New Transcriptome-Based SNP Markers for Noug (Guizotia abyssinica) and Their Conversion to KASP Markers for Population Genetics Analyses

 ,
,  , , , and
, , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. RNA Extraction, Transcriptome Sequencing and Assembly, and SNP Calling
2.3. Planting, Sampling, and DNA Extraction
2.4. Competitive Allele-Specific PCR (KASP) Assay Design and Genotyping
2.5. Statistical Analyses
3. Results
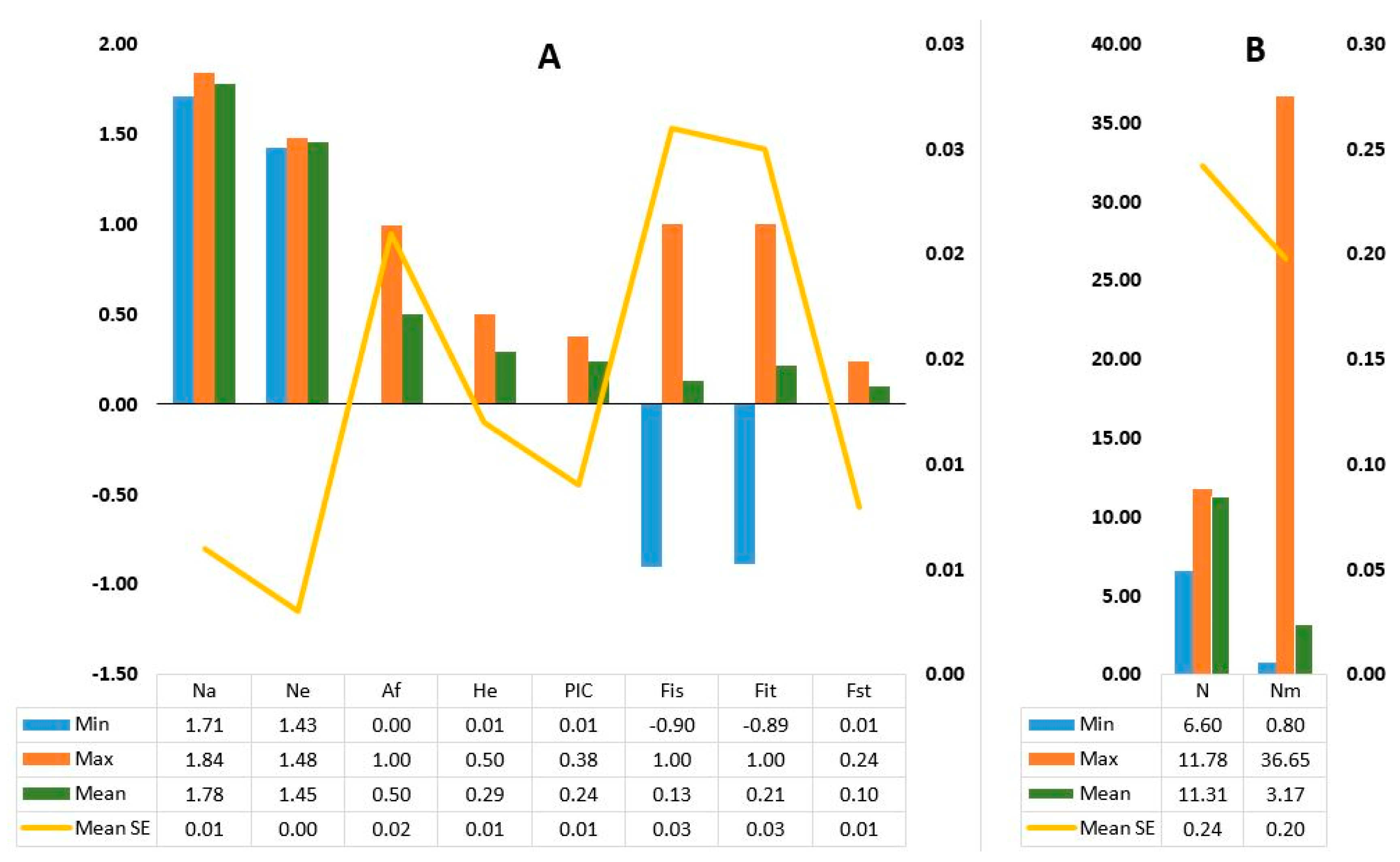
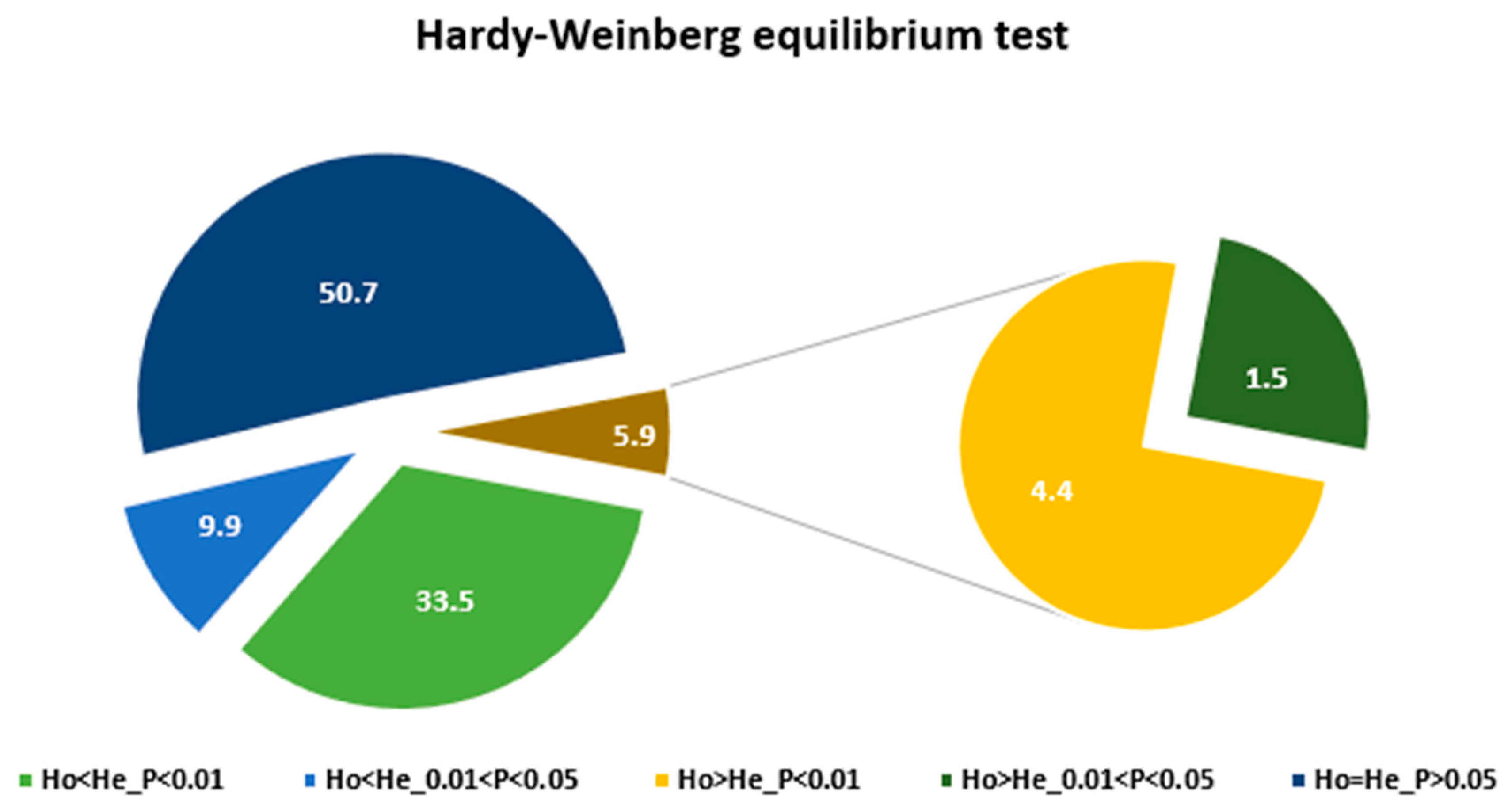
3.1. The KASP Genotyping and the SNP Loci
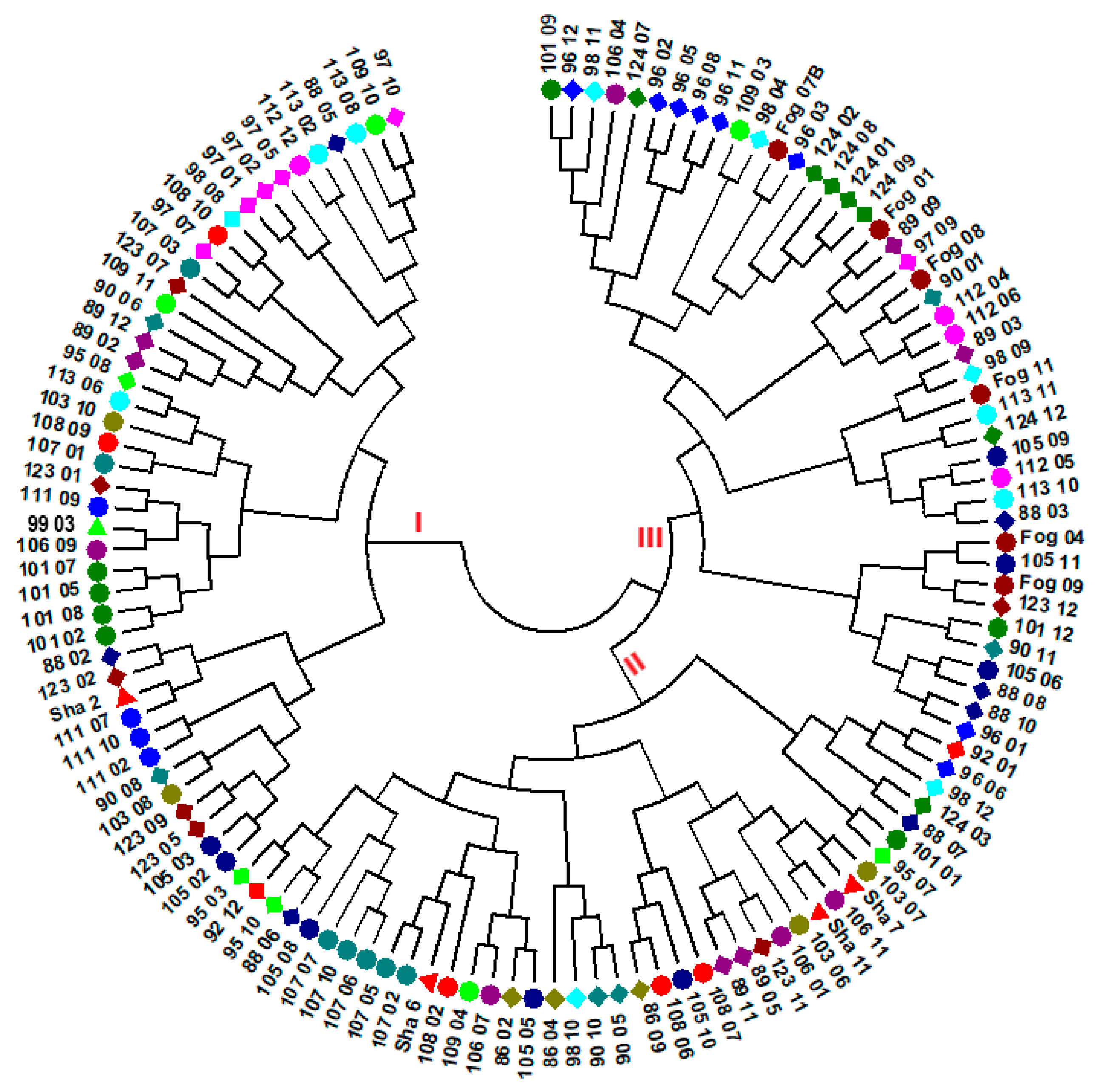
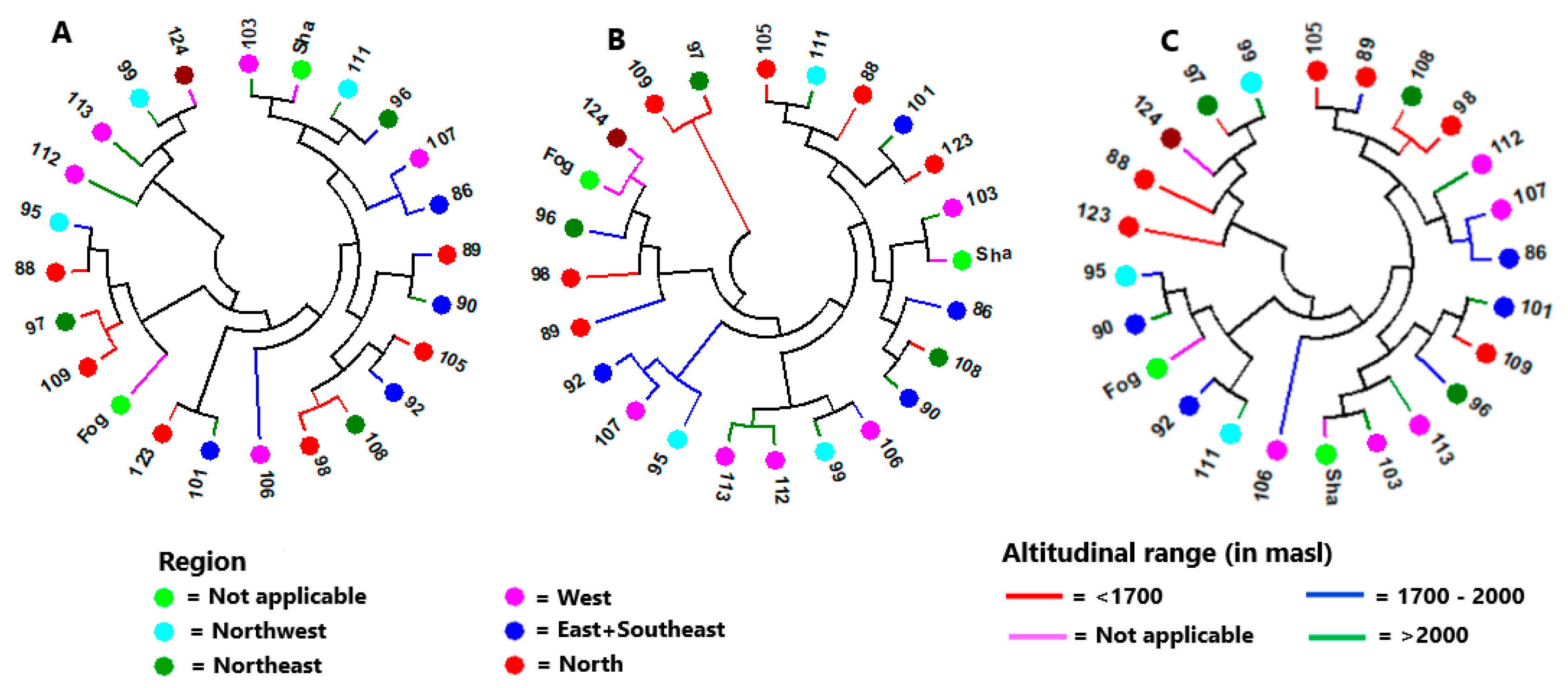
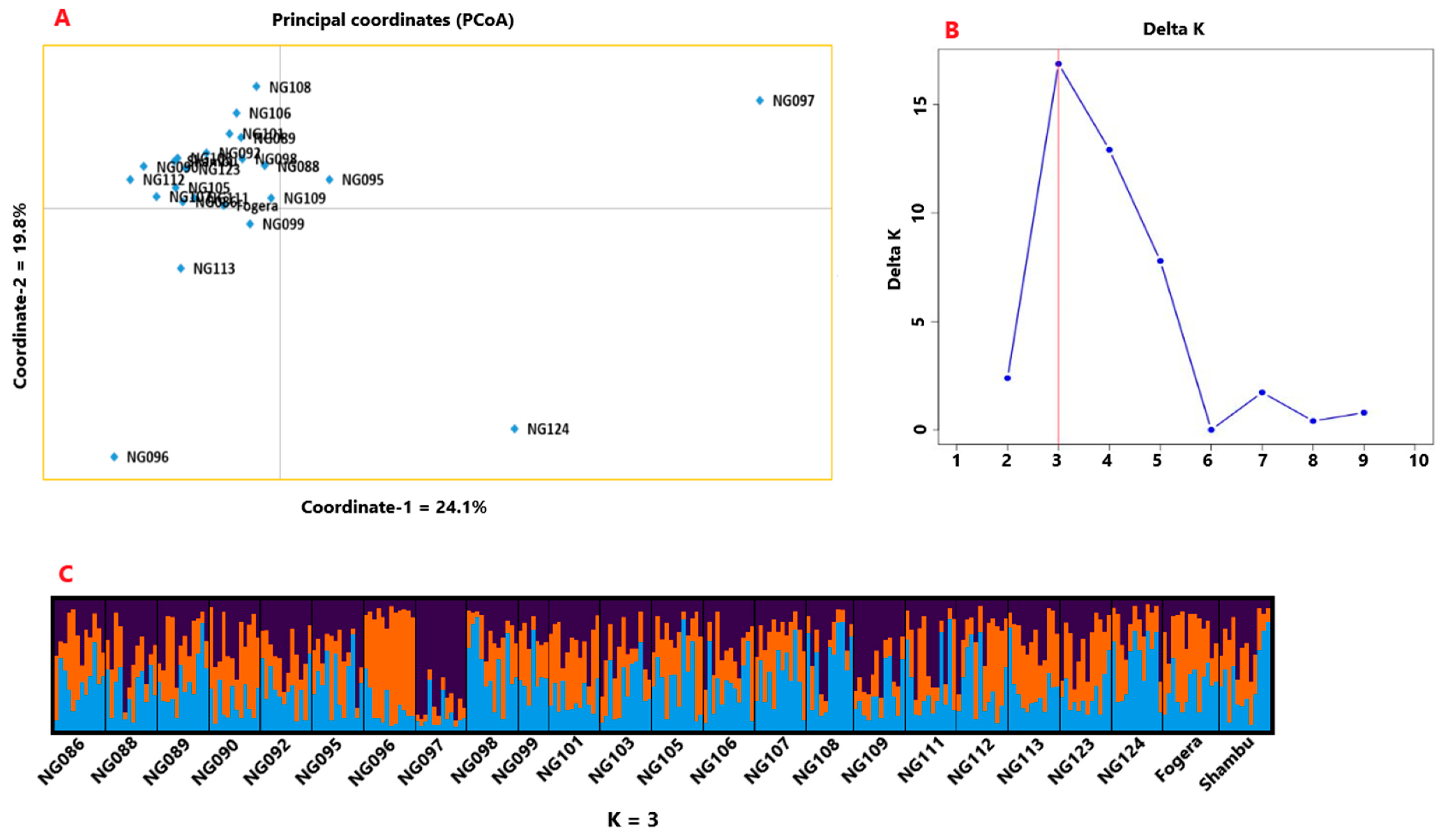
3.2. Genetic Diversity and Population Structure
4. Discussion
4.1. The SNP/KASP Markers
4.2. Genetic Variation within Accessions
4.3. Genetic Variation among Accessions and Population Structure
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dagne, K.; Heneen, W.K. The karyotype and nucleoli of Guizotia abyssinica (Compositae). Hereditas 2008, 117, 73–83. [Google Scholar] [CrossRef]
- Hiremath, S.C.; Murthy, H.N. Cytological studies in Guizotia (Asteraceae). Caryologia 1992, 45, 69–82. [Google Scholar] [CrossRef]
- Dagne, K. Karyotypes, C-banding and nucleolar numbers in Guizotia (Compositae). Plant Syst. Evol. 1995, 195, 121–135. [Google Scholar] [CrossRef]
- Hiremath, S.C.; Murthy, H.N.; Salimath, S.S. Quantitative nuclear DNA differences associated with genome evolution in Guizotia (Compositae). Genetica 1992, 85, 241–247. [Google Scholar] [CrossRef]
- Ohta, T. Further Examples of Evolution by Gene Duplication Revealed through DNA Sequence Comparisons. Genetics 1994, 138, 1331–1337. [Google Scholar]
- Meyerowitz, E.M. The first completely sequenced plant chromosomes, from the mustard Arabidopsis thaliana, reveal a dynamic genome that is constantly being rearranged. Nature 1999, 402, 731–732. [Google Scholar] [CrossRef]
- Bennetzen, J.L.; Ma, J.; Devos, K.M. Mechanisms of Recent Genome Size Variation in Flowering Plants. Ann. Bot. 2005, 95, 127–132. [Google Scholar] [CrossRef] [Green Version]
- Weiss, E.A. Oil Seed Crops; Longman Inc.: New York, NY, USA, 1983. [Google Scholar]
- Murthy, H.N.; Hiremath, S.C.; Salimath, S.S. Origin, evolution and genome differentiation in Guizotia abyssinica and its wild species. Theor. Appl. Genet. 1993, 87, 587–592. [Google Scholar] [CrossRef]
- Getinet, A.; Sharma, S.M. Niger (Guizotia abyssinica (L.F.)) Cass. In Promoting the Conservation and Use of Underutilized and Neglected Crops; International Plant Genetic Resources Institute (IPGRI): Rome, Italy, 1996. [Google Scholar]
- Geleta, M.; Ortiz, R. The importance of Guizotia abyssinica (niger) for sustainable food security in Ethiopia. Genet. Resour. Crop. Evol. 2013, 60, 1763–1770. [Google Scholar] [CrossRef]
- Geleta, M.; Stymne, S.; Bryngelsson, T. Variation and inheritance of oil content and fatty acid composition in niger (Guizotia abyssinica). J. Food Compos. Anal. 2011, 24, 995–1003. [Google Scholar] [CrossRef]
- Dagne, K.; Jonsson, A. Oil Content and Fatty Acid Composition of Seeds of Guizotia Cass (Compositae). J. Sci. Food Agric. 1997, 73, 274–278. [Google Scholar] [CrossRef]
- Ramadan, M.F.; Ramadan, M.F. Functional Properties, Nutritional Value, and Industrial Applications of Niger Oilseeds (Guizotia abyssinica Cass.). Crit. Rev. Food Sci. Nutr. 2012, 52, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Deme, T.; Haki, G.D.; Retta, N.; Woldegiorgis, A.Z.; Geleta, M. Mineral and Anti-Nutritional Contents of Niger Seed (Guizotia abyssinica (L.f.) Cass., Linseed (Linum usitatissimum L.) and Sesame (Sesamum indicum L.) Varieties Grown in Ethiopia. Foods 2017, 6, 27. [Google Scholar] [CrossRef] [PubMed]
- Geleta, M.; Asfaw, Z.; Bekele, E.; Teshome, A. Edible oil crops and their integration with the major cereals in North Shewa and South Welo, Central Highlands of Ethiopia: An ethnobotanical perspective. Hereditas 2002, 137, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Alemaw, G.; Alamayehu, N. Highland oilcrops: A two-decade research experience in Ethiopia. In Research Report No. 30; Institute of Agricultural Research: Addis Ababa, Ethiopia, 1997. [Google Scholar]
- Dempewolf, H.; Kane, N.C.; Ostevik, K.L.; Geleta, M.; Barker, M.S.; Lai, Z.; Stewart, M.L.; Bekele, E.; Engels, J.M.M.; Cronk, Q.C.B.; et al. Establishing genomic tools and resources for Guizotia abyssinica (L.f.) Cass.-the development of a library of expressed sequence tags, microsatellite loci, and the sequencing of its chloroplast genome. Mol. Ecol. Resour. 2010, 10, 1048–1058. [Google Scholar] [CrossRef] [PubMed]
- Geleta, M.; Bryngelsson, T.; Bekele, E.; Dagne, K. Genetic diversity of Guizotia abyssinica (L.f.) Cass. (Asteraceae) from Ethiopia as revealed by random amplified polymorphic DNA (RAPD). Genet. Resour. Crop. Evol. 2006, 54, 601–614. [Google Scholar] [CrossRef]
- Geleta, M.; Bryngelsson, T.; Bekele, E.; Dagne, K. Assessment of genetic diversity of Guizotia abyssinica (L.f.) Cass. (Asteraceae) from Ethiopia using amplified fragment length polymorphism. Plant Genet. Resour. 2008, 6, 41–51. [Google Scholar] [CrossRef]
- Petros, Y.; Merker, A.; Zeleke, H. Analysis of genetic diversity of Guizotia abyssinica from Ethiopia using inter simple sequence repeat markers. Hereditas 2007, 144, 18–24. [Google Scholar] [CrossRef]
- Dempewolf, H.; Tesfaye, M.; Teshome, A.; Bjorkman, A.D.; Andrew, R.L.; Scascitelli, M.; Black, S.; Bekele, E.; Engels, J.M.M.; Cronk, Q.C.B.; et al. Patterns of domestication in the Ethiopian oil-seed crop noug (Guizotia abyssinica). Evol. Appl. 2015, 8, 464–475. [Google Scholar] [CrossRef]
- Ertiro, B.T.; Ogugo, V.; Worku, M.; Das, B.; Olsen, M.; Labuschagne, M.; Semagn, K. Comparison of Kompetitive Allele Specific PCR (KASP) and genotyping by sequencing (GBS) for quality control analysis in maize. BMC Genom. 2015, 16, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Shi, Z.; Liu, S.; Noe, J.; Arelli, P.; Meksem, K.; Li, Z. SNP identification and marker assay development for high-throughput selection of soybean cyst nematode resistance. BMC Genom. 2015, 16, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Kabbaj, H.; Sall, A.T.; Al-Abdallat, A.; Geleta, M.; Amri, A.; Filali-Maltouf, A.; Belkadi, B.; Ortiz, R.; Bassi, F.M. Genetic Diversity within a Global Panel of Durum Wheat (Triticum durum) Landraces and Modern Germplasm Reveals the History of Alleles Exchange. Front. Plant Sci. 2017, 8, 1277. [Google Scholar] [CrossRef] [Green Version]
- Contreras-Soto, R.I.; Mora, F.; De Oliveira, M.A.R.; Higashi, W.; Scapim, C.A.; Schuster, I. A Genome-Wide Association Study for Agronomic Traits in Soybean Using SNP Markers and SNP-Based Haplotype Analysis. PLoS ONE 2017, 12, e0171105. [Google Scholar] [CrossRef] [PubMed]
- Qu, C.; Jia, L.; Fu, F.; Zhao, H.; Lu, K.; Wei, L.; Xu, X.; Liang, Y.; Li, S.; Wang, R.; et al. Genome-wide association mapping and Identification of candidate genes for fatty acid composition in Brassica napus L. using SNP markers. BMC Genom. 2017, 18, 232. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Gong, J.; Xiao, X.; Zhang, Z.; Li, J.; Liu, A.; Lu, Q.; Shang, H.; Shi, Y.; Ge, Q.; et al. GWAS Analysis and QTL Identification of Fiber Quality Traits and Yield Components in Upland Cotton Using Enriched High-Density SNP Markers. Front. Plant Sci. 2018, 9, 1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belaj, A.; De La Rosa, R.; Lorite, I.J.; Mariotti, R.; Cultrera, N.G.; Beuzón, C.R.; González-Plaza, J.J.; Muñoz-Merida, A.; Trelles, O.; Baldoni, L. Usefulness of a New Large Set of High Throughput EST-SNP Markers as a Tool for Olive Germplasm Collection Management. Front. Plant Sci. 2018, 9, 1320. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.; Hwang, I.S.; Lee, H.J.; Lee, J.M.; Seo, E.; Choi, D.; Oh, C.-S. Identification of a molecular marker tightly linked to bacterial wilt resistance in tomato by genome-wide SNP analysis. Theor. Appl. Genet. 2018, 131, 1017–1030. [Google Scholar] [CrossRef]
- Geleta, M.; Gustafsson, C.; Glaubitz, J.C.; Ortiz, R. High-Density Genetic Linkage Mapping of Lepidium Based on Genotyping-by-Sequencing SNPs and Segregating Contig Tag Haplotypes. Front. Plant Sci. 2020, 11, 448. [Google Scholar] [CrossRef]
- Palmé, A.E.; Hagenblad, J.; Solberg, S.Ø.; Aloisi, K.; Artemyeva, A.M. SNP Markers and Evaluation of Duplicate Holdings of Brassica oleracea in Two European Genebanks. Plants 2020, 9, 925. [Google Scholar] [CrossRef]
- Zanotto, S.; Vandenberg, A.; Khazaei, H. Development and validation of a robust KASP marker for zt2 locus in faba bean (Vicia faba). Plant Breed. 2019, 139, 375–380. [Google Scholar] [CrossRef]
- Islam, A.S.M.F.; Blair, M.W. Molecular Characterization of Mung Bean Germplasm from the USDA Core Collection Using Newly Developed KASP-based SNP Markers. Crop. Sci. 2018, 58, 1659–1670. [Google Scholar] [CrossRef]
- Grimm, K.D.S.; Porter, L.D. Development and Validation of KASP Markers for the Identification of Pea seedborne mosaic virus Pathotype P1 Resistance in Pisum sativum. Plant Dis. 2020, 104, 1824–1830. [Google Scholar] [CrossRef] [PubMed]
- Leal-Bertioli, S.C.M.; Cavalcante, U.; Gouveia, E.G.; Ballén-Taborda, C.; Shirasawa, K.; Guimarães, P.M.; Jackson, S.A.; Bertioli, D.J.; Moretzsohn, M.C. Identification of QTLs for Rust Resistance in the Peanut Wild Species Arachis magna and the Development of KASP Markers for Marker-Assisted Selection. G3 Genes. Genom. Genet. 2015, 5, 1403–1413. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.-W.; Lee, S.-B.; Lee, J.-Y.; Kwon, Y.-H.; Lee, S.-M.; Rolly, N.K.; Shin, D.; Cha, J.-G.; Park, D.-S.; Ko, J.-M.; et al. Development and Validation of KASP Markers for Stv-bi, a Rice Stripe Virus Resistance Gene in Rice (Oryza sativa L.). Plant Breed. Biotechnol. 2020, 8, 196–201. [Google Scholar] [CrossRef]
- Cheon, K.-S.; Baek, J.; Cho, Y.-I.; Jeong, Y.-M.; Lee, Y.-Y.; Oh, J.; Won, Y.J.; Kang, D.-Y.; Oh, H.; Kim, S.L.; et al. Single Nucleotide Polymorphism (SNP) Discovery and Kompetitive Allele-Specific PCR (KASP) Marker Development with Korean Japonica Rice Varieties. Plant Breed. Biotechnol. 2018, 6, 391–403. [Google Scholar] [CrossRef]
- Han, G.; Liu, S.; Jin, Y.; Jia, M.; Ma, P.; Liu, H.; Wang, J.; An, D.-G. Scale development and utilization of universal PCR-based and high-throughput KASP markers specific for chromosome arms of rye (Secale cereale L.). BMC Genom. 2020, 21, 1–14. [Google Scholar] [CrossRef]
- Burow, G.; Chopra, R.; Sattler, S.; Burke, J.; Acosta-Martinez, V.; Xin, Z. Deployment of SNP (CAPS and KASP) markers for allelic discrimination and easy access to functional variants for brown midrib genes bmr6 and bmr12 in Sorghum bicolor. Mol. Breed. 2019, 39, 115. [Google Scholar] [CrossRef]
- Wu, J.; Wang, Q.; Kang, Z.; Liu, S.; Li, H.; Mu, J.; Dai, M.; Han, D.; Zeng, Q.; Chen, X. Development and Validation of KASP-SNP Markers for QTL Underlying Resistance to Stripe Rust in Common Wheat Cultivar P10057. Plant Dis. 2017, 101, 2079–2087. [Google Scholar] [CrossRef] [Green Version]
- Fang, T.; Lei, L.; Li, G.; Powers, C.; Hunger, R.M.; Carver, B.F.; Yan, L. Development and deployment of KASP markers for multiple alleles of Lr34 in wheat. Theor. Appl. Genet. 2020, 133, 2183–2195. [Google Scholar] [CrossRef] [Green Version]
- Makhoul, M.; Rambla, C.; Voss-Fels, K.P.; Hickey, L.T.; Snowdon, R.J.; Obermeier, C. Overcoming polyploidy pitfalls: A user guide for effective SNP conversion into KASP markers in wheat. Theor. Appl. Genet. 2020, 133, 2413–2430. [Google Scholar] [CrossRef]
- Geleta, M.; Bryngelsson, T. Population genetics of self-incompatibility and developing self-compatible genotypes in Guizotia abyssinica (L.f.) Cass. (Asteraceae). Euphytica 2010, 176, 417–430. [Google Scholar] [CrossRef]
- Hodgins, K.A.; Lai, Z.; De Oliveira, L.O.; Still, D.W.; Scascitelli, M.; Barker, M.S.; Kane, N.C.; Dempewolf, H.; Kozik, A.; Kesseli, R.V.; et al. Genomics of Compositae crops: Reference transcriptome assemblies and evidence of hybridization with wild relatives. Mol. Ecol. Resour. 2013, 14, 166–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Subgroup, G.P.D.P. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, F.; Yang, R.; Boyle, T. POPGENE: Microsoft Window-Based Freeware for Population Genetic Analysis; Version 1.31; University of Alberta: Edmonton, AB, Canada, 1999. [Google Scholar]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research--an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [Green Version]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K.K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: New York, NY, USA, 1987. [Google Scholar]
- Tajima, F.; Nei, M. Estimation of evolutionary distance between nucleotide sequences. Mol. Biol. Evol. 1984, 1, 269–285. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar]
- Li, Y.-L.; Liu, J.-X. StructureSelector: A web-based software to select and visualize the optimal number of clusters using multiple methods. Mol. Ecol. Resour. 2017, 18, 176–177. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. Clumpak: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moragues, M.; Comadran, J.; Waugh, R.; Milne, I.; Flavell, A.J.; Russell, J.R. Effects of ascertainment bias and marker number on estimations of barley diversity from high-throughput SNP genotype data. Theor. Appl. Genet. 2010, 120, 1525–1534. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, R. Estimation of population parameters and recombination rates from single nucleotide polymorphisms. Genetics 2000, 154, 931–942. [Google Scholar] [PubMed]
- Chang, H.-W.; Cheng, Y.-H.; Chuang, L.-Y.; Yang, C.-H. SNP-RFLPing 2: An updated and integrated PCR-RFLP tool for SNP genotyping. BMC Bioinform. 2010, 11, 173. Available online: http://0-www-biomedcentral-com.brum.beds.ac.uk/1471-2105/11/173 (accessed on 19 November 2020). [CrossRef] [Green Version]
- Evans, J.; Kim, J.; Childs, K.L.; Vaillancourt, B.; Crisovan, E.; Nandety, A.; Gerhardt, D.J.; Richmond, T.A.; Jeddeloh, J.A.; Kaeppler, S.M.; et al. Nucleotide polymorphism and copy number variant detection using exome capture and next-generation sequencing in the polyploid grass Panicum virgatum. Plant J. 2014, 79, 993–1008. [Google Scholar] [CrossRef] [Green Version]
- Hildebrand, C.E.; Torney, D.V.; Wagner, R.P. Informativeness of polymorphic DNA markers. In The Human Genome Project: Decipering the Blueprint of Heredity (Cooper NG edited); University Science Books: Mill Valley, CA, USA, 1994; pp. 100–102. [Google Scholar]
- Shete, S.; Tiwari, H.; Elston, R.C. On Estimating the Heterozygosity and Polymorphism Information Content Value. Theor. Popul. Biol. 2000, 57, 265–271. [Google Scholar] [CrossRef]
- Singh, N.; Choudhury, D.R.; Singh, A.K.; Kumar, S.; Srinivasan, K.; Tyagi, R.K.; Singh, R. Comparison of SSR and SNP Markers in Estimation of Genetic Diversity and Population Structure of Indian Rice Varieties. PLoS ONE 2013, 8, e84136. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; He, H.; Zou, Y.; Chen, W.; Yu, R.; Liu, X.; Yang, Y.; Gao, Y.-M.; Xu, J.-L.; Fan, L.-M.; et al. Development and application of a set of breeder-friendly SNP markers for genetic analyses and molecular breeding of rice (Oryza sativa L.). Theor. Appl. Genet. 2011, 123, 869–879. [Google Scholar] [CrossRef]
- Geleta, M.; Bryngelsson, T.; Bekele, E.; Dagne, K. AFLP and RAPD analyses of genetic diversity of wild and/or weedy Guizotia (Asteraceae) from Ethiopia. Hereditas 2007, 144, 53–62. [Google Scholar] [CrossRef]
- Milligan, B.G.; McMurry, C.K. Dominant vs. codominant genetic markers in the estimation of male mating success. Mol. Ecol. 1993, 2, 275–283. [Google Scholar] [CrossRef]
- Bonnett, D.; Rebetzke, G.; Spielmeyer, W. Strategies for efficient implementation of molecular markers in wheat breeding. Mol. Breed. 2005, 15, 75–85. [Google Scholar] [CrossRef]
- Foll, M.; Gaggiotti, O.E. A Genome-Scan Method to Identify Selected Loci Appropriate for Both Dominant and Codominant Markers: A Bayesian Perspective. Genetics 2008, 180, 977–993. [Google Scholar] [CrossRef] [Green Version]
- Wright, S. The genetical structure of populations. Ann. Eugen. 1949, 15, 323–354. [Google Scholar] [CrossRef]
- Wright, S. The Interpretation of Population Structure by F-Statistics with Special Regard to Systems of Mating. Evolution 1965, 19, 395. Available online: http://www.jstor.com/stable/2406450 (accessed on 19 November 2020). [CrossRef]
- Nemomissa, S.; Bekele, E.; Dagne, K. Self-incompatibility system in the Ethiopian populations of Guizotia abyssinica (L.F.) Cass. (niger). SINET Ethiop. J. Sci. 1999, 22, 67–88. [Google Scholar] [CrossRef] [Green Version]
- Waters, E.R. The Molecular Evolution of the Small Heat-Shock Proteins in Plants. Genetics 1995, 141, 785–795. [Google Scholar]
- Hamrick, J.L.; Godt, M.J.W. Effects of life history traits on genetic diversity in plant species. Philos. Trans. R. Soc. B Biol. Sci. 1996, 351, 1291–1298. [Google Scholar] [CrossRef]
- Nybom, H. Comparison of different nuclear DNA markers for estimating intraspecific genetic diversity in plants. Mol. Ecol. 2004, 13, 1143–1155. [Google Scholar] [CrossRef]
- Gadissa, F.; Tesfaye, K.; Dagne, K.; Geleta, M. Genetic diversity and population structure analyses of Plectranthus edulis (Vatke) Agnew collections from diverse agro-ecologies in Ethiopia using newly developed EST-SSRs marker system. BMC Genet. 2018, 19, 92. [Google Scholar] [CrossRef]
- Ng’uni, D.; Geleta, M.; Hofvander, P.; Fatih, M.; Bryngelsson, T. Comparative genetic diversity and nutritional quality variation among some important Southern African sorghum accessions [Sorghum bicolor (L.) Moench]. Aust. J. Crop. Sci. 2012, 6, 56–64. [Google Scholar]
- Ng’Uni, D.; Geleta, M.; Bryngelsson, T. Genetic diversity in sorghum (Sorghum bicolor (L.) Moench) accessions of Zambia as revealed by simple sequence repeats (SSR). Hereditas 2011, 148, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Motlhaodi, T.; Geleta, M.; Bryngelsson, T.; Fatih, M.; Chite, S.; Ortiz, R. Genetic diversity in ex-situ conserved sorghum accessions of Botswana as estimated by microsatellite markers. Aust. J. Crop. Sci. 2014, 8, 35–43. [Google Scholar]
- Motlhaodi, T.; Geleta, M.; Chite, S.; Fatih, M.; Ortiz, R.; Bryngelsson, T. Genetic diversity in sorghum germplasm from Southern Africa as revealed by microsatellite markers and agromorphological traits. Genet. Resour. Crop. Evol. 2017, 64, 599–610. [Google Scholar] [CrossRef]
- Hegay, S.; Geleta, M.; Bryngelsson, T.; Gustavsson, L.; Hovmalm, H.P.; Ortiz, R. Comparing genetic diversity and population structure of common beans grown in Kyrgyzstan using microsatellites. Sci. J. Crop. Sci. 2012, 1, 63–75. [Google Scholar]
- Teshome, A.; Bryngelsson, T.; Dagne, K.; Geleta, M. Assessment of genetic diversity in Ethiopian field pea (Pisum sativum L.) accessions with newly developed EST-SSR markers. BMC Genet. 2015, 16, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Geleta, M.; Herrera, I.; Monzón, A.; Bryngelsson, T. Genetic Diversity of Arabica Coffee (Coffea arabica L.) in Nicaragua as Estimated by Simple Sequence Repeat Markers. Sci. World J. 2012, 2012, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Chombe, D.; Bekele, E.; Bryngelsson, T.; Teshome, A.; Geleta, M. Genetic structure and relationships within and between cultivated and wild korarima [Aframomum corrorima (Braun) P.C.M. Jansen] in Ethiopia as revealed by simple sequence repeat (SSR) markers. BMC Genet. 2017, 18, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Wright, S. Isolation by Distance. Genetics 1943, 28, 114–138. [Google Scholar]
- Ishikawa, M.; Naito, S.; Ohno, T. Effects of the tom1 mutation of Arabidopsis thaliana on the multiplication of tobacco mosaic virus RNA in protoplasts. J. Virol. 1993, 67, 5328–5338. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, T.; Ohta, T.; Takahashi, M.; Meshi, T.; Schmidt, R.; Dean, C.; Naito, S.; Ishikawa, M. TOM1, an Arabidopsis gene required for efficient multiplication of a tobamovirus, encodes a putative transmembrane protein. Proc. Natl. Acad. Sci. USA 2000, 97, 10107–10112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rannala, B.; Mountain, J.; Rannala, B.; Mountain, J.L. Detecting immigration by using multilocus genotypes. Proc. Natl. Acad. Sci. USA 1997, 94, 9197–9201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, N.; Villablanca, F.X.; Roderick, G.K. Determining the source of individuals: Multilocus genotyping in nonequilibrium population genetics. Tree 1999, 14, 17–21. [Google Scholar] [CrossRef] [Green Version]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Noug Contig | SNP Locus | SNP PINC c | Ref_Alt d,e | Missing Genotype | MAF f | Helianthus annuus Homologue: Accession Number and Aligned Region |
|---|---|---|---|---|---|---|
| CL3143Contig1 | 3143A a | 376 | G_C | GG | 0.131 | 17.8 kDa class I heat shock protein-like_LOC110904834: XM_022150704; 212..484 |
| 3143B a | 388 | C_A | AA | 0.120 | ||
| CCHT13019.b1F16.ab1 | 13,019A a | 30 | C_A | AA | 0.389 | Two-component response regulator-like PRR73_LOC110868813: XM_022118076; 1133..1903 |
| 13,019C b | 372 | T_A | AT | 0.448 | ||
| CCHT3719.b1M18.ab1 | 3719A a | 502 | A_G | AA | 0.239 | TPR2-like protein_LOC110930065: XM_022173292; 2716..3466 |
| CCHT4593.b1B22.ab1 | 4593B a | 274 | A_C | None | 0.389 | Calnexin homolog_LOC110865890: XM_022115225; 1115..1583 |
| CCHT4736.b1P07.ab1 | 4736 a | 498 | G_C | GG | 0.293 | Cytochrome P450 CYP82D47-like_LOC110879526: XM_022127994; 1210..1778 |
| CCHT7954.b1C22.ab1 | 7954A a | 387 | T_C | CC | 0.371 | Uncharacterized protein_LOC110878690: XM_022127042; 145..855 |
| CCHT8585.b1B11.ab1 | 8585 a | 280 | C_T | CC | 0.149 | Uncharacterized protein_ LOC110927216: XM_022170855; 1193..1751 |
| CCHT10160.b1P19.ab1 | 10,160 a | 328 | G_C | GG | 0.441 | UDP-arabinopyranose mutase 1-like_ LOC110915474: XM_022160180; 62..679 |
| CCHT13180.b1H07.ab1 | 13,180 a | 460 | G_A | None | 0.432 | 40S ribosomal protein S13_LOC110937753: XM_022180210; 4..544 |
| CCHT17789.b1J07.ab1 | 17,789 a | 698 | C_A | CC | 0.245 | Uncharacterized 38.1 kDa protein-like_LOC110867109: XM_022116245; 208..521 |
| CCHT20996.b1G18.ab1 | 20,996B a | 636 | C_T | CC | 0.470 | Heat shock 70 kDa protein 14 like_LOC110894223: XM_022141417, 93..831 |
| CCHT17807.b1N11.ab1 | 17,807 b | 350 | T_G | GT | 0.250 | Nucleobase-ascorbate transporter 6-like_LOC110941756: XM_022183418; 123..833 |
| CCHT17571.b1F02.ab1 | 17,571 b | 408 | T_A | AT | 0.470 | Probable ADP-ribosylation factor GTPase-activating protein AGD14_LOC110886873: XM_022134739; 92..748 |
| CCHT4779.b1F19.ab1 | 4779 b | 255 | A_C | AC | 0.185 | Probable E3 ubiquitin-protein ligase ARI1_LOC110929292: XM_022172423; 1130..1867 |
| Acc | PPL | I | Ho | He | uHe | F | Theta | EAED1 | EAED2 | EAED3 |
|---|---|---|---|---|---|---|---|---|---|---|
| NG086 a | 75.7 | 0.391 | 0.238 | 0.262 | 0.274 | 0.105 | 2.016 | 0.313 | 0.554 | 0.207 |
| NG088 a | 77.7 | 0.396 | 0.242 | 0.263 | 0.276 | 0.092 | 2.004 | 0.276 | 0.468 | 0.198 |
| NG089 a | 75.7 | 0.387 | 0.248 | 0.258 | 0.270 | 0.042 | 2.038 | 0.294 | 0.528 | 0.201 |
| NG090 a | 80.1 | 0.403 | 0.244 | 0.268 | 0.280 | 0.084 | 1.978 | 0.274 | 0.489 | 0.187 |
| NG092 a | 84.0 | 0.419 | 0.235 | 0.278 | 0.290 | 0.165 | 1.927 | 0.304 | 0.541 | 0.206 |
| NG095 a | 80.6 | 0.415 | 0.256 | 0.277 | 0.289 | 0.087 | 1.934 | 0.285 | 0.497 | 0.196 |
| NG096 a | 73.8 | 0.381 | 0.259 | 0.255 | 0.267 | 0.017 | 2.057 | 0.257 | 0.454 | 0.176 |
| NG097 a | 76.7 | 0.384 | 0.224 | 0.254 | 0.266 | 0.122 | 2.063 | 0.256 | 0.430 | 0.183 |
| NG098 a | 78.6 | 0.403 | 0.256 | 0.267 | 0.279 | 0.060 | 1.984 | 0.280 | 0.436 | 0.216 |
| NG099 a | 76.7 | 0.400 | 0.261 | 0.266 | 0.288 | 0.011 | 1.968 | n/c | n/c | n/c |
| NG101 a | 79.1 | 0.405 | 0.259 | 0.270 | 0.282 | 0.047 | 2.012 | 0.248 | 0.396 | 0.187 |
| NG103 a | 80.1 | 0.396 | 0.245 | 0.263 | 0.275 | 0.067 | 2.076 | 0.218 | 0.380 | 0.146 |
| NG105 a | 78.6 | 0.395 | 0.259 | 0.263 | 0.274 | 0.057 | 1.971 | 0.249 | 0.386 | 0.195 |
| NG106 a | 77.2 | 0.382 | 0.227 | 0.252 | 0.264 | 0.103 | 2.067 | 0.286 | 0.548 | 0.188 |
| NG107 a | 75.7 | 0.402 | 0.246 | 0.270 | 0.282 | 0.086 | 1.959 | 0.286 | 0.496 | 0.201 |
| NG108 a | 70.9 | 0.380 | 0.237 | 0.255 | 0.269 | 0.081 | 1.974 | 0.272 | 0.484 | 0.190 |
| NG109 a | 79.6 | 0.408 | 0.244 | 0.272 | 0.284 | 0.115 | 2.031 | 0.306 | 0.465 | 0.242 |
| NG111 a | 80.1 | 0.405 | 0.234 | 0.269 | 0.281 | 0.134 | 2.045 | 0.278 | 0.568 | 0.165 |
| NG112 a | 79.1 | 0.392 | 0.233 | 0.259 | 0.271 | 0.110 | 1.940 | 0.295 | 0.532 | 0.203 |
| NG113 a | 77.7 | 0.388 | 0.237 | 0.257 | 0.269 | 0.073 | 2.073 | 0.274 | 0.453 | 0.203 |
| NG123 a | 82.5 | 0.414 | 0.256 | 0.275 | 0.288 | 0.090 | 2.008 | 0.283 | 0.462 | 0.214 |
| NG124 b | 72.8 | 0.379 | 0.217 | 0.253 | 0.264 | 0.133 | 1.936 | 0.287 | 0.493 | 0.204 |
| Fogera c | 80.1 | 0.407 | 0.253 | 0.270 | 0.283 | 0.072 | 1.969 | 0.300 | 0.519 | 0.213 |
| Shambu c | 81.1 | 0.398 | 0.238 | 0.262 | 0.274 | 0.091 | 2.011 | 0.284 | 0.487 | 0.205 |
| Mean_Alt-1 | 77.8 | 0.397 | 0.245 | 0.264 | 0.277 | 0.088 | 2.005 | 0.275 | 0.447 | 0.205 |
| Mean_Alt-2 | 77.5 | 0.397 | 0.244 | 0.265 | 0.277 | 0.086 | 2.000 | 0.289 | 0.517 | 0.196 |
| Mean_Alt-3 | 79.0 | 0.398 | 0.245 | 0.265 | 0.278 | 0.075 | 2.013 | 0.265 | 0.470 | 0.182 |
| Mean_Reg-1 | 78.8 | 0.401 | 0.251 | 0.266 | 0.279 | 0.076 | 2.006 | 0.281 | 0.458 | 0.211 |
| Mean_Reg-2 | 73.8 | 0.382 | 0.240 | 0.255 | 0.267 | 0.073 | 2.031 | 0.262 | 0.456 | 0.183 |
| Mean_Reg-3 | 79.7 | 0.405 | 0.244 | 0.270 | 0.282 | 0.100 | 1.983 | 0.285 | 0.495 | 0.197 |
| Mean_Reg-4 | 77.4 | 0.391 | 0.236 | 0.260 | 0.272 | 0.093 | 2.010 | 0.285 | 0.507 | 0.199 |
| Mean_Reg-5 | 80.1 | 0.396 | 0.245 | 0.263 | 0.275 | 0.067 | 2.076 | 0.218 | 0.380 | 0.146 |
| Mean_Reg-6 | 79.1 | 0.407 | 0.250 | 0.271 | 0.286 | 0.077 | 1.982 | 0.282 | 0.533 | 0.181 |
| Mean_Landrace | 78.1 | 0.397 | 0.245 | 0.264 | 0.277 | 0.083 | 2.006 | 0.277 | 0.478 | 0.195 |
| Mean_Cultivar | 80.6 | 0.4025 | 0.245 | 0.266 | 0.278 | 0.081 | 1.99 | 0.292 | 0.503 | 0.209 |
| Mean_all | 78.1 | 0.397 | 0.244 | 0.264 | 0.277 | 0.085 | 2.002 | 0.278 | 0.481 | 0.197 |
| SE_all | 0.006 | 0.004 | 0.003 | 0.003 | 0.003 | 0.006 | 0.050 | 0.005 | 0.011 | 0.004 |
| Source of Variation | DF | Sum of Squares | Variance Components | Percentage of Variation | Fixation Indices | Probability (p) Value |
|---|---|---|---|---|---|---|
| Among accessions | 23 | 1078.2 | 1.019 Va | 4.52 | FST = 0.045 | Va & FST < 0.0001 |
| Among individuals within accessions | 259 | 5921.0 | 1.321 Vb | 5.86 | FIS = 0.061 | Vb & FIS < 0.0001 |
| Within individuals | 283 | 5722.0 | 20.219 Vc | 89.63 | FIT = 0.104 | Vc & FIT < 0.0001 |
| Total | 565 | 12,721.3 | 22.556 | |||
| a Among alt groups | 2 | 97.9 | 0.026 Va | 0.12 | FCT = 0.001 | Va & FCT = 0.1935 |
| Among accessions within alt groups | 18 | 800.1 | 1.015 Vb | 4.67 | FSC = 0.047 | Vb & FSC < 0.0001 |
| Within accessions | 471 | 9746.7 | 20.694 Vc | 95.21 | FST = 0.048 | Vc & FST < 0.0001 |
| Total | 491 | 10,644.8 | 21.735 | |||
| b Among regions-I | 5 | 217.0 | −0.027 Va | −0.12 | FCT = −0.001 | Va & FCT = 0.6715 |
| Among accessions within regions-I | 15 | 681.0 | 1.056 Vb | 4.86 | FSC = 0.048 | Vb & FSC < 0.0001 |
| Within accessions | 471 | 9746.7 | 20.694 Vc | 95.26 | FST = 0.047 | Vc & FST < 0.0001 |
| Total | 491 | 10,644.8 | 21.723 | |||
| c Among regions-II | 1 | 55.4 | 0.044 Va | 0.20 | FCT = 0.002 | Va & FCT = 0.047 |
| Among accessions within regions-II | 19 | 842.6 | 1.011 Vb | 4.65 | FSC = 0.047 | Vb & FSC < 0.0001 |
| Within accessions | 471 | 9746.7 | 20.694 Vc | 95.15 | FST = 0.048 | Vc & FST < 0.0001 |
| Total | 491 | 10,644.8 | 21.723 |
| Acc | 086 | 088 | 089 | 090 | 092 | 095 | 096 | 097 | 098 | 099 | 101 | 103 | 105 | 106 | 107 | 108 | 109 | 111 | 112 | 113 | 123 | 124 | Fog | Sha |
| 086 | 0.036 | |||||||||||||||||||||||
| 088 | 0.031 | 0.041 | ||||||||||||||||||||||
| 089 | 0.026 | 0.041 | 0.045 | |||||||||||||||||||||
| 090 | 0.031 | 0.055 | 0.041 | 0.052 | ||||||||||||||||||||
| 092 | 0.023 | 0.026 | 0.046 | 0.041 | 0.038 | |||||||||||||||||||
| 095 | 0.047 | 0.031 | 0.051 | 0.056 | 0.043 | 0.052 | ||||||||||||||||||
| 096 | 0.054 | 0.075 | 0.076 | 0.081 | 0.061 | 0.095 | 0.074 | |||||||||||||||||
| 097 | 0.069 | 0.066 | 0.065 | 0.082 | 0.065 | 0.064 | 0.124 | 0.076 | ||||||||||||||||
| 098 | 0.046 | 0.061 | 0.037 | 0.073 | 0.037 | 0.074 | 0.079 | 0.08 | 0.055 | |||||||||||||||
| 099 | 0.030 | 0.053 | 0.042 | 0.053 | 0.040 | 0.048 | 0.078 | 0.082 | 0.066 | 0.048 | ||||||||||||||
| 101 | 0.034 | 0.032 | 0.053 | 0.065 | 0.050 | 0.064 | 0.08 | 0.078 | 0.057 | 0.075 | 0.052 | |||||||||||||
| 103 | 0.024 | 0.031 | 0.031 | 0.031 | 0.017 | 0.041 | 0.075 | 0.069 | 0.052 | 0.027 | 0.036 | 0.035 | ||||||||||||
| 105 | 0.034 | 0.033 | 0.043 | 0.045 | 0.042 | 0.046 | 0.069 | 0.084 | 0.049 | 0.054 | 0.048 | 0.038 | 0.047 | |||||||||||
| 106 | 0.031 | 0.036 | 0.039 | 0.044 | 0.036 | 0.049 | 0.072 | 0.069 | 0.043 | 0.054 | 0.042 | 0.024 | 0.035 | 0.043 | ||||||||||
| 107 | 0.023 | 0.039 | 0.039 | 0.033 | 0.033 | 0.043 | 0.069 | 0.078 | 0.047 | 0.034 | 0.042 | 0.025 | 0.035 | 0.035 | 0.039 | |||||||||
| 108 | 0.038 | 0.057 | 0.059 | 0.057 | 0.031 | 0.056 | 0.103 | 0.091 | 0.042 | 0.068 | 0.068 | 0.043 | 0.051 | 0.043 | 0.038 | 0.056 | ||||||||
| 109 | 0.050 | 0.037 | 0.050 | 0.061 | 0.040 | 0.05 | 0.072 | 0.064 | 0.048 | 0.059 | 0.047 | 0.029 | 0.056 | 0.05 | 0.054 | 0.068 | 0.052 | |||||||
| 111 | 0.025 | 0.029 | 0.028 | 0.041 | 0.027 | 0.042 | 0.05 | 0.086 | 0.056 | 0.025 | 0.045 | 0.024 | 0.037 | 0.045 | 0.034 | 0.055 | 0.042 | 0.039 | ||||||
| 112 | 0.034 | 0.046 | 0.053 | 0.049 | 0.039 | 0.045 | 0.072 | 0.089 | 0.05 | 0.037 | 0.057 | 0.047 | 0.041 | 0.046 | 0.031 | 0.063 | 0.06 | 0.043 | 0.049 | |||||
| 113 | 0.038 | 0.059 | 0.053 | 0.041 | 0.050 | 0.057 | 0.066 | 0.075 | 0.072 | 0.023 | 0.061 | 0.03 | 0.062 | 0.055 | 0.033 | 0.07 | 0.061 | 0.046 | 0.041 | 0.051 | ||||
| 123 | 0.026 | 0.007 * | 0.020 | 0.039 | 0.018 | 0.044 | 0.057 | 0.055 | 0.034 | 0.037 | 0.027 | 0.019 | 0.021 | 0.025 | 0.023 | 0.043 | 0.047 | 0.006 * | 0.037 | 0.043 | 0.031 | |||
| 124 | 0.065 | 0.056 | 0.077 | 0.091 | 0.068 | 0.069 | 0.09 | 0.083 | 0.086 | 0.062 | 0.078 | 0.072 | 0.084 | 0.074 | 0.065 | 0.093 | 0.076 | 0.06 | 0.083 | 0.065 | 0.058 | 0.072 | ||
| Fog | 0.028 | 0.019 | 0.040 | 0.042 | 0.025 | 0.033 | 0.055 | 0.058 | 0.038 | 0.031 | 0.026 | 0.021 | 0.046 | 0.032 | 0.031 | 0.038 | 0.043 | 0.028 | 0.031 | 0.035 | 0.011 * | 0.038 | 0.033 | |
| Sha | 0.012 * | 0.023 | 0.023 | 0.036 | 0.019 | 0.04 | 0.054 | 0.065 | 0.03 | 0.034 | 0.031 | 0.007 * | 0.028 | 0.020 | 0.020 | 0.023 | 0.021 | 0.011 * | 0.03 | 0.032 | 0.010 * | 0.064 | 0.010 * | 0.028 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsehay, S.; Ortiz, R.; Johansson, E.; Bekele, E.; Tesfaye, K.; Hammenhag, C.; Geleta, M. New Transcriptome-Based SNP Markers for Noug (Guizotia abyssinica) and Their Conversion to KASP Markers for Population Genetics Analyses. Genes 2020, 11, 1373. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11111373
Tsehay S, Ortiz R, Johansson E, Bekele E, Tesfaye K, Hammenhag C, Geleta M. New Transcriptome-Based SNP Markers for Noug (Guizotia abyssinica) and Their Conversion to KASP Markers for Population Genetics Analyses. Genes. 2020; 11(11):1373. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11111373
Chicago/Turabian StyleTsehay, Sewalem, Rodomiro Ortiz, Eva Johansson, Endashaw Bekele, Kassahun Tesfaye, Cecilia Hammenhag, and Mulatu Geleta. 2020. "New Transcriptome-Based SNP Markers for Noug (Guizotia abyssinica) and Their Conversion to KASP Markers for Population Genetics Analyses" Genes 11, no. 11: 1373. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11111373