suki, B. Mechanical and Failure Properties of Extracellular Matrix Sheets as a Function of Structural Protein Composition. Biophys. J. 2008, 94, 1916–1929. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Guerin, G.; Wang, H.; Wang, Y.; Manners, I.; Winnik, M.A. Cylindrical block copolymer micelles and co-micelles of controlled length and architecture. Science 2007, 317, 644–647. [Google Scholar] [CrossRef] [Green Version]
- Arno, M.C.; Inam, M.; Coe, Z.; Cambridge, G.; Macdougall, L.J.; Keogh, R.; Dove, A.P.; O’Reilly, R.K. Precision Epitaxy for Aqueous 1D and 2D Poly(ε-caprolactone) Assemblies. J. Am. Chem. Soc. 2017, 139, 16980–16985. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Petzetakis, N.; Pitto-Barry, A.; Schiller, T.L.; Kirby, N.; Keddie, D.J.; Boyd, B.J.; O’Reilly, R.K.; Dove, A.P. Tuning the size of cylindrical micelles from poly (L-lactide)-b-poly (acrylic acid) diblock copolymers based on crystallization-driven self-assembly. Macromolecules 2013, 46, 9074–9082. [Google Scholar] [CrossRef]
- Shi, Z.; Wei, Y.; Zhu, C.; Sun, J.; Li, Z. Crystallization-Driven Two-Dimensional Nanosheet from Hierarchical Self-Assembly of Polypeptoid-Based Diblock Copolymers. Macromolecules 2018, 51, 6344–6351. [Google Scholar] [CrossRef]
- Tao, D.; Feng, C.; Cui, Y.; Yang, X.; Manners, I.; Winnik, M.A.; Huang, X. Monodisperse Fiber-like Micelles of Controlled Length and Composition with an Oligo(p-phenylenevinylene) Core via “Living” Crystallization-Driven Self-Assembly. J. Am. Chem. Soc. 2017, 139, 7136–7139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Wang, L.Q.; Wang, H.; Tu, K. Micellization Phenomena of Amphiphilic Block Copolymers Based on Methoxy Poly(ethylene glycol) and Either Crystalline or Amorphous Poly(caprolactone-b-lactide). Biomacromolecules 2006, 7, 2492–2500. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.X.; Xu, J.T.; Fan, Z.Q. Regulation of Micellar Morphology of PCL-b-PEO Block Copolymers by Crystallization Temperature. Macromol. Rapid Commun. 2008, 29, 467–471. [Google Scholar] [CrossRef]
- Chan, S.-C.; Kuo, S.W.; Lu, C.H.; Lee, H.F.; Chang, F.C. Syntheses and characterizations of the multiple morphologies formed by the self-assembly of the semicrystalline P4VP-b-PCL diblock copolymers. Polymer 2007, 48, 5059–5068. [Google Scholar] [CrossRef]
- Muñoz-Bonilla, A.; Cerrada, M.L.; Fernández-García, M.; Kubacka, A.; Ferrer, M.; Fernández-García, M. Biodegradable Polycaprolactone-Titania Nanocomposites: Preparation, Characterization and Antimicrobial Properties. Int. J. Mol. Sci. 2013, 14, 9249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Huo, H.; Li, J.; Shang, Y.; Chen, Y.; Funari, S.S.; Jiang, S. Crystallization behavior of poly(ε-caprolactone) and poly (ε-caprolactone)/LiClO4 complexes from the melt. CrystEngComm 2012, 14, 7972–7980. [Google Scholar] [CrossRef]
- Lee, K.Y.; Mooney, D.J. Alginate: Properties and biomedical applications. Prog. Polym. Sci. 2012, 37, 106–126. [Google Scholar] [CrossRef] [Green Version]
- Kuo, C.K.; Ma, P.X. Ionically crosslinked alginate hydrogels as scaffolds for tissue engineering: Part 1. Structure, gelation rate and mechanical properties. Biomaterials 2001, 22, 511–521. [Google Scholar] [CrossRef]
- Clogston, J.D.; Patri, A.K. Zeta Potential Measurement. In Characterization of Nanoparticles Intended for Drug Delivery; McNeil, S.E., Ed.; Humana Press: Totowa, NJ, USA, 2011; pp. 63–70. [Google Scholar]
- Cheng, K.-C.; Huang, C.F.; Wei, Y.; Hsu, S.H. Novel chitosan–cellulose nanofiber self-healing hydrogels to correlate self-healing properties of hydrogels with neural regeneration effects. NPG Asia Mater. 2019, 11, 25. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target (nm) | Lwa (nm) | Lnb (nm) | Lw/Ln |
|---|---|---|---|
| Seeds | 68 | 65 | 1.05 |
| 250 nm | 262 | 246 | 1.07 |
| 500 nm | 506 | 491 | 1.03 |
| 750 nm | 762 | 746 | 1.02 |
| 1000 nm | 1029 | 988 | 1.04 |
| 1500 nm | 1561 | 1532 | 1.02 |
| 2000 nm | 1984 | 1911 | 1.04 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Pearce, A.K.; Dove, A.P.; O’Reilly, R.K. Precise Tuning of Polymeric Fiber Dimensions to Enhance the Mechanical Properties of Alginate Hydrogel Matrices. Polymers 2021, 13, 2202. https://0-doi-org.brum.beds.ac.uk/10.3390/polym13132202
Li Z, Pearce AK, Dove AP, O’Reilly RK. Precise Tuning of Polymeric Fiber Dimensions to Enhance the Mechanical Properties of Alginate Hydrogel Matrices. Polymers. 2021; 13(13):2202. https://0-doi-org.brum.beds.ac.uk/10.3390/polym13132202
Chicago/Turabian StyleLi, Zehua, Amanda K. Pearce, Andrew P. Dove, and Rachel K. O’Reilly. 2021. "Precise Tuning of Polymeric Fiber Dimensions to Enhance the Mechanical Properties of Alginate Hydrogel Matrices" Polymers 13, no. 13: 2202. https://0-doi-org.brum.beds.ac.uk/10.3390/polym13132202