
Mimicked Mixing-Induced Heterogeneities of Industrial Bioreactors Stimulate Long-Lasting Adaption Programs in Ethanol-Producing Yeasts




Abstract
:1. Introduction
2. Materials and Methods
2.1. Strain, Maintenance and Seed Culture
2.2. Chemostat Setup
2.3. Stimulus–Response Design
2.4. Analytical Procedures
2.5. Processing of Next-Generation Sequencing Samples
2.6. Gene Expression Analysis
3. Results
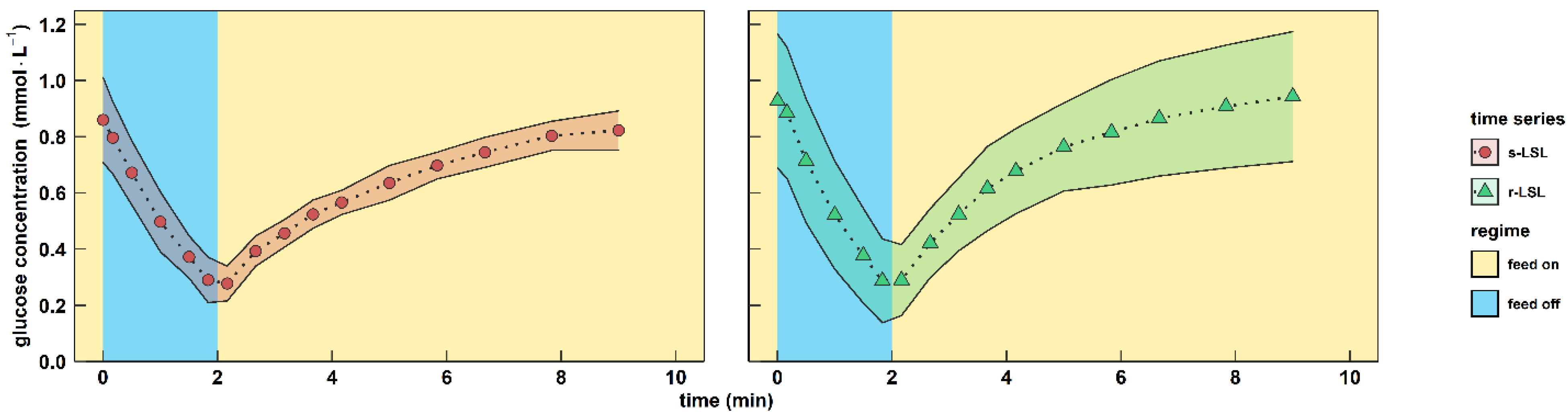
3.1. Characterization of Extracellular Glucose Profile
3.2. The Physiology of Dynamic and Steady-State Adaptation toward Short-Lived Famine Stimuli
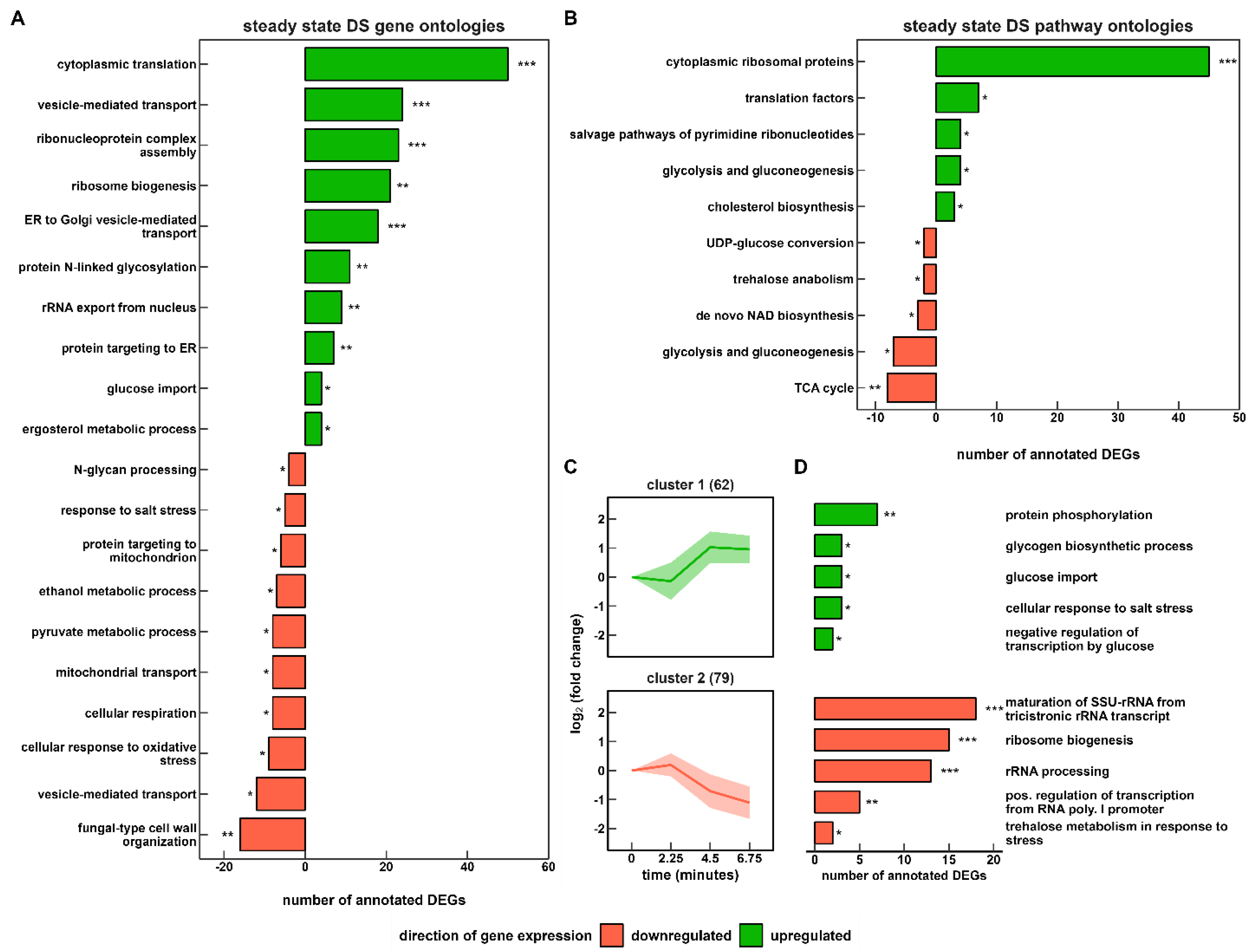
3.3. The Transcriptional Response to Single Starvation Exposure (s-LSL)
3.4. The Transcriptional Response to r-LSL
3.5. Comparing Transcriptional Responses of s-LSL and r-LSL
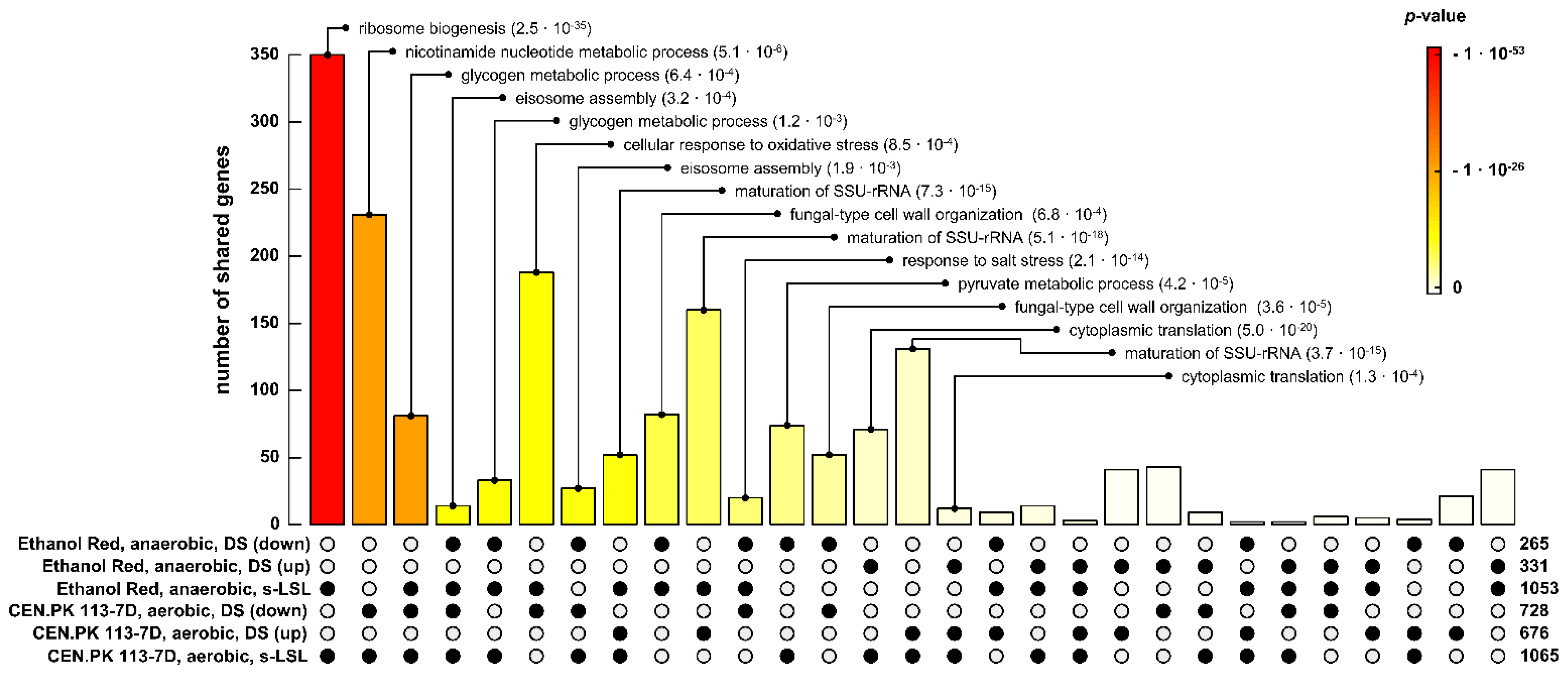
3.6. Comparing Different Strain Backgrounds and Production Scenarios to the Same Stimulus
4. Discussion
4.1. Fluctuating Glucose Supply—Threat or Not?
4.2. Not a Threat, but Still a New Habitat—How LSL Transitions Induce a New Growth Phenotype
4.3. Take-Away Message for Industrial Strain Engineers
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- López-Maury, L.; Marguerat, S.; Bähler, J. Tuning Gene Expression to Changing Environments: From Rapid Responses to Evolutionary Adaptation. Nat. Rev. Genet. 2008, 9, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Skoneczny, M. (Ed.) Stress Response Mechanisms in Fungi; Springer: Berlin/Heidelberg, Germany, 2018. [Google Scholar] [CrossRef]
- Boutte, C.C.; Crosson, S. Bacterial Lifestyle Shapes Stringent Response Activation. Trends Microbiol. 2013, 21, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Gasch, A.P. The Environmental Stress Response: A Common Yeast Response to Diverse Environmental Stresses. Yeast Stress Responses 2007, 1, 11–70. [Google Scholar] [CrossRef]
- Takors, R. Editorial: How Can We Ensure the Successful Transfer from Lab- to Large-Scale Production? Eng. Life Sci. 2016, 16, 587. [Google Scholar] [CrossRef]
- Crater, J.S.; Lievense, J.C. Scale-up of Industrial Microbial Processes. FEMS Microbiol. Lett. 2018, 365, fny138. [Google Scholar] [CrossRef]
- Enfors, S.O.; Jahic, M.; Rozkov, A.; Xu, B.; Hecker, M.; Jürgen, B.; Krüger, E.; Schweder, T.; Hamer, G.; O’Beirne, D.; et al. Physiological Responses to Mixing in Large Scale Bioreactors. J. Biotechnol. 2001, 85, 175–185. [Google Scholar] [CrossRef]
- Fowler, J.D.; Dunlop, E.H. Effects of Reactant Heterogeneity and Mixing on Catabolite Repression in Cultures of Saccharomyces cerevisiae. Biotechnol. Bioeng. 1989, 33, 1039–1046. [Google Scholar] [CrossRef]
- Wehrs, M.; Tanjore, D.; Eng, T.; Lievense, J.; Pray, T.R.; Mukhopadhyay, A. Engineering Robust Production Microbes for Large-Scale Cultivation. Trends Microbiol. 2019, 27, 524–537. [Google Scholar] [CrossRef]
- George, S.; Larsson, G.; Olsson, K.; Enfors, S.-O. Comparison of the Baker’s Yeast Process Performance in Laboratory and Production Scale. Bioprocess Eng. 1998, 18, 135–142. [Google Scholar] [CrossRef]
- Sarkizi Shams Hajian, C.; Haringa, C.; Noorman, H.; Takors, R. Predicting By-Product Gradients of Baker ’ s Yeast Production at Industrial Scale: A Practical Simulation Approach. Processes 2020, 8, 1554. [Google Scholar] [CrossRef]
- Haringa, C.; Deshmukh, A.T.; Mudde, R.F.; Noorman, H. Euler-Lagrange Analysis towards Representative down-Scaling of a 22 M3 Aerobic S. cerevisiae Fermentation. Chem. Eng. Sci. 2017, 170, 653–669. [Google Scholar] [CrossRef]
- Delvigne, F.; Goffin, P. Microbial Heterogeneity Affects Bioprocess Robustness: Dynamic Single-Cell Analysis Contributes to Understanding of Microbial Populations. Biotechnol. J. 2014, 9, 61–72. [Google Scholar] [CrossRef]
- Gutin, J.; Joseph-Strauss, D.; Sadeh, A.; Shalom, E.; Friedman, N. Genetic Screen of the Yeast Environmental Stress Response Dynamics Uncovers Distinct Regulatory Phases. Mol. Syst. Biol. 2019, 15, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Minden, S.; Aniolek, M.; Sarkizi, C.; Hajian, S.; Teleki, A.; Zerrer, T.; Delvigne, F.; van Gulik, W.; Deshmukh, A.; Noorman, H.; et al. Monitoring Intracellular Metabolite Dynamics in Saccharomyces cerevisiae during Industrially Relevant Famine Stimuli. Metabolites 2022, 12, 263. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.; Täuber, S.; Stute, B.; Grünberger, A.; von Lieres, E. Microfluidic Reproduction of Dynamic Bioreactor Environment Based on Computational Lifelines. Front. Chem. Eng. 2022, 4, 27. [Google Scholar] [CrossRef]
- Kuschel, M.; Takors, R. Simulated Oxygen and Glucose Gradients as a Prerequisite for Predicting Industrial Scale Performance a Priori. Biotechnol. Bioeng. 2020, 117, 2760–2770. [Google Scholar] [CrossRef] [PubMed]
- Nadal-Rey, G.; McClure, D.D.; Kavanagh, J.M.; Cassells, B.; Cornelissen, S.; Fletcher, D.F.; Gernaey, K.V. Development of Dynamic Compartment Models for Industrial Aerobic Fed-Batch Fermentation Processes. Chem. Eng. J. 2021, 420, 130402. [Google Scholar] [CrossRef]
- Löffler, M.; Simen, J.D.; Jäger, G.; Schäferhoff, K.; Freund, A.; Takors, R. Engineering, E. coli for Large-Scale Production—Strategies Considering ATP Expenses and Transcriptional Responses. Metab. Eng. 2016, 38, 73–85. [Google Scholar] [CrossRef]
- Ankenbauer, A.; Schäfer, R.A.; Viegas, S.C.; Pobre, V.; Voß, B.; Arraiano, C.M.; Takors, R. Pseudomonas Putida KT2440 Is Naturally Endowed to Withstand Industrial-Scale Stress Conditions. Microb. Biotechnol. 2020, 13, 1145–1161. [Google Scholar] [CrossRef]
- Michalowski, A.; Siemann-Herzberg, M.; Takors, R. Escherichia coli HGT: Engineered for High Glucose Throughput Even under Slowly Growing or Resting Conditions. Metab. Eng. 2017, 40, 93–103. [Google Scholar] [CrossRef]
- Ziegler, M.; Zieringer, J.; Döring, C.L.; Paul, L.; Schaal, C.; Takors, R. Engineering of a Robust Escherichia coli Chassis and Exploitation for Large-Scale Production Processes. Metab. Eng. 2021, 67, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Minden, S.; Aniolek, M.; Noorman, H.; Takors, R. Performing in Spite of Starvation: How Saccharomyces cerevisiae Maintains Robust Growth When Facing Famine Zones in Industrial Bioreactors. Microb. Biotechnol. 2023, 16, 148–168. [Google Scholar] [CrossRef]
- Nijkamp, J.F.; van den Broek, M.; Datema, E.; de Kok, S.; Bosman, L.; Luttik, M.A.H.; Dalen, J.P.; Vongsangnak, W.; Nielsen, J.; Heijne, W.H.M.; et al. De Novo Sequencing, Assembly and Analysis of the Genome of the Laboratory Strain Saccharomyces cerevisiae CEN.PK113-7D, a Model for Modern Industrial Biotechnology. Microb. Cell Factories 2012, 11, 36. [Google Scholar] [CrossRef] [PubMed]
- Parrou, J.L.; Francois, J. A Simplified Procedure for a Rapid and Reliable Assay of Both Glycogen and Trehalose in Whole Yeast Cells. Anal. Biochem. 1997, 248, 186–188. [Google Scholar] [CrossRef]
- Sasano, Y.; Kariya, T.; Usugi, S.; Sugiyama, M.; Harashima, S. Molecular Breeding of Saccharomyces cerevisiae with High RNA Content by Harnessing Essential Ribosomal RNA Transcription Regulator. AMB Express 2017, 7, 32. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, J. Development, Characterization, and Application of a Novel Scale-Down Apparatus for the Investigation of the Scale-Up Dependent CO2/HCO3—Stimulus in Corynebacterium Glutamicum. Ph.D. Thesis, University of Stuttgart, Stuttgart, Germany, 2015. Volume 278. [Google Scholar] [CrossRef]
- Löser, C.; Schröder, A.; Deponte, S.; Bley, T. Balancing the Ethanol Formation in Continuous Bioreactors with Ethanol Stripping. Eng. Life Sci. 2005, 5, 325–332. [Google Scholar] [CrossRef]
- Afgan, E.; Baker, D.; Batut, B.; Van Den Beek, M.; Bouvier, D.; Ech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy Platform for Accessible, Reproducible and Collaborative Biomedical Analyses: 2018 Update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef]
- RTSF–Michigan State University. FastQC Tutorial & FAQ. Available online: https://rtsf.natsci.msu.edu/genomics/tech-notes/fastqc-tutorial-and-faq/ (accessed on 18 May 2022).
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, L. TopHat2: Accurate Alignment of Transcriptomes in the Presence of Insertions, Deletions and Gene Fusions. Genome Biol. 2013, 14, 957–961. [Google Scholar] [CrossRef]
- Gronchi, N.; De Bernardini, N.; Cripwell, R.A.; Treu, L.; Campanaro, S.; Basaglia, M.; Foulquié-Moreno, M.R.; Thevelein, J.M.; Van Zyl, W.H.; Favaro, L.; et al. Natural Saccharomyces cerevisiae Strain Reveals Peculiar Genomic Traits for Starch-to-Bioethanol Production: The Design of an Amylolytic Consolidated Bioprocessing Yeast. Front. Microbiol. 2022, 12, 4226. [Google Scholar] [CrossRef]
- Howe, K.L.; Achuthan, P.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Ridwan Amode, M.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J.; et al. Ensembl 2021. Nucleic Acids Res. 2021, 49, D884–D891. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and Collaborative HTML5 Gene List Enrichment Analysis Tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Diaz, J.E.L.; Flamholz, Z.N.; Keenan, A.B.; Lachmann, A.; Wojciechowicz, M.L.; Cagan, R.L.; Ma’ayan, A. ModEnrichr: A Suite of Gene Set Enrichment Analysis Tools for Model Organisms. Nucleic Acids Res. 2019, 47, W183–W190. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Friedman, M.S.; Shedden, K.; Hankenson, K.D.; Woolf, P.J. GAGE: Generally Applicable Gene Set Enrichment for Pathway Analysis. BMC Bioinform. 2009, 10, 161. [Google Scholar] [CrossRef]
- Monteiro, P.T.; Oliveira, J.; Pais, P.; Antunes, M.; Palma, M.; Cavalheiro, M.; Galocha, M.; Godinho, C.P.; Martins, L.C.; Bourbon, N.; et al. YEASTRACT+: A Portal for Cross-Species Comparative Genomics of Transcription Regulation in Yeasts. Nucleic Acids Res. 2020, 48, D642–D649. [Google Scholar] [CrossRef]
- Wang, M.; Zhao, Y.; Zhang, B. Efficient Test and Visualization of Multi-Set Intersections. Sci. Rep. 2015, 5, 16923. [Google Scholar] [CrossRef]
- Boender, L.G.M.; De Hulster, E.A.F.; van Maris, A.J.A.; Dalen, J.P.; Pronk, J.T. Quantitative Physiology of Saccharomyces cerevisiae at Near-Zero Specific Growth Rates. Appl. Environ. Microbiol. 2009, 75, 5607–5614. [Google Scholar] [CrossRef]
- Warner, J.R. The Economics of Ribosome Biosynthesis in Yeast. Trends Biochem. Sci. 1999, 24, 437–440. [Google Scholar] [CrossRef]
- Wang, G.; Wu, B.; Zhao, J.; Haringa, C.; Xia, J.; Chu, J.; Zhuang, Y.; Zhang, S.; Heijnen, J.J.; van Gulik, W.; et al. Power Input Effects on Degeneration in Prolonged Penicillin Chemostat Cultures: A Systems Analysis at Flux, Residual Glucose, Metabolite, and Transcript Levels. Biotechnol. Bioeng. 2018, 115, 114–125. [Google Scholar] [CrossRef]
- Nissen, T.L.; Schulze, U.; Nielsen, J.; Villadsen, J. Flux Distributions in Anaerobic, Glucose-Limited Continuous Cultures of Saccharomyces cerevisiae. Microbiology 1997, 143, 203–218. [Google Scholar] [CrossRef] [PubMed]
- Pais, T.M.; Foulquié-Moreno, M.R.; Hubmann, G.; Duitama, J.; Swinnen, S.; Goovaerts, A.; Yang, Y.; Dumortier, F.; Thevelein, J.M. Comparative Polygenic Analysis of Maximal Ethanol Accumulation Capacity and Tolerance to High Ethanol Levels of Cell Proliferation in Yeast. PLoS Genet. 2013, 9, e1003548. [Google Scholar] [CrossRef] [PubMed]
- Steensels, J.; Snoek, T.; Meersman, E.; Nicolino, M.P.; Voordeckers, K.; Verstrepen, K.J. Improving Industrial Yeast Strains: Exploiting Natural and Artificial Diversity. FEMS Microbiol. Rev. 2014, 38, 947–995. [Google Scholar] [CrossRef]
- HERSKOWITZ, I. Life Cycle of the Budding Yeast Saccharomyces cerevisiae IRA. Microbiol. Rev. 1988, 52, 536–553. [Google Scholar] [CrossRef] [PubMed]
- Cherry, J.M.; Hong, E.L.; Amundsen, C.; Balakrishnan, R.; Binkley, G.; Chan, E.T.; Christie, K.R.; Costanzo, M.C.; Dwight, S.S.; Engel, S.R.; et al. Saccharomyces Genome Database: The genomics resource of budding yeast. Nucleic Acids Res. 2012, 40, D700–D705. [Google Scholar] [CrossRef] [PubMed]
- Lang, M.J.; Martinez-Marquez, J.Y.; Prosser, D.C.; Ganser, L.R.; Buelto, D.; Wendland, B.; Duncan, M.C. Glucose Starvation Inhibits Autophagy via Vacuolar Hydrolysis and Induces Plasma Membrane Internalization by Down-Regulating Recycling. J. Biol. Chem. 2014, 289, 16736–16747. [Google Scholar] [CrossRef]
- Thevelein, J.M. Regulation of Trehalose Mobilization in Fungi. Microbiol. Rev. 1984, 48, 42–59. [Google Scholar] [CrossRef]
- Kohlwein, S.D.; Veenhuis, M.; van der Klei, I.J. Lipid Droplets and Peroxisomes: Key Players in Cellular Lipid Homeostasis or a Matter of Fat-Store’em up or Burn’em Down. Genetics 2013, 193, 1–50. [Google Scholar] [CrossRef]
- Herrero, P.; Galíndez, J.; Ruiz, N.; Martínez-Campa, C.; Moreno, F. Transcriptional Regulation of the Saccharomyces cerevisiae HXK1, HXK2 and GLK1 Genes. Yeast 1995, 11, 137–144. [Google Scholar] [CrossRef]
- Castelli, L.M.; Lui, J.; Campbell, S.G.; Rowe, W.; Zeef, L.A.H.; Holmes, L.E.A.; Hoyle, N.P.; Bone, J.; Selley, J.N.; Sims, P.F.G.; et al. Glucose Depletion Inhibits Translation Initiation via EIF4A Loss and Subsequent 48S Preinitiation Complex Accumulation, While the Pentose Phosphate Pathway Is Coordinately up-Regulated. Mol. Biol. Cell 2011, 22, 3379–3393. [Google Scholar] [CrossRef] [PubMed]
- Antonin, W.; Meyer, H.-A.; Hartmann, E. Interactions between Spc2p and Other Components of the Endoplasmic Reticulum Translocation Sites of the YeastSaccharomyces cerevisiae. J. Biol. Chem. 2000, 275, 34068–34072. [Google Scholar] [CrossRef] [PubMed]
- Sutterlin, C.; Doering, T.L.; Schimmoller, F.; Schroder, S.; Riezman, H. Specific Requirements for the ER to Golgi Transport of GPI-Anchored Proteins in Yeast. J. Cell Sci. 1997, 110, 2703–2714. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, F.; Ludovicio, P.; Leao, C. Sugar Metabolism in Yeasts: An Overview of Aerobic and Anaerobic Glucose Catabolism. Biodivers. Ecophysiol. Yeasts 2006, 62, 101–122. [Google Scholar]
- Helbig, A.O.; De Groot, M.J.L.; Van Gestel, R.A.; Mohammed, S.; De Hulster, E.A.F.; Luttik, M.A.H.; Daran-Lapujade, P.; Pronk, J.T.; Heck, A.J.R.; Slijper, M. A Three-Way Proteomics Strategy Allows Differential Analysis of Yeast Mitochondrial Membrane Protein Complexes under Anaerobic and Aerobic Conditions. Proteomics 2009, 9, 4787–4798. [Google Scholar] [CrossRef]
- David, P.S.; Poyton, R.O. Effects of a Transition from Normoxia to Anoxia on Yeast Cytochrome c Oxidase and the Mitochondrial Respiratory Chain: Implications for Hypoxic Gene Induction. Biochim. Biophys. Acta Bioenerg. 2005, 1709, 169–180. [Google Scholar] [CrossRef]
- Sazegari, S.; Niazi, A.; Zinati, Z.; Eskandari, M.H. Mining Transcriptomic Data to Identify Saccharomyces cerevisiae Signatures Related to Improved and Repressed Ethanol Production under Fermentation. PLoS ONE 2022, 17, e259476. [Google Scholar] [CrossRef]
- Fazio, A.; Jewett, M.C.; Daran-Lapujade, P.; Mustacchi, R.; Usaite, R.; Pronk, J.T.; Workman, C.T.; Nielsen, J. Transcription Factor Control of Growth Rate Dependent Genes in Saccharomyces cerevisiae: A Three Factor Design. BMC Genom. 2008, 9, 341. [Google Scholar] [CrossRef] [PubMed]
- Gasch, A.P.; Yu, F.B.; Hose, J.; Escalante, L.E.; Place, M.; Bacher, R.; Kanbar, J.; Ciobanu, D.; Sandor, L.; Grigoriev, I.V.; et al. Single-Cell RNA Sequencing Reveals Intrinsic and Extrinsic Regulatory Heterogeneity in Yeast Responding to Stress. PLoS Biol. 2017, 15, e2004050. [Google Scholar] [CrossRef]
- Gutin, J.; Sadeh, A.; Rahat, A.; Aharoni, A.; Friedman, N. Condition-specific Genetic Interaction Maps Reveal Crosstalk between the CAMP/PKA and the HOG MAPK Pathways in the Activation of the General Stress Response. Mol. Syst. Biol. 2015, 11, 829. [Google Scholar] [CrossRef]
- Hohmann, S. Osmotic Stress Signaling and Osmoadaptation in Yeasts. Microbiol. Mol. Biol. Rev. 2002, 66, 300–372. [Google Scholar] [CrossRef]
- Hanlon, S.E.; Rizzo, J.M.; Tatomer, D.C.; Lieb, J.D.; Buck, M.J. The Stress Response Factors Yap6, Cin5, Phd1, and Skn7 Direct Targeting of the Conserved Co-Repressor Tup1-Ssn6 in S. cerevisiae. PLoS ONE 2011, 6, e29590. [Google Scholar] [CrossRef]
- Bardwell, L.; Cook, J.G.; Voora, D.; Baggott, D.M.; Martinez, A.R.; Thorner, J. Repression of Yeast Ste12 Transcription Factor by Direct Binding of Unphosphorylated Kss1 MAPK and Its Regulation by the Ste7 MEK. Genes Dev. 1998, 12, 2887–2898. [Google Scholar] [CrossRef]
- Ratnakumar, S.; Kacherovsky, N.; Arms, E.; Young, E.T. Snf1 Controls the Activity of Adr1 through Dephosphorylation of Ser230. Genetics 2009, 182, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Livas, D.; Almering, M.J.H.; Daran, J.M.; Pronk, J.T.; Gancedo, J.M. Transcriptional Responses to Glucose in Saccharomyces cerevisiae Strains Lacking a Functional Protein Kinase A. BMC Genomics 2011, 12, 405. [Google Scholar] [CrossRef] [PubMed]
- Iyer, V.R.; Horak, C.E.; Scafe, C.S.; Botstein, D.; Snyder, M.; Brown, P.O. Genomic Binding Sites of the Yeast Cell-Cycle Transcription Factors SBF and MBF. Nature 2001, 409, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Murakami, H.; Aiba, H.; Nakanishi, M.; Murakami-Tonami, Y. Regulation of Yeast Forkhead Transcription Factors and FoxM1 by Cyclin-Dependent and Polo-like Kinases. Cell Cycle 2010, 9, 3233–3242. [Google Scholar] [CrossRef]
- Tsuchiya, D.; Yang, Y.; Lacefield, S. Positive Feedback of NDT80 Expression Ensures Irreversible Meiotic Commitment in Budding Yeast. PLoS Genet. 2014, 10, e1004398. [Google Scholar] [CrossRef] [PubMed]
- Huber, A.; French, S.L.; Tekotte, H.; Yerlikaya, S.; Stahl, M.; Perepelkina, M.P.; Tyers, M.; Rougemont, J.; Beyer, A.L.; Loewith, R. Sch9 Regulates Ribosome Biogenesis via Stb3, Dot6 and Tod6 and the Histone Deacetylase Complex RPD3L. EMBO J. 2011, 30, 3052–3064. [Google Scholar] [CrossRef]
- Lempiäinen, H.; Shore, D. Growth Control and Ribosome Biogenesis. Curr. Opin. Cell Biol. 2009, 21, 855–863. [Google Scholar] [CrossRef]
- Shenhar, G.; Kassir, Y. A Positive Regulator of Mitosis, Sok2, Functions as a Negative Regulator of Meiosis in Saccharomyces cerevisiae. Mol. Cell. Biol. 2001, 21, 1603–1612. [Google Scholar] [CrossRef] [PubMed]
- Charizanis, C.; Juhnke, H.; Krems, B.; Entian, K.-D. The Oxidative Stress Response Mediated via Pos9/Skn7 Is Negatively Regulated by the Ras/PKA Pathway in Saccharomyces cerevisiae. Mol. Gen. Genet. MGG 1999, 261, 740–752. [Google Scholar] [CrossRef] [PubMed]
- Broach, J.R. Nutritional Control of Growth and Development in Yeast. Genetics 2012, 192, 73–105. [Google Scholar] [CrossRef] [PubMed]
- Busti, S.; Coccetti, P.; Alberghina, L.; Vanoni, M. Glucose Signaling-Mediated Coordination of Cell Growth and Cell Cycle in Saccharomyces cerevisiae. Sensors 2010, 10, 6195–6240. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Bu, P.; Zeng, J.; Vancura, A. Increased Heme Synthesis in Yeast Induces a Metabolic Switch from Fermentation to Respiration Even under Conditions of Glucose Repression. J. Biol. Chem. 2017, 292, 16942–16954. [Google Scholar] [CrossRef]
- Johnston, N.R.; Nallur, S.; Gordon, P.B.; Smith, K.D.; Strobel, S.A. Genome-Wide Identification of Genes Involved in General Acid Stress and Fluoride Toxicity in Saccharomyces cerevisiae. Front. Microbiol. 2020, 11, 1410. [Google Scholar] [CrossRef]
- Haringa, C.; Tang, W.; Deshmukh, A.T.; Xia, J.; Reuss, M.; Heijnen, J.; Mudde, R.F.; Noorman, H. Euler-Lagrange Computational Fluid Dynamics for (Bio)Reactor Scale down: An Analysis of Organism Lifelines. Eng. Life Sci. 2016, 16, 652–663. [Google Scholar] [CrossRef]
- Bergdahl, B.; Heer, D.; Sauer, U.; Hahn-Hägerdal, B.; Van Niel, E.W. Dynamic Metabolomics Differentiates between Carbon and Energy Starvation in Recombinant Saccharomyces cerevisiae Fermenting Xylose. Biotechnol. Biofuels 2012, 5, 34. [Google Scholar] [CrossRef]
- Saini, P.; Beniwal, A.; Kokkiligadda, A.; Vij, S. Response and Tolerance of Yeast to Changing Environmental Stress during Ethanol Fermentation. Process Biochem. 2018, 72, 1–12. [Google Scholar] [CrossRef]
- Osiro, K.O.; Brink, D.P.; Borgström, C.; Wasserstrom, L.; Carlquist, M.; Gorwa-Grauslund, M.F. Assessing the Effect of D-Xylose on the Sugar Signaling Pathways of Saccharomyces cerevisiae in Strains Engineered for Xylose Transport and Assimilation. FEMS Yeast Res. 2018, 18, fox096. [Google Scholar] [CrossRef]
- Gasch, A.P.; Spellman, P.T.; Kao, C.M.; Carmel-Harel, O.; Eisen, M.B.; Storz, G.; Botstein, D.; Brown, P.O. Genomic Expression Programs in the Response of Yeast Cells to Environmental Changes. TMolecular Biol. Cell 2000, 11, 4241–4257. [Google Scholar] [CrossRef] [PubMed]
- Brauer, J.M.; Huttenhower, C.; Airoldi, E.M.; Rosenstein, R.; Matese, J.C.; Gresham, D.; Boer, V.M.; Troyanskaya, O.G.; Botstein, D. Coordination of Growth Rate, Cell Cycle, Stress Response, and Metabolic Activity in Yeast. Mol. Biol. Cell 2008, 19, 308–317. [Google Scholar] [CrossRef] [PubMed]
- De Nadal, E.; Ammerer, G.; Posas, F. Controlling Gene Expression in Response to Stress. Nat. Rev. Genet. 2011, 12, 833–845. [Google Scholar] [CrossRef]
- De Wever, V.; Reiter, W.; Ballarini, A.; Ammerer, G.; Brocard, C. A Dual Role for PP1 in Shaping the Msn2-Dependent Transcriptional Response to Glucose Starvation. EMBO J. 2005, 24, 4115–4123. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, P.; Shu, W.; Li, C.; Li, H.; Liu, S.; Xia, J.; Noorman, H. Dynamic Response of Aspergillus niger to Single Pulses of Glucose with High and Low Concentrations. Bioresour. Bioprocess. 2019, 6, 16. [Google Scholar] [CrossRef]
- Woolford, J.L.; Baserga, S.J. Ribosome Biogenesis in the Yeast Saccharomyces cerevisiae. Genetics 2013, 195, 643–681. [Google Scholar] [CrossRef]
- Metzl-Raz, E.; Kafri, M.; Yaakov, G.; Soifer, I.; Gurvich, Y.; Barkai, N. Principles of Cellular Resource Allocation Revealed by Condition-Dependent Proteome Profiling. Elife 2017, 6, e28034. [Google Scholar] [CrossRef]
- Remigi, P.; Ferguson, G.C.; McConnell, E.; De Monte, S.; Rogers, D.W.; Rainey, P.B. Ribosome Provisioning Activates a Bistable Switch Coupled to Fast Exit from Stationary Phase. Mol. Biol. Evol. 2019, 36, 1056–1070. [Google Scholar] [CrossRef]
- Mattanovich, D.; Gasser, B.; Hohenblum, H.; Sauer, M. Stress in Recombinant Protein Producing Yeasts. J. Biotechnol. 2004, 113, 121–135. [Google Scholar] [CrossRef]
- Gasch, A.P. Comparative Genomics of the Environmental Stress Response in Ascomycete Fungi. Yeast 2007, 24, 961–976. [Google Scholar] [CrossRef]
- Brion, C.; Pflieger, D.; Souali-Crespo, S.; Friedrich, A.; Schacherer, J. Differences in Environmental Stress Response among Yeasts Is Consistent with Species-Specific Lifestyles. Mol. Biol. Cell 2016, 27, 1694–1705. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, J.; Luo, X.; Capaldi, A.P. Integrated TORC1 and PKA Signaling Control the Temporal Activation of Glucose-Induced Gene Expression in Yeast. Nat. Commun. 2019, 10, 3558. [Google Scholar] [CrossRef] [PubMed]
- Görner, W.; Durchschlag, E.; Wolf, J.; Brown, E.L.; Ammerer, G.; Ruis, H.; Schüller, C. Acute Glucose Starvation Activates the Nuclear Localization Signal of a Stress-Specific Yeast Transcription Factor. EMBO J. 2002, 21, 135–144. [Google Scholar] [CrossRef]
- Martínez-Pastor, M.T.; Marchler, G.; Schüller, C.; Marchler-Bauer, A.; Ruis, H.; Estruch, F. The Saccharomyces cerevisiae Zinc Finger Proteins Msn2p and Msn4p Are Required for Transcriptional Induction through the Stress Response Element (STRE). EMBO J. 1996, 15, 2227–2235. [Google Scholar] [CrossRef]
- Plank, M. Interaction of TOR and PKA Signaling in S. cerevisiae. Biomolecules 2022, 12, 210. [Google Scholar] [CrossRef]
- Winkler, A.; Arkind, C.; Mattison, C.P.; Burkholder, A.; Knoche, K.; Ota, I. Heat Stress Activates the Yeast High-Osmolarity Glycerol Mitogen-Activated Protein Kinase Pathway, and Protein Tyrosine Phosphatases Are Essential under Heat Stress. Eukaryot. Cell 2002, 1, 163–173. [Google Scholar] [CrossRef]
- Panadero, J.; Pallotti, C.; Rodríguez-Vargas, S.; Randez-Gil, F.; Prieto, J.A. A Downshift in Temperature Activates the High Osmolarity Glycerol (HOG) Pathway, Which Determines Freeze Tolerance in Saccharomyces cerevisiae. J. Biol. Chem. 2006, 281, 4638–4645. [Google Scholar] [CrossRef]
- Bilsland, E.; Molin, C.; Swaminathan, S.; Ramne, A.; Sunnerhagen, P. Rck1 and Rck2 MAPKAP Kinases and the HOG Pathway Are Required for Oxidative Stress Resistance. Mol. Microbiol. 2004, 53, 1743–1756. [Google Scholar] [CrossRef]
- Krantz, M.; Nordlander, B.; Valadi, H.; Johansson, M.; Gustafsson, L.; Hohmann, S. Anaerobicity Prepares Saccharomyces cerevisiae Cells for Faster Adaptation to Osmotic Shock. Eukaryot. Cell 2004, 3, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- James, T.C.; Campbell, S.; Donnelly, D.; Bond, U. Transcription Profile of Brewery Yeast under Fermentation Conditions. J. Appl. Microbiol. 2003, 94, 432–448. [Google Scholar] [CrossRef]
- Sharifian, H.; Lampert, F.; Stojanovski, K.; Regot, S.; Vaga, S.; Buser, R.; Lee, S.S.; Koeppl, H.; Posas, F.; Pelet, S.; et al. Parallel Feedback Loops Control the Basal Activity of the HOG MAPK Signaling Cascade. Integr. Biol. 2015, 7, 412–422. [Google Scholar] [CrossRef]
- Ball, W.J.; Atkinson, D.E. Adenylate Energy Charge in Saccharomyces cerevisiae during Starvation. J. Bacteriol. 1975, 121, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Caligaris, M.; Nicastro, R.; Hu, Z.; Tripodi, F.; Hummel, J.E.; Deprez, M.-A.; Winderickx, J.; Rospert, S.; Coccetti, P.; Dengjel, J.; et al. Snf1/AMPK Fine-Tunes TORC1 Signaling in Response to Glucose Starvation. Elife 2022, 12, e84319. [Google Scholar] [CrossRef]
- Zaman, S.; Lippman, S.I.; Schneper, L.; Slonim, N.; Broach, J.R. Glucose Regulates Transcription in Yeast through a Network of Signaling Pathways. Mol. Syst. Biol. 2009, 5, 245. [Google Scholar] [CrossRef] [PubMed]
- Shashkova, S.; Welkenhuysen, N.; Hohmann, S. Molecular Communication: Crosstalk between the Snf1 and Other Signaling Pathways. FEMS Yeast Res. 2015, 15, fov026. [Google Scholar] [CrossRef]
- Nicastro, R.; Tripodi, F.; Gaggini, M.; Castoldi, A.; Reghellin, V.; Nonnis, S.; Tedeschi, G.; Coccetti, P. Snf1 Phosphorylates Adenylate Cyclase and Negatively Regulates Protein Kinase A-Dependent Transcription in Saccharomyces cerevisiae. J. Biol. Chem. 2015, 290, 24715–24726. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A Nutrient and Energy Sensor That Maintains Energy Homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Coccetti, P.; Nicastro, R.; Tripodi, F. Conventional and Emerging Roles of the Energy Sensor Snf1/AMPK in Saccharomyces cerevisiae. Microb. Cell 2018, 5, 482–494. [Google Scholar] [CrossRef]
- Zampar, G.G.; Kümmel, A.; Ewald, J.; Jol, S.; Niebel, B.; Picotti, P.; Aebersold, R.; Sauer, U.; Zamboni, N.; Heinemann, M. Temporal System-Level Organization of the Switch from Glycolytic to Gluconeogenic Operation in Yeast. Mol. Syst. Biol. 2013, 9, 651. [Google Scholar] [CrossRef]
- Knijnenburg, T.A.; de Winde, J.H.; Daran, J.-M.; Daran-Lapujade, P.; Pronk, J.T.; Reinders, M.J.T.; Wessels, L.F.A. Exploiting Combinatorial Cultivation Conditions to Infer Transcriptional Regulation. BMC Genom. 2007, 8, 25. [Google Scholar] [CrossRef]
- MacGilvray, M.E.; Shishkova, E.; Place, M.; Wagner, E.R.; Coon, J.J.; Gasch, A.P. Phosphoproteome Response to Dithiothreitol Reveals Unique Versus Shared Features of Saccharomyces cerevisiae Stress Responses. J. Proteome Res. 2020, 19, 3405–3417. [Google Scholar] [CrossRef]
- Shore, D.; Zencir, S.; Albert, B. Transcriptional Control of Ribosome Biogenesis in Yeast: Links to Growth and Stress Signals. Biochem. Soc. Trans. 2021, 49, 1589–1599. [Google Scholar] [CrossRef]
- Cipollina, C.; van den Brink, J.; Daran-Lapujade, P.; Pronk, J.T.; Porro, D.; de Winde, J.H. Saccharomyces cerevisiae SFP1: At the Crossroads of Central Metabolism and Ribosome Biogenesis. Microbiology 2008, 154, 1686–1699. [Google Scholar] [CrossRef]
- Ashe, M.P.; De Long, S.K.; Sachs, A.B. Glucose Depletion Rapidly Inhibits Translation Initiation in Yeast. Mol. Biol. Cell 2000, 11, 833–848. [Google Scholar] [CrossRef] [PubMed]
- Huch, S.; Nissan, T. Interrelations between Translation and General MRNA Degradation in Yeast. Wiley Interdiscip. Rev. RNA 2014, 5, 747–763. [Google Scholar] [CrossRef]
- Brengues, M.; Parker, R. Accumulation of Polyadenylated MRNA, Pab1p, EIF4E, and EIF4G with P-Bodies in Saccharomyces cerevisiae. Mol. Biol. Cell 2007, 18, 2592–2602. [Google Scholar] [CrossRef] [PubMed]
- Bresson, S.; Tuck, A.; Staneva, D.; Tollervey, D. Nuclear RNA Decay Pathways Aid Rapid Remodeling of Gene Expression in Yeast. Mol. Cell 2017, 65, 787–800.e5. [Google Scholar] [CrossRef]
- Barraza, C.E.; Solari, C.A.; Rinaldi, J.; Ojeda, L.; Rossi, S.; Ashe, M.P.; Portela, P. A Prion-like Domain of Tpk2 Catalytic Subunit of Protein Kinase A Modulates P-Body Formation in Response to Stress in Budding Yeast. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118884. [Google Scholar] [CrossRef]
- Ramachandran, V.; Shah, K.H.; Herman, P.K. The CAMP-Dependent Protein Kinase Signaling Pathway Is a Key Regulator of P Body Foci Formation. Mol. Cell 2011, 43, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.F.; Tatchell, K. Characterization of Saccharomyces cerevisiae Genes Encoding Subunits of Cyclic AMP-Dependent Protein Kinase. Mol. Cell. Biol. 1987, 7, 2653–2663. [Google Scholar] [CrossRef] [PubMed]
- Müller, D.; Exler, S.; Aguilera-Vázquez, L.; Guerrero-Martín, E.; Reuss, M. Cyclic AMP Mediates the Cell Cycle Dynamics of Energy Metabolism in Saccharomyces cerevisiae. Yeast 2003, 20, 351–367. [Google Scholar] [CrossRef]
- Pelechano, V.; Chávez, S.; Pérez-Ortín, J.E. A Complete Set of Nascent Transcription Rates for Yeast Genes. PLoS ONE 2010, 7, e15442. [Google Scholar] [CrossRef]
- Beullens, M.; Mbonyi, K.; Geerts, L.; Gladines, D.; Detremerie, K.; Jans, A.W.H.; Thevelein, J.M. Studies on the Mechanism of the Glucose-induced CAMP Signal in Glycolysis and Glucose Repression Mutants of the Yeast Saccharomyces cerevisiae. Eur. J. Biochem. 1988, 172, 227–231. [Google Scholar] [CrossRef]
- Botman, D.; O’Toole, T.G.; Goedhart, J.; Bruggeman, F.J.; van Heerden, J.H.; Teusink, B. A New FRET Biosensor Enlightens CAMP Signalling in Budding Yeast. bioRxiv 2019. [Google Scholar] [CrossRef]
- Peebo, K.; Neubauer, P. Application of Continuous Culture Methods to Recombinant Protein Production in Microorganisms. Microorganisms 2018, 6, 56. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Yan, Q.; Jones, J.A.; Tang, Y.J.; Fong, S.S.; Koffas, M.A.G. Metabolic Burden: Cornerstones in Synthetic Biology and Metabolic Engineering Applications. Trends Biotechnol. 2016, 34, 652–664. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, S.; Bailey, J.E. Plasmid Presence Changes the Relative Levels of Many Host Cell Proteins and Ribosome Components in Recombinant Escherichia coli. Biotechnol. Bioeng. 1991, 37, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Wright, N.R.; Wulff, T.; Palmqvist, E.A.; Jørgensen, T.R.; Workman, C.T.; Sonnenschein, N.; Rønnest, N.P.; Herrgård, M.J. Fluctuations in Glucose Availability Prevent Global Proteome Changes and Physiological Transition during Prolonged Chemostat Cultivations of Saccharomyces cerevisiae. Biotechnol. Bioeng. 2020, 117, 2074–2088. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Chomvong, K.; Acosta-Sampson, L.; Estrela, R.; Galazka, J.M.; Kim, S.R.; Jin, Y.-S.; Cate, J.H. Leveraging Transcription Factors to Speed Cellobiose Fermentation by Saccharomyces cerevisiae. Biotechnol. Biofuels 2014, 7, 126. [Google Scholar] [CrossRef]
- Michael, D.G.; Maier, E.J.; Brown, H.; Gish, S.R.; Fiore, C.; Brown, R.H.; Brent, M.R. Model-Based Transcriptome Engineering Promotes a Fermentative Transcriptional State in Yeast. Proc. Natl. Acad. Sci. USA 2016, 113, E7428–E7437. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, R.A.; Bourbon-Melo, N.; Sá-Correia, I. The Cell Wall and the Response and Tolerance to Stresses of Biotechnological Relevance in Yeasts. Front. Microbiol. 2022, 13, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Creamer, D.R.; Hubbard, S.J.; Ashe, M.P.; Grant, C.M. Yeast Protein Kinase A Isoforms: A Means of Encoding Specificity in the Response to Diverse Stress Conditions? Biomolecules 2022, 12, 958. [Google Scholar] [CrossRef] [PubMed]
- Soetaert, K.; Petzoldt, T.; Setzer, R.W. Solving Differential Equations in R: Package DeSolve. J. Stat. Softw. 2010, 33, 1–25. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Dimension | Steady State RS | Steady State DS | % Change | Welch Test (p-Value) |
|---|---|---|---|---|---|
| D | h−1 | 0.098 ± 0.003 | 0.099 ± 0.003 | +0.5 | n.s. |
| DMB | g·l−1 | 5.06 ± 0.04 | 4.72 ± 0.17 | −6.8 | n.s. |
| YDMB/glucose | molC·molC−1 | 0.114 ± 0.001 | 0.106 ± 0.003 | −6.9 | 0.05 |
| qglucose | mmolC·gDMB−1·h−1 | 32.1 ± 0.8 | 34.7 ± 1.2 | +8.0 | 0.04 |
| qglucose,max 1 | mmolC·gDMB−1·h−1 | 71.50 | 64.80 | −9.4 | n.a. |
| KS 1 | mmolC·l−1 | 6.19 | 5.47 | −11.6 | n.a. |
| Yethanol/glucose | molC·molC−1 | 0.488 ± 0.033 | 0.468 ± 0.026 | −4.0 | n.s. |
| qethanol | mmolC·gDMB−1·h−1 | 16.7 ± 1.3 | 17.3 ± 1.4 | +3.6 | n.s. |
| qcarbon dioxide | mmolC·gDMB−1·h−1 | 7.68 ± 0.25 | 8.47 ± 0.18 | +10.3 | 0.01 |
| qglycerole | mmolC·gDMB−1·h−1 | 2.70 ± 0.09 | 2.99 ± 0.12 | +11 | n.s. |
| qacetic acid | mmolC·gDMB−1·h−1 | 0.04 ± 0.00 | 0.04 ± 0.00 | −2.1 | 0.03 |
| qsuccinic acid | mmolC·gDMB−1·h−1 | 2.9 × 10−2 ± 4.7 × 10−3 | 3.6 × 10−2 ± 1.4 × 10−3 | +25.7 | n.s. |
| qunknown carbon | mmolC·gDMB−1·h−1 | 1.06 ± 0.53 | 1.81 ± 0.91 | +71.2 | n.s. |
| cglycogen | mmolC·gDMB−1 | 2.87 ± 0.16 | 1.65 ± 0.12 | −42 | 1.0 × 10−3 |
| ctrehalose | mmolC·gDMB−1 | 1.84 ± 0.19 | 1.25 ± 0.52 | −32 | n.s. |
| cRNA | mg·gDMB−1 | 64.3 ± 2.5 | 80.7 ± 3.1 | +25 | 2.0 × 10−3 |
| C-recovery | molC·molC−1 | 0.98 ± 0.02 | 0.98 ± 0.02 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Minden, S.; Aniolek, M.; Noorman, H.; Takors, R. Mimicked Mixing-Induced Heterogeneities of Industrial Bioreactors Stimulate Long-Lasting Adaption Programs in Ethanol-Producing Yeasts. Genes 2023, 14, 997. https://0-doi-org.brum.beds.ac.uk/10.3390/genes14050997
Minden S, Aniolek M, Noorman H, Takors R. Mimicked Mixing-Induced Heterogeneities of Industrial Bioreactors Stimulate Long-Lasting Adaption Programs in Ethanol-Producing Yeasts. Genes. 2023; 14(5):997. https://0-doi-org.brum.beds.ac.uk/10.3390/genes14050997
Chicago/Turabian StyleMinden, Steven, Maria Aniolek, Henk Noorman, and Ralf Takors. 2023. "Mimicked Mixing-Induced Heterogeneities of Industrial Bioreactors Stimulate Long-Lasting Adaption Programs in Ethanol-Producing Yeasts" Genes 14, no. 5: 997. https://0-doi-org.brum.beds.ac.uk/10.3390/genes14050997