
Temporal Changes of Adsorbed Layer Thickness and Electrophoresis of Polystyrene Sulfate Latex Particles after Long Incubation of Oppositely Charged Polyelectrolytes with Different Charge Densities


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
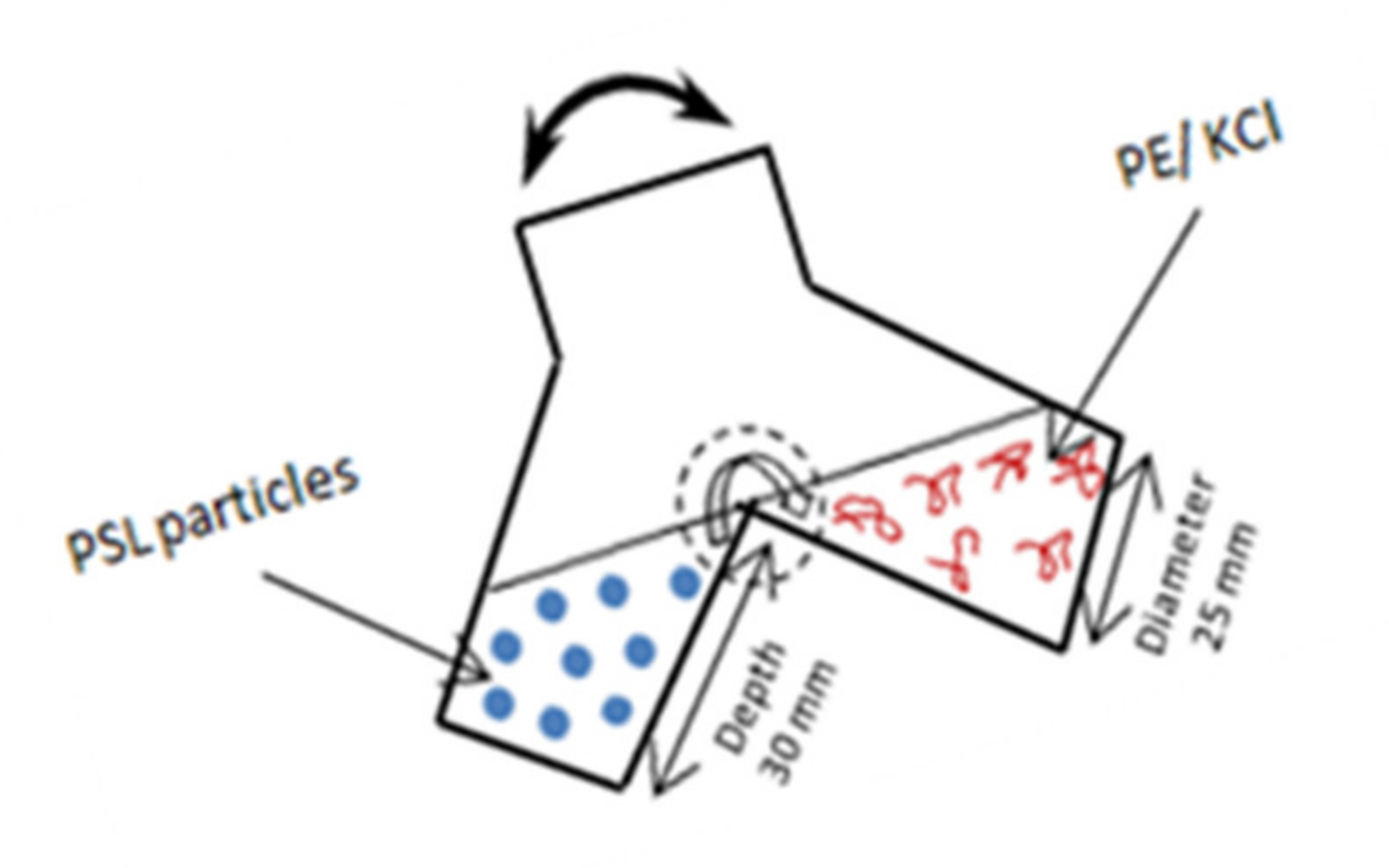
2.3. Experimental Procedure
2.3.1. Experimental Measurements of Hydrodynamic Layer Thickness of Adsorbed Polyelectrolytes
2.3.2. Electrophoretic Mobility Measurements
2.3.3. Viscosity Measurements
3. Results and Discussion
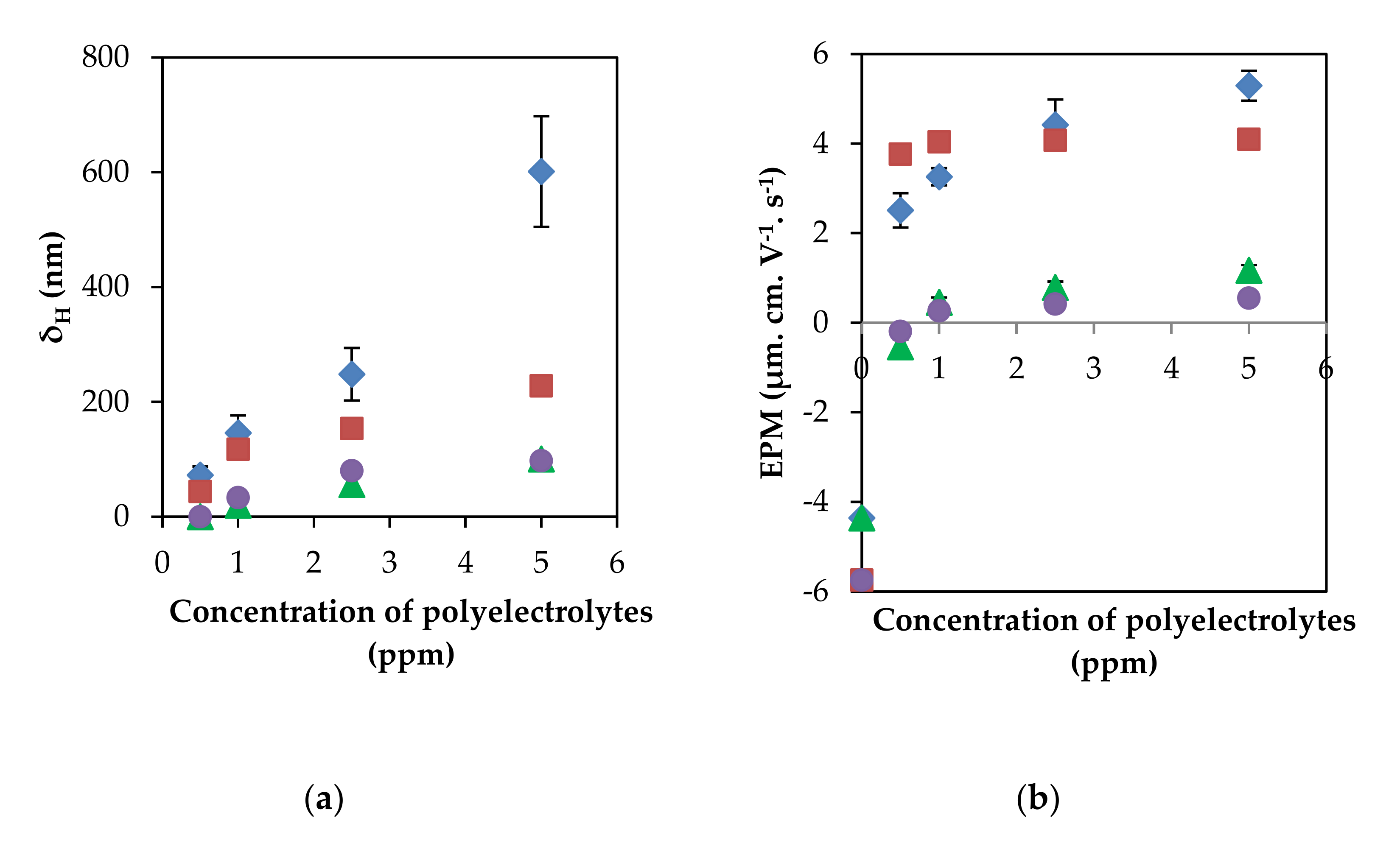
3.1. Contributions of Electrostatic and Non-Electrostatic Interactions on Adsorption of Polyelectrolytes with Different Charge Densities onto PSL Particles at Different Ionic Strengths
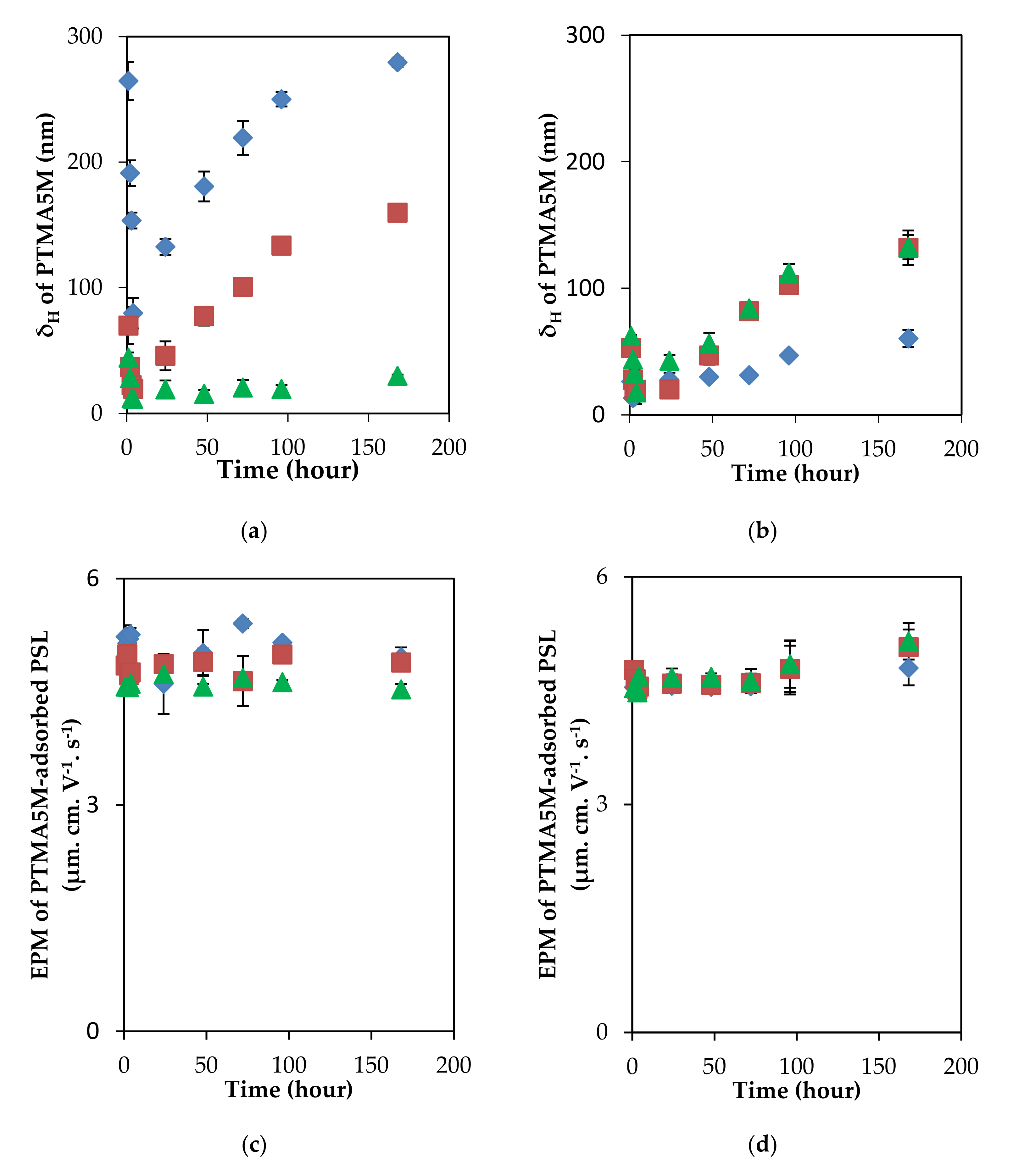
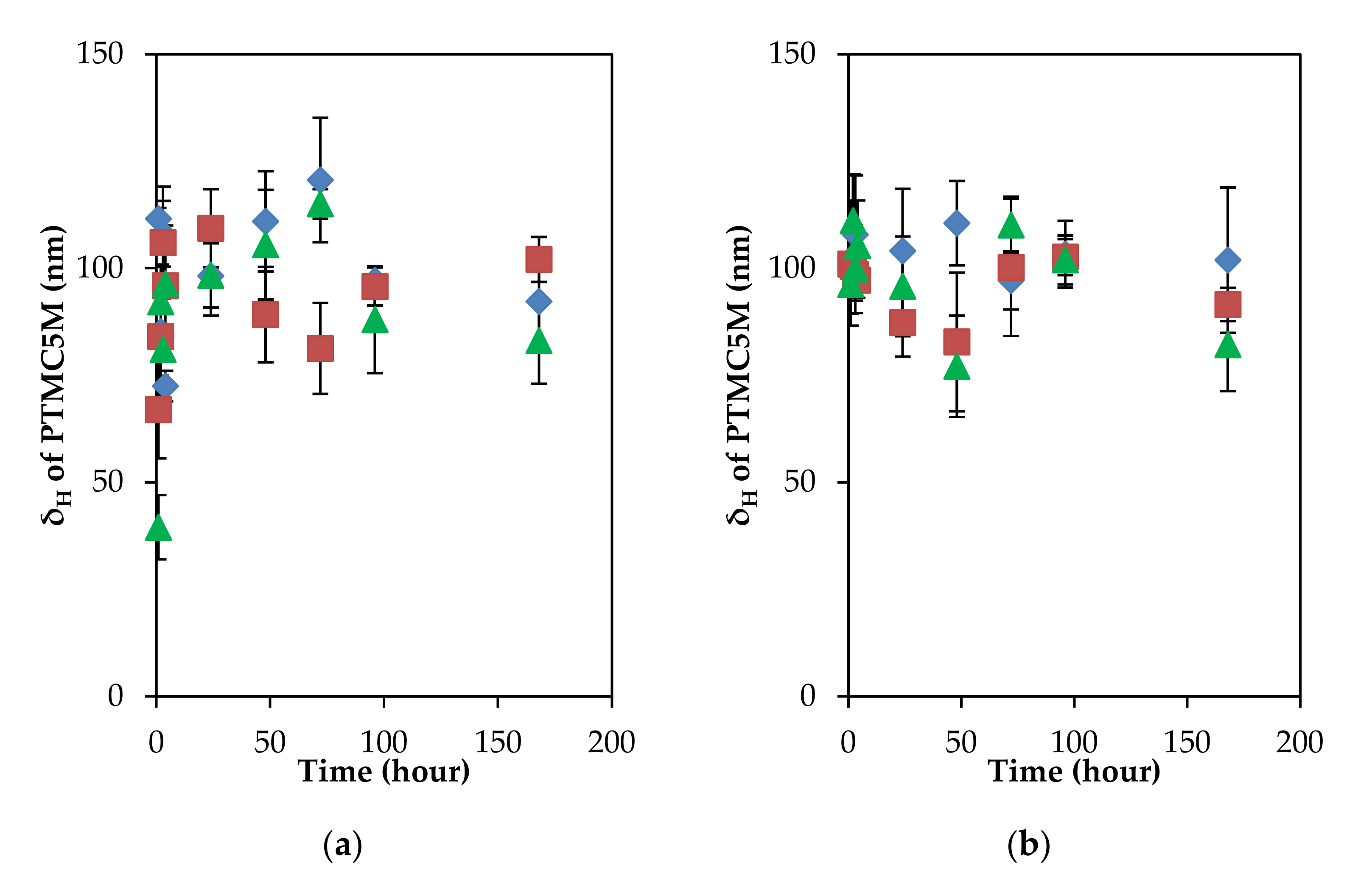
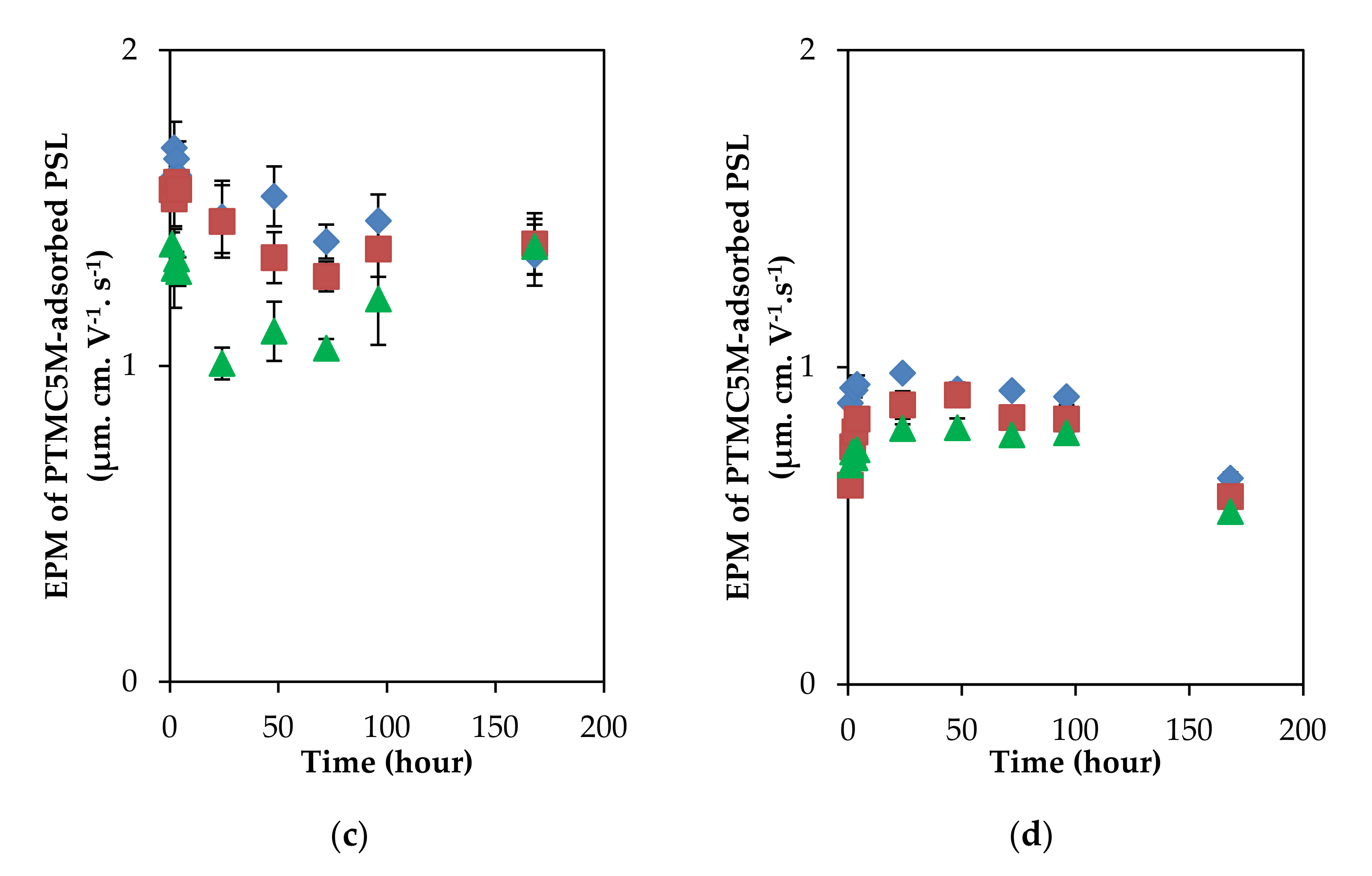
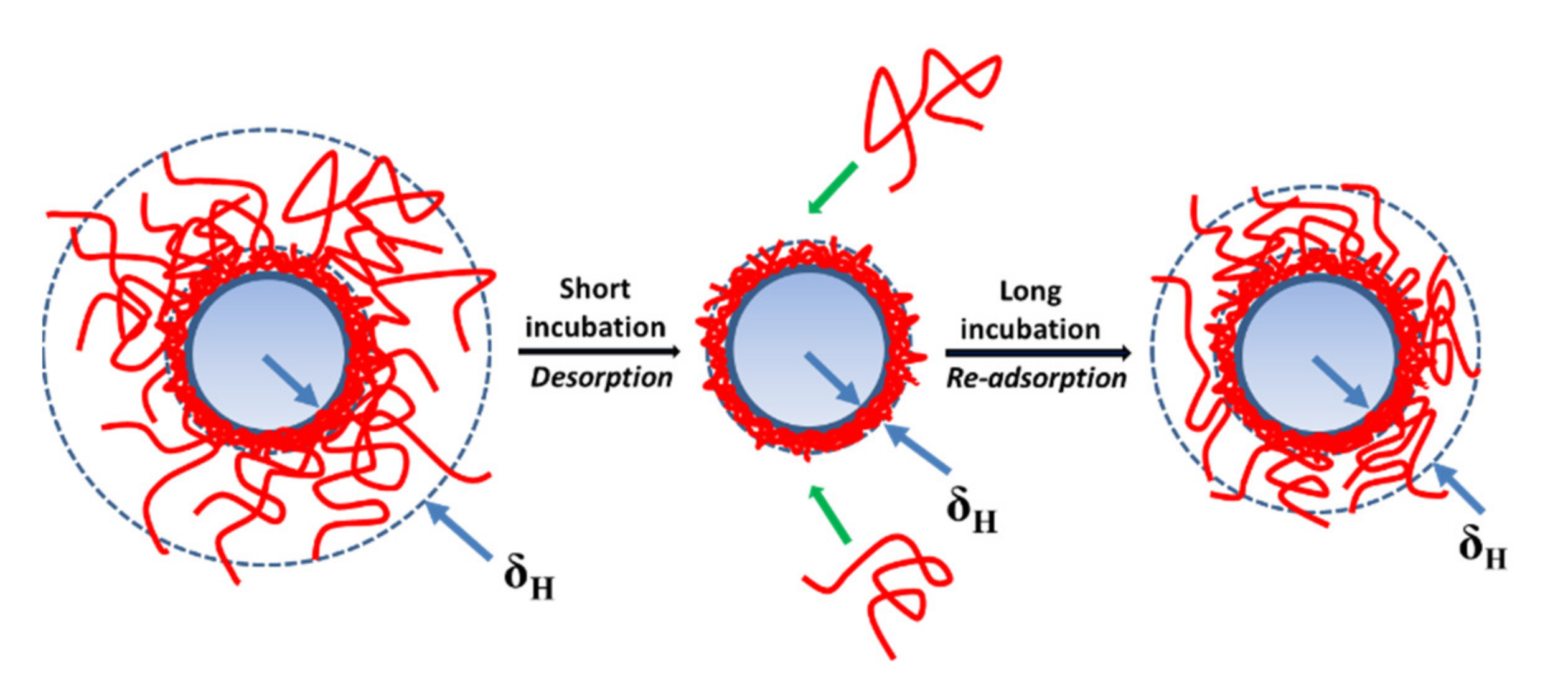
3.2. Relaxation Behaviors of Adsorbed Layer of Polyelectrolytes with Different Charge Density on the PSL Particles in a Long Incubation
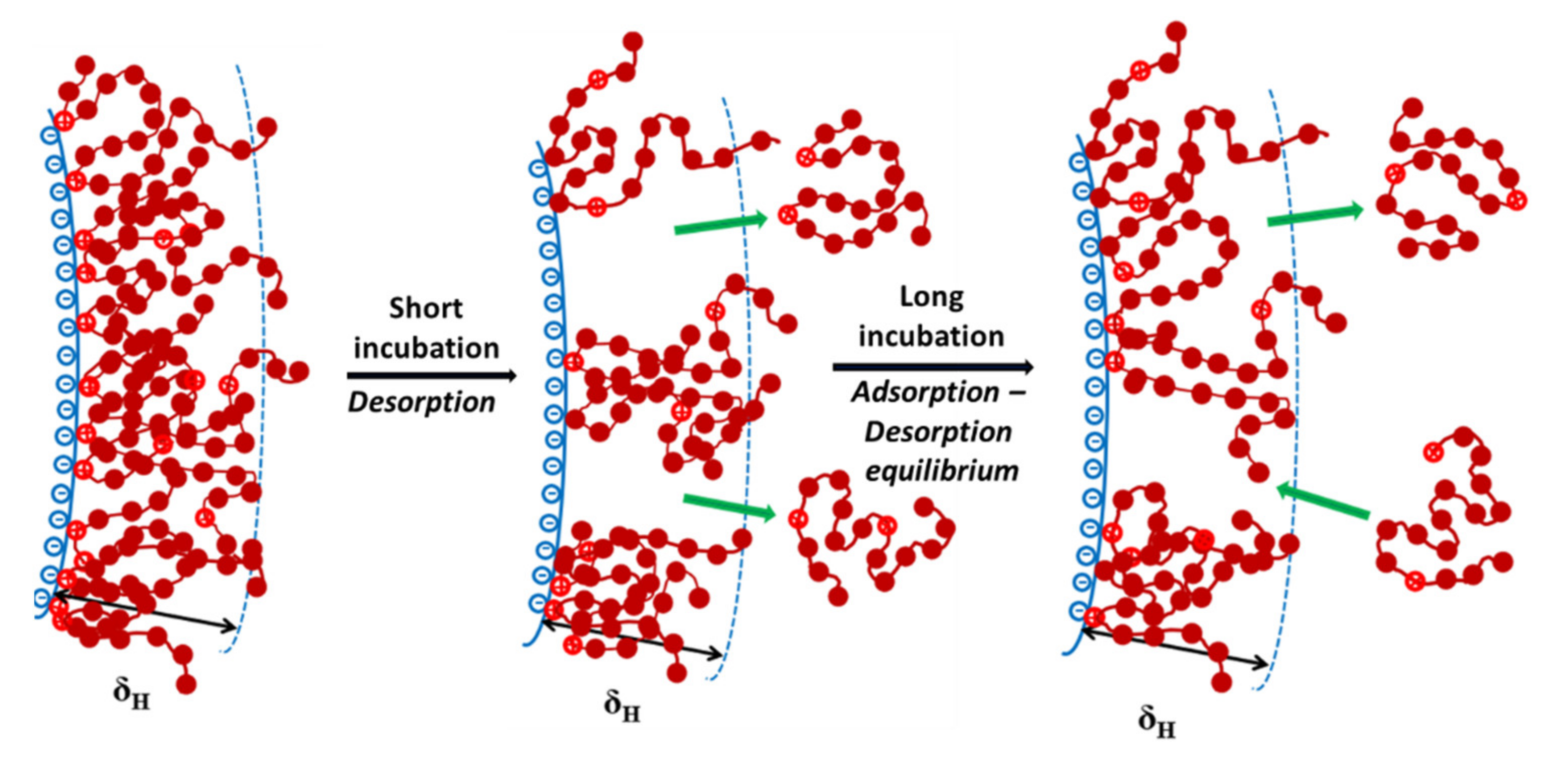
3.3. Adsorption/Desorption Concepts of Polyelectrolytes with Different Charge Density on the PSL Particles in a Long Incubation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Moody, G.M. Polymeric Flocculants. In Handbook of Industrial Water Soluble Polymers; John Wiley & Sons: Hoboken, NJ, USA, 2007; pp. 134–173. [Google Scholar]
- Nasser, M.S.; Twaiq, F.A.; Onaizi, S.A. Effect of polyelectrolytes on the degree of flocculation of papermaking suspensions. Sep. Purif. Technol. 2013, 103, 43–52. [Google Scholar] [CrossRef]
- Dickinson, E.; Eriksson, L. Particle flocculation by adsorbing polymers. Adv. Colloid Interface Sci. 1991, 34, 1–29. [Google Scholar] [CrossRef]
- Rojas, O.J.; Claesson, P.M.; Muller, D.; Neuman, R.D. The Effect of Salt Concentration on Adsorption of Low-Charge-Density Polyelectrolytes and Interactions between Polyelectrolyte-Coated Surfaces. J. Colloid Interface Sci. 1998, 205, 77–88. [Google Scholar] [CrossRef] [Green Version]
- Shin, Y.; Roberts, J.E.; Santore, M.M. The relationship between polymer/substrate charge density and charge overcompensation by adsorbed polyelectrolyte layers. J. Colloid Interface Sci 2002, 247, 220–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, L.; Adachi, Y. Brownian flocculation of negatively charged latex particles with low charge density polycation at various ionic strengths. Colloids Surfaces A Physicochem. Eng. Asp. 2014, 454, 128–134. [Google Scholar] [CrossRef]
- Hasan, A.; Fatehi, P. Flocculation of kaolin particles with cationic lignin polymers. Sci. Rep. 2019, 9, 2672. [Google Scholar] [CrossRef]
- Lindquist, G.M.; Stratton, R.A. The role of polyelectrolyte charge density and molecular weight on the adsorption and flocculation of colloidal silica with polyethylenimine. J. Colloid Interface Sci. 1976, 55, 45–59. [Google Scholar] [CrossRef] [Green Version]
- Aoki, K.; Adachi, Y. Kinetics of polyelectrolyte adsorption onto polystyrene latex particle studied using electrophoresis: Effects of molecular weight and ionic strength. J. Colloid Interface Sci. 2006, 300, 69–77. [Google Scholar] [CrossRef]
- Linse, P. Adsorption of Weakly Charged Polyelectrolytes at Oppositely Charged Surfaces. Macromolecules 1996, 29, 326–336. [Google Scholar] [CrossRef]
- Tjipangandjara, K.F.; Somasundaran, P. Effects of changes in adsorbed polyacrylic acid conformation on alumina flocculation. Colloids Surfaces 1991, 55, 245–255. [Google Scholar] [CrossRef]
- Ishiduki, K.; Esumi, K. Adsorption Characteristics of Poly(acrylic acid) and Poly(vinyl pyrrolidone) on Alumina from Their Mixtures in Aqueous Solution. J. Colloid Interface Sci. 1997, 185, 274–277. [Google Scholar] [CrossRef]
- Yu, X.; Somasundaran, P. Role of Polymer Conformation in Interparticle-Bridging Dominated Flocculation. J. Colloid Interface Sci. 1996, 177, 283–287. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, S.; Bratskaya, S.; Jaeger, W.; Paulke, B.R. Effect of charge density, molecular weight, and hydrophobicity on polycations adsorption and flocculation of polystyrene latices and silica. J. Appl. Polym. Sci. 2006, 101, 3422–3429. [Google Scholar] [CrossRef]
- Lapointe, M.; Barbeau, B. Understanding the roles and characterizing the intrinsic properties of synthetic vs. natural polymers o improve clarification through interparticle Bridging: A review. Sep. Purif. Technol. 2020, 231, 115893. [Google Scholar] [CrossRef]
- Kawaguchi, M. Sequential polymer adsorption: Competition and displacement process. Adv. Colloid Interface Sci 1990, 32, 1–41. [Google Scholar] [CrossRef]
- Feng, L.; Stuart, M.C.; Adachi, Y. Dynamics of polyelectrolyte adsorption and colloidal flocculation upon mixing studied using mono-dispersed polystyrene latex particles. Adv. Colloid Interface Sci 2015, 226, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Brynda, M.; Chodanowski, P.; Stoll, S. Polyelectrolyte–particle complex formation. Polyelectrolyte linear charge density and ionic concentration effects. Monte Carlo simulations. Colloid Polym. Sci. 2002, 280, 789–797. [Google Scholar] [CrossRef]
- De Witt, J.A.; Van de Ven, T.G.M. Kinetics and reversibility of the adsorption of poly(vinyl alcohol) onto polystyrene latex particles. Langmuir 1992, 8, 788–793. [Google Scholar] [CrossRef]
- Einarson, M.; Aksberg, R.; Ödberg, L.; Berg, J.C. Adsorption and reconformation of a series of cationic polyacrylamides on charged surfaces. Colloids Surfaces 1991, 53, 183–191. [Google Scholar] [CrossRef]
- Kohay, H.; Bilkis, I.I.; Mishael, Y.G. Effect of polycation charge density on polymer conformation at the clay surface and consequently on pharmaceutical binding. J. Colloid Interface Sci. 2019, 552, 517–527. [Google Scholar] [CrossRef]
- Rojas, O.J.; Ernstsson, M.; Neuman, R.D.; Claesson, P.M. Effect of Polyelectrolyte Charge Density on the Adsorption and Desorption Behavior on Mica. Langmuir 2002, 18, 1604–1612. [Google Scholar] [CrossRef]
- Plunkett, M.A.; Claesson, P.M.; Ernstsson, M.; Rutland, M.W. Comparison of the Adsorption of Different Charge Density Polyelectrolytes: A Quartz Crystal Microbalance and X-ray Photoelectron Spectroscopy Study. Langmuir 2003, 19, 4673–4681. [Google Scholar] [CrossRef]
- Barany, S.; Nagy, M.; Skvarla, J. Electrokinetic potential of polystyrene particles in polyelectrolyte and polyelectrolyte mixtures solutions. Colloids Surfaces A Physicochem. Eng. Asp. 2012, 413, 200–207. [Google Scholar] [CrossRef]
- Dukhin, S.S.; Zimmermann, R.; Werner, C. Charge density distribution at interfaces between polyelectrolyte layers and aqueous solutions—Experimental access and limitations of traditional electrokinetics. J. Colloid Interface Sci. 2008, 328, 186. [Google Scholar] [CrossRef]
- Adachi, Y.; Kusaka, Y.; Kobayashi, A. Transient behavior of adsorbing/adsorbed polyelectrolytes on the surface of colloidal particles studied by means of trajectory analysis of Brownian motion. Colloids Surfaces A Physicochem. Eng. Asp. 2011, 376, 9–13. [Google Scholar] [CrossRef]
- Doan, T.H.Y.; Adachi, Y. Relaxation of adsorbed layer thickness and electrophoresis of polystyrene latex particles after overshooting of polyelectrolytes with different charge density. Colloids Surfaces A Physicochem. Eng. Asp. 2020, 603, 125208. [Google Scholar] [CrossRef]
- Kusaka, Y.; Adachi, Y. Determination of hydrodynamic diameter of small flocs by means of direct movie analysis of Brownian motion. Colloids Surfaces A Physicochem. Eng. Asp. 2007, 306, 166–170. [Google Scholar] [CrossRef]
- Holde, K.E. Viscosity. In Physical Biochemistry; Prentice-Hall Inc.: Hoboken, NJ, USA, 1971; pp. 166–168. [Google Scholar]
- Adachi, Y.; Feng, L.; Kobayashi, M. Kinetics of flocculation of polystyrene latex particles in the mixing flow induced with high charge density polycation near the isoelectric point. Colloids Surfaces A Physicochem. Eng. Asp. 2015, 471, 38–44. [Google Scholar] [CrossRef]
- Yu, X.; Somasundaran, P. Enhanced flocculation with double flocculants. Colloids Surfaces A Physicochem. Eng. Asp. 1993, 81, 17–23. [Google Scholar] [CrossRef]
- Gregory, J.; Barany, S. Adsorption and flocculation by polymers and polymer mixtures. Adv. Colloid Interface Sci 2011, 169, 1–12. [Google Scholar] [CrossRef]
- Iruthayaraj, J.; Poptoshev, E.; Vareikis, A.; Makuška, R.; van der Wal, A.; Claesson, P.M. Adsorption of Low Charge Density Polyelectrolyte Containing Poly(ethylene oxide) Side Chains on Silica: Effects of Ionic Strength and pH. Macromolecules 2005, 38, 6152–6160. [Google Scholar] [CrossRef]
- Pefferkorn, E.; Elaissari, A. Adsorption—desorption processes in charged polymer/colloid systems; structural relaxation of adsorbed macromolecules. J. Colloid Interface Sci. 1990, 138, 187–194. [Google Scholar] [CrossRef]
- Matsumoto, Y.A. and T. Dynamics of initial stage flocculation of polystyrene latex spheres with polyelectrolytes. Colloids Surfaces A Physicochem. Eng. Asp. 1996, 113, 229–236. [Google Scholar]
- Van de Steeg, H.G.M.; Cohen Stuart, M.A.; De Keizer, A.; Bijsterbosch, B.H. Polyelectrolyte adsorption: A subtle balance of forces. Langmuir 1992, 8, 2538–2546. [Google Scholar] [CrossRef]
- Popa, I.; Gillies, G.; Papastavrou, G.; Borkovec, M. Attractive and Repulsive Electrostatic Forces between Positively Charged Latex Particles in the Presence of Anionic Linear Polyelectrolytes. J. Phys. Chem. B 2010, 114, 3170–3177. [Google Scholar] [CrossRef]
- Lyklema, J.; Deschenes, L. The first step in layer-by-layer deposition: Electrostatics and/or non-electrostatics? Adv. Colloid Interface Sci 2011, 168, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Gärdlund, L.; Wågberg, L.; Norgren, M. New insights into the structure of polyelectrolyte complexes. J. Colloid Interface Sci. 2007, 312, 237–246. [Google Scholar] [CrossRef]
- Shubin, V.; Linse, P. Effect of Electrolytes on Adsorption of Cationic Polyacrylamide on Silica: Ellipsometric Study and Theoretical Modeling. J. Phys. Chem. 1995, 99, 1285–1291. [Google Scholar] [CrossRef]
- Bolto, B.; Gregory, J. Organic polyelectrolytes in water treatment. Water Res. 2007, 41, 2301–2324. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, H.; Tamotsu, K. Approximate analytic expression for the electrophoretic mobility of colloidal particles with surface-charge layers. J. Colloid Interface Sci. 1989, 130, 281–282. [Google Scholar] [CrossRef]
- Ohshima, H. Approximate Analytic Expression for the Electrophoretic Mobility of Moderately Charged Cylindrical Colloidal Particles. Langmuir 2015, 31, 13633–13638. [Google Scholar] [CrossRef]
- Ohshima, H. Approximate Analytic Expression for the Electrophoretic Mobility of a Spherical Colloidal Particle. J. Colloid Interface Sci. 2001, 239, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, H.; Nakamura, M.; Kondo, T. Electrophoretic mobility of colloidal particles coated with a layer of adsorbed polymers. Colloid Polym. Sci. 1992, 270, 873–877. [Google Scholar] [CrossRef]
- Ohshima, H.; Kondo, T. Electrophoresis of large colloidal particles with surface charge layers. Position of the slipping plane and surface layer thickness. Colloid Polym. Sci. 1986, 264, 1080–1084. [Google Scholar]
- Ohshima, H.; Kondo, T. Electrophoretic mobility and Donnan potential of a large colloidal particle with a surface charge layer. J. Colloid Interface Sci. 1987, 116, 305–311. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polyelectrolytes | Relative Viscosity | Hydrodynamic Diameter (nm) | ||
|---|---|---|---|---|
| 0.1 mM KCl | 10 mM KCl | 1 M KCl | 1 M KCl | |
| PTMA5M, ρ = 100% | 1.0260 ± 0.0012 | 1.0089 ± 0.0009 | 1.0030 ± 0.0005 | 155.46 ± 8.97 |
| PTMC5M, ρ = 4% | 1.0063 ± 0.0008 | 1.0062 ± 0.0007 | 1.0038 ± 0.0004 | 171.91 ± 6.69 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doan, T.H.Y.; Pham, T.D.; Hunziker, J.; Hoang, T.H. Temporal Changes of Adsorbed Layer Thickness and Electrophoresis of Polystyrene Sulfate Latex Particles after Long Incubation of Oppositely Charged Polyelectrolytes with Different Charge Densities. Polymers 2021, 13, 2394. https://0-doi-org.brum.beds.ac.uk/10.3390/polym13152394
Doan THY, Pham TD, Hunziker J, Hoang TH. Temporal Changes of Adsorbed Layer Thickness and Electrophoresis of Polystyrene Sulfate Latex Particles after Long Incubation of Oppositely Charged Polyelectrolytes with Different Charge Densities. Polymers. 2021; 13(15):2394. https://0-doi-org.brum.beds.ac.uk/10.3390/polym13152394
Chicago/Turabian StyleDoan, Thi Hai Yen, Tien Duc Pham, Johan Hunziker, and Thu Ha Hoang. 2021. "Temporal Changes of Adsorbed Layer Thickness and Electrophoresis of Polystyrene Sulfate Latex Particles after Long Incubation of Oppositely Charged Polyelectrolytes with Different Charge Densities" Polymers 13, no. 15: 2394. https://0-doi-org.brum.beds.ac.uk/10.3390/polym13152394