Decoding the Role of Satellite DNA in Genome Architecture and Plasticity—An Evolutionary and Clinical Affair

 , , and
, , and {kind=link}
{kind=link}
Abstract
:1. Introduction
2. SatDNA Features and Organization in the Genome and Chromosomes: Emerging Technologies and Changing Concepts
3. Modulating Genome Architecture with SatDNAs
3.1. Repetitive Sequences, Chromosome Instability and Disease
3.1.1. Remodeling Genome Architecture Through Robertsonian Translocations from a SatDNA Perspective
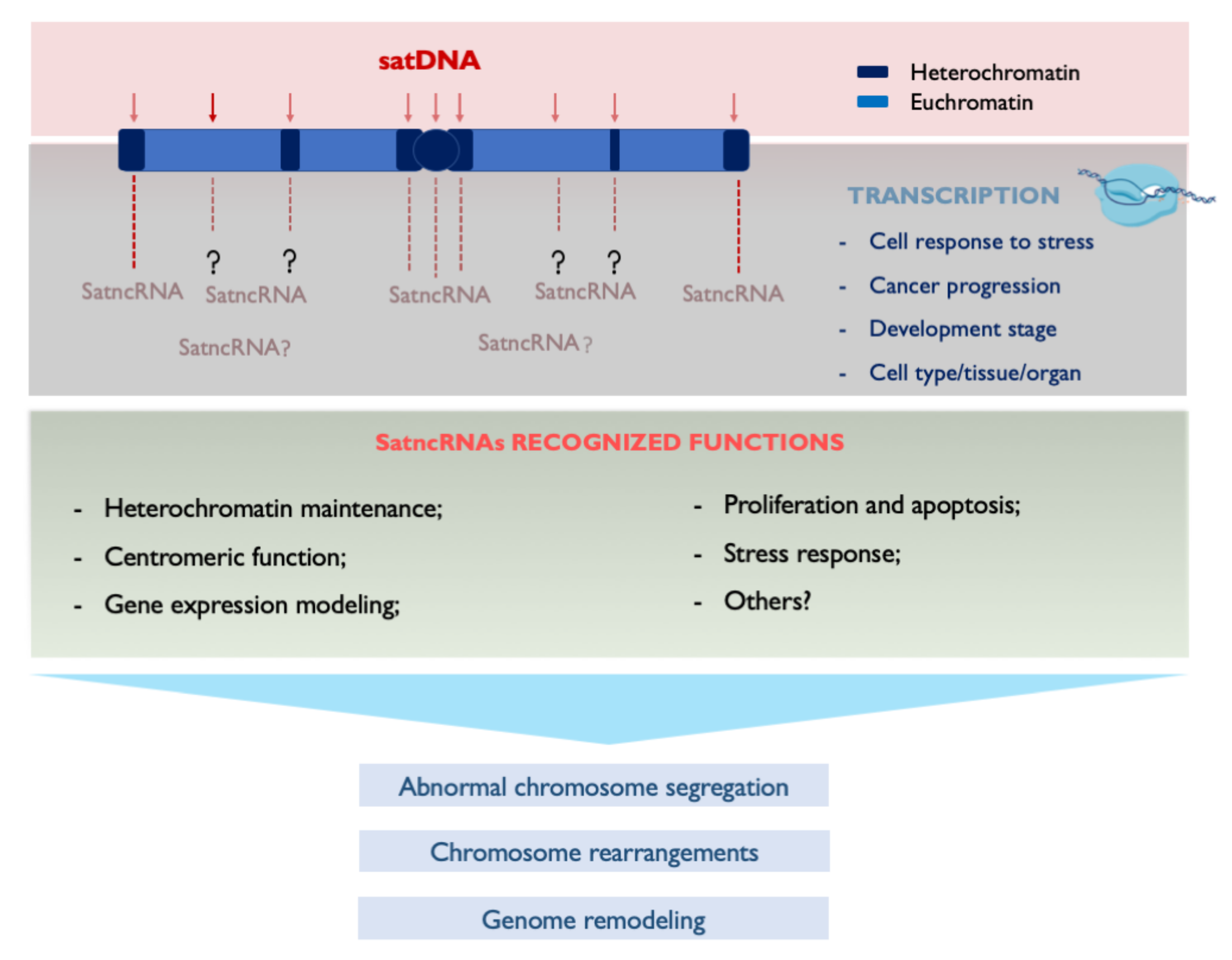
4. Transcribing SatDNAs: Targeting Genomic Functions
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Charlesworth, B.; Sniegowski, P.; Stephan, L.W. The evolutionary dynamics of repetitive DNA in eukaryotes. Nature 1994, 371, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Plohl, M.; Mestrovic, N.; Mravinac, B. Satellite DNA evolution. Genome Dyn. 2012, 7, 126–152. [Google Scholar] [CrossRef]
- López-Flores, I.; Garrido-Ramos, M.A. The Repetitive DNA Content of Eukaryotic Genomes. Genome Dyn. 2012, 7, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Brajković, J.; Feliciello, I.; Bruvo-Mađarić, B.; Ugarković, D. Satellite DNA-like elements associated with genes within euchromatin of the beetle Tribolium castaneum. G3 Genes Genomes Genet. 2012, 2, 931–941. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, G.C.; Küttler, H.; Moreira-Filho, O.; Heslop-Harrison, J.S. The 1.688 repetitive DNA of Drosophila: Concerted evolution at different genomic scales and association with genes. Mol. Biol. Evol. 2012, 29, 7–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larracuente, A.M. The organization and evolution of the Responder satellite in species of the Drosophila melanogaster group: Dynamic evolution of a target of meiotic drive. BMC Evol. Biol. 2014, 14, 233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlek, M.; Gelfand, Y.; Plohl, M.; Meštrović, N. Genome-wide analysis of tandem repeats in Tribolium castaneum genome reveals abundant and highly dynamic tandem repeat families with satellite DNA features in euchromatic chromosomal arms. DNA Res. 2015, 22, 387–401. [Google Scholar] [CrossRef] [Green Version]
- Feliciello, I.; Akrap, I.; Ugarkovic, D. Satellite DNA modulates gene expression in the beetle Tribolium castaneum after heat stress. PLoS Genet. 2015, 11, e1005466. [Google Scholar] [CrossRef] [Green Version]
- Volpe, T.A.; Kidner, C.; Hall, I.M.; Teng, G.; Grewal, S.I.; Martienssen, R.A. Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi. Science 2002, 297, 1833–1837. [Google Scholar] [CrossRef] [Green Version]
- Martienssen, R.A. Maintenance of heterochromatin by RNA interference of tandem repeats. Nat. Genet. 2003, 35, 213–214. [Google Scholar] [CrossRef]
- Allshire, R.C.; Madhani, H.D. Ten Principles of Heterochromatin Formation and Function. Nat. Rev. Mol. Cell Biol. 2017, 19, 229–244. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Ramos, M.A. Satellite DNA: An Evolving Topic. Genes 2017, 8, 230. [Google Scholar] [CrossRef] [PubMed]
- Ugarković, Đ. Evolution of α-Satellite DNA. In Encyclopedia of Life Sciences; John Wiley & Sons, Ltd.: Chichester, UK, 2013. [Google Scholar]
- Adega, F.; Guedes-Pinto, H.; Chaves, R. Satellite DNA in the karyotype evolution of domestic animals—Clinical considerations. Cytogenet. Genome Res. 2009, 126, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Herrera, A.; Castresana, J.; Robinson, T.J. Is mammalian chromosomal evolution driven by regions of genome fragility? Genome Biol. 2006, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farré, M.; Bosch, M.; López-Giráldez, F.; Ponsà, M.; Ruiz-Herrera, A. Assessing the role of tandem repeats in shaping the genomic architecture of great apes. PLoS ONE 2011, 6, e27239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vieira-da-Silva, A.; Louzada, S.; Adega, F.; Chaves, R. A high-resolution comparative chromosome map of Cricetus cricetus and Peromyscus eremicus reveal the involvement of constitutive heterochromatin in breakpoint regions. Cytogenet. Genome Res. 2015, 145, 59–67. [Google Scholar] [CrossRef] [PubMed]
- De La Fuente, R.; Baumann, C.; Viveiros, M.M. ATRX contributes to epigenetic asymmetry and silencing of major satellite transcripts in the maternal genome of the mouse embryo. Development 2015, 142, 1806–1817. [Google Scholar] [CrossRef] [Green Version]
- Giunta, S.; Funabiki, H. Integrity of the human centromere DNA repeats is protected by CENP-A, CENP-C, and CENP-T. Proc. Natl. Acad. Sci. USA 2017, 11, 1928–1933. [Google Scholar] [CrossRef] [Green Version]
- Chaves, R.; Adega, F.; Heslop-Harrison, J.S.; Guedes-Pinto, H.; Wienberg, J. Complex satellite DNA reshuffling in the polymorphic t (1; 29) Robertsonian translocation and evolutionarily derived chromosomes in cattle. Chromosome Res. 2003, 11, 641–648. [Google Scholar] [CrossRef]
- Escudeiro, A.; Ferreira, D.; Mendes-da-Silva, A.; Heslop-Harrison, J.S.; Adega, F.; Chaves, R. Bovine satellite DNAs—A history of the evolution of complexity and impact in the Bovidae Family. Eur. Zool. J. 2019, 86, 20–37. [Google Scholar] [CrossRef]
- Mravinac, B.; Plohl, M.; Mestrović, N.; Ugarković, D. Sequence of PRAT Satellite DNA ‘Frozen’ in Some Coleopteran Species. J. Mol. Evol. 2002, 54, 774–783. [Google Scholar] [CrossRef] [PubMed]
- Plohl, M.; Petrović, V.; Luchetti, A.; Ricci, A.; Satović, E.; Passamonti, M.; Mantovani, B. Long-term conservation vs high sequence divergence: The case of an extraordinarily old satellite DNA in bivalve mollusks. Heredity 2010, 104, 543–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaves, R.; Ferreira, D.; Mendes-da-Silva, A.; Meles, S.; Adega, F. FA-SAT Is an Old Satellite DNA Frozen in Several Bilateria Genomes. Genome Biol. Evol. 2017, 9, 3073–3087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pezer, Z.; Brajković, J.; Feliciello, I.; Ugarkovć, D. Satellite DNA-Mediated Effects on Genome Regulation. Genome Dyn. 2012, 7, 153–169. [Google Scholar] [CrossRef] [PubMed]
- Grenfell, A.W.; Heald, R.; Strzelecka, M. Mitotic noncoding RNA processing promotes kinetochore and spindle assembly in Xenopus. J. Cell Biol. 2016, 214, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, D.; Meles, S.; Escudeiro, A.; Mendes-da-Silva, A.; Adega, F.; Chaves, R. Satellite non-coding RNAs: The emerging players in cells, cellular pathways and cancer. Chromosome Res. 2015, 23, 479–493. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, D.; Escudeiro, A.; Adega, F.; Anjo, S.I.; Manadas, B.; Chaves, R. FA-SAT ncRNA interacts with PKM2 protein: Depletion of this complex induces a switch from cell proliferation to apoptosis. Cell Mol. Life Sci. 2019. [Google Scholar] [CrossRef]
- Masumoto, H.; Masukata, H.; Muro, Y.; Nozaki, N.; Okazaki, T. Ahumancentromereantigen(CENP-B) interacts with a short specific sequence in alphoid DNA, a human centromeric satellite. J. Cell Biol. 1989, 109, 1963–1973. [Google Scholar] [CrossRef]
- Muro, Y.; Masumoto, H.; Yoda, K.; Nozaki, N.; Ohashi, M.; Okazaki, T. Centromereprotein Bassembles human centromeric α-satellite DNA at the 17-bp sequence, CENP-B box. J. Cell Biol. 1992, 116, 585–596. [Google Scholar] [CrossRef] [Green Version]
- Fachinetti, D.; Han, J.S.; McMahon, M.A.; Ly, P.; Abdullah, A.; Wong, A.J.; Cleveland, D.W. DNA sequence-specific binding of CENP-B enhances the fidelity of human centromere function. Dev. Cell 2015, 33, 314–327. [Google Scholar] [CrossRef] [Green Version]
- Kipling, D.; Warburton, P.E. Centromeres, CENP-B and Tigger too. Trends Genet. 1997, 13, 141–145. [Google Scholar] [CrossRef]
- Kim, Y.B.; Oh, J.H.; McIver, L.J.; Rashkovetsky, E.; Michalak, K.; Garner, H.R.; Kang, L.; Nevo, E.; Korol, A.B.; Michalak, P. Divergence of Drosophila melanogaster repeatomes in response to a sharp microclimate contrast in Evolution Canyon, Israel. Proc. Natl. Acad. Sci. USA 2014, 111, 10630–10635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Ruano, F.J.; López-León, M.D.; Cabrero, J.; Camacho, J.P.M. High-Throughput Analysis of the Satellitome Illuminates Satellite DNA Evolution. Sci. Rep. 2016, 6, 28333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belyayev, A.; Josefiová, J.; Jandová, M.; Kalendar, R.; Krak, K.; Mandák, B. Natural History of a Satellite DNA Family: From the Ancestral Genome Component to Species-Specific Sequences, Concerted and Non-Concerted Evolution. Int. J. Mol. Sci. 2019, 20, 1201. [Google Scholar] [CrossRef] [Green Version]
- Kit, S.J. Equilibrium Sedimentation in Density Gradients of DNA Preparations from Animal Tissues. J. Mol. Biol. 1961, 3, 711–716. [Google Scholar] [CrossRef]
- Orgel, L.E.; Crick, F.H.C. Selfish DNA: The Ultimate Parasite. Nature 1980, 284, 604–607. [Google Scholar] [CrossRef]
- Plohl, M.; Luchetti, A.; Mestrovic, N.; Mantovani, B. Satellite DNAs between selfishness and functionality: Structure, genomics and evolution of tandem repeats in centromeric (hetero) chromatin. Gene 2008, 409, 72–82. [Google Scholar] [CrossRef]
- Willard, H.F. Chromosome-specific organization of human α satellite DNA. Am. J. Hum. Genet. 1985, 37, 524–532. [Google Scholar]
- Alexandrov, I.A.; Medvedev, L.; Mashkova, T.D.; Kisselev, L.L.; Romanova, L.Y.; Yurov, Y.B. Definition of a new α satellite suprachromosomal family characterized by monomeric organization. Nucleic Acids Res. 1993, 21, 2209–2215. [Google Scholar] [CrossRef]
- McNulty, S.M.; Sullivan, B.A. α satellite DNA biology: Finding function in the recesses of the genome. Chromosome Res. 2018, 26, 115–138. [Google Scholar] [CrossRef]
- Waye, J.S.; Willard, H.F. Chromosome-specific α satellite DNA: Nucleotide sequence analysis of the 2.0 kilobasepair repeat from the human X chromosome. Nucleic Acids Res. 1985, 13, 2731–2743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlahovic, I.; Gluncic, M.; Rosandic, M.; Ugarkovic, Ð.; Paar, V. Regular Higher Order Repeat Structures in Beetle Tribolium castaneum Genome. Genome Biol Evol. 2017, 9, 2668–2680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araújo, N.P.; de Lima, L.G.; Dias, G.B.; Kuhn, G.C.S.; de Melo, A.L.; Yonenaga-Yassuda, Y.; Stanyon, R.; Svartman, M. Identification and characterization of a subtelomeric satellite DNA in Callitrichini monkeys. DNA Res. 2017, 24, 377–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Utsunomia, R.; Ruiz-Ruano, F.J.; Silva, D.M.Z.A.; Serrano, É.A.; Rosa, I.F.; Scudeler, P.E.S.; Hashimoto, D.T.; Oliveira, C.; Camacho, J.P.M.; Foresti, F. A Glimpse into the Satellite DNA Library in Characidae Fish (Teleostei, Characiformes). Front. Genet. 2017, 8, 103. [Google Scholar] [CrossRef] [Green Version]
- Adega, F.; Chaves, R.; Guedes-Pintos, H. Chromosomal evolution and phylogenetic analysis in Tayassu pecari and Pecari tajacu (Tayassuidae): Tales from constitutive heterochromatin. J. Genet. 2007, 86, 19–26. [Google Scholar] [CrossRef]
- Henikoff, S.; Dalal, Y. Centromeric chromatin: What makes it unique? Curr. Opin. Genet. Dev. 2005, 15, 177–184. [Google Scholar] [CrossRef]
- Paço, A.; Adega, F.; Guedes-Pinto, H.; Chaves, R. Hidden heterochromatin Characterization in the Rodentia species Cricetus cricetus, Peromyscus eremicus (Cricetidae) and Praomys tullbergi (Muridae). Genet. Mol. Biol. 2009, 32, 58–68. [Google Scholar] [CrossRef]
- Ugarković, Ð.; Plohl, M. Variation in satellite DNA profiles—Causes and effects. EMBO J. 2002, 21, 5955–5959. [Google Scholar] [CrossRef] [Green Version]
- Louzada, S.; Vieira-da-Silva, A.; Mendes-da-Silva, A.; Kubickova, S.; Rubes, J.; Adega, F.; Chaves, R. A Novel Satellite DNA Sequence in the Peromyscus Genome (PMSat): Evolution via Copy Number Fluctuation. Mol. Phylogenet. Evol. 2015, 92, 193–203. [Google Scholar] [CrossRef]
- Gall, J.G.; Cohen, E.G.; Polan, M.L. Repetitive DNA sequences in Drosophila. Chromosoma 1971, 33, 319–344. [Google Scholar] [CrossRef]
- Lohe, A.R.; Brutlag, D.L. Identical satellite DNA sequences in sibling species of Drosophila. J. Mol. Biol. 1987, 194, 161–170. [Google Scholar] [CrossRef]
- Wei, K.H.-C.; Lower, S.E.; Caldas, I.V.; Sless, T.J.S.; Barbash, D.A.; Clark, A.G. Variable rates of simple satellite gains across the Drosophila phylogeny. Mol. Biol. Evol. 2018, 35, 925–941. [Google Scholar] [CrossRef] [PubMed]
- Ferree, P.M.; Barbash, D.A. Species-specific heterochromatin prevents mitotic chromosome segregation to cause hybrid lethality in Drosophila. PLoS Biol. 2009, 7, e1000234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferree, P.M.; Prasad, S. How can satellite DNA divergence cause reproductive isolation? Let us count the chromosomal ways. Genet Res Int 2012, 430136. [Google Scholar] [CrossRef] [Green Version]
- Walsh, J.B. Persistence of tandem arrays: Implications for satellite and simple-sequence DNAs. Genetics 1987, 115, 553–567. [Google Scholar]
- Adega, F.; Chaves, R.; Guedes-Pinto, H. Suiformes orthologous satellite DNAs as a hallmark of collared and white-lipped peccaries (Tayassuidae) evolutionary rearrangements. Micron 2008, 39, 1281–1287. [Google Scholar] [CrossRef]
- Dover, G.A. Molecular drive in multigene families: How biological novelties arise, spread and are assimilated. Trends Genet. 1986, 2, 159–165. [Google Scholar] [CrossRef]
- Elder, J.F., Jr.; Turner, B.J. Concerted evolution of repetitive DNA sequences in eukaryotes. Q. Rev. Biol. 1995, 70, 297–320. [Google Scholar] [CrossRef]
- Lewin, H.A.; Robinson, G.E.; Kress, W.J.; Baker, W.J.; Coddington, J.; Crandall, K.A.; Durbin, R.; Edwards, S.V.; Forest, F.; Gilbert, M.T.P.; et al. Earth BioGenome Project: Sequencing life for the future of life. Proc. Natl. Acad. Sci. USA 2018, 115, 4325–4333. [Google Scholar] [CrossRef] [Green Version]
- Treangen, T.J.; Salzberg, S.L. Repetitive DNA and next-generation sequencing: Computational challenges and solutions. Nat. Rev. Genet. 2011, 13, 36–46. [Google Scholar] [CrossRef]
- Van Dijk, E.L.; Jaszczyszyn, Y.; Naquin, D.; Thermes, C. The Third Revolution in Sequencing Technology. Trends Genet. 2018, 34, 666–681. [Google Scholar] [CrossRef] [PubMed]
- Sedlazeck, F.J.; Lee, H.; Darby, C.A.; Schartz, M.C. Piercing the dark matter: Bioinformatics of long-range sequencing and mapping. Nat. Rev. Genet. 2018, 19, 329–346. [Google Scholar] [CrossRef] [PubMed]
- Novák, P.; Neumann, P.; Pech, J.; Steinhaisl, J.; Macas, J. RepeatExplorer: A Galaxy-based web server for genome-wide characterization of eukaryotic repetitive elements from next-generation sequence reads. Bioinformatics 2013, 29, 792–793. [Google Scholar] [CrossRef] [Green Version]
- Lower, S.S.; McGurk, M.P.; Clark, A.G.; Barbash, D.A. Satellite DNA Evolution: Old Ideas, New Approaches. Curr. Opin. Genet. Dev. 2018, 49, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Salmela, L.; Rivals, E. LoRDEC: Accurate and efficient long read error correction. Bioinformatics 2014, 30, 3506–3514. [Google Scholar] [CrossRef]
- Cook, D.E.; Zdraljevic, S.; Tanny, R.E.; Seo, B.; Riccardi, D.D.; Noble, L.M.; Rockman, M.V.; Alkema, M.J.; Braendle, C.; Kammenga, J.E.; et al. The Genetic Basis of Natural Variation in Caenorhabditis elegans Telomere Length. Genetics 2016, 204, 371–383. [Google Scholar] [CrossRef] [Green Version]
- Wei, K.H.; Grenier, J.K.; Barbash, D.A.; Clark, A.G. Correlated Variation and Population Differentiation in Satellite DNA Abundance among Lines of Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 2014, 111, 18793–18798. [Google Scholar] [CrossRef] [Green Version]
- Silva, D.M.Z.A.; Utsunomia, R.; Ruiz-Ruano, F.J.; Daniel, S.N.; Porto-Foresti, F.; Hashimoto, D.T.; Oliveira, C.; Camacho, J.P.M.; Foresti, F. High-throughput analysis unveils a highly shared satellite DNA library among three species of fish genus Astyanax. Sci. Rep. 2017, 7, 12726. [Google Scholar] [CrossRef] [Green Version]
- Utsunomia, R.; Silva, D.M.Z.A.; Ruiz-Ruano, F.J.; Goes, C.A.G.; Melo, S.; Ramos, L.P.; Oliveira, C.; Porto-Foresti, F.; Foresti, F.; Hashimoto, D.T. Satellitome landscape analysis of Megaleporinus macrocephalus (Teleostei, Anostomidae) reveals intense accumulation of satellite sequences on the heteromorphic sex chromosome. Sci. Rep. 2019, 9, 5856. [Google Scholar] [CrossRef] [Green Version]
- Palacios-Gimenez, O.M.; Dias, G.B.; De Lima, L.G.; Ramos, É.; Martins, C.; Cabral-de-Mello, D.C. High-throughput analysis of the satellitome revealed enormous diversity of satellite DNAs in the neo-Y chromosome of the cricket Eneoptera surinamensis. Sci. Rep. 2017, 7, 6422. [Google Scholar] [CrossRef] [Green Version]
- Usai, G.; Mascagni, F.; Natali, L.; Giordani, T.; Cavallini, A. Comparative Genome-Wide Analysis of Repetitive DNA in the Genus Populus L. Tree Genet. Genomes 2017, 13, 96. [Google Scholar] [CrossRef] [Green Version]
- Vozdova, M.; Kubickova, S.; Cernohorska, H.; Fröhlich, J.; Rubes, J. Satellite DNA Sequences in Canidae and Their Chromosome Distribution in Dog and Red Fox. Cytogenet. Genome Res. 2017, 150, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Miga, K.H. Completing the human genome: The progress and challenge of satellite DNA assembly. Chromosome Res. 2015, 23, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Olsen, H.E.; Paten, B.; Akeson, M. The Oxford Nanopore MinION: Delivery of nanopore sequencing to the genomics community. Genome Biol. 2016, 17, 239. [Google Scholar] [CrossRef] [Green Version]
- Kuderna, L.F.K.; Lizano, E.; Julià, E.; Gomez-Garrido, J.; Serres-Armero, A.; Kuhlwilm, M.; Alandes, R.A.; Alvarez-Estape, M.; Juan, D.; Simon, H.; et al. Selective Single Molecule Sequencing and Assembly of a Human Y Chromosome of African Origin. Nat. Commun. 2019, 10, 4. [Google Scholar] [CrossRef] [Green Version]
- Miga, K.H. Centromeric Satellite DNAs: Hidden Sequence Variation in the Human Population. Genes 2019, 10, 352. [Google Scholar] [CrossRef] [Green Version]
- Jain, M.; Koren, S.; Miga, K.H.; Quick, J.; Rand, A.C.; Sasani, T.A.; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T.; et al. Nanopore Sequencing and Assembly of a Human Genome with Ultra-Long Reads. Nat. Biotechnol. 2018, 36, 338–345. [Google Scholar] [CrossRef] [Green Version]
- Miga, K.H.; Koren, S.; Rhie, A.; Vollger, M.R.; Gershman, A.; Bzikadze, A.; Brooks, S.; Howe, E.; Porubsky, D.; Logsdon, G.A.; et al. Telomere-to-Telomere Assembly of a Complete Human X Chromosome. bioRxiv 2019, 735928. [Google Scholar] [CrossRef]
- Muller, H.; Gil, J., Jr.; Drinnenberg, I.A. The impact of centromeres on spatial genome architecture. Trends Genet. 2019, 35, 565–578. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.H.; Chavan, A.; Palladino, J.; Wei, X.; Martins, N.M.C.; Santinello, B.; Chen, C.C.; Erceg, J.; Beliveau, B.J.; Wu, C.T.; et al. Islands of retroelements are major components of Drosophila centromeres. PLoS Biol. 2019, 17, e3000241. [Google Scholar] [CrossRef] [Green Version]
- Weissensteiner, M.H.; Pang, A.W.C.; Bunikis, I.; Höijer, I.; Vinnere-Petterson, O.; Suh, A.; Wolf, J.B.W. Combination of Short-Read, Long-Read, and Optical Mapping Assemblies Reveals Large-Scale Tandem Repeat Arrays with Population Genetic Implications. Genome Res. 2017, 27, 697–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Ruano, F.J.; Cuadrado, Á.; Montiel, E.E.; Camacho, J.P.; López-León, M.D. Next Generation Sequencing and FISH Reveal Uneven and Nonrandom Microsatellite Distribution in Two Grasshopper Genomes. Chromosoma 2015, 124, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Skinner, B.M.; Sargent, C.A.; Churcher, C.; Hunt, T.; Herrero, J.; Loveland, J.E.; Dunn, M.; Louzada, S.; Fu, B.; Chow, W.; et al. The Pig X and Y Chromosomes: Structure, Sequence, and Evolution. Genome Res. 2016, 26, 130–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louzada, S.; Komatsu, J.; Yang, F. Fluorescence in situ hybridization onto DNA fibres generated using molecular combing. In Fluorescence In Situ Hybridization (FISH); Liehr, T., Ed.; Springer: Berlin/Heidelberg, Germany, 2017; pp. 275–293. [Google Scholar]
- Giunta, S. Centromere Chromosome Orientation Fluorescent In Situ Hybridization (Cen-CO-FISH) Detects Sister Chromatid Exchange at the Centromere in Human Cells. Bio-protocol 2018, 8. [Google Scholar] [CrossRef]
- Saha, A.; Mourad, M.; Kaplan, M.H.; Chefetz, I.; Malek, S.N.; Buckanovich, R.; Markovitz, D.M.; Contreras-Galindo, R. The Genomic Landscape of Centromeres in Cancers. bioRxiv 2019, 505800. [Google Scholar] [CrossRef] [Green Version]
- White, T.B.; McCoy, A.M.; Streva, V.A.; Fenrich, J.; Deininger, P.L. A droplet digital PCR detection method for rare L1 insertions in tumors. Mobile DNA 2014, 5, 30. [Google Scholar] [CrossRef] [Green Version]
- Kishikawa, T.; Otsuka, M.; Yoshikawa, T.; Ohno, M.; Yamamoto, K.; Yamamoto, N.; Yamamoto, N.; Kotani, A.; Koike, K. Quantitation of circulating satellite RNAs in pancreatic cancer patients. JCI Insight 2016, 1, e86646. [Google Scholar] [CrossRef] [Green Version]
- Deakin, J.E.; Potter, S.; O’Neill, R.; Ruiz-Herrera, A.; Cioffi, M.B.; Eldridge, M.D.B.; Fukui, K.; Marshall Graves, J.A.; Griffin, D.; Grutzner, F.; et al. Chromosomics: Bridging the Gap between Genomes and Chromosomes. Genes 2019, 10, 627. [Google Scholar] [CrossRef] [Green Version]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [Green Version]
- Palacios-Gimenez, O.M.; Bardella, V.B.; Lemos, B.; Cabral-de-Mello, D.C. Satellite DNAs are conserved and differentially transcribed among Gryllus cricket species. DNA Res. 2018, 25, 137–147. [Google Scholar] [CrossRef] [Green Version]
- Kowar, T.; Zakrzewski, F.; Macas, J.; Kobližková, A.; Viehoever, P.; Weisshaar, B.; Schmidt, T. Repeat Composition of CenH3-chromatin and H3K9me2-marked heterochromatin in Sugar Beet (β vulgaris). BMC Plant Biol. 2016, 16, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, H.; Liu, Y.; Liu, C.; Shi, Q.; Huang, Y.; Han, F. Centromere satellite repeats have undergone rapid changes in polyploid wheat subgenomes. Plant Cell 2019, 31, 2035–2051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khost, D.E.; Eickbush, D.G.; Larracuente, A.M. Single-Molecule Sequencing Resolves the Detailed Structure of Complex Satellite DNA Loci in Drosophila. Genome Res. 2017, 27, 709–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Lima, L.G.; Svartman, M.; Kuhn, C.S. Dissecting the Satellite DNA Landscape in Three Cactophilic Drosophila Sequenced Genomes. G3 Genes Genomes Genet. 2017, 7, 2831–2843. [Google Scholar] [CrossRef] [Green Version]
- Flynn, J.M.; Long, M.; Wing, R.A.; Clark, A.G. Evolutionary dynamics of abundant 7 bp satellites in the genome of Drosophila virilis. bioRxiv 2019, 693077. [Google Scholar] [CrossRef] [Green Version]
- Hartley, G.; O’neill, R.J. Centromere Repeats: Hidden Gems of the Genome. Genes 2019, 10, 223. [Google Scholar] [CrossRef] [Green Version]
- Ferguson-Smith, M.A.; Trifonov, V. Mammalian karyotype evolution. Nat. Rev. Genet. 2007, 8, 950–962. [Google Scholar] [CrossRef]
- Spielmann, M.; Lupiáñez, D.G.; Mundlos, S. Structural variation in the 3D genome. Nat. Rev. Genet. 2018, 19, 453–467. [Google Scholar] [CrossRef] [Green Version]
- Dobzhansky, T. Genetics of the Evolutionary Process; Columbia University Press: New York, NY, USA, 1970. [Google Scholar]
- Wichman, H.; Payne, C.; Ryder, O.; Hamilton, M.; Maltbie, M.; Baker, R. Genomic distribution of heterochromatic sequences in equids: Implications to rapid chromosomal evolution. J. Hered. 1991, 82, 369–377. [Google Scholar] [CrossRef]
- Rossi, M.; Redi, C.; Viale, G.; Massarini, A.; Capanna, E. Chromosomal distribution of the major satellite DNA of South American rodents of the genus Ctenomys. Cytogenet. Cell Genet. 1995, 69, 179–184. [Google Scholar] [CrossRef]
- Chaves, R.; Guedes-Pinto, H.; Heslop-Harrison, J.S. Phylogenetic relationships and the primitive X chromosome inferred from chromosomal and satellite DNA analysis in Bovidae. Proc. Biol. Sci. 2005, 272, 2009–2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adega, F.; Chaves, R.; Guedes-Pinto, H.; Heslop-Harrison, J. Physical organization of the 1.709 satellite IV DNA family in Bovini and Tragelaphini tribes of the Bovidae: Sequence and chromosomal evolution. Cytogenet. Genome Res. 2006, 114, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Maynard Smith, J.; Burian, R.; Kaufman, S.; Alberch, P.; Campbell, J.; Goodwin, B. Developmental constraints and evolution. Q. Rev. Biol. 1985, 60, 265–287. [Google Scholar] [CrossRef]
- Farré, M.; Robinson, T.J.; Ruiz-Herrera, A. An Integrative Breakage Model of genome architecture, reshuffling and evolution: The Integrative Breakage Model of genome evolution, a novel multidisciplinary hypothesis for the study of genome plasticity. Bioessays 2015, 37, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Kehrer-Sawatzki, H.; Cooper, D.N. Molecular mechanisms of chromosomal rearrangement during primate evolution. Chromosome Res. 2008, 16, 41–56. [Google Scholar] [CrossRef]
- Carbone, L.; Harris, R.A.; Vessere, G.M.; Mootnick, A.R.; Humphray, S.; Rogers, J.; Kim, S.K.; Wall, J.D.; Martin, D.; Jurka, J.; et al. Evolutionary breakpoints in the gibbon suggest association between cytosine methylation and karyotype evolution. PLoS Genet. 2009, 5, e1000538. [Google Scholar] [CrossRef]
- Zhao, H.; Bourque, G. Recovering genome rearrangements in the mammalian phylogeny. Genome Res. 2009, 19, 934–942. [Google Scholar] [CrossRef] [Green Version]
- Paço, A.; Chaves, R.; Vieira-da-Silva, A.; Adega, F. The involvement of repetitive sequences in the remodelling of karyotypes: The Phodopus genomes (Rodentia, Cricetidae). Micron 2013, 46, 27–34. [Google Scholar] [CrossRef]
- Bourque, G. Transposable elements in gene regulation and in the evolution of vertebrate genomes. Curr. Opin. Genet. Dev. 2009, 19, 607–612. [Google Scholar] [CrossRef]
- Longo, M.S.; Carone, D.M.; Green, E.D.; O’Neill, M.J.; O’Neill, R.J.; NISC Comparative Sequencing Program. Distinct retroelement classes define evolutionary breakpoints demarcating sites of evolutionary novelty. BMC Genomics 2009, 10, 334. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Perez, J.L.; Widmann, T.J.; Adams, I.R. The impact of transposable elements on mammalian development. Development 2016, 143, 4101–4114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platt, R.N.; Vandewege, M.W.; Ray, D.A. Mammalian transposable elements and their impacts on genome evolution. Chromosome Res. 2018, 26, 25–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, J.A.; Eichler, E.E. Primate segmental duplications: Crucibles of evolution, diversity and disease. Nat. Rev. Genet. 2006, 7, 552–564. [Google Scholar] [CrossRef] [PubMed]
- Louzada, S.; Paço, A.; Kubickova, S.; Adega, F.; Guedes-Pinto, H.; Rubes, J.; Chaves, R. Different evolutionary trails in the related genomes Cricetus cricetus and Peromyscus eremicus (Rodentia, Cricetidae) uncovered by orthologous satellite DNA repositioning. Micron 2008, 39, 1149–1155. [Google Scholar] [CrossRef] [PubMed]
- Mashkova, T.; Oparina, N.; Alexandrov, I.; Zinovieva, O.; Marusina, A.; Yurov, Y.; Lacroix, M.H.; Kisselev, L. Unequal cross-over is involved in human α satellite DNA rearrangements on a border of the satellite domain. FEBS Lett. 1998, 441, 451–457. [Google Scholar] [CrossRef] [Green Version]
- Kelkar, Y.D.; Tyekucheva, S.; Chiaromonte, F.; Makova, K.D. The genome-wide determinants of human and chimpanzee microsatellite evolution. Genome Res. 2008, 18, 30–38. [Google Scholar] [CrossRef] [Green Version]
- Sankoff, D. The where and wherefore of evolutionary breakpoints. J. Biol. 2009, 8, 66. [Google Scholar] [CrossRef]
- Catasti, P.; Chen, X.; Mariappan, S.V.; Bradbury, E.M.; Gupta, G. DNA repeats in the human genome. Genetica 1999, 106, 15–36. [Google Scholar] [CrossRef]
- Yang, D.; Okamoto, K. Structural insights into G-quadruplexes: Towards new anticancer drugs. Future Med. Chem. 2010, 2, 619–646. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Dai, J.; Veliath, E.; Jones, R.A.; Yang, D.Z. Structure of a two-G-tetrad intramolecular G-quadruplex formed by a variant human telomeric sequence in K+ solution: Insights into the interconversion of human telomeric G-quadruplex structures. Nucleic Acids Res. 2010, 38, 1009–1021. [Google Scholar] [CrossRef]
- Zhao, J.; Bacolla, A.; Wang, G.; Vasquez, K.M. Non-B DNA structure-induced genetic instability and evolution. Cell. Mol. Life Sci. 2010, 67, 43–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black, E.M.; Giunta, S. Repetitive Fragile Sites: Centromere Satellite DNA as a Source of Genome Instability in Human Diseases. Genes 2018, 9, 615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khodaverdian, V.Y.; Hanscom, T.; Yu, A.M.; Yu, T.L.; Mak, V.; Brown, A.J.; Roberts, S.A.; McVey, M. Secondary structure forming sequences drive SD-MMEJ repair of DNA double-strand breaks. Nucleic Acids Res. 2017, 45, 12848–12861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Sotero-Caio, C.G.; Cabral-de-Mello, D.C.; Calixto, M.D.S.; Valente, G.T.; Martins, C.; Loreto, V.; de Souza, M.J.; Santos, N. Centromeric enrichment of LINE-1 retrotransposons and its significance for the chromosome evolution of Phyllostomid bats. Chromosome Res. 2017, 25, 313–325. [Google Scholar] [CrossRef]
- Klein, S.J.; O’Neill, R.J. Transposable elements: Genome innovation, chromosome diversity, and centromere conflict. Chromosome Res. 2018, 26, 5–23. [Google Scholar] [CrossRef] [Green Version]
- Mayorov, V.I.; Adkison, L.R.; Vorobyeva, N.V.; Khrapov, E.A.; Kholodhov, N.G.; Rogozin, I.B.; Nesterova, T.B.; Protopopov, A.I.; Sablina, O.V.; Graphodatsky, A.S.; et al. Organization and chromosomal localization of a B1-like containing repeat of Microtus subarvalis. Mamm. Genome 1996, 7, 593. [Google Scholar] [CrossRef]
- Marchal, J.A.; Acosta, M.J.; Bullejos, M.; Puerma, E.; Díaz de la Guardia, R.; Sánchez, A. Distribution of L1-retroposons on the giant sex chromosomes of Microtus cabrerae (Arvicolidae, Rodentia): Functional and evolutionary implications. Chromosome Res. 2006, 14, 177–186. [Google Scholar] [CrossRef]
- Escudeiro, A.; Adega, F.; Robinson, T.J.; Heslop-Harrison, J.S.; Chaves, R. Conservation, divergence and functions of centromeric satellite DNA families in the Bovidae. Genome Biol. Evol. 2019, 11, 1152–1165. [Google Scholar] [CrossRef]
- Wessler, S.R. Transposable elements and the evolution of eukaryotic genomes. Proc. Natl. Acad. Sci. USA 2006, 103, 17600–17601. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.K.; Simmons, J.M. Gross chromosome rearrangements mediated by transposable elements in Drosophila melanogaster. Bioessays 1994, 16, 269–275. [Google Scholar] [CrossRef]
- Gray, Y.H.M. It takes two transposons to tango: Transposable-element-mediated chromosomal rearrangements. Trends Genet. 2000, 16, 461–468. [Google Scholar] [CrossRef]
- Hedges, D.J.; Deininger, P.L. Inviting instability: Transposable elements, double-strand breaks, and the maintenance of genome integrity. Mutat. Res. 2007, 616, 46–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbone, L.; Harris, R.A.; Gnerre, S.; Veeramah, K.R.; Lorente-Galdos, B.; Huddleston, J.; Meyer, T.J.; Herrero, J.; Roos, C.; Aken, B.; et al. Gibbon genome and the fast karyotype evolution of small apes. Nature 2014, 513, 195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, J.A.; Baertsch, R.; Kent, W.J.; Haussler, D.; Eichler, E.E. Hotspots of mammalian chromosomal evolution. Genome Biol. 2004, 5, R23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordaux, R.; Batzer, M.A. The impact of retrotransposons on human genome evolution. Nat. Rev. Genet. 2009, 10, 691–703. [Google Scholar] [CrossRef] [Green Version]
- Warren, I.; Naville, M.; Chalopin, D.; Levin, P.; Berger, C.; Galiana, D.; Volff, J.N. Evolutionary impact of transposable elements on genomic diversity and lineage-specific innovation in vertebrates. Chromosome Res. 2015, 23, 505–531. [Google Scholar] [CrossRef]
- Meštrović, N.; Mravinac, B.; Pavlek, M.; Vojvoda-Zeljko, T.; Šatović, E.; Plohl, M. Structural and functional liaisons between transposable elements and satellite DNAs. Chrom. Res. 2015, 23, 583–596. [Google Scholar] [CrossRef]
- Paço, A.; Adega, F.; Chaves, R. Line-1 Retrotransposons: From “parasite” sequences to Functional Elements. J. Appl. Genet. 2015, 56, 133–145. [Google Scholar] [CrossRef]
- Slamovits, C.H.; Rossi, M.S. Satellite DNA: Agent of chromosomal evolution in mammals. A review. J. Neotrop. Mammal. 2002, 9, 297–308. [Google Scholar]
- Chaves, R.; Louzada, S.; Meles, S.; Wienberg, J.; Adega, F. Praomys tullbergi (Muridae, Rodentia) genome architecture decoded by comparative chromosome painting with Mus and Rattus. Chromosome Res. 2012, 20, 673–683, Epub 31 July 2012. [Google Scholar] [CrossRef]
- Chaves, R.; Heslop-Harrison, J.S.; Guedes-Pinto, H. Centromeric heterochromatin in the cattle rob (1;29) translocation: α-satellite I sequences, In-Situ MspI digestion patterns, chromomycin staining and C-bands. Chromosome Res. 2000, 8, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Di Meo, G.P.; Perucatti, A.; Chaves, R.; Adega, F.; De Lorenzi, L.; Molteni, L.; De Giovanni, A.; Incarnato, D.; Guedes-Pinto, H.; Eggen, A.; et al. Cattle rob(1;29) originating from complex chromosome rearrangements as revealed by both banding and FISH-mapping techniques. Chromosome Res. 2006, 14, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Smalec, B.M.; Heider, T.N.; Flynn, B.L.; O’Neil, R.J. A centromere satellite concomitant with extensive karyotypic diversity across the Peromyscus genus defies predictions of molecular drive. Chromosome Res. 2019, 27, 237–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adega, F.; Matoso, S.R.; Kjöllerström, H.; Vercammen, P.; Raudsepp, T.; Collares-Pereira, M.J.; Fernandes, C.; Oom, M.; Chaves, R. Comparative chromosome painting in genets (Carnivora, Viverridae, Genetta), the only known feliforms with a highly rearranged karyotype. Cytogenet. Genome Res. 2018, 156, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Caceres, M.; Ranz, J.M.; Barbadilla, A.; Long, M.; Ruiz, A. Generation of a widespread Drosophila inversion by a transposable element. Science 1999, 285, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Rojo, V.; Martínez-Lage, A.; Giovannotti, M.; González-Tizón, A.M.; Cerioni, P.N.; Barucchi, V.C.; Galán, P.; Olmo, E.; Naveira, H. Evolutionary dynamics of two satellite DNA families in rock lizards of the genus Iberolacerta (Squamata, Lacertidae): Different histories but common traits. Chromosome Res. 2015, 23, 441–461. [Google Scholar] [CrossRef] [PubMed]
- Dela Paz, J.S.; Stronghill, P.E.; Douglas, S.J.; Saravia, S.; Hasenkampf, C.A.; Riggs, C.D. Chromosome fragile sites in Arabidopsis harbor matrix attachment regions that may be associated with ancestral chromosome rearrangement events. PLoS Genet. 2012, 8, e1003136. [Google Scholar] [CrossRef]
- Chaves, R.; Santos, S.; Guedes-Pinto, H. Comparative analysis (Hippotragini versus Caprini, Bovidae) of X-chromosome’s constitutive heterochromatin by in situ restriction endonuclease digestion: X-chromosome constitutive heterochromatin evolution. Genetica 2004, 121, 315–325. [Google Scholar] [CrossRef] [Green Version]
- Biscotti, M.A.; Olmo, E.; Heslop-Harrison, J.S. Repetitive DNA in Eukaryotic Genomes. Chromosome Res. 2015, 23, 415–420. [Google Scholar] [CrossRef]
- Santos, S.; Chaves, R.; Guedes-Pinto, H. Chromosomal localization of the major satellite DNA family (FA-SAT) in the domestic cat. Cytogenet. Genome Res. 2004, 107, 119–122. [Google Scholar] [CrossRef] [Green Version]
- Ting, D.T.; Lipson, D.; Paul, S.; Brannigan, B.W.; Akhavanfard, S.; Coffman, E.J.; Contino, G.; Deshpande, V.; Iafrate, A.J.; Letovsky, S.; et al. Aberrant Overexpression of Satellite Repeats in Pancreatic and Other Epithelial Cancers. Science 2011, 331, 593–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Q.; Pao, G.; Huynh, A.; Suh, H.; Tonnu, N.; Nederlof, P.M.; Gage, F.H.; Verma, I.M. BRCA1 tumour suppression occurs via heterochromatin-mediated silencing. Nature 2011, 477, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.S.; Liu, L.L.; Ganesan, S.; Michor, F.; De, S. Nuclear topology modulates the mutational landscapes of cancer genomes. Nat. Struct. Mol. Biol. 2017, 24, 1000–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, C.; Jeong, H.H.; Hsieh, Y.C.; Klein, H.U.; Bennett, D.A.; De Jager, P.L.; Liu, Z.; Shulman, J.M. Tau Activates Transposable Elements in Alzheimer’s Disease. Cell Rep. 2018, 23, 2874–2880. [Google Scholar] [CrossRef]
- Francastel, C.; Magdinier, F. DNA methylation in satellite repeats disorders. Essays Biochem. 2019. [Google Scholar] [CrossRef]
- Payer, L.M.; Burns, K.H. Transposable elements in human genetic disease. Nat. Rev. Genet. 2019, 20, 760–772. [Google Scholar] [CrossRef]
- Fournier, A.; Mcleer-florin, A.; Lefebvre, C.; Duley, S.; Debernardi, A.; Rousseaux, S.; De Fraipont, F.; Figeac, M.; Kerckaert, J.; De Vos, J.; et al. 1q12 chromosome translocations form aberrant heterochromatic foci associated with changes in nuclear architecture and gene expression in B cell lymphoma. EMBO Mol. Med. 2010, 2, 159–171. [Google Scholar] [CrossRef]
- Ducos, A.; Revay, T.; Kovacs, A.; Hidas, A.; Pinton, A.; Bonnet-Garnier, A.; Molteni, L.; Slota, E.; Switonski, M.; Arruga, M.V.; et al. Cytogenetic screening of livestock populations in Europe: An overview. Cytogenet. Genome Res. 2008, 120, 26–41. [Google Scholar] [CrossRef]
- Iannuzzi, L.; King, W.; Di Berardino, D. Chromosome evolution in domestic bovids as revealed by chromosome banding and FISH-mapping techniques. Cytogenet. Genome Res. 2009, 126, 49–62. [Google Scholar] [CrossRef]
- Therman, E.; Susman, B.; Denniston, C. The nonrandom participation of human acrocentric chromosomes in Robertsonian translocations. Ann. Hum. Genet. 1989, 53, 49–65. [Google Scholar] [CrossRef]
- Page, S.L.; Shin, J.C.; Han, J.Y.; Choo, K.A.; Shaffer, L.G. Breakpoint diversity illustrates distinct mechanisms for Robertsonian translocation formation. Hum. Mol. Genet. 1996, 5, 1279–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garagna, S.; Marziliano, N.; Zuccotti, M.; Searle, J.B.; Capanna, E.; Redi, C.A. Pericentromeric organization at the fusion point of mouse Robertsonian translocation chromosomes. Proc. Natl. Acad. Sci. USA 2001, 98, 171–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustavsson, I. Cytogenetics, distribution and phenotypic effects of a translocation in Swedish cattle. Hereditas 1969, 63, 68–169. [Google Scholar] [CrossRef] [PubMed]
- Dyrendahl, I.; Guatavsson, I. Sexual functions, semen characteristics and fertility of bulls carrying the 1/29 chromosome translocation. Hereditas 1979, 90, 281–289. [Google Scholar] [CrossRef]
- Rangel-Figueiredo, T.; Iannuzzi, L. A cattle breed close to 58 diploid number due to high frequency of rob (1; 29). Hereditas 1991, 115, 73–78. [Google Scholar] [CrossRef]
- Rangel-Figueiredo, T.; Iannuzzi, L. Frequency and distribution of rob (1; 29) in three Portuguese cattle breeds. Hereditas 1993, 119, 233–237. [Google Scholar] [CrossRef]
- Rubes, J.; Kubickova, S.; Pagacova, E.; Cernohorska, H.; Di Berardino, D.; Antoninova, M.; Vahala, J.; Robinson, T.J. Phylogenomic study of spiral-horned antelope by cross-species chromosome painting. Chromosome Res. 2008, 16, 935–947. [Google Scholar] [CrossRef]
- Gravholt, C.H.; Friedrich, U.; Caprani, M.; Jørgensen, A.L. Breakpoints in Robertsonian translocations are localized to satellite III DNA by fluorescence in situ hybridization. Genomics 1992, 14, 924–930. [Google Scholar] [CrossRef]
- Wang, B.; Nie, B.; Tang, D.; Li, R.; Liu, X.; Song, J.; Wang, W.; Liu, Z. Analysis of meiotic segregation patterns and interchromosomal effects in sperm from 13 robertsonian translocations. Balk. J. Med. Genet. 2017, 20, 43–50. [Google Scholar] [CrossRef]
- Kim, S.R.; Shaffer, L.G. Robertsonian translocations: Mechanisms of formation, aneuploidy, and uniparental disomy and diagnostic considerations. Genet. Test. 2002, 6, 163–168. [Google Scholar] [CrossRef]
- Jarmuz-Szymczak, M.; Janiszewska, J.; Szyfter, K.; Shaffer, L.G. Narrowing the localization of the region breakpoint in most frequent Robertsonian translocations. Chromosome Res. 2014, 22, 517–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choo, K.H.; Vissel, B.; Brown, R.; Filby, R.G.; Earle, E. Homologous α satellite sequences on human acrocentric chromosomes with selectivity for chromosomes 13, 14 and 21: Implications for recombination between nonhomologues and Robertsonian translocations. Nucleic Acids Res. 1988, 16, 1273–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akgun, E.; Zahn, J.; Baumes, S.; Brown, G.; Liang, F.; Romanienko, P.J.; Lewis, S.; Jasin, M. Palindrome resolution and recombination in the mammalian germ line. Mol. Cell. Biol. 1997, 17, 5559–5570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandyopadhyay, R.; Heller, A.; Knox-DuBois, C.; McCaskill, C.; Berend, S.A.; Page, S.L.; Shaffer, L.G. Parental origin and timing of de novo Robertsonian translocation formation. Am. J. Hum. Genet. 2002, 71, 1456–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H. Toward better understanding of artifacts in variant calling from high-coverage samples. Bioinformatics 2014, 30, 2843–2851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biscotti, M.A.; Canapa, A.; Forconi, M.; Olmo, E.; Barucca, M. Transcription of tandemly repetitive DNA: Functional roles. Chromosome Res. 2015, 23, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Mills, W.K.; Lee, Y.C.G.; Kochendoerfer, A.M.; Dunleavy, E.M.; Karpen, G.H. RNA from a simple-tandem repeat is required for sperm maturation and male fertility in Drosophila melanogaster. Elife 2019, 8, 48940. [Google Scholar] [CrossRef]
- McNulty, S.M.; Sullivan, L.L.; Sullivan, B.A. Human Centromeres Produce Chromosome-Specific and Array-Specific α Satellite Transcripts that Are Complexed with CENP-A and CENP-C. Dev. Cell 2017, 42, 226–240. [Google Scholar] [CrossRef]
- Liu, H.; Qu, Q.; Warrington, R.; Rice, A.; Cheng, N.; Yu, H. Mitotic transcription installs Sgo1 at centromeres to coordinate chromosome segregation. Mol. Cell 2015, 59, 426–436. [Google Scholar] [CrossRef] [Green Version]
- Camacho, O.V.; Galan, C.; Swist-Rosowska, K.; Ching, R.; Gamalinda, M.; Karabiber, F.; De La Rosa-Velazquez, I.; Engist, B.; Koschorz, B.; Shukeir, N. Major satellite repeat RNA stabilize heterochromatin retention of Suv39h enzymes by RNA-nucleosome association and RNA: DNA hybrid formation. Elife 2017, 6, e25293. [Google Scholar] [CrossRef]
- Johnson, W.L.; Yewdell, W.T.; Bell, J.C.S.; McNulty, M.; Duda, Z.; O’Neill, R.J.; Sullivan, B.A.; Straight, A.F. RNA-dependent stabilization of SUV39H1 at constitutive heterochromatin. Elife 2017, 6, e25299. [Google Scholar] [CrossRef] [PubMed]
- Shirai, A.; Kawaguchi, T.; Shimojo, H.; Muramatsu, D.; Ishida-Yonetani, M.; Nishimura, Y.; Kimura, H.; Nakayama, J.I.; Shinkai, Y. Impact of nucleic acid and methylated H3K9 binding activities of Suv39h1 on its heterochromatin assembly. Elife 2017, 6, e25317. [Google Scholar] [CrossRef] [PubMed]
- Chan, F.L.; Marshall, O.J.; Saffery, R.; Kim, B.W.; Earle, E.; Choo, K.H.; Wong, L.H. Active transcription and essential role of RNA polymerase II at the centromere during mitosis. Proc. Natl. Acad. Sci. USA 2012, 109, 1979–1984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, F.L.; Wong, L.H. Transcription in the maintenance of centromere chromatin identity. Nucleic Acids Res. 2012, 40, 11178–11188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grenfell, A.W.; Strzelecka, M.; Heald, R. Transcription brings the complex(ity) to the centromere. CellCycle 2017, 16, 235–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smurova, K.; De Wulf, P. Centromere and pericentromere transcription: Roles and regulation…in sickness and in health. Front. Genet. 2018, 9, 674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouzinba-Segard, H.; Guais, A.; Francastel, C. Accumulation of small murine minor satellite transcripts leads to impaired centromeric architecture and function. Proc. Natl. Acad. Sci. USA 2006, 103, 8709–8714. [Google Scholar] [CrossRef] [Green Version]
- Rošič, S.; Köhler, F.; Erhardt, S. Repetitive centromeric satellite RNA is essential for kinetochore formation and cell division. J. Cell Biol. 2014, 207, 335–349. [Google Scholar] [CrossRef] [Green Version]
- Chan, D.Y.L.; Moralli, D.; Khoja, S.; Monaco, Z.L. Noncoding Centromeric RNA Expression Impairs Chromosome Stability in Human and Murine Stem Cells. Dis. Markers 2017, 7506976. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Gilbert, D.M. Proliferation-dependent and cell cycle regulated transcription of mouse pericentric heterochromatin. J. Cell Biol. 2007, 179, 411–421. [Google Scholar] [CrossRef] [Green Version]
- Maison, C.; Bailly, D.; Roche, D.; Montes de Oca, R.; Probst, A.V.; Vassias, I.; Dingli, F.; Lombard, B.; Loew, D.; Quivy, J.P.; et al. Sumoylation promotes de novo targeting of HP1alpha to pericentric heterochromatin. Nat. Genet. 2011, 43, 220–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vourc’h, C.; Biamonti, G. Transcription of Satellite DNAs in Mammals. Prog. Mol. Subcell. Biol. 2011, 51, 95–118. [Google Scholar] [CrossRef] [PubMed]
- Jolly, C.; Metz, A.; Govin, J.; Vigneron, M.; Turner, B.M.; Khochbin, S.; Vourc’h, C. Stress-induced transcription of satellite III repeats. J. Cell Biol. 2004, 164, 25–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzi, N.; Denegri, M.; Chiodi, I.; Corioni, M.; Valgardsdottir, R.; Cobianchi, F.; Riva, S.; Biamonti, G. Transcriptional activation of a constitutive heterochromatic domain of the human genome in response to heat shock. Mol. Biol. Cell 2004, 15, 543–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biamonti, G. Nuclear stress bodies: A heterochromatin affair? Nat. Rev. Mol. Cell Biol. 2004, 5, 493–498. [Google Scholar] [CrossRef]
- Biamonti, G.; Vourc’h, C. Nuclear stress bodies. Cold Spring Harb Perspect. Biol. 2010, 2. [Google Scholar] [CrossRef]
- Goenka, A.; Sengupta, S.; Pandey, R.; Parihar, R.; Mohanta, G.C.; Mukerji, M.; Ganesh, S. Human satellite-III non-coding RNAs modulate heat-shock-induced transcriptional repression. J. Cell Sci. 2016, 129, 3541–3552. [Google Scholar] [CrossRef] [Green Version]
- Valgardsdottir, R.; Chiodi, I.; Giordano, M.; Rossi, A.; Bazzini, S.; Ghigna, C.; Riva, S.; Biamonti, G. Transcription of Satellite III non-coding RNAs is a general stress response in human cells. Nucleic Acids Res. 2008, 36, 423–434. [Google Scholar] [CrossRef]
- Kuhn, G.C. Satellite DNA transcripts have diverse biological roles in Drosophila’. Heredity 2015, 115, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Usakin, L.; Abad, J.; Vagin, V.V.; de Pablos, B.; Villasante, A.; Gvozdev, V.A. Transcription of the 1.688 satellite DNA family is under the control of RNA interference machinery in Drosophila melanogaster ovaries. Genetics 2007, 176, 1343–1349. [Google Scholar] [CrossRef] [Green Version]
- Menon, D.U.; Coarfa, C.; Xiao, W.; Gunaratne, P.H.; Meller, V.H. siRNAs from an X-linked satellite repeat promote X-chromosome recognition in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 2014, 111, 16460–16465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, D.; Escudeiro, A.; Adega, F.; Chaves, R. DNA Methylation Patterns of a Satellite Non-coding Sequence—FA-SAT in Cancer Cells: Its Expression Cannot Be Explained Solely by DNA Methylation. Front. Genet. 2019, 10, 101. [Google Scholar] [CrossRef] [PubMed]
- Sana, J.; Faltejskova, P.; Svoboda, M.; Slaby, O. Novel classes of non-coding RNAs and cancer. J. Transl. Med. 2012, 10, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eymery, A.; Horard, B.; El Atifi-Borel, M.; Fourel, G.; Berger, F.; Vitte, A.L.; Van den Broeck, A.; Brambilla, E.; Fournier, A.; Callanan, M.; et al. A transcriptomic analysis of human centromeric and pericentric sequences in normal and tumor cells. Nucleic Acids Res. 2009, 37, 6340–6354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrlich, M.; Sanchez, C.; Shao, C.; Nishiyama, R.; Kehrl, J.; Kuick, R.; Kubota, T.; Hanash, S.M. ICF, an immunodeficiency syndrome: DNA methyltransferase 3B involvement, chromosome anomalies, and gene dysregulation. Autoimmunity 2008, 41, 253–271. [Google Scholar] [CrossRef] [Green Version]
- Saksouk, N.; Simboeck, E.; Dejardin, J. Constitutive heterochromatin formation and transcription in mammals. Epigenet. Chromatin 2015, 8, 3. [Google Scholar] [CrossRef] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Louzada, S.; Lopes, M.; Ferreira, D.; Adega, F.; Escudeiro, A.; Gama-Carvalho, M.; Chaves, R. Decoding the Role of Satellite DNA in Genome Architecture and Plasticity—An Evolutionary and Clinical Affair. Genes 2020, 11, 72. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11010072
Louzada S, Lopes M, Ferreira D, Adega F, Escudeiro A, Gama-Carvalho M, Chaves R. Decoding the Role of Satellite DNA in Genome Architecture and Plasticity—An Evolutionary and Clinical Affair. Genes. 2020; 11(1):72. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11010072
Chicago/Turabian StyleLouzada, Sandra, Mariana Lopes, Daniela Ferreira, Filomena Adega, Ana Escudeiro, Margarida Gama-Carvalho, and Raquel Chaves. 2020. "Decoding the Role of Satellite DNA in Genome Architecture and Plasticity—An Evolutionary and Clinical Affair" Genes 11, no. 1: 72. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11010072