Identification of Genic SSRs Provide a Perspective for Studying Environmental Adaptation in the Endemic Shrub Tetraena mongolica

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. RNA Preparation, cDNA Library Construction, and Sequencing
2.3. De novo Assembly and Sequence Clustering
2.4. Identification of Potential Polymorphic Genic SSRs
2.5. Location Prediction and Functional Annotation of the Identified SSRs
2.6. SSR Polymorphic Validation
2.7. Data Availability
3. Results
3.1. Sequencing Outputs and de novo Assembly
3.2. Identification of Polymorphic SSRs
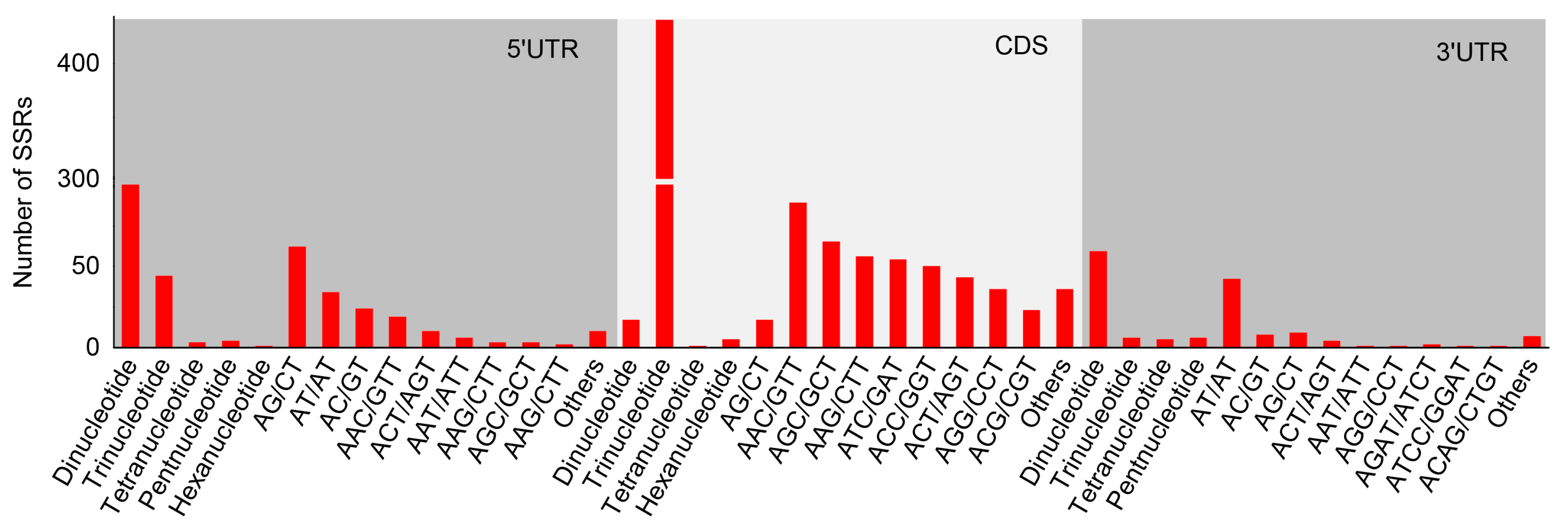
3.3. Location Prediction and Frequency Analysis of the Polymorphic SSRs
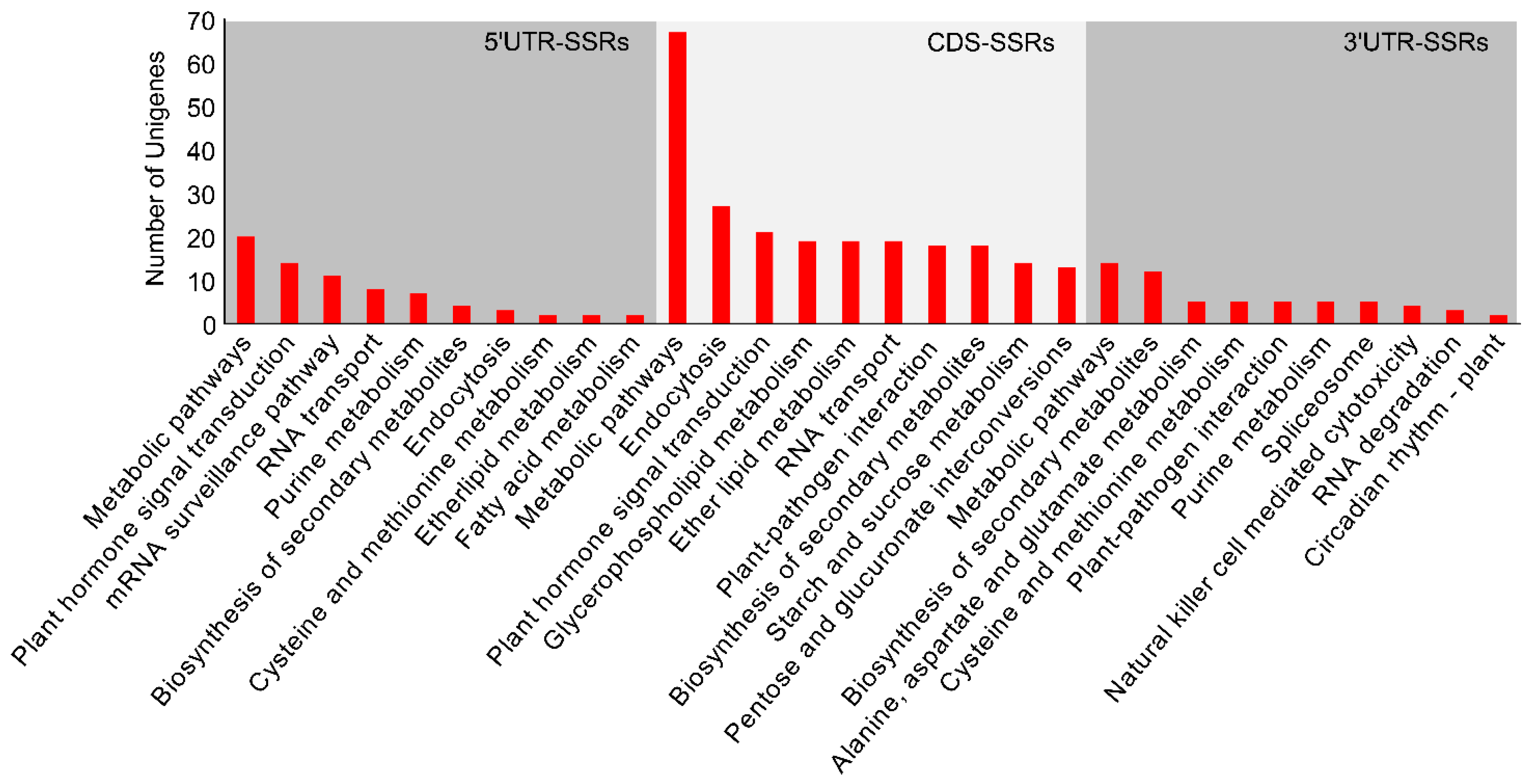
3.4. Functional Annotation of the Polymorphic SSR-Containing Sequences
3.5. Validation of the Polymorphic SSRs
4. Discussion
4.1. Identifying and General Profiling of Genic SSRs in T. mongolica Transcriptomes
4.2. Probable Functions of Genic SSRs in the Environmental Adaptation of T. mongolica
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Guo, X.H.; Ci, Z.L.; Sun, J.; Yang, J. Chromosome variations during the tissue culture of Tetraena mongolia Maxim. J. Inn. Mong. Agric. Univ. (Nat. Sci. Edn.) 2001, 22, 55–59. [Google Scholar]
- Zhang, Y.J.; Yang, C. Comparative analysis of genetic diversity in the endangered shrub Tetraena mongolica and its related congener Zygophyllum xanthoxylon. Acta Phytoecol. Sin. 2000, 24, 425–429. [Google Scholar] [CrossRef]
- Chen, N.M.; Feng, J.C.; Song, B.; Tang, S.; He, J.Q.; Zhou, Y.J.; Shi, S.; Xu, X.J. De novo transcriptome sequencing and identification of genes related to salt and PEG stress in Tetraena mongolica Maxim. Trees 2019, 33, 1639–1656. [Google Scholar] [CrossRef]
- Dang, Z.H.; Zheng, L.L.; Wang, J.; Gao, Z.; Wu, S.B.; Qi, Z.; Wang, Y.C. Transcriptomic profiling of the salt-stress response in the wild recretohalophyte Reaumuria trigyna. BMC Genom. 2013, 14, 29. [Google Scholar] [CrossRef] [Green Version]
- Ge, X.J.; Yu, Y.; Zhao, N.X.; Chen, H.S.; Qi, W.Q. Genetic variation in the endangered Inner Mongolia endemic shrub Tetraena mongolica Maxim (Zygophyllaceae). Biol. Conserv. 2003, 111, 427–434. [Google Scholar] [CrossRef]
- Wang, G.L.; Lin, Q.Q.; Xu, Y.N. Tetraena mongolica Maxim can accumulate large amounts of triacylglycerol in phloem cells and xylem parenchyma of stems. Phytochemistry 2007, 68, 2112–2117. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Yang, C.; Cheng, W.L. A comparison of ITS sequences of nuclear ribosomal DNA between endemic species Tetraena mongolica Maxim in Ordos Platean and three species of zygophyllaceae in the same habitat. Acta Sci. Natl. Univ. Neimong. 2003, 34, 420–424. [Google Scholar] [CrossRef]
- Ma, X.; Chang, J.Y.; Li, Z.H.; Zhai, W.; Yu, X.X.; Feng, Y.L. The complete chloroplast genome of Tetraena mongolica (Zygophyllaceae), an endangered shrub endemic to China. Mitochondrial DNA B 2019, 4, 1030–1031. [Google Scholar] [CrossRef] [Green Version]
- Duan, Y.Z.; Du, Z.Y.; Wang, H.T. Chloroplast genome characteristics of endangered relict plant Tetraena mongolica in the arid region of northwest China. Bull. Bot. Res. 2019, 39, 653–663. [Google Scholar] [CrossRef]
- Xu, Q.; Jiang, C.Q.; Liu, S.R.; Guo, Q.S. Study on Pollination ecology of endangered plant Tetraena mongolica population. Forest Res. 2003, 16, 391–397. [Google Scholar] [CrossRef]
- Jiang, S.; Ren, X.H.; Gu, S.; Li, J.; Xu, K. Wood anatomy of Tetraena mongolica Maxim (Zygophyllaceae). J. Trop. Subtrop. Bot. 2008, 16, 466–471. [Google Scholar] [CrossRef]
- Xu, Q.; Guo, Q.S.; Liu, S.R.; Jiang, C.Q.; Hao, Y.G. A study on the relationship between fruiting characteristics and reproductive age, habitat of endangered species Tetraena mongolica. Sci. Silv. Sin. 2003, 39, 26–32. [Google Scholar] [CrossRef]
- Li, X.; Wang, Y.C.; Zheng, R. Water parameters of xeric shrubs in west erdos region (I). Chin. J. Desert Res. 2005, 25, 581–586. [Google Scholar] [CrossRef]
- Shi, S.L.; Wang, Y.C.; Zhou, J.H. The change of water parameters in Tetraena mongolica with season and habitat. Acta Ecol. Sin. 2008, 28, 6079–6089. [Google Scholar] [CrossRef]
- Shi, S.L.; Wang, Y.C.; Zhou, J.H. The seasonal change of endogenous phytohormone and differentiation of populations in Tetraena mongolica in different habitats. Acta Ecol. Sin. 2009, 29, 2252–2262. [Google Scholar] [CrossRef]
- Shi, S.L.; Wang, Y.C.; Li, X.; Zhou, H.B. Variety of antioxidant system of Tetraena mongolica in different growth stages and habitats. Chin. J. Desert Res. 2012, 32, 771–779. [Google Scholar]
- Shi, G.R.; Ding, L.L.; Liu, Q.; TANG, S.A.; Duan, H.Q. Chemical constituents contained in Tetraena mongolica. China J. Chin. Mater. Med. 2012, 37, 1579–1580. [Google Scholar] [CrossRef]
- Zhi, Y.B.; Yang, C.; Wang, Z.L.; Yao, Y.P.; Gao, T.Y.; Hao, X.; Liu, J.P. Characteristic analysis of amino acid contents of Tetraena mongolica Maxim. Acta Sci. Natl. Univ. Neimongol 2005, 36, 306–312. [Google Scholar] [CrossRef]
- Zhi, Y.B.; Yang, C.; Wang, Z.S.; An, S.Q.; Wang, Z.L.; Li, H.L.; Su, Z.A.; Wang, Q. The endangered characteristics and mechanism of the endemic relict shrub Tetraena mongolica Maxim. Acta Ecol. Sin. 2008, 28, 767–776. [Google Scholar] [CrossRef]
- Ge, X.J.; Hwang, C.C.; Liu, Z.H.; Huang, C.C.; Huang, W.H.; Huang, K.H.; Wang, W.K.; Chiang, T.Y. Conservation genetics and phylogeography of endangered and endemic shrub Tetraena mongolica (Zygophyllaceae) in Inner Mongolia, China. BMC Genet. 2011, 12, 1. [Google Scholar] [CrossRef] [Green Version]
- Zhen, J.H.; Liu, G.H. Population structure characteristics of Tetraena mongolica in different habitats. Acta Ecol. Sin. 2008, 28, 1829–1841. [Google Scholar] [CrossRef]
- Wu, Z.G.; Wei, W.; Cheng, K.; Zheng, L.L.; Ma, C.M.; Wang, Y.C. Insecticidal activity of triterpenoids and volatile oil from the stems of Tetraena mongolica. Pestic. Biochem. Phys. 2020. [Google Scholar] [CrossRef]
- Wu, Z.G.; Wei, W.; Xu, H.Y.; Zheng, L.L.; Ma, C.M.; Wang, Y.C. Constituents from the leaves of Tetraena mongolica and their protective activity in HEK 293t cells damaged by CdCl2. J. Nat. Prod. 2019, 82, 2707–2712. [Google Scholar] [CrossRef]
- Zhi, Y.B.; Sun, Z.L.; Sun, P.; Zhao, K.; Guo, Y.N.; Zhang, D.J.; Zhang, B.W. How much genetic variation is stored in the endangered and fragmented shrub Tetraena mongolica Maxim? PeerJ 2018, 6, e5645. [Google Scholar] [CrossRef] [Green Version]
- Varshney, R.K.; Graner, A.; Sorrells, M.E. Genic microsatellite markers in plants: Features and applications. Trends Biotechnol. 2005, 23, 48–55. [Google Scholar] [CrossRef]
- Nachimuthu, V.V.; Muthurajan, R.; Duraialaguraja, S.; Sivakami, R.; Pandian, B.A.; Ponniah, G.; Gunasekaran, K.; Swaminathan, M.; Suji, K.K.; Sabariappan, R. Analysis of population structure and genetic diversity in rice germplasm using SSR markers: An initiative towards association mapping of agronomic traits in Oryza Sativa. Rice 2015, 8, 30. [Google Scholar] [CrossRef] [Green Version]
- Pan, L.; Huang, T.; Yang, Z.F.; Tang, L.; Cheng, Y.J.; Wang, J.P.; Ma, X.; Zhang, X.Q. EST-SSR marker characterization based on RNA-sequencing of Lolium multiflorum and cross transferability to related species. Mol. Breed. 2018, 38, 80. [Google Scholar] [CrossRef]
- Qu, J.B.; Huang, C.Y.; Zhang, J.X. Genome-wide functional analysis of SSR for an edible mushroom Pleurotus ostreatus. Gene 2016, 575, 524–530. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.C.; Korol, A.B.; Fahima, T.; Nevo, E. Microsatellites within genes: Structure, function, and evolution. Mol. Biol. Evol. 2004, 21, 991–1007. [Google Scholar] [CrossRef]
- Usdin, K. The biological effects of simple tandem repeats: Lessons from the repeat expansion diseases. Genome Res. 2008, 18, 1011–1019. [Google Scholar] [CrossRef] [Green Version]
- Potaman, V.N.; Bissler, J.J.; Hashem, V.I.; Oussatcheva, E.A.; Lu, L.; Shlyakhtenko, L.S.; Lyubchenko, Y.L.; Matsuura, T.; Ashizawa, T.; Leffak, M.; et al. Unpaired structures in SCA10 (ATTCT)n·(AGAAT)n repeats. J. Mol. Biol. 2003, 326, 1095–1111. [Google Scholar] [CrossRef]
- Vetcher, A.A.; Napierala, M.; Wells, R.D. Sticky DNA: Effect of the polypurine·polypyrimidine sequence. J. Biol. Chem. 2002, 227, 39228–39234. [Google Scholar] [CrossRef] [Green Version]
- Potaman, V.N.; Oussatcheva, E.A.; Lyubchenko, Y.L.; Shlyakhtenko, L.S.; Bidichandani, S.I.; Ashizawa, T.; Sinden, R.R. Length-dependent structure formation in Friedreich ataxia (GAA)n·(TTC)n repeats at neutral pH. Nucleic Acids Res. 2004, 32, 1224–1231. [Google Scholar] [CrossRef] [Green Version]
- Kashi, Y.; King, D.G. Simple sequence repeats as advantageous mutators in evolution. Trends Genet. 2006, 22, 253–259. [Google Scholar] [CrossRef] [Green Version]
- Rocha, E.P.C.; Matic, I.; Taddei, F. Over-representation of repeats in stress response genes: A strategy to increase versatility under stressful conditions? Nucleic Acids Res. 2002, 30, 1886–1894. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.C.; Korol, A.B.; Fahima, T.; Beiles, A.; Nevo, E. Microsatellites: Genomic distribution, putative functions and mutational mechanisms: A review. Mol. Ecol. 2002, 11, 2453–2465. [Google Scholar] [CrossRef]
- Ekblom, R.; Galindo, J. Applications of next generation sequencing in molecular ecology of non-model organisms. Heredity 2011, 107, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Gayral, P.; Weinert, L.; Chiari, Y.; Tsagkogeorga, G.; Ballenghien, M.; Galtier, N. Next-generation sequencing of transcriptomes: A guide to RNA isolation in nonmodel animals. Mol. Ecol. Resour. 2011, 11, 650–661. [Google Scholar] [CrossRef]
- Xia, E.H.; Yao, Q.Y.; Zhang, H.B.; Jiang, J.J.; Zhang, L.P.; Gao, L.Z. CandiSSR: An efficient pipeline used for identifying candidate polymorphic SSRs based on multiple assembled sequences. Front. Plant Sci. 2016, 6, 1171. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.Y.; Zhang, M.Y.; Hu, Y.M.; Zhuang, X.; Xu, W.Q.; Li, P.F.; Wang, Z.S. Mining and characterization of novel EST-SSR markers of Parrotia subaequalis (Hamamelidaceae) from the first Illumina-based transcriptome datasets. PLoS ONE 2019, 14, e0215874. [Google Scholar] [CrossRef]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3-new capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.J.; Muse, S.V. PowerMarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef] [Green Version]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl. Acids. Symp. Ser. 1999, 41, 95–98. [Google Scholar] [CrossRef]
- Thiel, T.; Michalek, W.; Varshney, R.K.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef]
- Gao, L.F.; Tang, J.F.; Li, H.W.; Jia, J.Z. Analysis of microsatellites in major crops assessed by computational and experimental approaches. Mol. Breed. 2003, 12, 245–261. [Google Scholar] [CrossRef]
- Temnykh, S.; DeClerck, G.; Lukashova, A.; Lipovich, L.; Cartinhour, S.; McCouch, S. Computational and experimental analysis of microsatellites in rice (Oryza sativa L.): Frequency, length variation, transposon associations, and genetic marker potential. Genome Res. 2001, 11, 1441–1452. [Google Scholar] [CrossRef] [Green Version]
- Xia, E.H.; Li, F.D.; Tong, W.; Li, P.H.; Wu, Q.; Zhao, H.J.; Ge, R.P.; Li, R.P.; Li, Y.Y.; Zhang, Z.Z.; et al. Tea plant information archive: A comprehensive genomics and bioinformatics platform for tea plant. Plant Biotechnol. J. 2019, 17, 1938–1953. [Google Scholar] [CrossRef]
- Morgante, M.; Hanafey, M.; Powell, W. Microsatellites are preferentially associated with nonrepetitive DNA in plant genomes. Nat. Genet. 2002, 30, 194–200. [Google Scholar] [CrossRef]
- Cordeiro, G.M.; Casu, R.; McIntyre, C.L.; Manners, J.M.; Henry, R.J. Microsatellite markers from sugarcane (Saccharum spp.) ESTs cross transferable to erianthus and sorghum. Plant Sci. 2001, 160, 1115–1123. [Google Scholar] [CrossRef]
- Metzgar, D.; Bytof, J.; Wills, C. Selection against frameshift mutations limits microsatellite expansion in coding DNA. Genome Res. 2000, 10, 72–80. [Google Scholar] [CrossRef]
- Lawson, M.J.; Zhang, L.Q. Distinct patterns of SSR distribution in the Arabidopsis thaliana and rice genomes. Genome Biol. 2006, 7, R14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonah, H.; Deshmukh, R.K.; Sharma, A.; Singh, V.P.; Gupta, D.K.; Gacche, R.N.; Rana, J.C.; Singh, N.K.; Sharma, T.R. Genome-wide distribution and organization of microsatellites in plants: An insight into marker development in Brachypodium. PLoS ONE 2011, 6, e21298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yotoko, K.S.C.; Dornelas, M.C.; Togni, P.D.; Fonseca, T.C.; Salzano, F.M.; Bonatto, S.L.; Freitas, L.B. Does variation in genome sizes reflect adaptive or neutral processes? New clues from Passiflora. PLoS ONE 2011, 6, e18212. [Google Scholar] [CrossRef] [Green Version]
- Gao, C.H.; Ren, X.D.; Mason, A.S.; Li, J.N.; Wang, W.; Xiao, M.L.; Fu, D.H. Revisiting an important component of plant genomes: Microsatellites. Funct. Plant Biol. 2013, 40, 645–661. [Google Scholar] [CrossRef]
- Bao, J.S.; Corke, H.; Sun, M. Microsatellites in starch-synthesizing genes in relation to starch physicochemical properties in waxy rice (Oryza sativa L.). Theor. Appl. Genet. 2002, 105, 898–905. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.Z.; Pan, W.; Xu, B.H.; Li, B.L.; Zhang, D.Q. Polymorphic simple sequence repeat (SSR) loci within cellulose synthase (PtoCesA) genes are associated with growth and wood properties in Populus tomentosa. New Phytol. 2013, 197, 763–776. [Google Scholar] [CrossRef]
- Ranum, L.P.W.; Cooper, T.A. RNA-mediated neuromuscular disorders. Annu. Rev. Neurosci. 2006, 29, 259–277. [Google Scholar] [CrossRef]
- Ranum, L.P.W.; Day, J.W. Myotonic dystrophy: Clinical and molecular parallels between myotonic dystrophy type 1 and type 2. Curr. Neurol. Neurosci. Rep. 2002, 2, 465–470. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | Longitude (E) | Latitude (N) | Altitude (m) | Habitats | Soil Water Content (%) * |
|---|---|---|---|---|---|
| D1 | 106°53′43″ | 39°21′57″ | 1212.5 | Foothills | 5.48 |
| D2 | 106°53′52″ | 39°22′30″ | 1185.6 | Tableland | 5.24 |
| D3 | 106°53′31″ | 39°29′52″ | 1216.5 | Foothills | 4.93 |
| D4 | 107°05′45″ | 40°14′58″ | 1150.5 | Tableland | 3.89 |
| D5 | 106°52′07″ | 40°08′03″ | 1036.9 | Sandy Land | 2.43 |
| D6 | 106°55′07″ | 40°08′02″ | 1049.5 | Piedmont Plain | 2.43 |
| CR (No.) | CN (nt) | Q20 (%) | GC (%) | Ug (No.) | ML (bp) | N50 (bp) | |
|---|---|---|---|---|---|---|---|
| D1 | 53,284,254 | 7,992,638,100 | 97.22% | 43.76% | 80,409 | 791 | 1499 |
| D2 | 55,473,386 | 8,321,007,900 | 97.19% | 43.99% | 80,829 | 824 | 1579 |
| D3 | 64,363,372 | 9,654,505,800 | 96.88% | 45.15% | 77,641 | 786 | 1516 |
| D4 | 52,017,954 | 7,802,693,100 | 96.94% | 44.27% | 84,673 | 851 | 1600 |
| D5 | 63,352,430 | 9,502,864,500 | 97.36% | 43.72% | 92,301 | 794 | 1534 |
| D6 | 54,704,688 | 8,205,703,200 | 97.31% | 44.54% | 73,977 | 788 | 1489 |
| All | 119,603 | 1098 | 1843 |
| Gene ID | PS (5′–3′) | RM | AS | Ta (°C) | PF | NA | HO | HE | PIC |
|---|---|---|---|---|---|---|---|---|---|
| CL3279.Ct4 | F: GTAGTACTACTGCTGCATCGTATCCT R: CAACCCTATCTTCATCATCATCG | TGC | 101–113 | 54 | protoporphyrinogen oxidase, Vitis vinifera | 4 | 0.436 | 0.510 | 0.458 |
| CL4993.Ct1 | F: ACTCCTCTCATCCATCCATTAAG R: GGAGTTTAACGCTGTCATTGTG | TC | 102–114 | 55 | - | 6 | 0.500 | 0.695 | 0.644 |
| CL6111.Ct3 | F: TGGAGTCTGAAGGCAGTGAG R: ACTTGAACTTCTTGATTCCACC | TAG | 118–124 | 55 | - | 3 | 0.167 | 0.590 | 0.508 |
| CL8609.Ct3 | F: GCATTAGAGGAGCGAATCGAAG R: GCCTCGCTTCTCATTTCTCAAC | GA | 164–174 | 59 | - | 6 | 0.417 | 0.617 | 0.584 |
| CL8025.Ct4 | F: CATCGCCGCCTTTCATAGAC R: GACGCTTAGAATTGGAAGATGATG | TC | 175–181 | 55 | cyclin-dependent kinase G-2-like, Citrus sinensis | 3 | 0.354 | 0.472 | 0.422 |
| Ug20261 | F: GGGGAAAGATGCTGTTATGGAG R: TAGCATCCGAGCCACTACCAC | AGG | 186–195 | 59 | - | 4 | 0.500 | 0.625 | 0.552 |
| CL6305.Ct2 | F: CGCTTGCTTTAACGACGAACC R: TGTGGTGGGTCGGATGATGTT | GCA | 176–188 | 55 | serine/threonine-protein kinase RIO1-like isoform X1, Citrus sinensis | 5 | 0.279 | 0.686 | 0.621 |
| CL7264.Ct1 | F: GTTGTGGCGGCGTAGTTTATG R: AACTCGCAAACCAAGAGCATAAC | TG | 193–205 | 58 | - | 5 | 0.333 | 0.774 | 0.728 |
| CL9244.Ct2 | F: CTGAGATTTGTTGGTGGGTTTG R: CCAGTATCTCCGAACCACCTCT | AGG | 373–382 | 56 | Glutaredoxin 4 isoform 1, Theobroma cacao | 3 | 0.438 | 0.551 | 0.457 |
| CL8609.Ct2 | F: GGAGCTGAATTAGAGCATTAGAGG R: GAAATCTCTCTTGTTCAATCCACC | GA | 202–212 | 55 | - | 6 | 0.458 | 0.663 | 0.626 |
| Ug13288 | F: AGCATTACATTATCCCTTCCTCAC R: CAGAGACGGTGTCGTATTGGA | TAA | 240–258 | 55 | peptide chain release factor 1-like, Glycine max | 5 | 0.533 | 0.710 | 0.649 |
| Ug19883 | F: GAGTTATGAATGACGCTACACGAG R: GCCTGCTTTGCGTTTCTTC | TGC | 351–360 | 55 | - | 3 | 0.319 | 0.274 | 0.240 |
| Ug13288 | F: CATCGCCGCCTTTCATAGAC R: GACGCTTAGAATTGGAAGATGATG | TAA | 242–263 | 55 | peptide chain release factor 1-like, Glycine max | 7 | 0.575 | 0.758 | 0.713 |
| Ug31697 | F: CAACAGAAAGCACCAACCCAG R: GCATCCACCCTGTTCAGCAT | CTC | 241–253 | 60 | - | 5 | 0.729 | 0.770 | 0.722 |
| Ug4409 | F: CATCGGCCTCTGCTCATACAC R: CGCTTCAGGCTCTCATATTCAG | TCA | 272–275 | 55 | - | 2 | 0.292 | 0.399 | 0.317 |
| CL12118.Ct3 | F: CAGAGAGAATAATAGCAGCCATAG R: CCCAAGCATCCAACAATAAC | AG | 289–299 | 55 | Ethylene-responsive transcription factor, Morus notabilis | 5 | 0.313 | 0.349 | 0.331 |
| Ug31697 | F: GGAGGTGATGGAGAAGGTGAGA R: CAACCCATCACAATCTCACATCA | GA | 302–312 | 59 | - | 4 | 0.458 | 0.527 | 0.459 |
| Mean | 4.471 | 0.418 | 0.587 | 0.531 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dang, Z.; Huang, L.; Jia, Y.; Lockhart, P.J.; Fong, Y.; Tian, Y. Identification of Genic SSRs Provide a Perspective for Studying Environmental Adaptation in the Endemic Shrub Tetraena mongolica. Genes 2020, 11, 322. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030322
Dang Z, Huang L, Jia Y, Lockhart PJ, Fong Y, Tian Y. Identification of Genic SSRs Provide a Perspective for Studying Environmental Adaptation in the Endemic Shrub Tetraena mongolica. Genes. 2020; 11(3):322. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030322
Chicago/Turabian StyleDang, Zhenhua, Lei Huang, Yuanyuan Jia, Peter J. Lockhart, Yang Fong, and Yunyun Tian. 2020. "Identification of Genic SSRs Provide a Perspective for Studying Environmental Adaptation in the Endemic Shrub Tetraena mongolica" Genes 11, no. 3: 322. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11030322